CO2 Methanation over Hydrotalcite-Derived Nickel/Ruthenium and Supported Ruthenium Catalysts

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
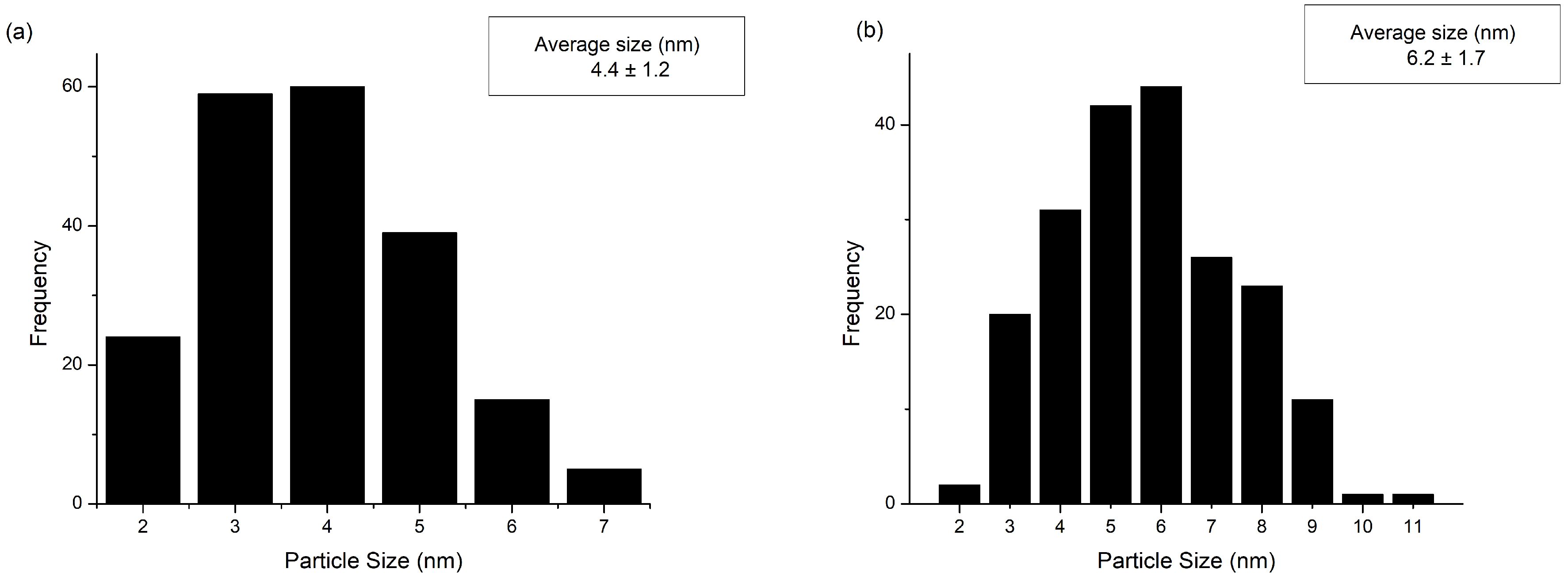
2.1. Catalysts Characterization
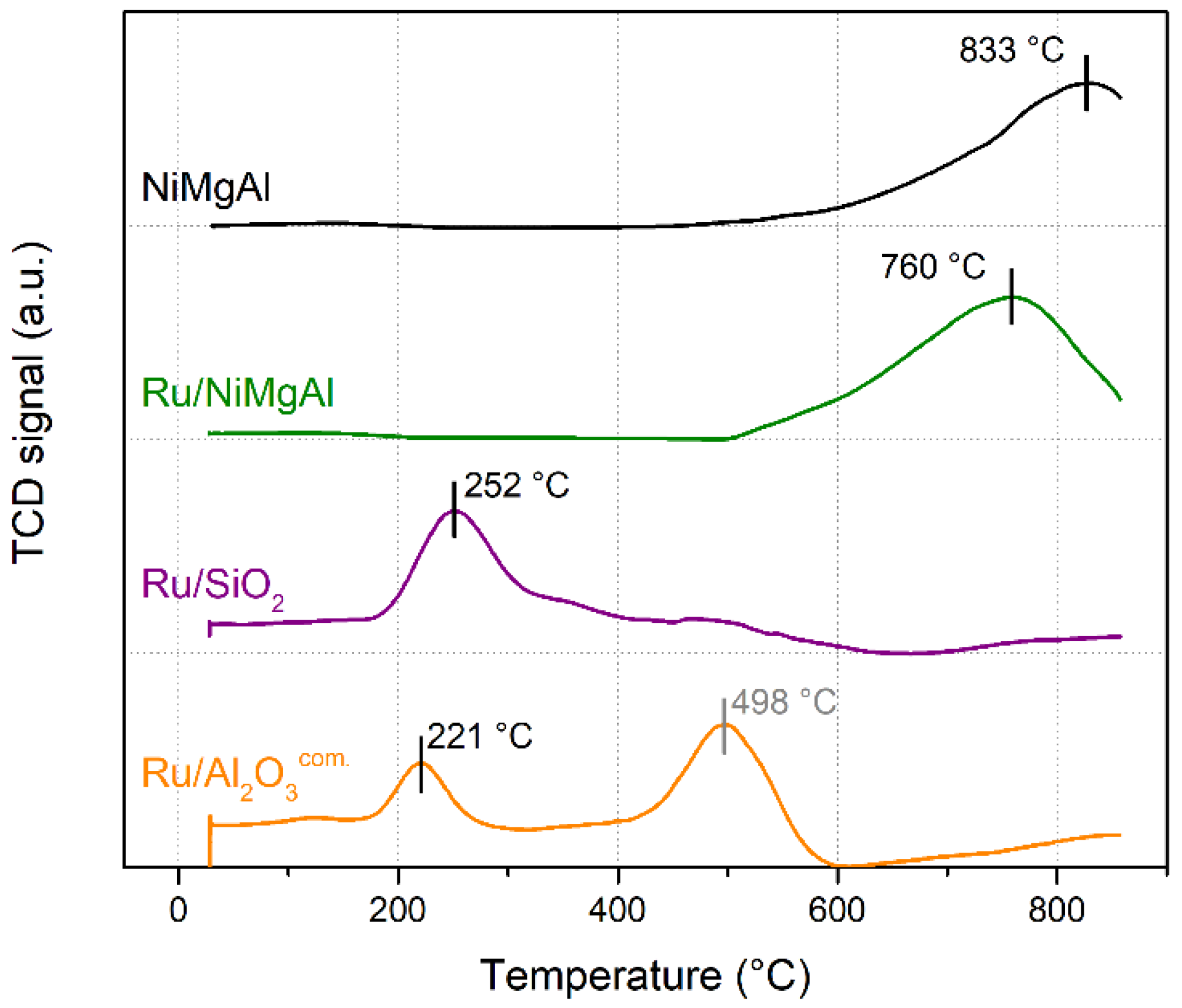
2.1.1. Temperature Programmed Reduction
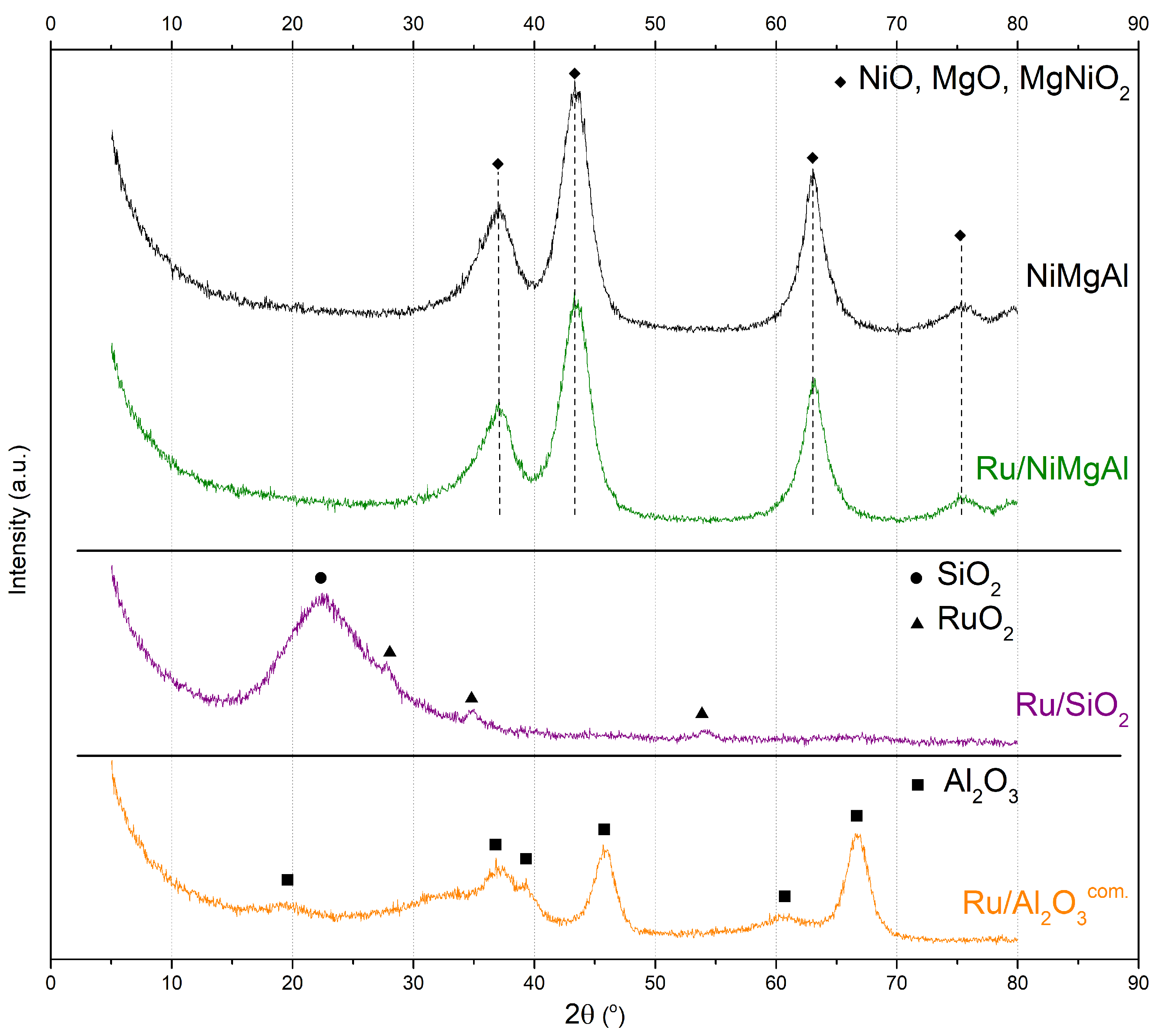
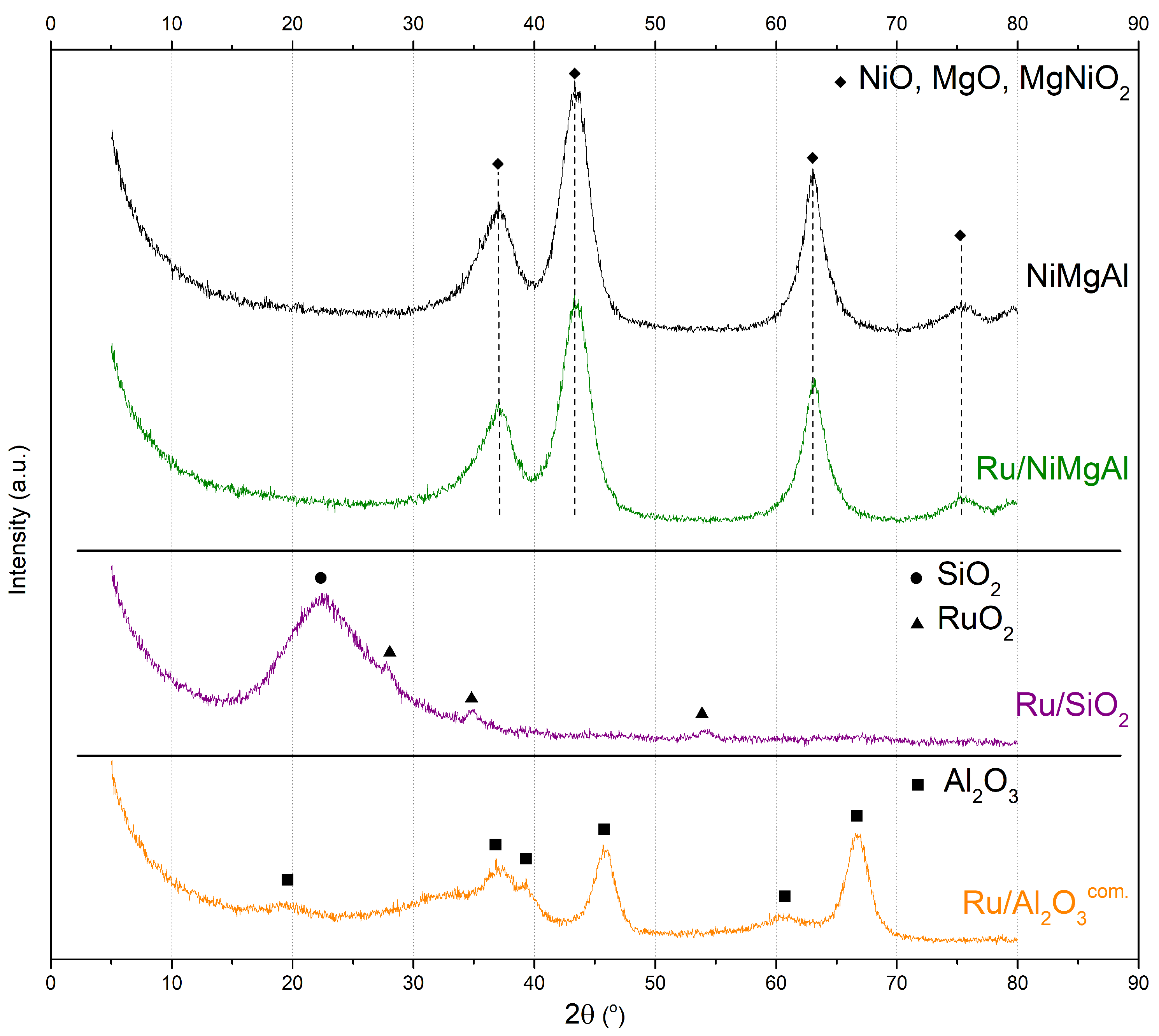
2.1.2. X-ray Diffraction
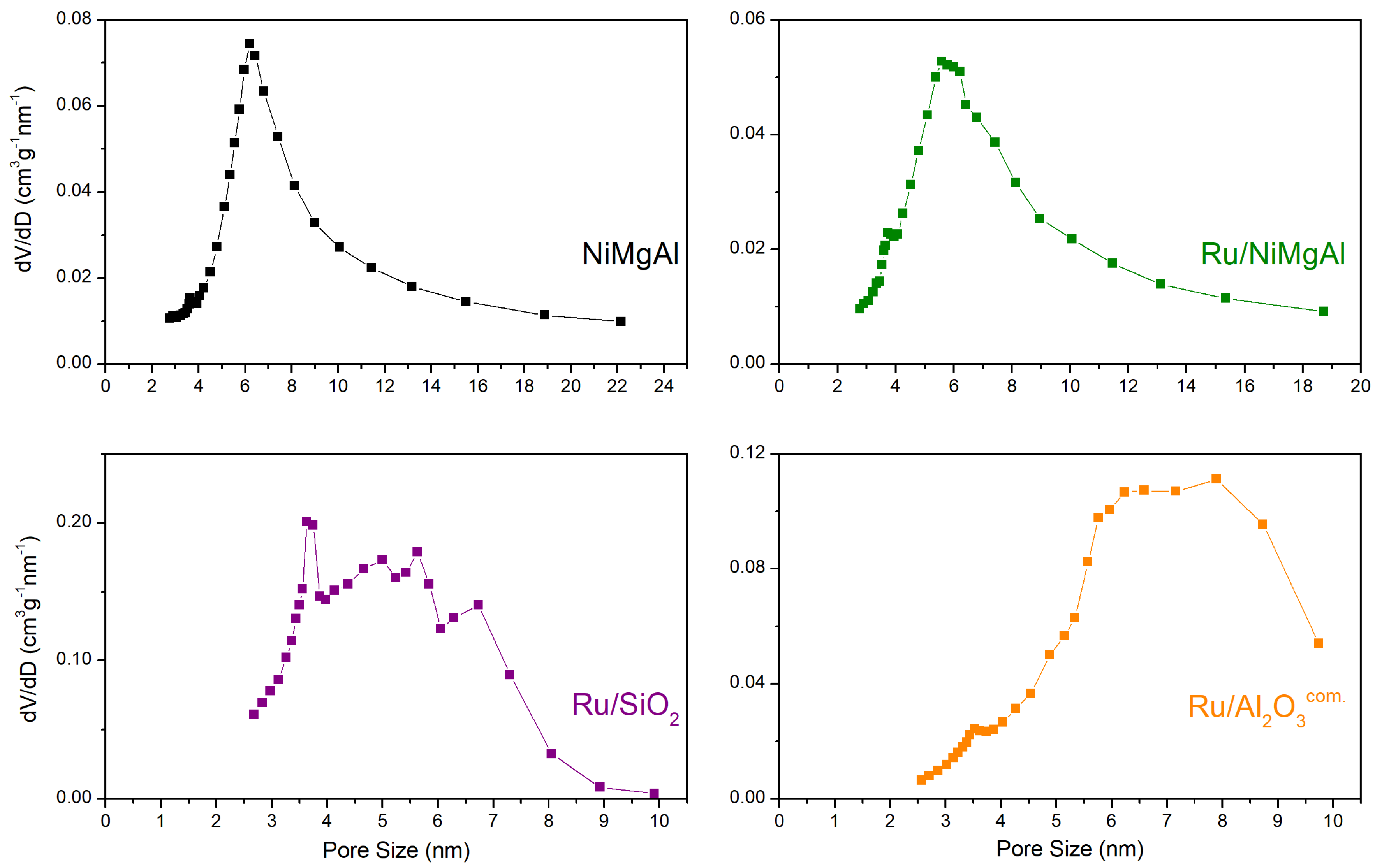
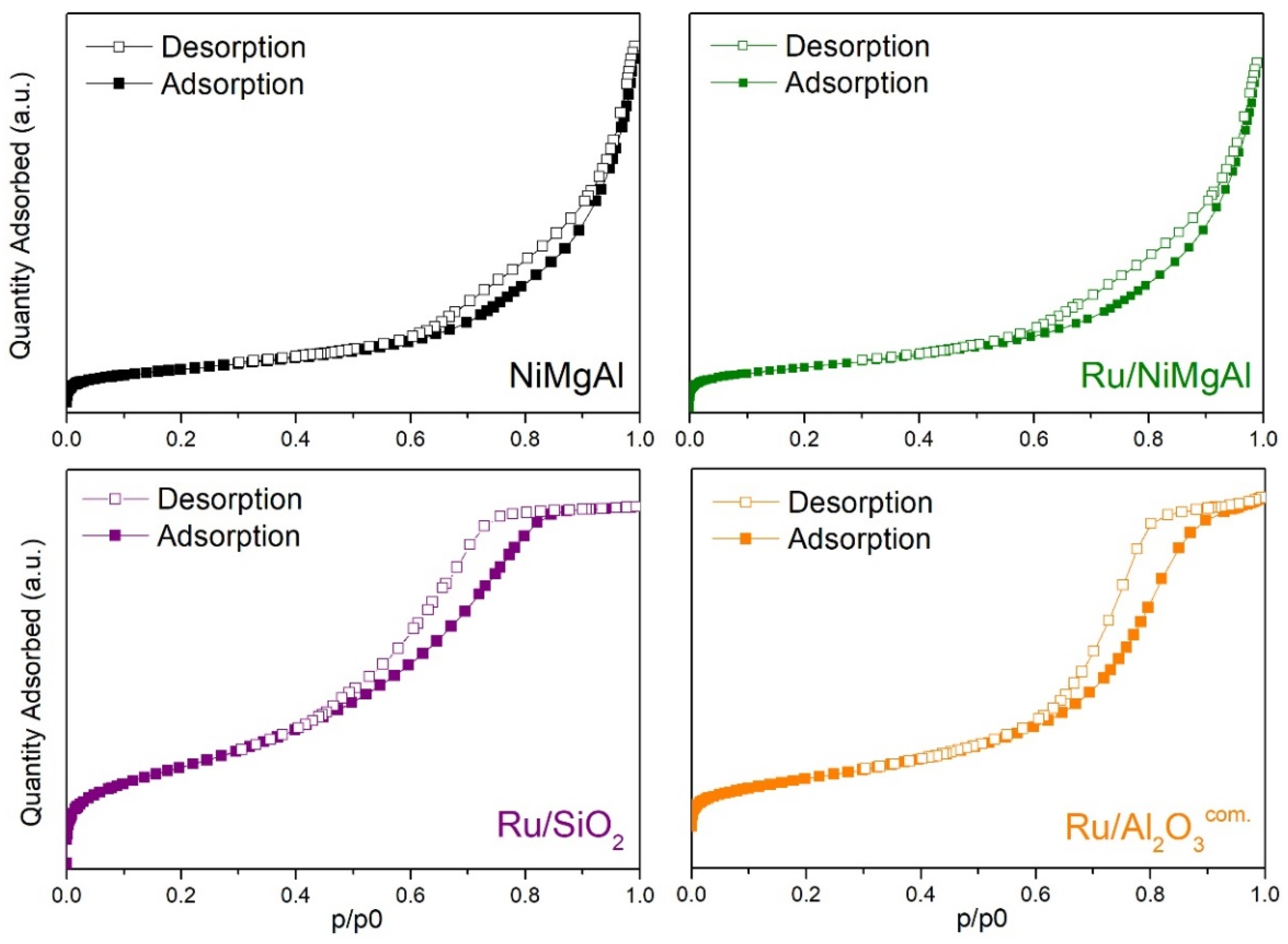
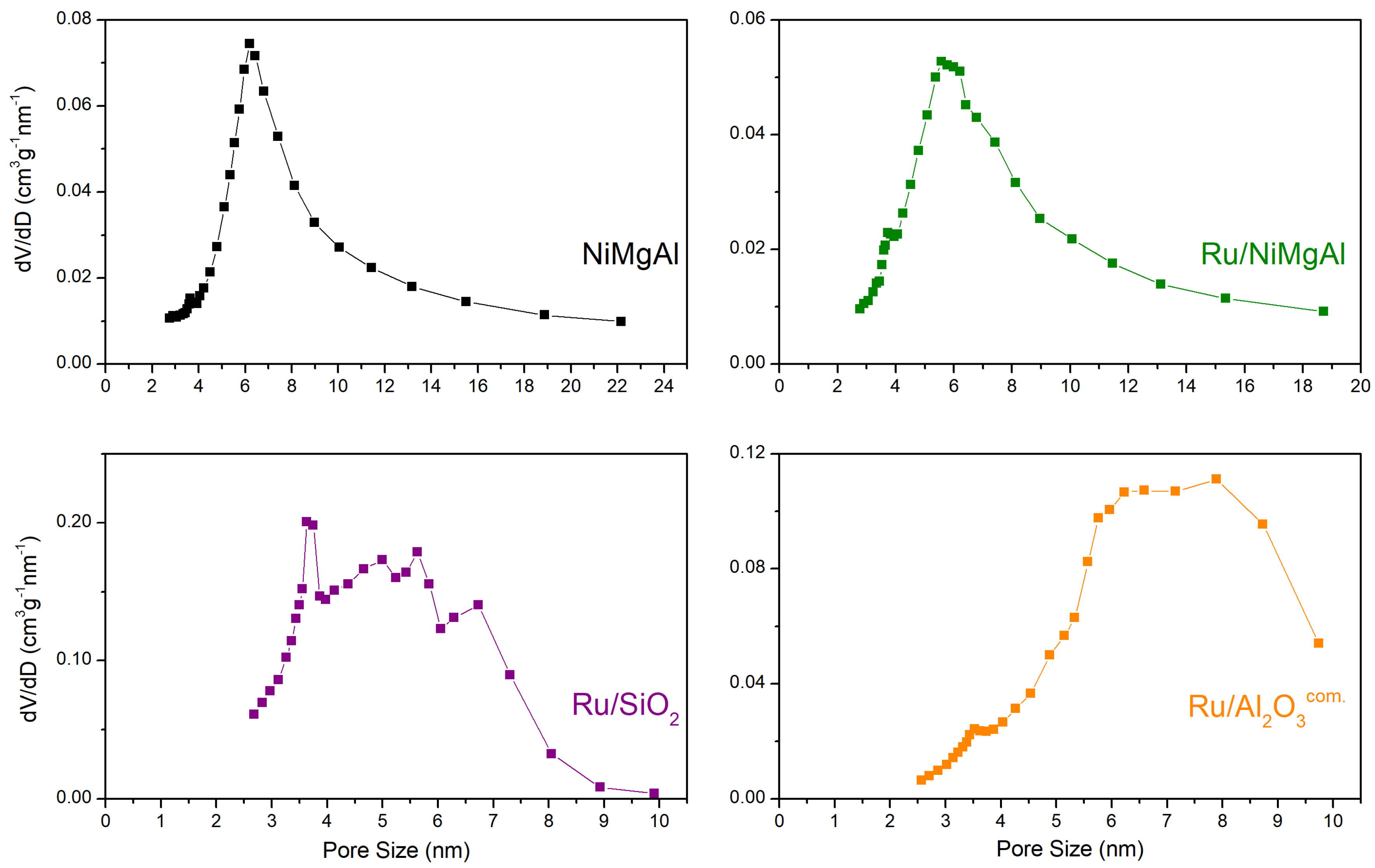
2.1.3. N2 Physisorption at −196 °C
2.1.4. Inductively Coupled Plasma Optical Emission Spectrometry
2.1.5. H2 and CO Chemisorption
2.2. Catalysts Assessment
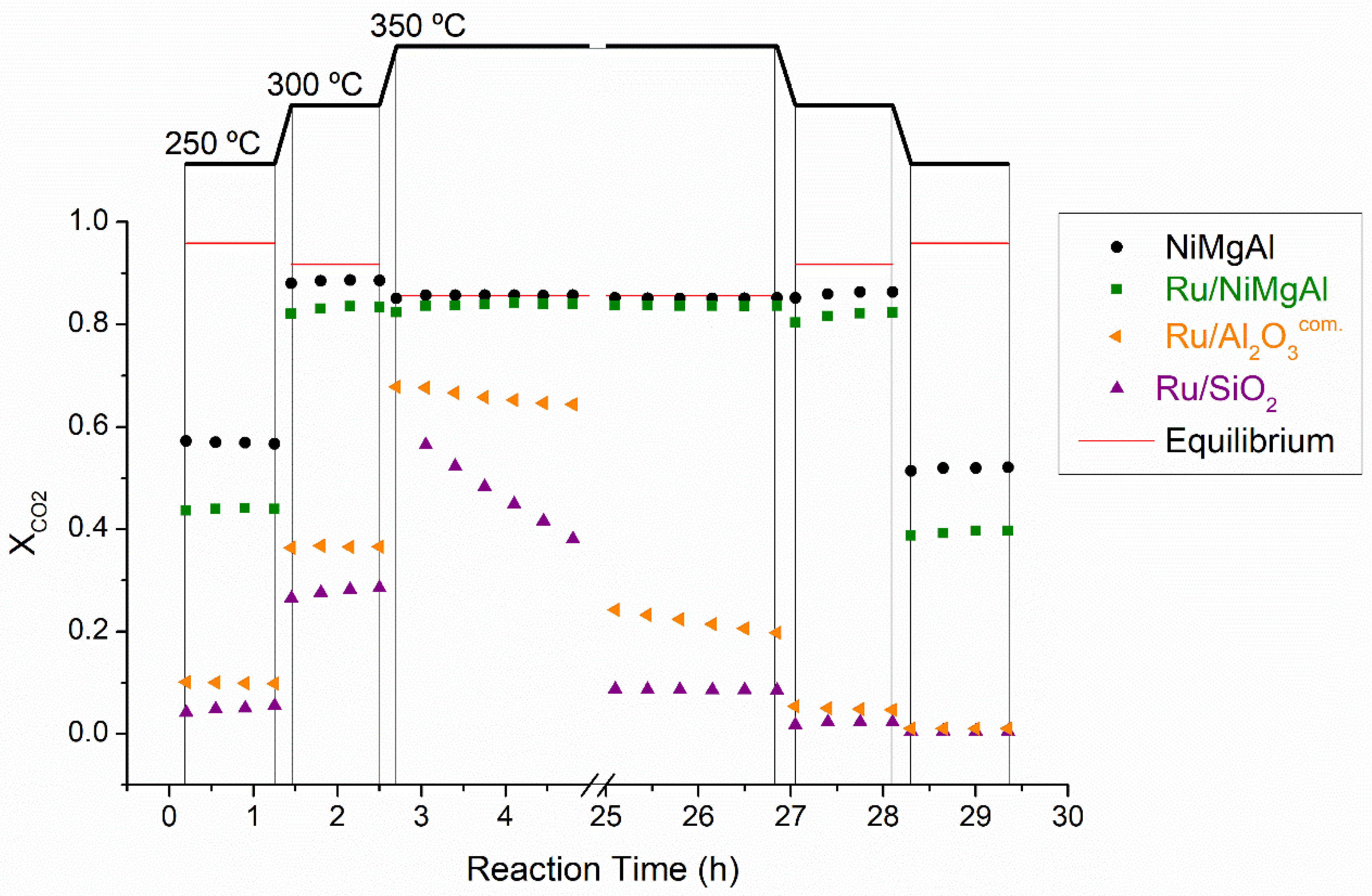
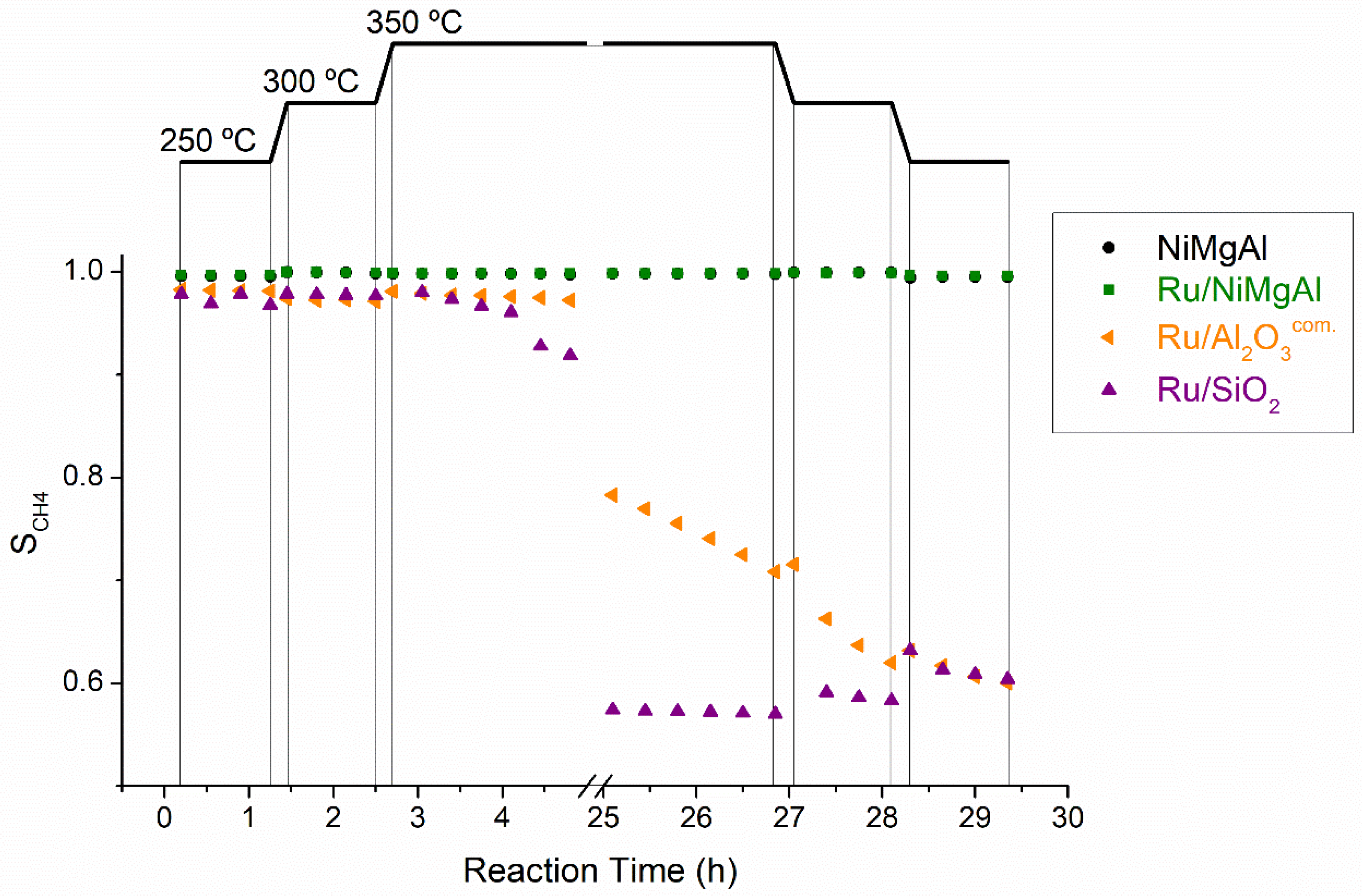
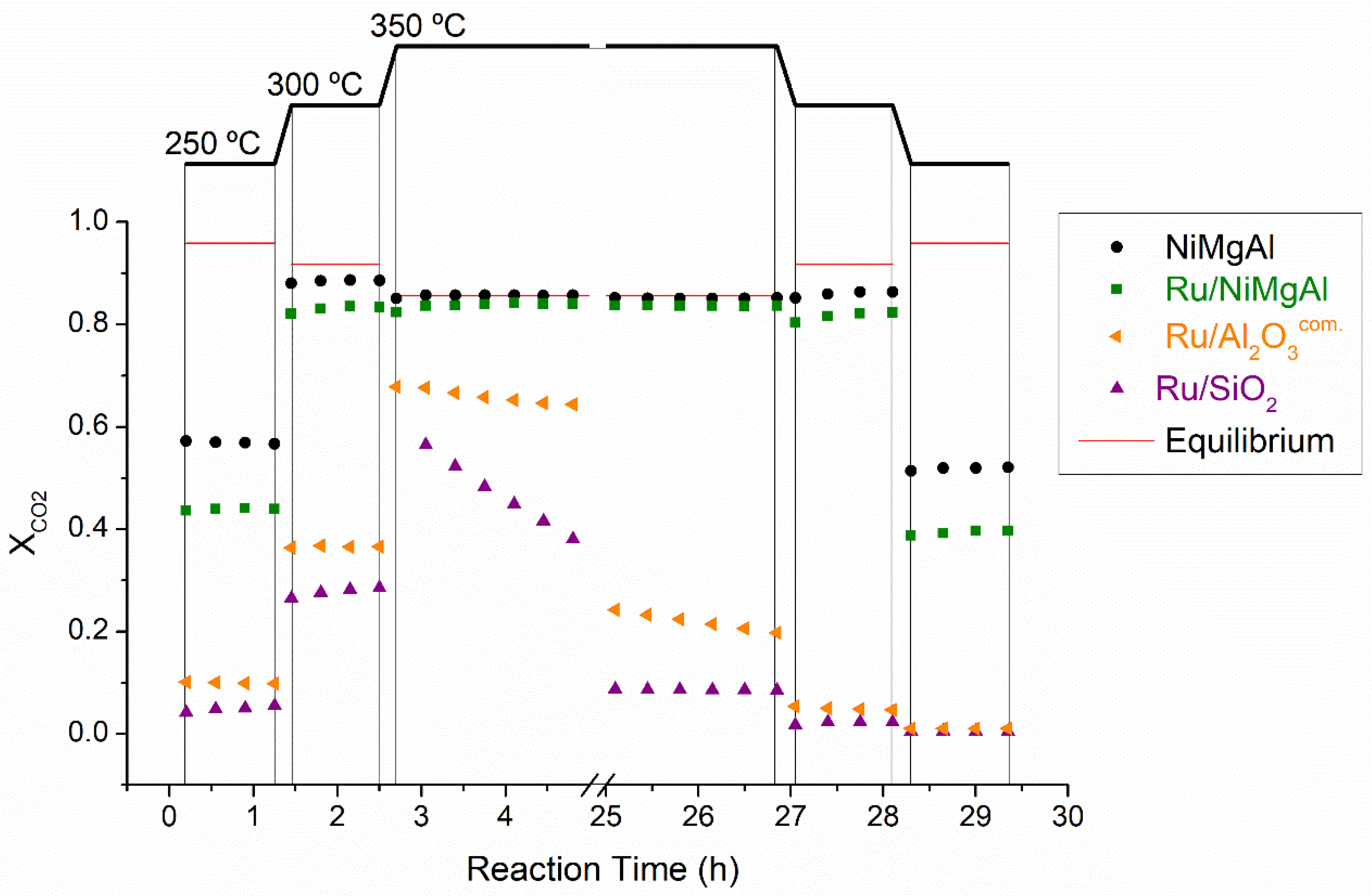
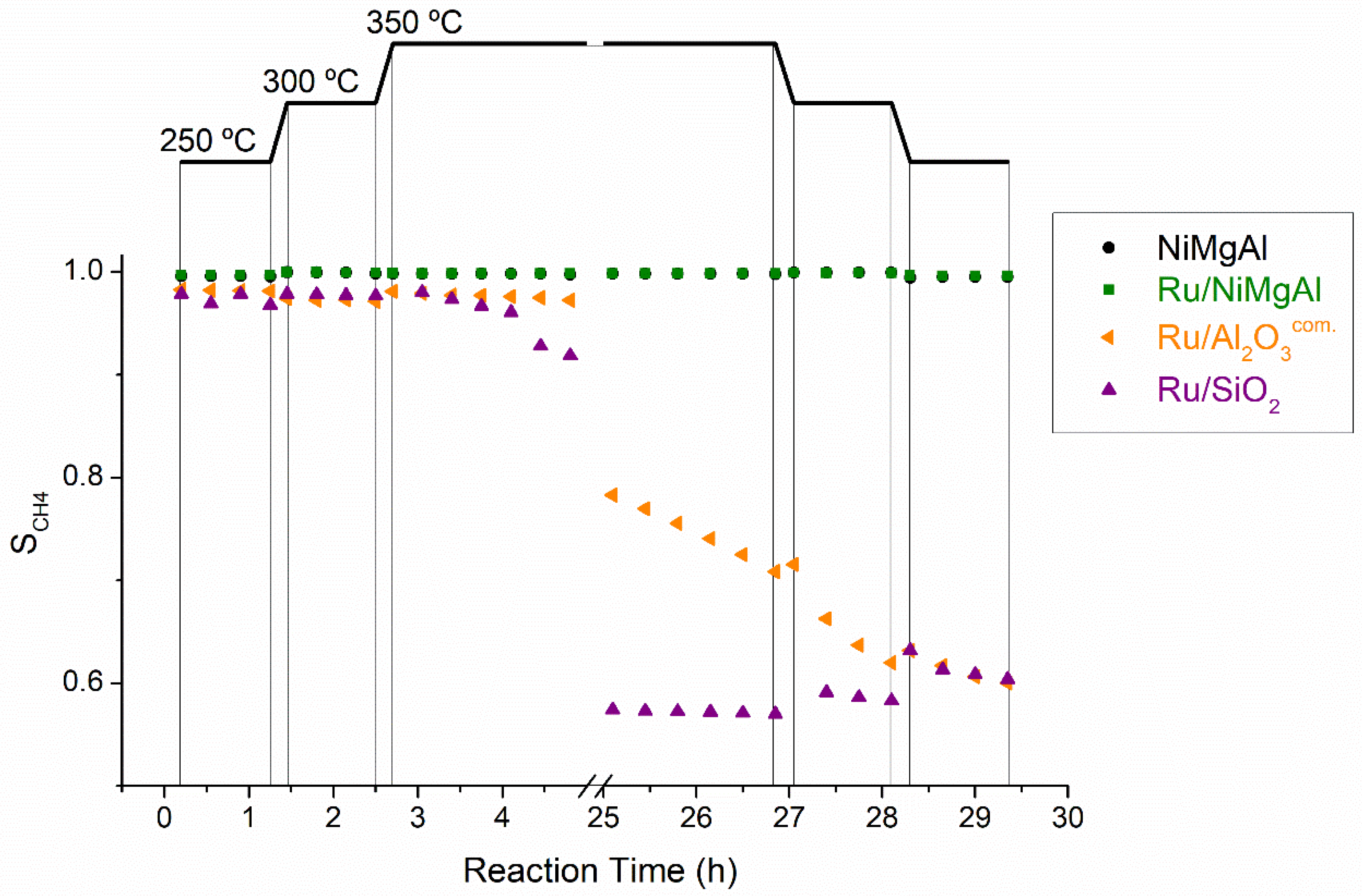
2.2.1. Screening Tests
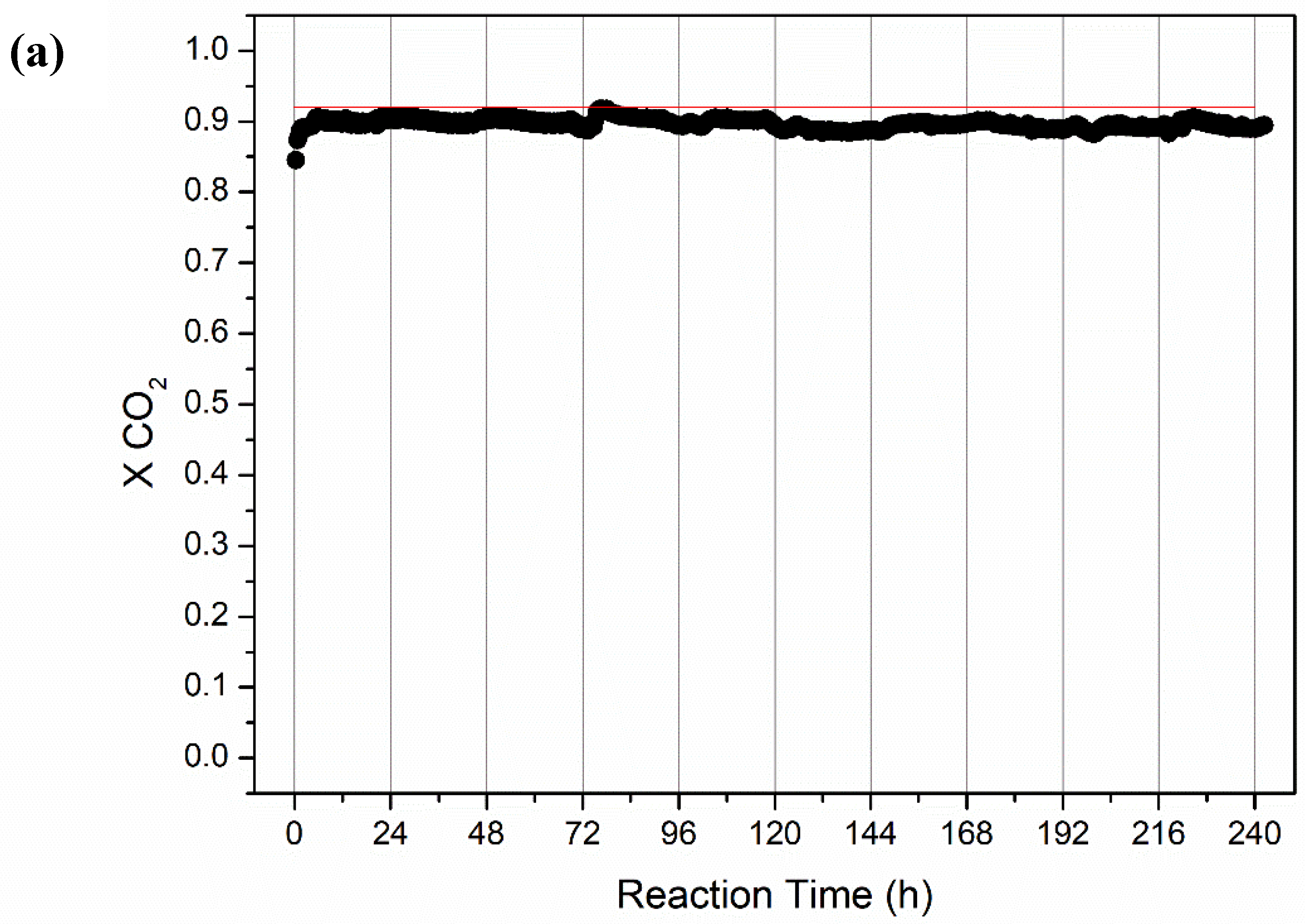
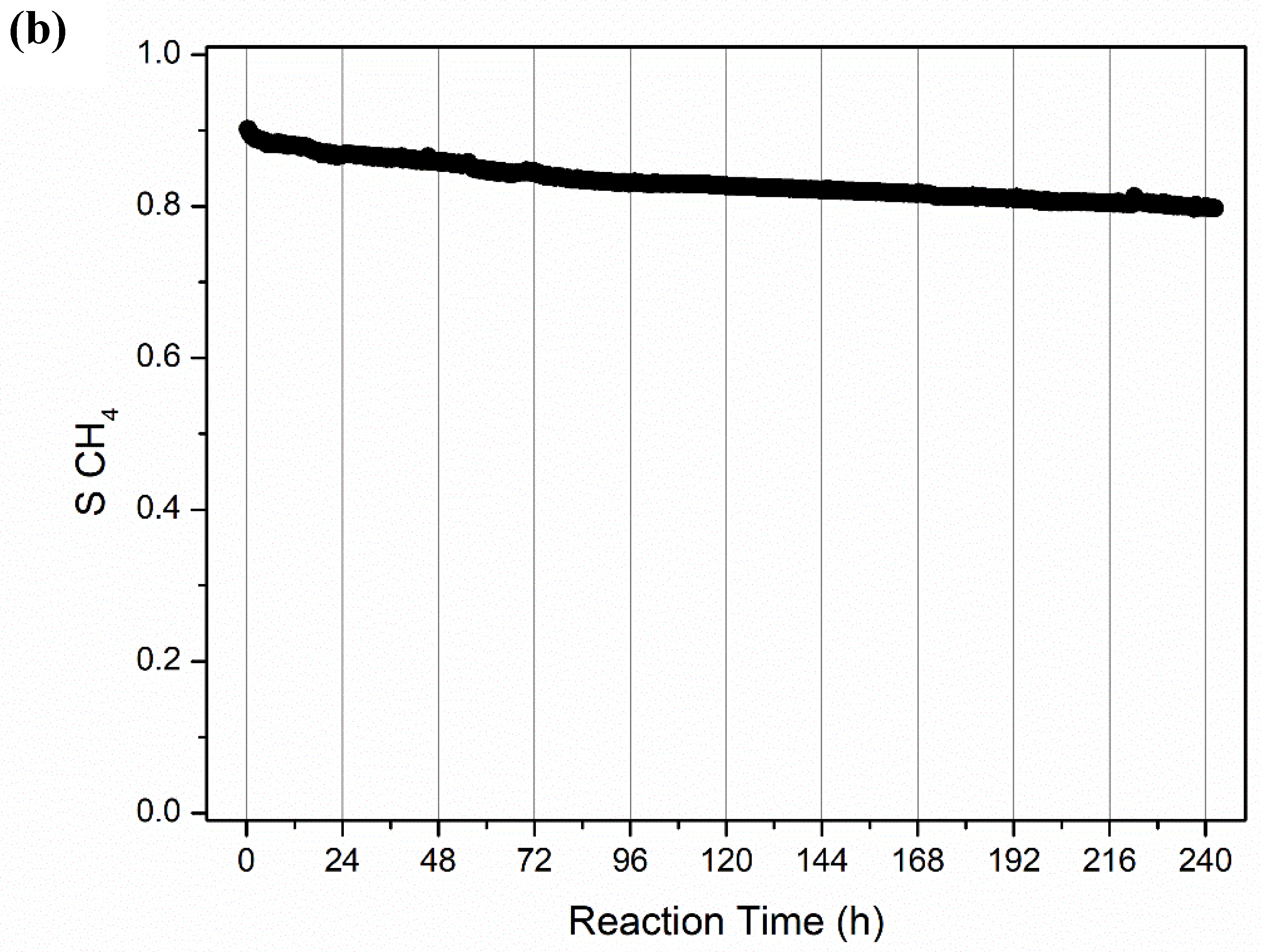
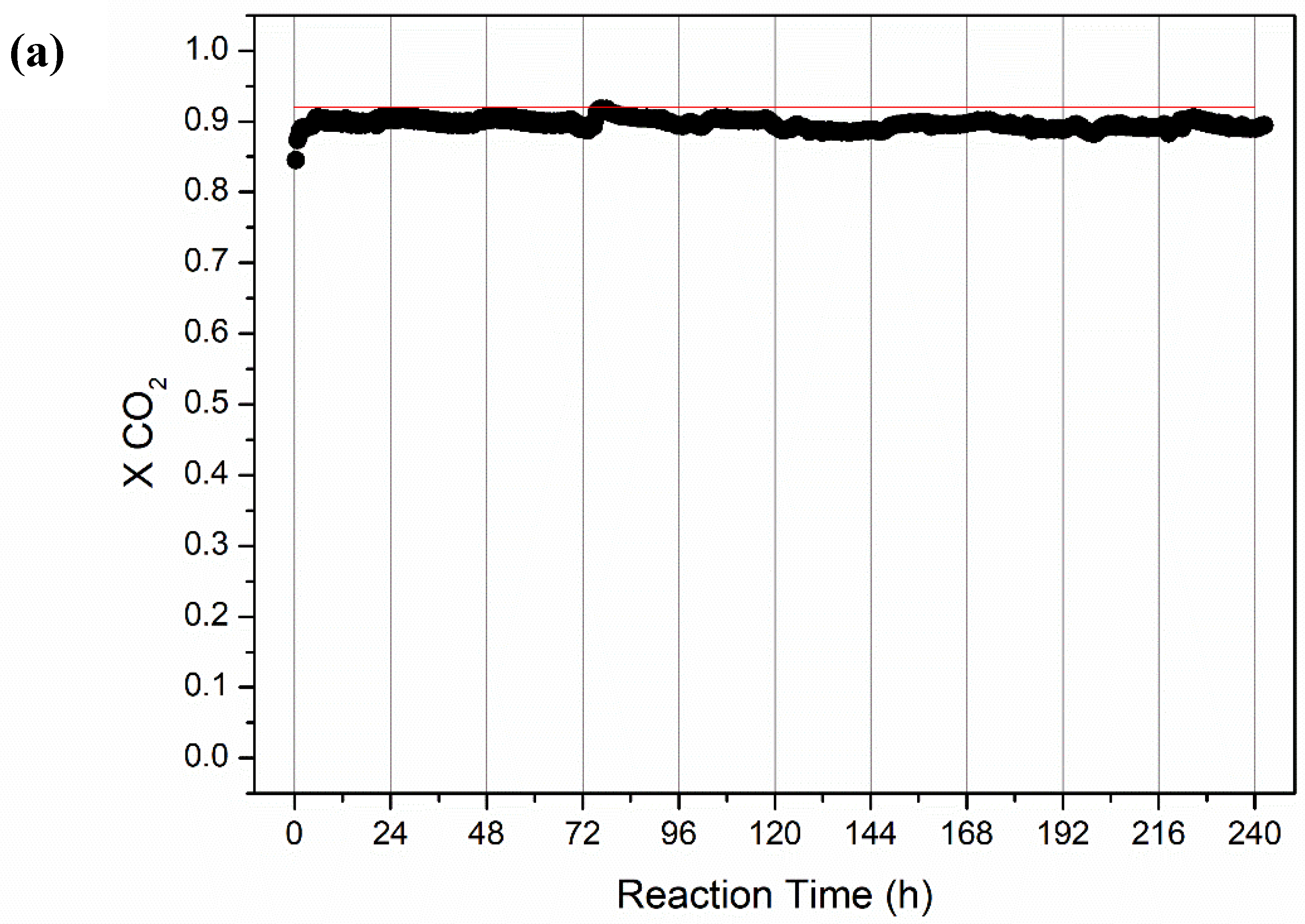
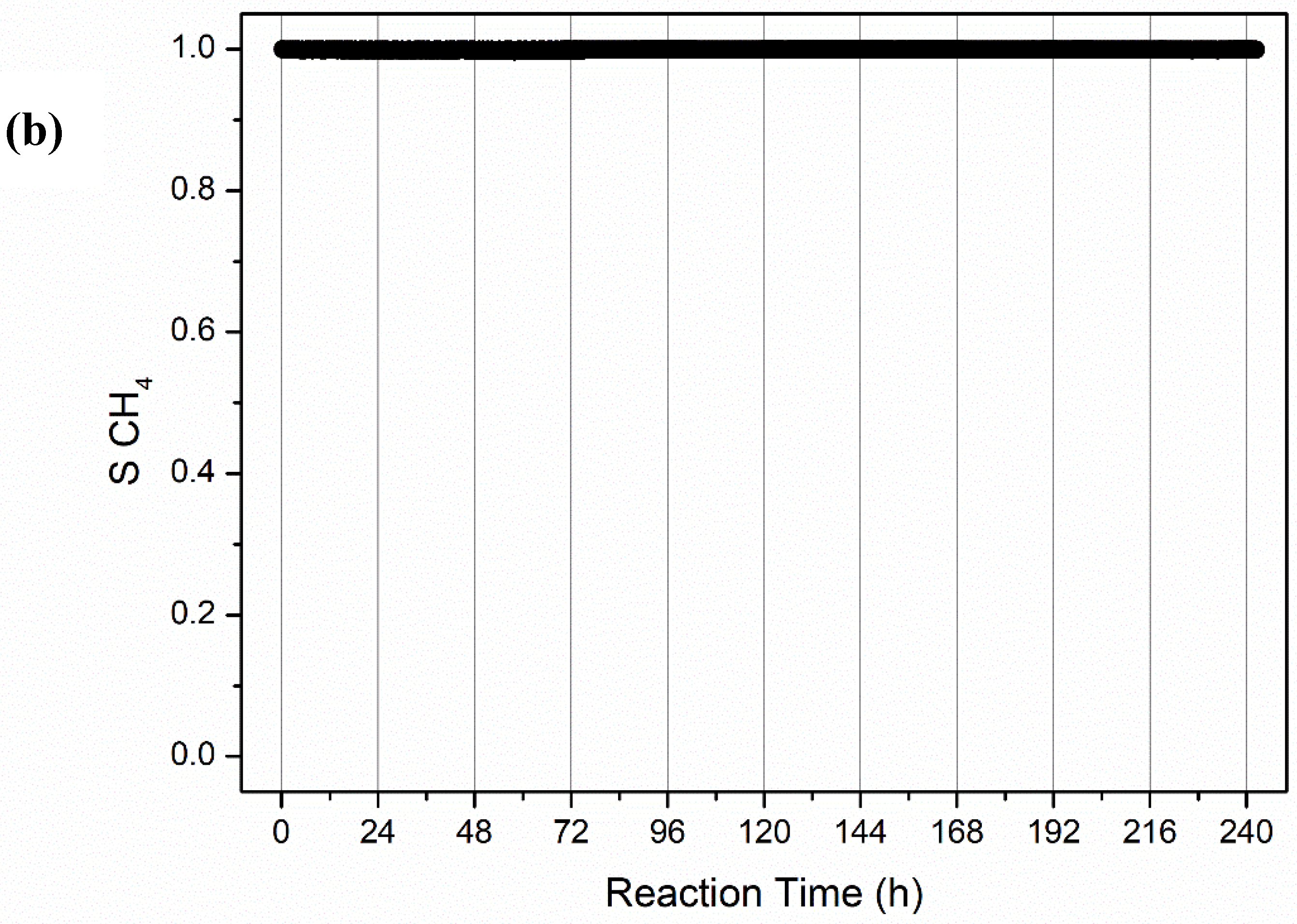
2.2.2. Long-Term Stability Tests
3. Experimental
3.1. Catalysts Synthesis
3.2. Catalysts Characterization
3.3. Catalysts Assessment
3.3.1. Experimental Setup
3.3.2. Experimental Procedure
3.3.3. Catalyst Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Nomenclature
| Symbols | |
| DM | Metal dispersion (%) |
| CH4 outlet molar flow rate (mol·s−1) | |
| CO outlet molar flow rate (mol·s−1) | |
| CO2 inlet molar flow rate (mol·s−1) | |
| M | Molecular weight of the metal (g·mol−1) |
| mcat | Catalyst mass (g) |
| NA | Avogadro’s number (mol−1) |
| nm | Quantity of chemisorbed molecules (mol·gcat−1) |
| ns | Number of atoms at surface, per unit area (m−2) |
| s | Chemisorption stoichiometry |
| CH4 selectivity | |
| SM | Metal surface area (m2·gcat−1) |
