Direct Synthesis of Hydrogen Peroxide under Semi-Batch Conditions over Un-Promoted Palladium Catalysts Supported by Ion-Exchange Sulfonated Resins: Effects of the Support Morphology

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
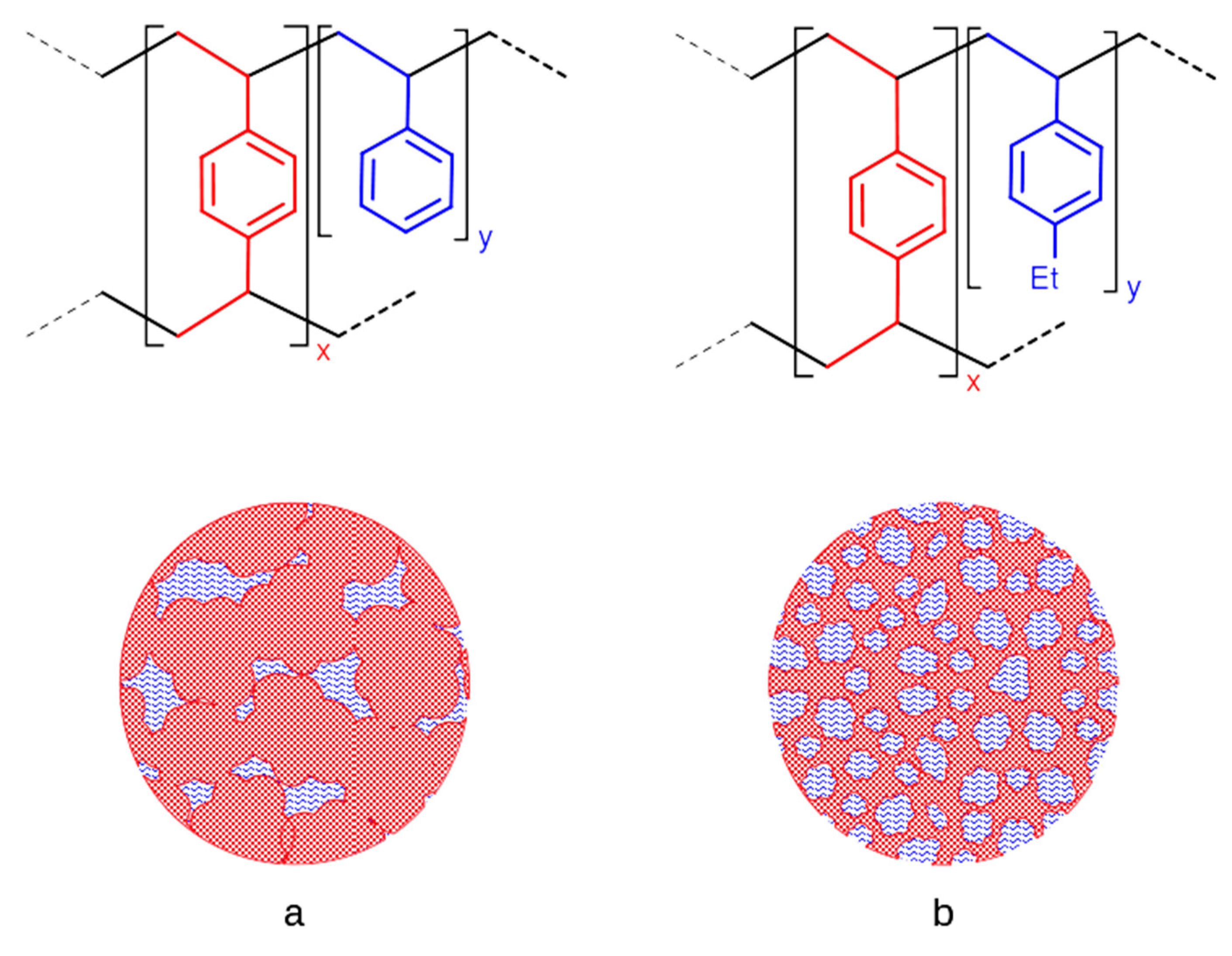
2.1. Catalyst Preparation
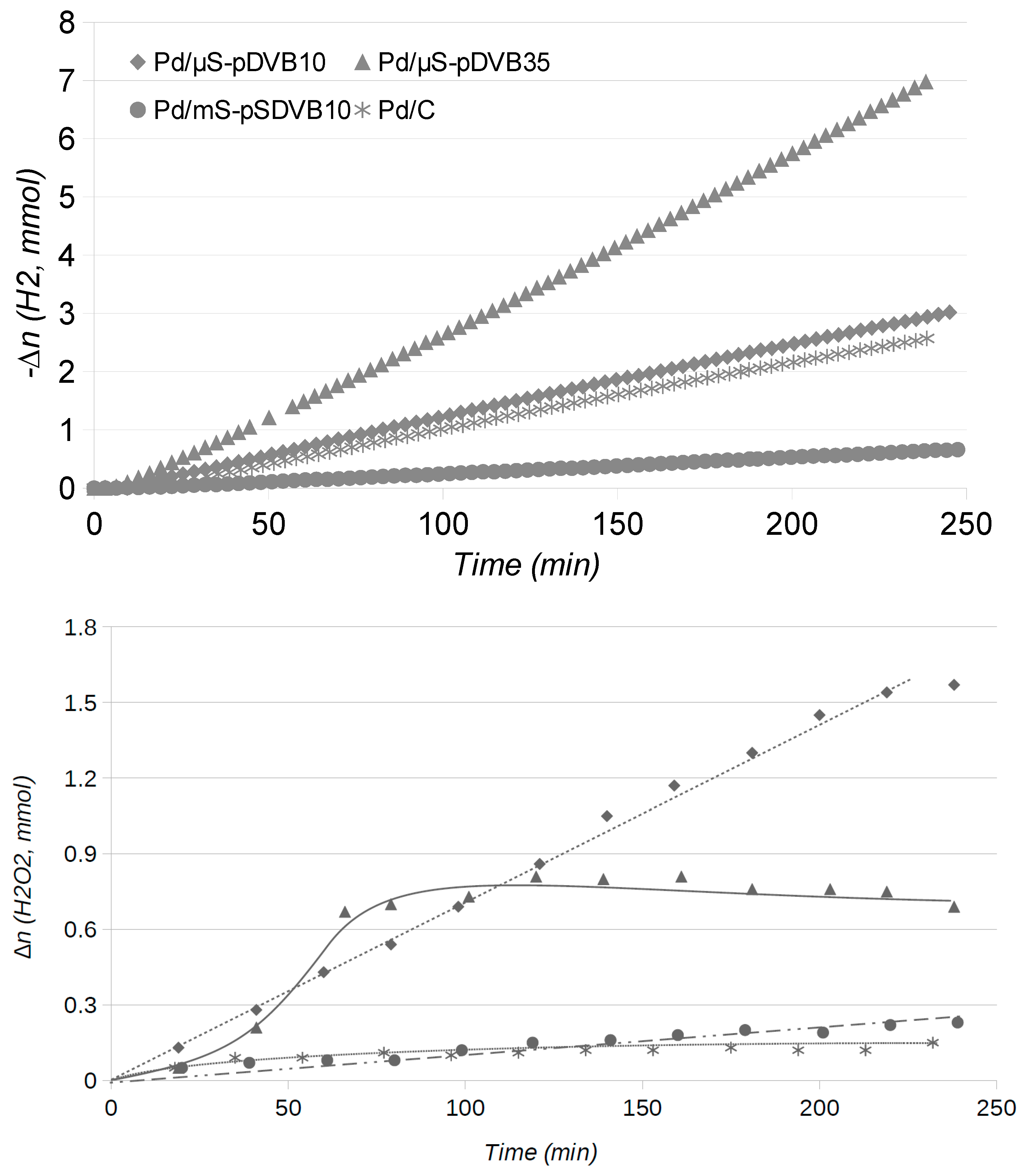
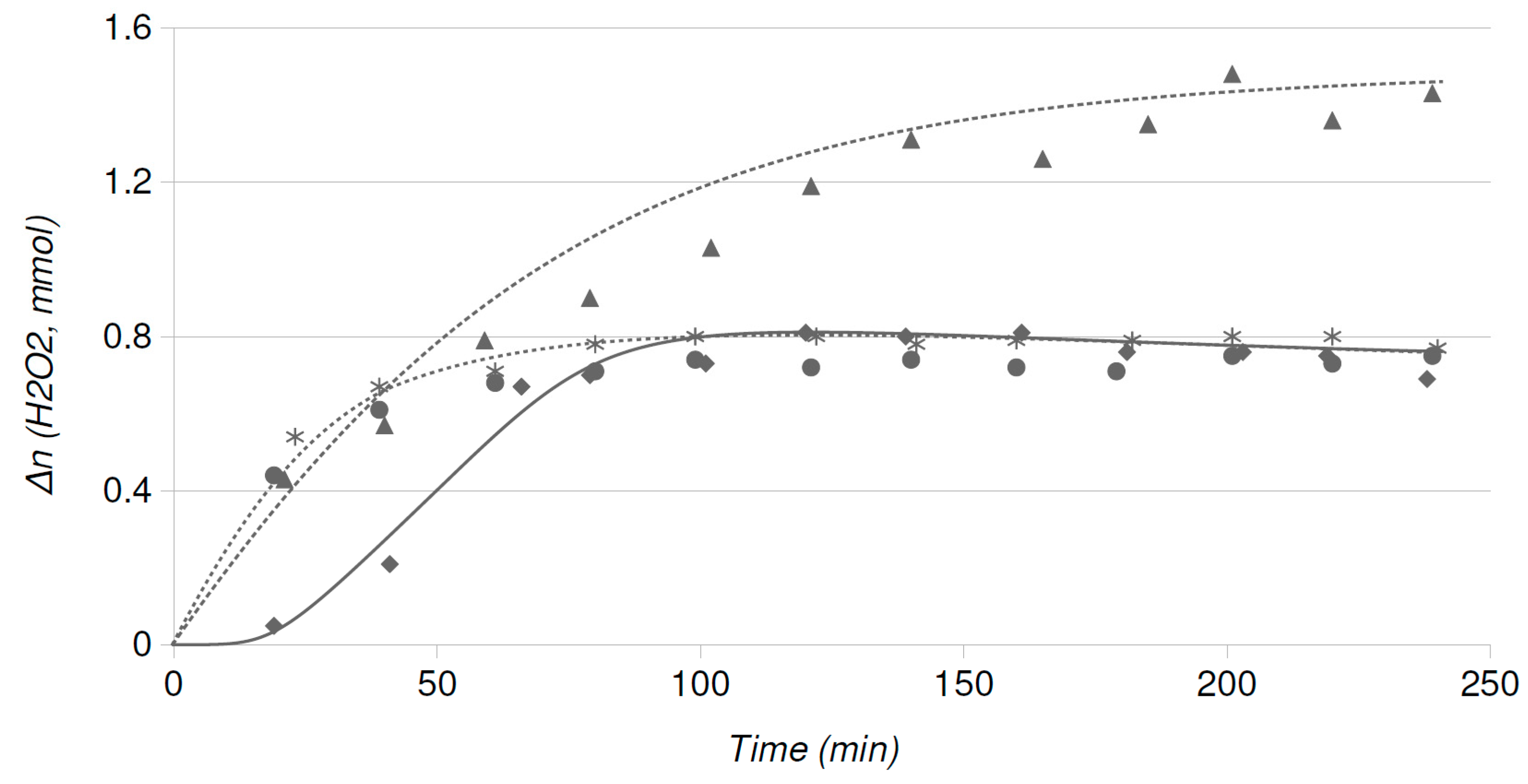
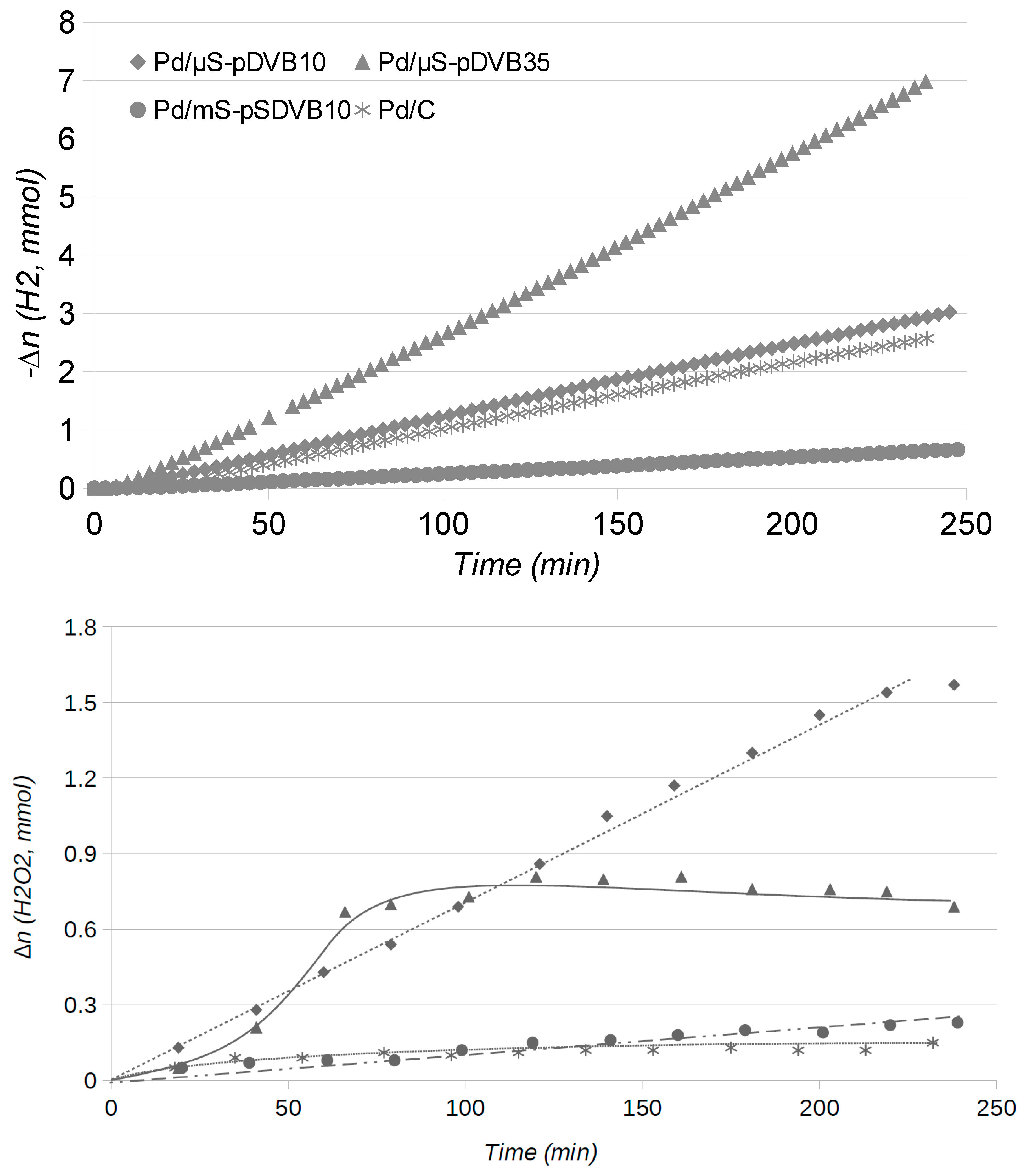
2.2. Catalytic Runs: Fresh Catalysts
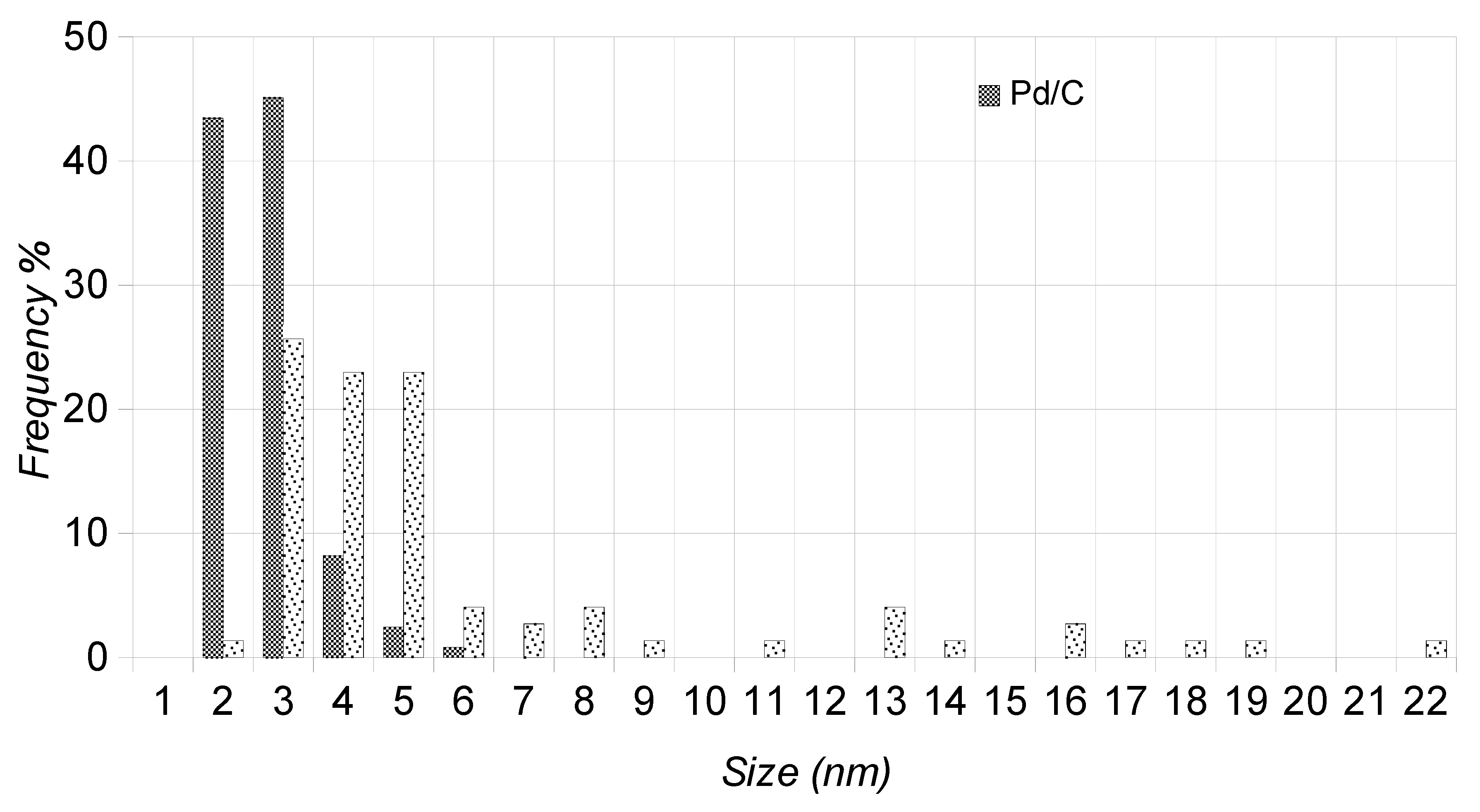
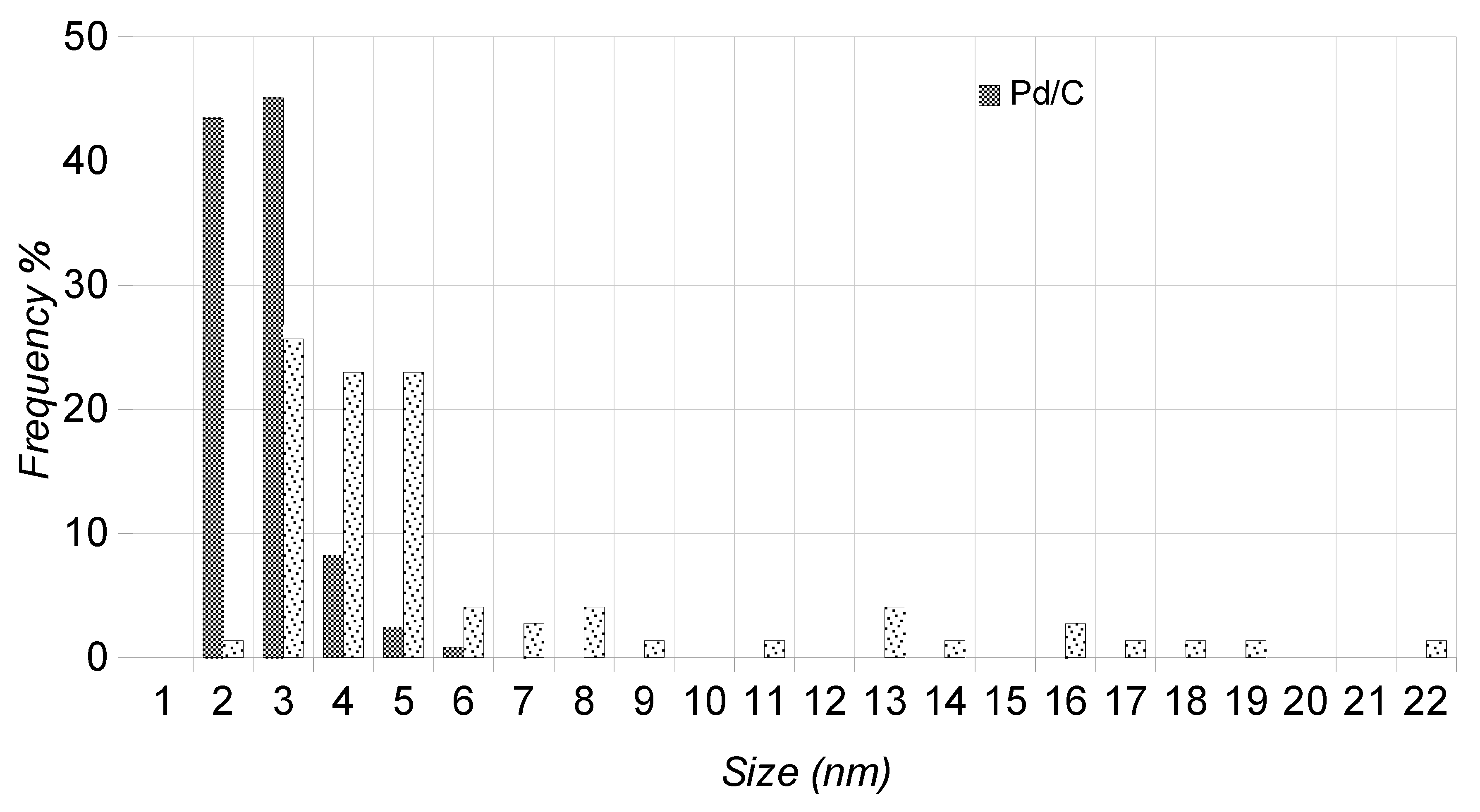
2.3. TEM Characterization
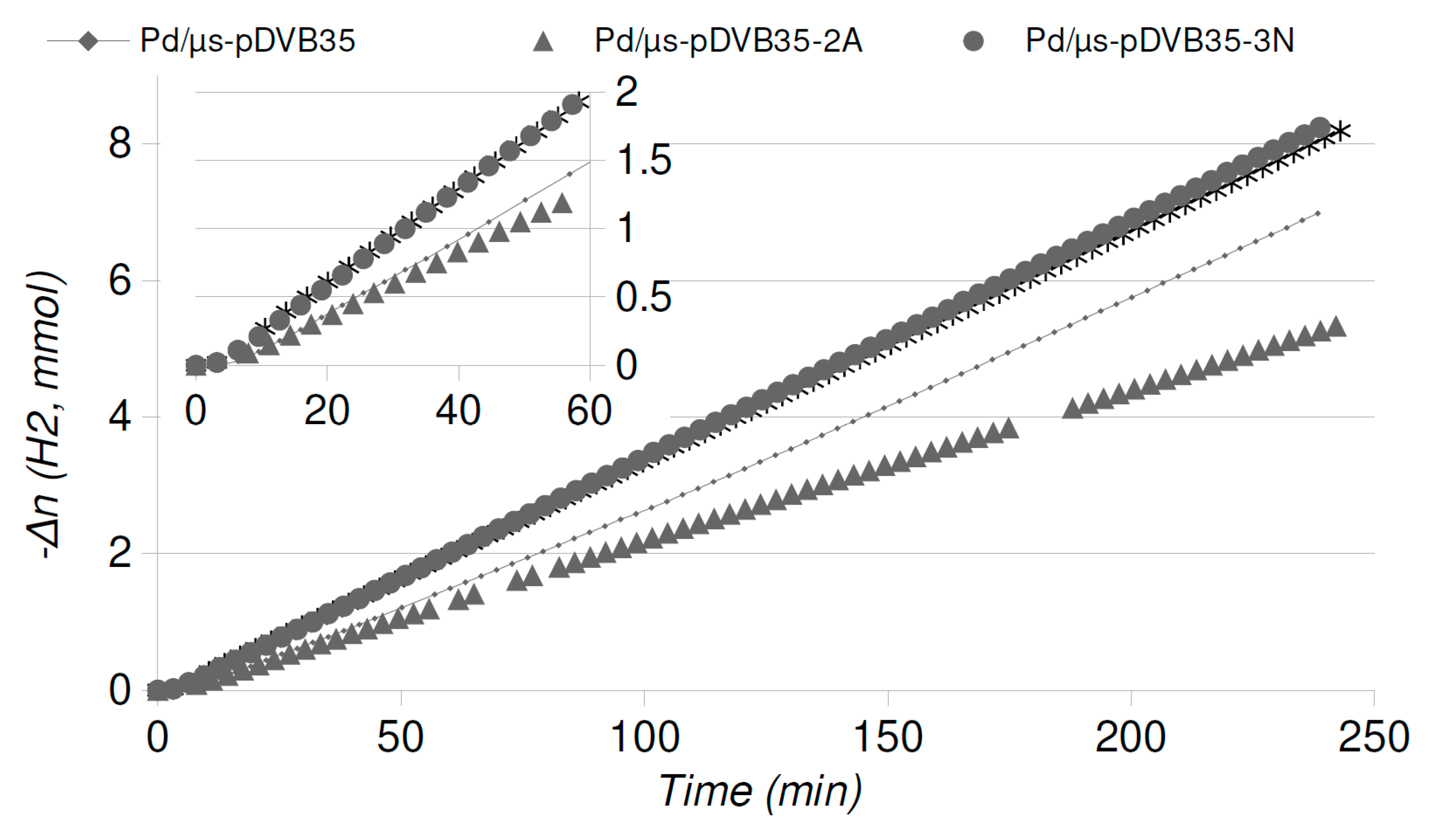
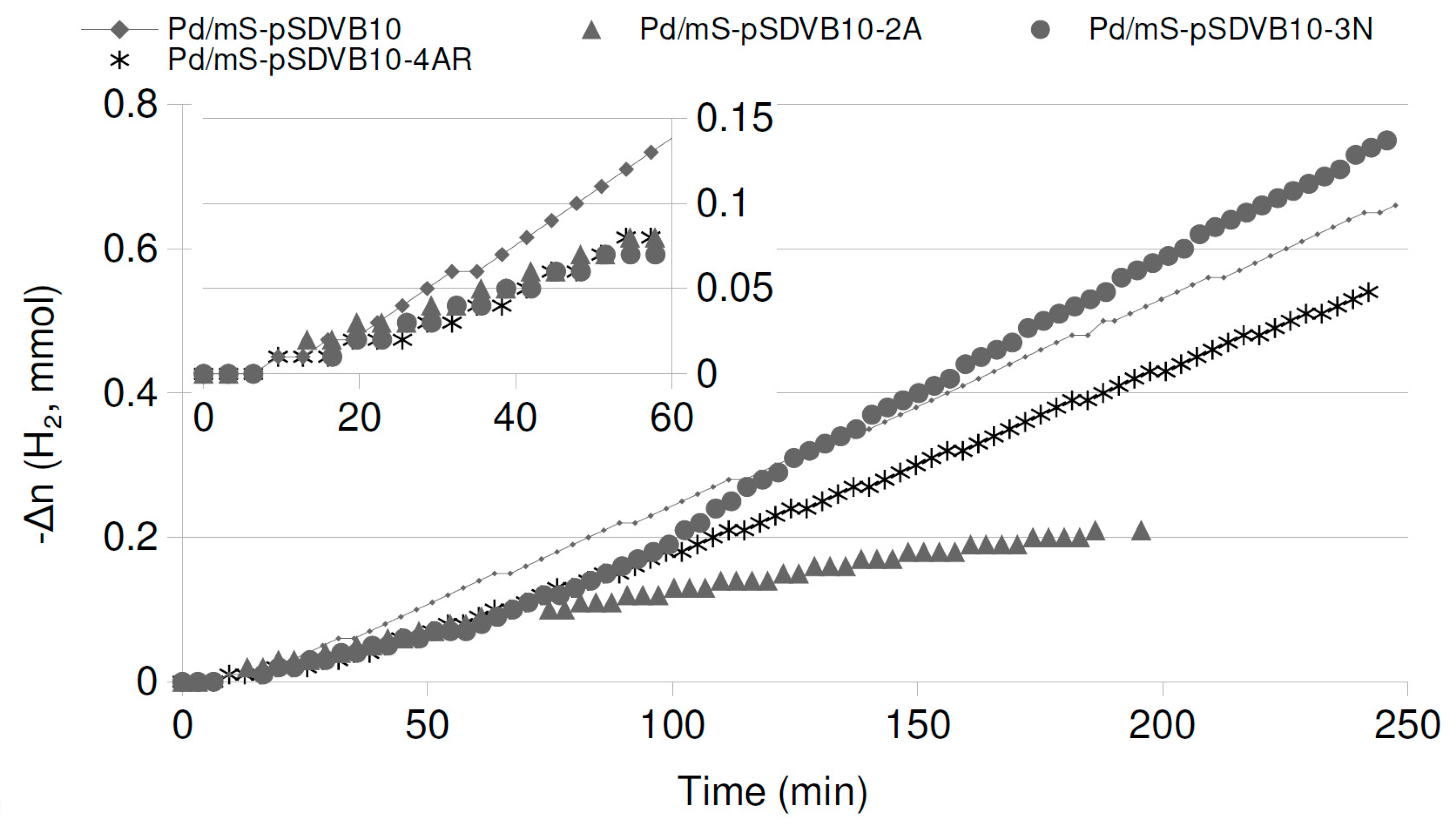
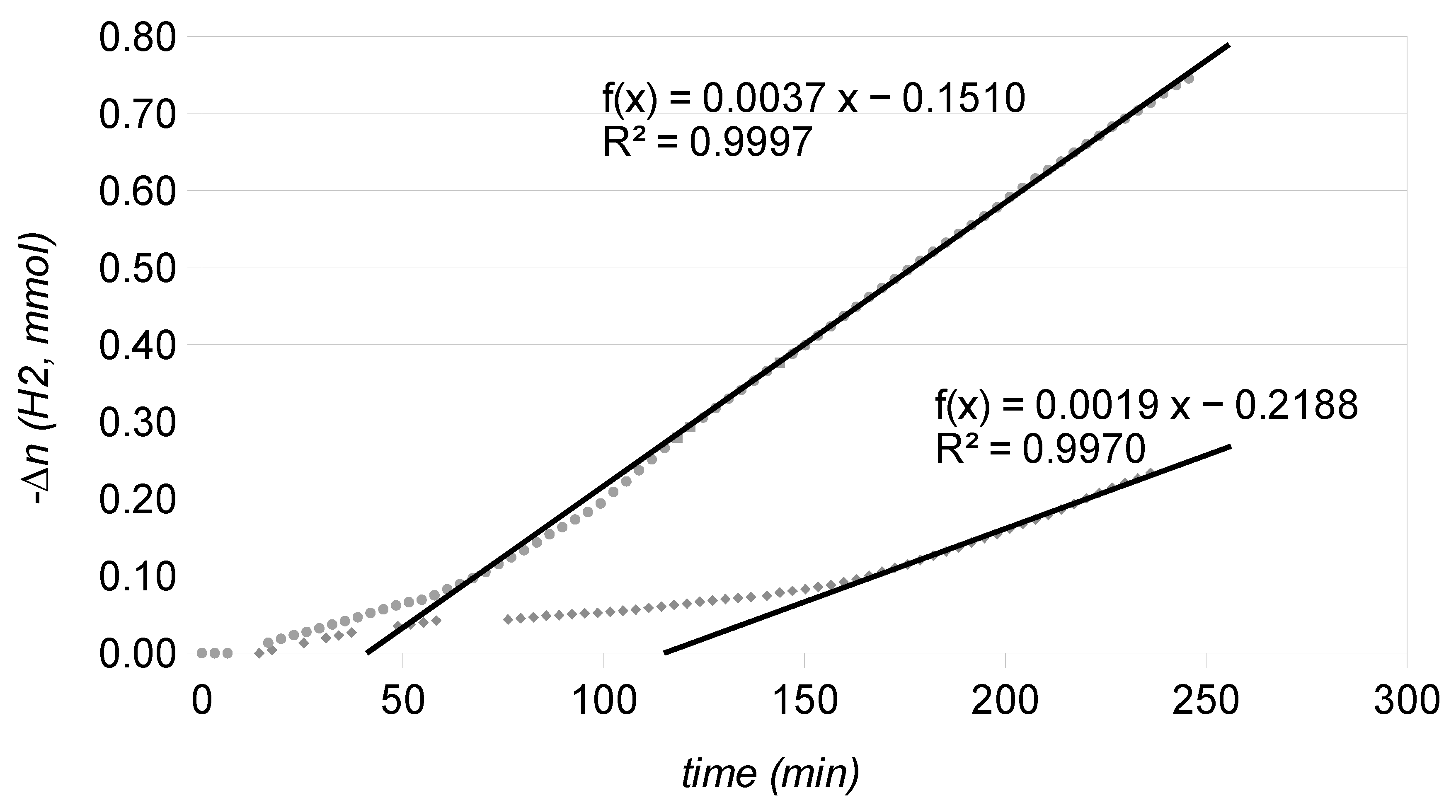
2.4. Catalytic Runs: Aged Catalysts
3. Experimental
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Goor, G.; Glenneberg, J.; Jacobi, S. Hydrogen Peroxide. In Ullmann’s Encyclopedia of Industrial Chemistry; American Cancer Society: Atlanta, GA, USA, 2007; ISBN 978-3-527-30673-2. [Google Scholar]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.K.; Freakley, S.J.; Lewis, R.J.; Pritchard, J.C.; Hutchings, G.J. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Today 2015, 248, 3–9. [Google Scholar] [CrossRef]
- Dittmeyer, R.; Grunwaldt, J.-D.; Pashkova, A. A review of catalyst performance and novel reaction engineering concepts in direct synthesis of hydrogen peroxide. Catal. Today 2015, 248, 149–159. [Google Scholar] [CrossRef]
- Bassler, P.; Weidenbach, M.; Goebbel, H. He New Hppo Process for Propylene Oxide: From Joint Development to Worldscale Production. Chem. Eng. Trans. 2010, 21, 571–576. [Google Scholar]
- Brieva, G.B.; Martin, J.M.C.; Fierro, J.L.G.; Argaiz, M.M.; Garaffa, R.; Janssens, F. Process to Obtain Hydrogen Peroxide, and Catalyst Supports for the Same Process. U.S. Patent 9,610,573, 4 April 2017. [Google Scholar]
