Synthesis and Organocatalytic Asymmetric Nitro-aldol Initiated Cascade Reactions of 2-Acylbenzonitriles Leading to 3,3-Disubstituted Isoindolinones

 , and
, and
Abstract
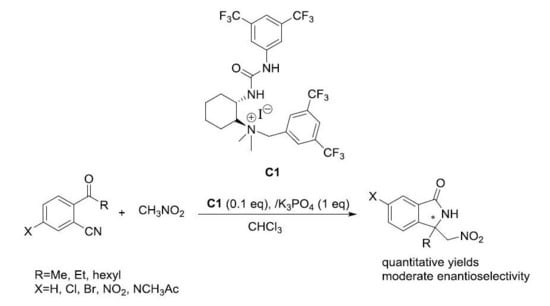
:
1. Introduction
2. Results
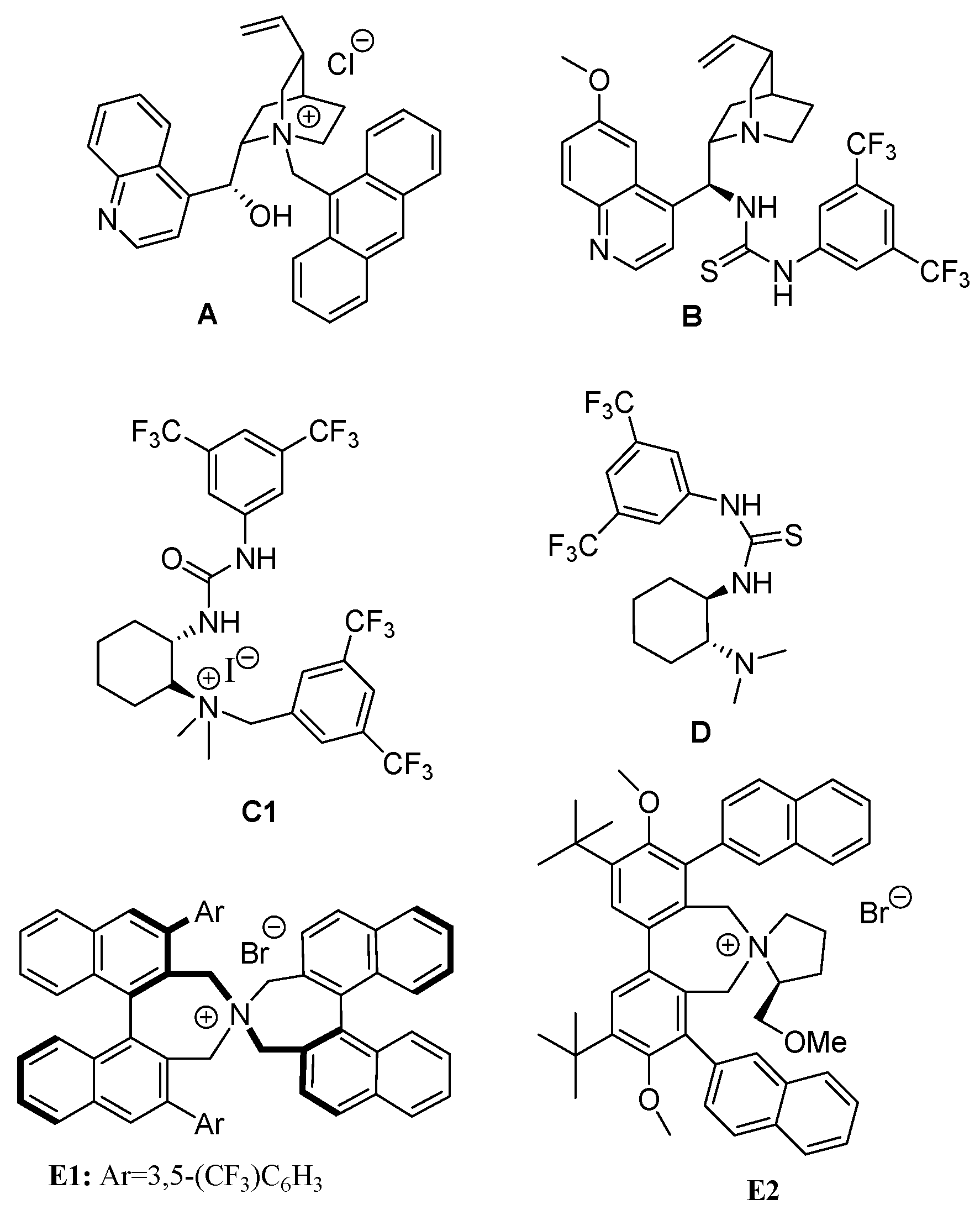
2.1. Synthesis of 2-acylbenzonitriles by Suzuki-Miyaura Type Cross-Coupling Reactions
2.2. Wittig/Oxidation Strategy in the Synthesis of 2-acylbenzonitriles
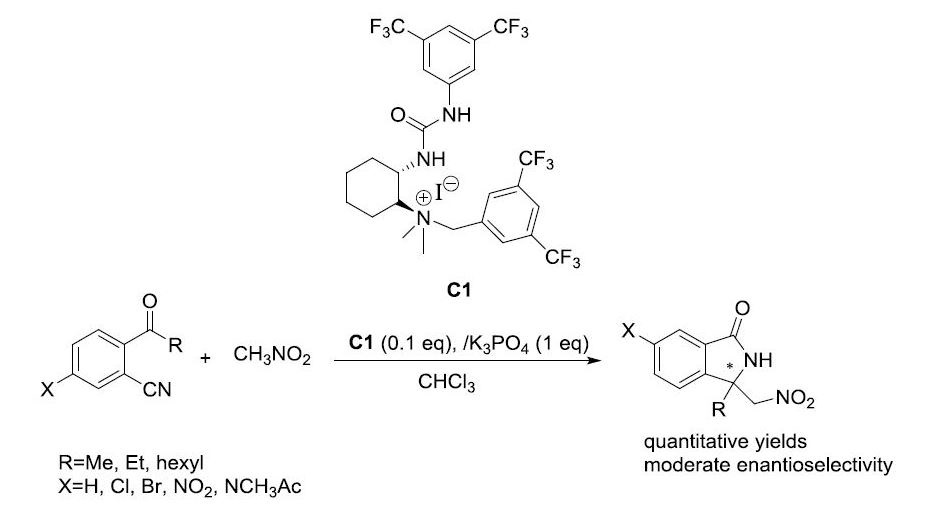
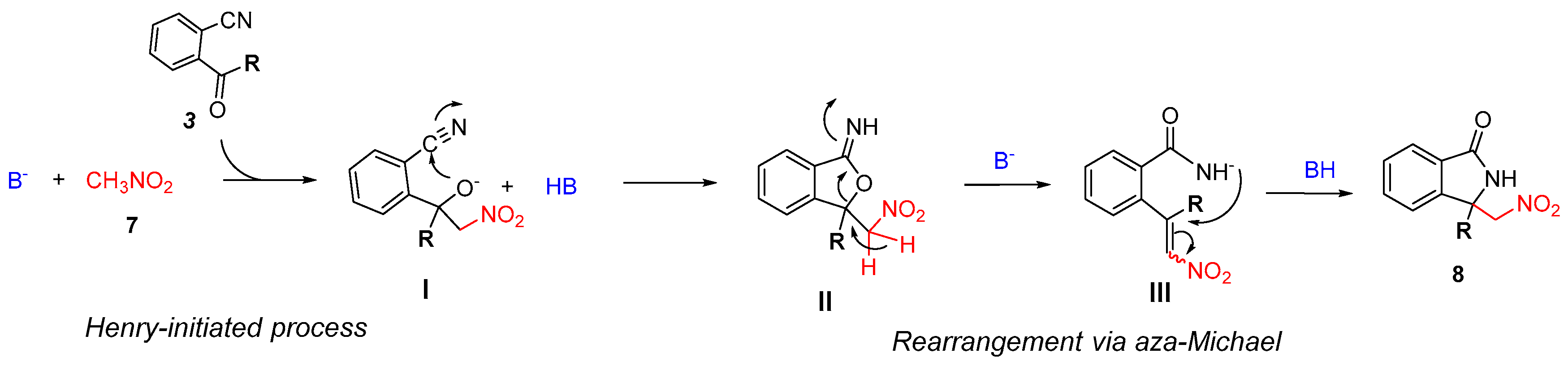
2.3. Asymmetric Henry-Initiated Cascade Reactions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chanda, T.; Zhao, J.C.-G. Recent Progress in Organocatalytic Asymmetric Domino Transformations. Adv. Synth. Catal. 2018, 360, 2–79. [Google Scholar] [CrossRef]
- Grossmann, A.; Enders, D. N-Heterocyclic Carbene Catalyzed Domino Reactions. Angew. Chem. Int. Ed. 2012, 51, 314–325. [Google Scholar] [CrossRef]
- Fogg, D.E.; dos Santos, E.N. Tandem catalysis: A taxonomy and illustrative review. Coord. Chem. Rev. 2004, 248, 2365–2379. [Google Scholar] [CrossRef]
- Tietze, L.F.; Beifuss, U. Sequential Transformations in Organic Chemistry: A Synthetic Strategy with a Future. Angew. Chem. Int. Ed. Engl. 1993, 32, 131–163. [Google Scholar] [CrossRef]
- Riant, O.; Hannedouche, J. Asymmetric catalysis for the construction of quaternary carbon centres: Nucleophilic addition on ketones and ketimines. Org. Biomol. Chem. 2007, 5, 873–888. [Google Scholar] [CrossRef]
- Bella, M.; Gasperi, T. Organocatalytic Formation of Quaternary Stereocenters. Synthesis 2009, 10, 1583–1614. [Google Scholar] [CrossRef]
- Li, Z.; Jangra, H.; Chen, Q.; Mayer, P.; Ofial, A.R.; Zipse, H.; Mayr, H. Kinetics and Mechanism of Oxirane Formation by Darzens Condensation of Ketones: Quantification of the Electrophilicities of Ketones. J. Am. Chem. Soc. 2018, 140, 5500–5515. [Google Scholar] [CrossRef]