| TOF | Turnover frequency (h−1) |
| CO2 conversion | |
| WHSV | Weight hourly space velocity (mLN,CO2·g−1·h−1) |
| y | Metal content |
| CH4 yield | |
| Abbreviations | |
| BET | Brunauer-Emmet-Teller |
| BJH | Barret-Joyner-Halenda |
| FID | Flame ionization detector |
| ICP-OES | Inductively Coupled Plasma Optical Emission Spectrometry |
| TPR | Temperature programmed reduction |
| PtM | Power-to-Methane |
| RWGS | Reverse water-gas shift |
| TCD | Thermal conductivity detector |
| TEM | Transmission Electron Microscopy |
| TOC | Total organic carbon |
| XRD | Powder X-ray diffraction |
| Subscripts and superscripts | |
| cat | Catalyst |
| in | Reactor inlet |
| M | Metal |
| m | Monolayer |
| N | Normal temperature and pressure |
| out | Reactor outlet |
| s | Surface |
| STP | Standard temperature and pressure |
References
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Meylan, F.D.; Moreau, V.; Erkman, S. CO2 utilization in the perspective of industrial ecology, an overview. J. CO2 Util. 2015, 12, 101–108. [Google Scholar] [CrossRef]
- Miguel, C.V.; Soria, M.A.; Mendes, A.; Madeira, L.M. A sorptive reactor for CO2 capture and conversion to renewable methane. Chem. Eng. J. 2017, 322, 590–602. [Google Scholar] [CrossRef]
- Ghaib, K.; Ben-Fares, F.Z. Power-to-Methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Miguel, C.V.; Mendes, A.; Madeira, L. An overview of the Portuguese energy sector and perspectives for power-to-gas implementation. Energies 2018, 11, 3259. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable power-to-gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Faria, A.C.; Miguel, C.V.; Madeira, L.M. Thermodynamic analysis of the CO2 methanation reaction with in situ water removal for biogas upgrading. J. CO2 Util. 2018, 26, 271–280. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Miguel, C.V.; Mendes, A.; Madeira, L.M. Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J. CO2 Util. 2018, 25, 128–136. [Google Scholar] [CrossRef]
- Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315–321. [Google Scholar] [CrossRef]
- Gabrovska, M.; Edreva-Kardjieva, R.; Crişan, D.; Tzvetkov, P.; Shopska, M.; Shtereva, I. Ni-Al layered double hydroxides as catalyst precursors for CO2 removal by methanation. React. Kinet. Mech. Catal. 2012, 105, 79–99. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Gálvez, M.E.; da Costa, P. Novel Ni-La-hydrotalcite derived catalysts for CO2 methanation. Catal. Commun. 2016, 83, 5–8. [Google Scholar] [CrossRef]
- He, L.; Lin, Q.; Liu, Y.; Huang, Y. Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2 methanation: Cooperative effect between Ni nanoparticles and a basic support. J. Energy Chem. 2014, 23, 587–592. [Google Scholar] [CrossRef]
- Zhen, W.; Li, B.; Lu, G.; Ma, J. Enhancing catalytic activity and stability for CO2 methanation on Ni-Ru/γ-Al2O3 via modulating impregnation sequence and controlling surface active species. RSC Adv. 2014, 4, 16472–16479. [Google Scholar] [CrossRef]
- Silva, J.M.; Trujillano, R.; Rives, V.; Soria, M.A.; Madeira, L.M. High temperature CO2 sorption over modified hydrotalcites. Chem. Eng. J. 2017, 325, 25–34. [Google Scholar] [CrossRef]
- Miguel, C.V.; Trujillano, R.; Rives, V.; Vicente, M.A.; Ferreira, A.F.P.; Rodrigues, A.E.; Mendes, A.; Madeira, L.M. High temperature CO2 sorption with gallium-substituted and promoted hydrotalcites. Sep. Purif. Technol. 2014, 127, 202–211. [Google Scholar] [CrossRef]
- Rocha, C.; Soria, M.A.; Madeira, L.M. Doping of hydrotalcite-based sorbents with different interlayer anions for CO2 capture. Sep. Purif. Technol. 2019, 235, 116140. [Google Scholar] [CrossRef]