- Blanco-Brieva, G.; Cano-Serrano, E.; Campos-Martin, J.M.; Fierro, J.L.G. Direct synthesis of hydrogen peroxide solution with palladium-loaded sulfonic acid polystyrene resins. Chem. Commun. 2004, 10, 1184–1185. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; Montiel-Argaiz, M.; Desmedt, F.; Miquel, P.; Campos-Martin, J.M.; Fierro, J.L.G. Direct synthesis of hydrogen peroxide with no ionic halides in solution. RSC Adv. 2016, 6, 99291–99296. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; Montiel-Argaiz, M.; Desmedt, F.; Miquel, P.; Campos-Martin, J.M.; Fierro, J.L.G. Effect of the Acidity of the Groups of Functionalized Silicas on the Direct Synthesis of H2O2. Top. Catal. 2017, 60, 1151–1155. [Google Scholar] [CrossRef]
- Schröder, V.; Emonts, B.; Janßen, H.; Schulze, H.-P. Explosion Limits of Hydrogen/Oxygen Mixtures at Initial Pressures up to 200 bar. Chem. Eng. Technol. 2004, 27, 847–851. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.; Ntainjua, E.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Switching Off Hydrogen Peroxide Hydrogenation in the Direct Synthesis Process. Science 2009, 323, 1037–1041. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. Strategies for designing supported gold–palladium bimetallic catalysts for the direct synthesis of hydrogen peroxide. Acc. Chem. Res. 2013, 47, 845–854. [Google Scholar] [CrossRef]
- Villa, A.; Freakley, S.J.; Schiavoni, M.; Edwards, J.K.; Hammond, C.; Veith, G.M.; Wang, W.; Wang, D.; Prati, L.; Dimitratos, N.; et al. Depressing the hydrogenation and decomposition reaction in H2O2 synthesis by supporting AuPd on oxygen functionalized carbon nanofibers. Catal. Sci. Technol. 2016, 6, 694–697. [Google Scholar] [CrossRef]
- Samanta, C. Direct synthesis of hydrogen peroxide from hydrogen and oxygen: An overview of recent developments in the process. Appl. Catal. Gen. 2008, 350, 133–149. [Google Scholar] [CrossRef]
- Liu, Q.; Bauer, J.C.; Schaak, R.E.; Lunsford, J.H. Supported palladium nanoparticles: An efficient catalyst for the direct formation of H2O2 from H2 and O2. Angew. Chem. Int. Ed. 2008, 47, 6221–6224. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, D.P.; Lunsford, J.H. Evidence for the role of colloidal palladium in the catalytic formation of H2O2 from H2 and O2. J. Catal. 2002, 206, 173–176. [Google Scholar] [CrossRef]