- Di Mola, A.; Di Martino, M.; Capaccio, V.; Pierri, G.; Palombi, L.; Tedesco, C.; Massa, A. Synthesis of 2-Acetylbenzonitriles and Their Reactivity in Tandem Reactions with Carbon and Hetero Nucleophiles: Easy Access to 3,3-Disubstituted Isoindolinones. Eur. J. Org. Chem. 2018, 2018, 1699–1708. [Google Scholar] [CrossRef]
- Sanchez, J.M.; Busto, E.; Gotor-Fernandez, V.; Gotor, V. Highly Stereoselective Chemoenzymatic Synthesis of the 3H-Isobenzofuran Skeleton. Access to Enantiopure 3-Methylphthalides. Org. Lett. 2012, 14, 1444–1447. [Google Scholar] [CrossRef]
- Mochalov, S.S.; Fedotov, A.N.; Trofimova, E.V.; Zefirov, N.S. o-Acylbenzonitriles: Synthesis and Heterocyclization under Acid Hydrolysis of the Cyano Group. Russ. J. Org. Chem. 2018, 54, 403–413. [Google Scholar] [CrossRef]
- Ogawa, D.; Hyodo, K.; Suetsugu, M.; Li, J.; Inoue, Y.; Fujisawa, M.; Iwasaki, M.; Takagi, K.; Nishihara, Y. Palladium-catalyzed and copper-mediated cross-coupling reaction of aryl- or alkenylboronic acids with acid chlorides under neutral conditions: Efficient synthetic methods for diaryl ketones and chalcones at room temperature. Tetrahedron 2013, 69, 2565–2571. [Google Scholar] [CrossRef]
- Ishiyama, T.; Kizaki, K.; Hayashi, T.; Suzuki, A.; Miyaura, M. Palladium-Catalyzed Carbonylative Cross-Coupling Reaction of Arylboronic Acids with Aryl Electrophiles: Synthesis of Biaryl Ketones. J. Org. Chem. 1998, 63, 4726–4731. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Q.; Li, H.; Guo, H.; He, W. Ag/C nanoparticles catalysed aerobic oxidation of diaryl and aryl(hetero) methylenes into ketones. Nano Res. 2017, 10, 3261–3267. [Google Scholar] [CrossRef]
- Haddach, M.; McCarthy, J.R. A new method for the synthesis of ketones: The palladium-catalyzed cross-coupling of acid chlorides with arylboronic acids. Tetrahedron Lett. 1999, 40, 3109–3112. [Google Scholar] [CrossRef]
- Urawa, Y.; Ogura, K. A convenient method for preparing aromatic ketones from acyl chlorides and arylboronic acids via Suzuki–Miyaura type coupling reaction. Tetrahedron Lett. 2003, 44, 271–273. [Google Scholar] [CrossRef]