- Wang, S.; Schrunk, E.T.; Mahajan, H.; Farrauto, R.J. The role of ruthenium in CO2 capture and catalytic conversion to fuel by dual function materials (DFM). Catalysts 2017, 7, 88. [Google Scholar] [CrossRef]
- Duyar, M.S.; Treviño, M.A.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168–169, 370–376. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Baddour, F.G.; Snowden-Swan, L.; Super, J.D.; van Allsburg, K.M. Estimating precommercial heterogeneous catalyst price: a simple step-based method. Org. Process Res. Dev. 2018, 22, 1599–1605. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Motak, M.; Grzybek, T.; Gálvez, M.E.; da Costa, P. The influence of lanthanum incorporation method on the performance of nickel-containing hydrotalcite-derived catalysts in CO2 methanation reaction. Catal. Today 2018, 307, 205–211. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Li, S.; Wang, S.; Ma, X. Hydrogen production from ethanol steam reforming over nickel based catalyst derived from Ni/Mg/Al hydrotalcite-like compounds. Int. J. Hydrogen Energy 2010, 35, 6699–6708. [Google Scholar] [CrossRef]
- Nawfal, M.; Gennequin, C.; Labaki, M.; Nsouli, B.; Aboukaïs, A.; Abi-Aad, E. Hydrogen production by methane steam reforming over Ru supported on Ni-Mg-Al mixed oxides prepared via hydrotalcite route. Int. J. Hydrogen Energy 2015, 40, 1269–1277. [Google Scholar] [CrossRef]
- Takehira, K. “Intelligent” reforming catalysts: Trace noble metal-doped Ni/Mg(Al)O derived from hydrotalcites. J. Nat. Gas Chem. 2009, 18, 237–259. [Google Scholar] [CrossRef]
- Lanza, R.; Järås, S.G.; Canu, P. Partial oxidation of methane over supported ruthenium catalysts. Appl. Catal. A Gen. 2007, 325, 57–67. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Murata, K.; Okabe, K.; Inaba, M.; Takahara, I. Performance and characterization of Ru/Al2O3 and Ru/SiO2 catalysts modified with Mn for Fischer-Tropsch synthesis. Appl. Catal. A Gen. 2008, 340, 203–211. [Google Scholar] [CrossRef]