- Gemo, N.; Biasi, P.; Canu, P.; Menegazzo, F.; Pinna, F.; Samikannu, A.; Kordas, K.; Salmi, T.O.; Mikkola, J.-P. Reactivity Aspects of SBA15-Based Doped Supported Catalysts: H2O2 Direct Synthesis and Disproportionation Reactions. Top. Catal. 2013, 56, 540–549. [Google Scholar] [CrossRef]
- Garcia-Serna, J.; Moreno, T.; Biasi, P.; Cocero, M.J.; Mikkola, J.-P.; Salmi, T.O. Engineering in direct synthesis of hydrogen peroxide: Targets, reactors and guidelines for operational conditions. Green Chem. 2014, 16, 2320–2343. [Google Scholar] [CrossRef]
- Huerta, I.; Biasi, P.; Garcia-Serna, J.; Cocero, M.J.; Mikkola, J.-P.; Salmi, T. Effect of low hydrogen to palladium molar ratios in the direct synthesis of H2O2 in water in a trickle bed reactor. Catal. Today 2015, 248, 91–100. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Wrabetz, S.; Amakawa, K.; Teschner, D.; Freni, M.; Centi, G.; Perathoner, S.; Haevecker, M.; et al. Dynamics of Palladium on Nanocarbon in the Direct Synthesis of H2O2. ChemSusChem 2014, 7, 179–194. [Google Scholar] [CrossRef]
- Arrigo, R.; Schuster, M.E.; Abate, S.; Giorgianni, G.; Centi, G.; Perathoner, S.; Wrabetz, S.; Pfeifer, V.; Antonietti, M.; Schloegl, R. Pd Supported on Carbon Nitride Boosts the Direct Hydrogen Peroxide Synthesis. ACS Catal. 2016, 6, 6959–6966. [Google Scholar] [CrossRef]
- Zecca, M.; Centomo, P.; Corain, B. Metal Nanoclusters Supported on Cross-Linked Functional Polymers: A Class of Emerging Metal Catalysts. In Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control; Elsevier Science: Amsterdam, The Netherlands, 2008; pp. 201–232. ISBN 978-0-444-53057-8. [Google Scholar]
- Bortolus, M.; Centomo, P.; Zecca, M.; Sassi, A.; Jeřábek, K.; Maniero, A.L.; Corain, B. Characterisation of Solute Mobility in Hypercross-Linked Resins in Solvents of Different Polarity: Two Promising Supports for Catalysis. Chem. Eur. J. 2012, 18, 4706–4713. [Google Scholar] [CrossRef]
- Centomo, P.; Zecca, M.; Kralik, M.; Gasparovicova, D.; Jeřábek, K.; Canton, P.; Corain, B. Cross-linked poly-vinyl polymers versus polyureas as designed supports for catalytically active M-0 nanoclusters Part II. Pd-0/cross-linked poly-vinyl polymers versus Pd-0/EnCat (TM) 30NP in mild hydrogenation reactions. J. Mol. Catal. Chem. 2009, 300, 48–58. [Google Scholar] [CrossRef]