- Blangetti, M.; Rosso, H.; Prandi, C.; Deagostino, A.; Venturello, P. Suzuki-Miyaura Cross-Coupling in Acylation Reactions, Scope and Recent Developments. Molecules 2013, 18, 1188–1213. [Google Scholar] [CrossRef]
- Urawa, Y.; Naka, H.; Miyazawa, M.; Souda, S.; Ogura, K. Investigations into the Suzuki–Miyaura coupling aiming at multikilogram synthesis of E2040 using (o-cyanophenyl)boronic esters. J. Organometal. Chem. 2002, 653, 269–278. [Google Scholar] [CrossRef]
- Bunce, R.A.; Johnson, L.B. Acylation and alkylation of 2-and 4-methylbenzonitrile. Org. Prep. Proced. Int. 1999, 31, 407–412. [Google Scholar] [CrossRef]
- Kobayashi, K.; Ezaki, K.; Nozawa, I. Synthesis of 2-Substituted 3-Alkylidene-2,3-dihydro-1H-isoindol-1-imines through Cyclization of [1-(2-Cyanophenyl)alkylidene]aminide Intermediates Generated from the Reaction of 2-(1-Azidoalkyl)benzonitriles with NaH. Helv. Chim. Acta 2014, 97, 1624–1629. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hashimoto, K.; Shiokawa, T.; Morkawa, O.; Konishi, H. Synthesis of (Z)-2-(2H-Isoquinolin-1-ylidene)acetamides by Iodine-Mediated Cyclization of (Z)-3-Amino-3-(2-vinylphenyl)propenamides. Synthesis 2007, 2007, 824–828. [Google Scholar] [CrossRef]
- Se Song, Y.; Lee, C.H.; Lee, K.J. Application of baylis-hillman methodology in a new synthesis of 3-oxo-2,3-dihydro-1H-isoindoles. J. Heterocycl. Chem. 2003, 40, 939–941. [Google Scholar] [CrossRef]
- Angelin, M.; Rahm, M.; Fischer, A.; Brinck, T.; Ramström, O. Diastereoselective one-pot tandem synthesis of 3-substituted isoindolinones: A mechanistic investigation. J. Org. Chem. 2010, 75, 5882–5887. [Google Scholar] [CrossRef] [PubMed]
- Massa, A.; Roscigno, A.; De Caprariis, P.; Filosa, R.; Di Mola, A. Trimethylchlorosilane and Silicon Tetrachloride in Two Novel Methodologies for the Efficient and Mild Aldol Addition of β-Keto Esters and Malonates to Aldehydes. Adv. Synth. Catal. 2010, 352, 3348–3354. [Google Scholar] [CrossRef]