- Okal, J.; Kępiński, L. Sintering of colloidal Ru/γ-Al2O3 catalyst in hydrogen. Catal. Lett. 2009, 128, 331–336. [Google Scholar] [CrossRef]
- Schmal, M. Heterogeneous Catalysis and Its Industrial Applications; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Farrauto, R.J.; Karp, S.; Jeon, J.H.; Schrunk, E.T.; Wang, S. Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM). J. CO2 Util. 2018, 27, 390–397. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1052–1069. [Google Scholar] [CrossRef] [Green Version]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Springer: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Guo, X.; Traitangwong, A.; Hu, M.; Zuo, C.; Meeyoo, V.; Peng, Z.; Li, C. Carbon dioxide methanation over nickel-based catalysts supported on various mesoporous material. Energy Fuels 2018, 32, 3681–3689. [Google Scholar] [CrossRef]
- Danaci, S.; Protasova, L.; Lefevere, J.; Bedel, L.; Guilet, R.; Marty, P. Efficient CO2 methanation over Ni/Al2O3 coated structured catalysts. Catal. Today 2016, 273, 234–243. [Google Scholar] [CrossRef]
- Vrijburg, W.L.; Moioli, E.; Chen, W.; Zhang, M.; Terlingen, B.J.P.; Zijlstra, B.; Filot, I.A.W.; Züttel, A.; Pidko, E.A.; Hensen, E.J.M. Efficient base-metal NiMn/TiO2 catalyst for CO2 methanation. ACS Catal. 2019, 9, 7823–7839. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Zhang, X.; Rui, N.; Hu, X.; Liu, C.J. Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity. Appl. Catal. B Environ. 2019, 244, 159–169. [Google Scholar] [CrossRef]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Wang, J.; Yao, N.; Liu, B.; Cen, J.; Li, X. Deposition of carbon species on the surface of metal: As a poison or a promoter for the long-term stability of Ni/SiO2 methanation catalyst? Chem. Eng. J. 2017, 322, 339–345. [Google Scholar] [CrossRef]