- Centomo, P.; Zecca, M.; Di Noto, V.; Lavina, S.; Bombi, G.G.; Nodari, L.; Salviulo, G.; Ingoglia, R.; Milone, C.; Galvagno, S.; et al. Characterization of Synthetic Iron Oxides and their Performance as Support for Au Catalysts. ChemCatChem 2010, 2, 1143–1149. [Google Scholar] [CrossRef]
- Okay, O. Macroporous copolymer networks. Prog. Polym. Sci. 2000, 25, 711–779. [Google Scholar] [CrossRef]
- Widdecke, H. Design and Industrial Application of Polymeric Acid Catalysts. In Syntheses and Separations Using Functional Polymers; Sherrington, D., Hodge, P., Eds.; Wiley: Chichester, UK, 1988; pp. 149–180. [Google Scholar]
- Guyot, A.; Hodge, P.; Sherrington, D.C.; Widdecke, H. Recent studies aimed at the development of polymer-supported reactants with improved accessibility and capacity. React. Polym. 1992, 16, 233–259. [Google Scholar] [CrossRef]
- Burato, C.; Centomo, P.; Rizzoli, M.; Biffis, A.; Campestrini, S.; Corain, B. Functional Resins as Hydrophilic Supports for Nanoclustered Pd(0) and Pd(0)-Au(0) Catalysts Designed for the Direct Synthesis of Hydrogen Peroxide. Adv. Synth. Catal. 2006, 348, 255–259. [Google Scholar] [CrossRef]
- Burato, C.; Campestrini, S.; Han, Y.-F.; Canton, P.; Centomo, P.; Canu, P.; Corain, B. Chemoselective and re-usable heterogeneous catalysts for the direct synthesis of hydrogen peroxide in the liquid phase under non-explosive conditions and in the absence of chemoselectivity enhancers. Appl. Catal. Gen. 2009, 358, 224–231. [Google Scholar] [CrossRef]
- Sterchele, S.; Biasi, P.; Centomo, P.; Canton, P.; Campestrini, S.; Salmi, T.; Zecca, M. Pd-Au and Pd-Pt catalysts for the direct synthesis of hydrogen peroxide in absence of selectivity enhancers. Appl. Catal. Gen. 2013, 468, 160–174. [Google Scholar] [CrossRef]
- Centomo, P.; Meneghini, C.; Sterchele, S.; Trapananti, A.; Aquilanti, G.; Zecca, M. EXAFS in situ: The effect of bromide on Pd during the catalytic direct synthesis of hydrogen peroxide. Catal. Today 2015, 248, 138–141. [Google Scholar] [CrossRef]
- Centomo, P.; Meneghini, C.; Sterchele, S.; Trapananti, A.; Aquilanti, G.; Zecca, M. In Situ X-ray Absorption Fine Structure Spectroscopy of a Palladium Catalyst for the Direct Synthesis of Hydrogen Peroxide: Leaching and Reduction of the Metal Phase in the Presence of Bromide Ions. ChemCatChem 2015, 7, 3712–3718. [Google Scholar] [CrossRef]
- Gemo, N.; Sterchele, S.; Biasi, P.; Centomo, P.; Canu, P.; Zecca, M.; Shchukarev, A.; Kordás, K.; Olavi Salmi, T.; Mikkola, J.-P. The influence of catalyst amount and Pd loading on the H2O2 synthesis from hydrogen and oxygen. Catal. Sci. Technol. 2015, 5, 3545–3555. [Google Scholar] [CrossRef]
- Sterchele, S.; Biasi, P.; Centomo, P.; Campestrini, S.; Shchukarev, A.; Rautio, A.-R.; Mikkola, J.-P.; Salmi, T.; Zecca, M. The effect of the metal precursor-reduction with hydrogen on a library of bimetallic Pd-Au and Pd-Pt catalysts for the direct synthesis of H2O2. Catal. Today 2015, 248, 40–47. [Google Scholar] [CrossRef]
- Sterchele, S.; Biasi, P.; Centomo, P.; Shchukarev, A.; Kordás, K.; Rautio, A.-R.; Mikkola, J.-P.; Salmi, T.; Canton, P.; Zecca, M. Influence of Metal Precursors and Reduction Protocols on the Chloride-Free Preparation of Catalysts for the Direct Synthesis of Hydrogen Peroxide without Selectivity Enhancers. ChemCatChem 2016, 8, 1564–1574. [Google Scholar] [CrossRef]
- Biasi, P.; Mikkola, J.-P.; Sterchele, S.; Salmi, T.; Gemo, N.; Shchukarev, A.; Centomo, P.; Zecca, M.; Canu, P.; Rautio, A.-R.; et al. Revealing the role of bromide in the H2O2 direct synthesis with the catalyst wet pretreatment method (CWPM). AIChE J. 2017, 63, 32–42. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, S.; Liu, F.; Du, Y.; Liu, S.; Ji, Y.; Yokoi, T.; Tatsumi, T.; Xiao, F.-S. Superhydrophobic nanoporous polymers as efficient adsorbents for organic compounds. Nano Today 2009, 4, 135–142. [Google Scholar] [CrossRef]
- Paljevac, M.; Krajnc, P.; Hanková, L.; Holub, L.; Droumaguet, B.L.; Grande, D.; Jeřábek, K. Two-step syneretic formation of highly porous morphology during copolymerization of hydroxyethyl methacrylate and ethylene glycol dimethylacrylate. Mater. Today Commun. 2016, 7, 16–21. [Google Scholar] [CrossRef]