- Eckle, S.; Anfang, H.G.; Behm, R.J. Reaction intermediates and side products in the methanation of CO and CO2 over supported Ru catalysts in H2-rich reformate gases. J. Phys. Chem. C 2011, 115, 1361–1367. [Google Scholar] [CrossRef]
- Ren, J.; Guo, H.; Yang, J.; Qin, Z.; Lin, J.; Li, Z. Insights into the mechanisms of CO2 methanation on Ni(111) surfaces by density functional theory. Appl. Surf. Sci. 2015, 351, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.M.; Dumesic, J.A.; Hill, C.G. Criteria for stable Ni particle size under methanation reaction conditions: Nickel transport and particle size growth via nickel carbonyl. J. Catal. 1981, 68, 152–165. [Google Scholar] [CrossRef]
- Munnik, P.; Velthoen, M.E.Z.; de Jongh, P.E.; de Jong, K.P.; Gommes, C.J. Nanoparticle growth in supported nickel catalysts during methanation reaction—Larger is better. Angew. Chem. Int. Ed. 2014, 53, 9493–9497. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, M.; Kolb, M.; Mirodatos, C. CO Hydrogenation on a nickel catalyst - I. Kinetics and modeling of a low temperature sintering process. J. Catal. 1994, 148, 9–21. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Carbon deposition in steam reforming and methanation. Catal. Rev. Sci. Eng. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Agnelli, M.; Swaan, H.M.; Marquez-Alvarez, C.; Martin, G.A.; Mirodatos, C. CO hydrogenation on a nickel catalyst: II. A mechanistic study by transient kinetics and infrared spectroscopy. J. Catal. 1998, 175, 117–128. [Google Scholar] [CrossRef]