- Sterchele, S.; Centomo, P.; Zecca, M.; Hanková, L.; Jeřábek, K. Dry- and swollen-state morphology of novel high surface area polymers. Microporous Mesoporous Mater. 2014, 185, 26–29. [Google Scholar] [CrossRef]
- Hanková, L.; Holub, L.; Meng, X.; Xiao, F.-S.; Jeřábek, K. Role of water as a coporogen in the synthesis of mesoporous poly(divinylbenzenes). J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Hanková, L.; Holub, L.; Jeřábek, K. Formation of porous polymer morphology by microsyneresis during divinylbenzene polymerization. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 774–781. [Google Scholar] [CrossRef]
- Liu, F.; Meng, X.; Zhang, Y.; Ren, L.; Nawaz, F.; Xiao, F.-S. Efficient and stable solid acid catalysts synthesized from sulfonation of swelling mesoporous polydivinylbenzenes. J. Catal. 2010, 271, 52–58. [Google Scholar] [CrossRef]
- Hanková, L.; Holub, L.; Jeřábek, K. Relation between functionalization degree and activity of strongly acidic polymer supported catalysts. React. Funct. Polym. 2006, 66, 592–598. [Google Scholar] [CrossRef]
- Albright, L.; Jakovac, I.J. Catalysis by Functionalized Porous Organic Polymers. Rohm Haas Bull. 1985, IE-287, 4–6. [Google Scholar]
- Kanungo, S.; Paunovic, V.; Schouten, J.C.; Neira D’Angelo, M.F. Facile Synthesis of Catalytic AuPd Nanoparticles within Capillary Microreactors Using Polyelectrolyte Multilayers for the Direct Synthesis of H2O2. Nano Lett. 2017, 17, 6481–6486. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, H.; Guo, C.; Zhou, Z.; Zheng, N. Simplifying the Creation of Hollow Metallic Nanostructures: One-Pot Synthesis of Hollow Palladium/Platinum Single-Crystalline Nanocubes. Angew. Chem. Int. Ed. 2009, 48, 4808–4812. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.M.; Silva, F.P.; Vono, L.L.R.; Kiyohara, P.K.; Duarte, E.L.; Itri, R.; Landers, R.; Machado, G. Superparamagnetic nanoparticle-supported palladium: A highly stable magnetically recoverable and reusable catalyst for hydrogenation reactions. Green Chem. 2007, 9, 379–385. [Google Scholar] [CrossRef]
- Venkatesan, R.; Prechtl, M.H.; Scholten, J.D.; Pezzi, R.P.; Machado, G.; Dupont, J. Palladium nanoparticle catalysts in ionic liquids: Synthesis, characterisation and selective partial hydrogenation of alkynes to Z-alkenes. J. Mater. Chem. 2011, 21, 3030–3036. [Google Scholar] [CrossRef]
- Biffis, A.; Landes, H.; Jeřábek, K.; Corain, B. Metal palladium dispersed inside macroporous ion-exchange resins: The issue of the accessibility to gaseous reactants. J. Mol. Catal. A Chem. 2000, 151, 283–288. [Google Scholar] [CrossRef]
- Jeřábek, K.; Hanková, L.; Holub, L. Working-state morphologies of ion exchange catalysts and their influence on reaction kinetics. J. Mol. Catal. A Chem. 2010, 333, 109–113. [Google Scholar] [CrossRef]

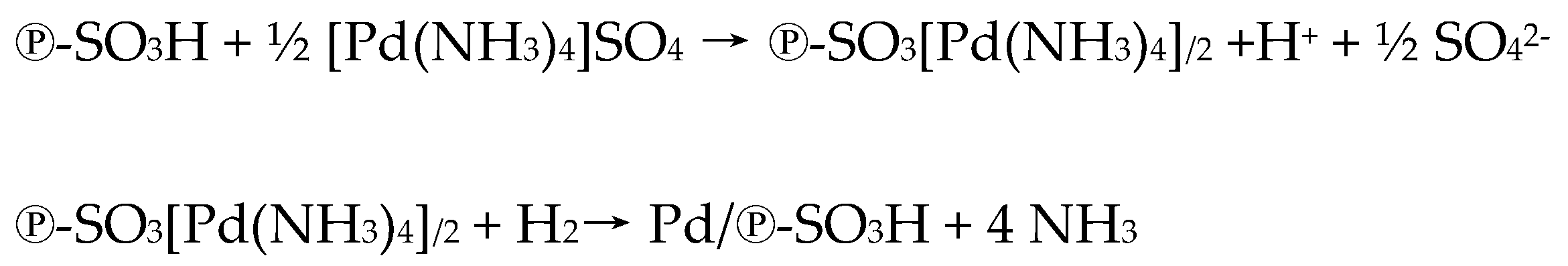





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
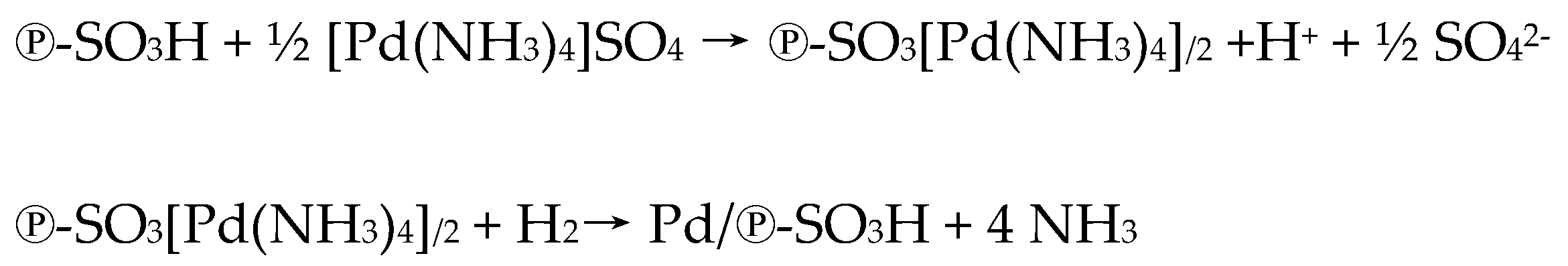{kind=link}
| Ion-Exchange Capacity of the Support | Pd (%, w/w) | ||
|---|---|---|---|
| M+ | M2+ | ||
| Pd/µS-pSDVB10 | 5.0 | 2.5 | 1.0 |
| Pd/µS-pDVB10 a | 2.0 | 1.0 | 1.0 |
| Pd/µS-pDVB35 a | 3.2 | 1.6 | 3.6 |
| Age (weeks) a | Storage Atmosphere | Re-Activation | |
|---|---|---|---|
| Pd/mS-pSDVB10-2A Pd/µS-pDVB35-2A | 2 | Air (closed desiccator) | none |
| Pd/mS-pSDVB10-3N Pd/µS-pDVB35-3N | 3 | Nitrogen (dry-box) | none |
| Pd/mS-pSDVB10-4AR Pd/µS-pDVB35-4AR | 4 | Air (laboratory) | H2, 506 kPa, 60 °C |
| Pd/µS-pDVB10 | Pd/µS-pDVB35 | Pd/mS-pSDVB10 | Pd/C | |||
|---|---|---|---|---|---|---|
| 1 | H2 feed rate (µmol·h−1) a | 2452 | ||||
| 2 | nPd (µmol) | 9.92 | 34.34 | 9.98 | 9.44 | |
| 3 | (µmol·h−1) b | 792 | 1674 | 162 | 714 | |
| 4 | (µmol·h−1) c | 739 | 1757 | 161 | 646 | |
| 5 | (µmol·h−1) d | 414 | 720 | <54 | <60 | |
| 6 | (µmol·h−1) e | 396 | 174 | 58 | 39 | |
| 7 | (h−1) | 80’ f | 79.8 | 48.7 | 16.2 | 75.6 |
| 240’ g | 75.5 | 51.2 | 16.1 | 75.6 | ||
| 8 | (h−1) | 80’ h | 41.7 | 21.0 | <5.4 | <6.4 |
| 240’ i | 39.9 | 5.1 | 5.8 | 4.1 | ||
| 9 | C (%) | 80’ j | 32.3 | 68.3 | 6.6 | 29.1 |
| 240’ k | 30.1 | 71.7 | 6.6 | 26.3 | ||
| 10 | S (%) | 80’ l | 52.2 | 43.1 | <33 | <8.5 |
| 240’ m | 53.6 | 10.0 | 36.0 | 5.4 | ||
| Catalyst | Pd/µS-pDVB35-2A | Pd/µS-pDVB35-3N | Pd/µS-pDVB35-4AR | ||
|---|---|---|---|---|---|
| #1 | H2 feed rate (µmol·h−1) a | 2452 | |||