- Gierlich, H.H.; Fremery, M.; Skov, A.; Rostrup-Nielsen, J.R. Deactivation phenomena of a Ni-based catalyst for high temperature methanation. Stud. Surf. Sci. Catal. 1980, 6, 459–469. [Google Scholar]
- Coenen, K.; Gallucci, F.; Hensen, E.; Annaland, M.v. CO2 and H2O chemisorption mechanism on different potassium-promoted sorbents for SEWGS processes. J. CO2 Util. 2018, 25, 180–193. [Google Scholar] [CrossRef]
- Yong, Z.; Mata, V.; Rodrigues, A.E. Adsorption of carbon dioxide onto hydrotalcite-like compounds (HTlcs) at high temperatures. Ind. Eng. Chem. Res. 2001, 40, 204–209. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Z.; Wu, J.; Yi, X.; Zheng, A.; Umar, A.; O’Hare, D.; Wang, Q. Comprehensive investigation of CO2 adsorption on Mg-Al-CO3 LDH-derived mixed metal oxides. J. Mater. Chem. A 2013, 1, 12782–12790. [Google Scholar] [CrossRef]
- Vaidya, P.D.; Rodrigues, A.E. Kinetics of steam reforming of ethanol over a Ru/Al2O3 catalyst. Ind. Eng. Chem. Res. 2006, 45, 6614–6618. [Google Scholar] [CrossRef]
- Rouquerol, J.; Rouquerol, F.; Llewellyn, P.; Maurin, G.; Sing, K.S.W. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: London, UK, 2012. [Google Scholar]
- Figueiredo, J.L.; Ribeiro, F.R. Catálise Heterogénea; Fundação Calouste Gulbenkian: Lisboa, Portugal, 2007. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Surface Area (m²·g−1) | Pore Volume (cm³·g−1) | Average Pore Size (nm) |
|---|---|---|---|
| NiMgAl | 212 | 0.472 | 8.2 |
| Ru/NiMgAl | 182 | 0.379 | 7.6 |
| Ru/SiO2 | 464 | 0.723 | 4.9 |
| Ru/Al2O3com. | 252 | 0.539 | 6.6 |
| Catalyst | Ru (wt.%) | Ni (wt.%) | Ni/Al (molar) | (Ni + Mg)/Al (molar) |
|---|---|---|---|---|
| NiMgAl | - | 44.02 | 1.7 | 2.1 |
| Ru/NiMgAl | 0.39 | 42.26 | 1.7 | 2.1 |
| Ru/Al2O3com. | 1.08 | - | - | |
| Ru/SiO2 | 1.15 (a) | - | - |
| Catalyst | Metal Dispersion DM (%) | Metal Surface Area SM (m2·gcat−1) |