| #2 | nPd (µmol) | 36.0 | 35.3 | 33.1 | |
| #3 | (µmol·h−1) | 80’ b | 1386 (0.83) | 2124 (1.27) | 2070 (1.24) |
| 240’ c | 1320 | 2088 | 2034 | ||
| #4 | (µmol·h−1) d | 80’ | 554 (0.77) | 532 (0.74) | 585 (0.81) |
| 240’ | 359 | 188 | 193 | ||
| #5 | (h−1) e | 80’ | 38.5 (0.79) | 60.2 (1.23) | 62.5 (1.28) |
| 240’ | 36.7 | 59.2 | 61.5 | ||
| #6 | (h−1) f | 80’ | 15.4 (0.73) | 15.1 (0.72) | 17.7 (0.84) |
| 240’ | 10.0 | 5.3 | 5.8 | ||
| #7 | C (%) g | 80’ | 56.5 | 86.6 | 84.4 |
| 240’ | 53.8 | 85.2 | 83.0 | ||
| #8 | S (%) h | 80’ | 40.0 | 25.0 | 28.3 |
| 240’ | 27.2 | 9.0 | 9.5 | ||
| Catalyst | Pd/mS-pSDVB10-2A | Pd/mS-pSDVB10-3N | Pd/mS-pSDVB10-4AR | ||
|---|---|---|---|---|---|
| #1 | H2 feed rate (µmol·h−1) a | 2452 | |||
| #2 | nPd (µmol) | 9.63 | 9.42 | 9.57 | |
| #3 | (µmol·h−1) | 240’ b | 53.0 (0.32) | 220 (1.36) | 155 (0.97) |
| #4 | (µmol·h−1) c | 240’ | 17.6 (0.31) | 102 (1.76) | 55.6 (0.96) |
| #5 | (h−1) d | 240’ | 5.5 (0.34) | 23.4 (1.45) | 16.2 (1.01) |
| #6 | (h−1) e | 240’ | 1.8 (0.31) | 10.8 (1.86) | 5.8 (1.00) |
| #7 | C (%) f | 240’ | 2.2 | 9.0 | 6.3 |
| #8 | S (%) g | 240’ | 33.2 | 46.4 | 35.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frison, F.; Dalla Valle, C.; Evangelisti, C.; Centomo, P.; Zecca, M. Direct Synthesis of Hydrogen Peroxide under Semi-Batch Conditions over Un-Promoted Palladium Catalysts Supported by Ion-Exchange Sulfonated Resins: Effects of the Support Morphology. Catalysts 2019, 9, 124. https://doi.org/10.3390/catal9020124
Frison F, Dalla Valle C, Evangelisti C, Centomo P, Zecca M. Direct Synthesis of Hydrogen Peroxide under Semi-Batch Conditions over Un-Promoted Palladium Catalysts Supported by Ion-Exchange Sulfonated Resins: Effects of the Support Morphology. Catalysts. 2019; 9(2):124. https://doi.org/10.3390/catal9020124
Chicago/Turabian StyleFrison, Francesco, Chiara Dalla Valle, Claudio Evangelisti, Paolo Centomo, and Marco Zecca. 2019. "Direct Synthesis of Hydrogen Peroxide under Semi-Batch Conditions over Un-Promoted Palladium Catalysts Supported by Ion-Exchange Sulfonated Resins: Effects of the Support Morphology" Catalysts 9, no. 2: 124. https://doi.org/10.3390/catal9020124