|---|---|---|
| NiMgAl | 12 | 34.3 |
| Ru/NiMgAl | 10 | 28.7 |
| Ru/SiO2 | 2 | 0.1 |
| Ru/Al2O3com. | 100 | 4.0 |
| Catalyst | T (°C) | WHSV mLN,CO2·g−1·h−1 | TOF (h−1) | Ref | ||
|---|---|---|---|---|---|---|
| NiMgAl | 250 | 2400 | 65.4 | 0.57 | 0.57 | This work |
| 300 | 2400 | 101.9 | 0.88 | 0.88 | ||
| 350 | 2400 | 98.5 | 0.85 | 0.85 | ||
| Ru/NiMgAl | 250 | 2400 | 63.6 | 0.44 | 0.44 | This work |
| 300 | 2400 | 120.7 | 0.83 | 0.83 | ||
| 350 | 2400 | 121.2 | 0.84 | 0.83 | ||
| METH 134 (nickel-based; commercial) | 250 | 2383 | - | 0.07 | 0.07 | [11] |
| 350 | 2383 | - | 0.63 | 0.63 | ||
| 350 | 14,545 | - | 0.33 | 0.33 | ||
| (10 wt.%) Ni/ZSM-5 | 250 | - | 27.3 | 0.19 | - | [35] |
| (15 wt.%) Ni/ZSM-5 | 250 | - | 27.2 | 0.27 | - | |
| (12 wt.%) Ni/Al2O3 | 350 | 1500 | - | 0.85 | - | [36] |
| Ni/TiO2 | 250 | - | 39.6 | - | - | [37] |
| NiMn/TiO2 | 250 | - | 212.4 | - | - | |
| Ni/ZrO2-P | 235 | - | 255.6 | - | - | [38] |
| Ni/ZrO2-C | 235 | - | 162.0 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, J.A.; Faria, A.C.; Soria, M.A.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. CO2 Methanation over Hydrotalcite-Derived Nickel/Ruthenium and Supported Ruthenium Catalysts. Catalysts 2019, 9, 1008. https://doi.org/10.3390/catal9121008
Martins JA, Faria AC, Soria MA, Miguel CV, Rodrigues AE, Madeira LM. CO2 Methanation over Hydrotalcite-Derived Nickel/Ruthenium and Supported Ruthenium Catalysts. Catalysts. 2019; 9(12):1008. https://doi.org/10.3390/catal9121008
Chicago/Turabian StyleMartins, Joana A., A. Catarina Faria, Miguel A. Soria, Carlos V. Miguel, Alírio E. Rodrigues, and Luís M. Madeira. 2019. "CO2 Methanation over Hydrotalcite-Derived Nickel/Ruthenium and Supported Ruthenium Catalysts" Catalysts 9, no. 12: 1008. https://doi.org/10.3390/catal9121008