- Petronzi, C.; Collarile, S.; Croce, G.; Filosa, R.; De Caprariis, P.; Peduto, A.; Palombi, L.; Intintoli, V.; Di Mola, A.; Massa, A. Synthesis and Reactivity of the 3-Substituted Isoindolinone Framework to Assemble Highly Functionalized Related Structures. Eur. J. Org. Chem. 2012, 2012, 5357–5365. [Google Scholar] [CrossRef]
- Perillo, M.; Di Mola, A.; Filosa, R.; Palombi, L.; Massa, A. Cascade reactions of glycine Schiff bases and chiral phase transfer catalysts in the synthesis of α-amino acids 3-substituted phthalides or isoindolinones. RSC Adv. 2014, 9, 4239–4246. [Google Scholar] [CrossRef]
- Li, T.; Zhou, S.; Wang, J.; Luis Acena, J.; Soloshonok, V.A.; Liu, H. Asymmetric synthesis of α-(1-oxoisoindolin-3-yl)glycine: Synthetic and mechanistic challenges. Chem. Commun. 2015, 9, 1624–1626. [Google Scholar] [CrossRef] [PubMed]
- Di Mola, A.; Tiffner, M.; Scorzelli, F.; Palombi, L.; Filosa, R.; De Caprariis, P.; Waser, M.; Massa, A. Bifunctional phase-transfer catalysis in the asymmetric synthesis of biologically active isoindolinones. Beilstein J. Org. Chem. 2015, 11, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Capaccio, V.; Capobianco, A.; Stanzione, A.; Pierri, G.; Tedesco, C.; Di Mola, A.; Massa, A.; Palombi, L. Organocatalytic Heterocyclization Driven by Dynamic Kinetic Resolution: Enantioselective Access to Multi-heteroatomic Cyclic Structures Mediated by Cinchona Alkaloid-based Catalysts. Adv. Synth. Catal. 2017, 359, 2874–2880. [Google Scholar] [CrossRef]
- Liu, L.; Qiang, J.; Bai, S.H.; Sung, H.L.; Miao, C.B.; Li, J. Direct Access to Isoindolin-1-one Scaffolds by Copper-Catalyzed Divergent Cyclizations of 2-Formylbenzonitrile and Diaryliodonium Salts. Adv. Synth. Catal. 2017, 359, 1283–1289. [Google Scholar] [CrossRef]
- Liu, L.; Bai, S.H.; Li, Y.; Wang, L.X.; Hu, Y.; Sung, H.L.; Li, J.J. Synthesis of 2,3-diarylisoindolin-1-one by copper-catalyzed cascade annulation of 2-formylbenzonitriles, arenes, and diaryliodonium Salts. J. Org. Chem. 2017, 82, 11084–11090. [Google Scholar] [CrossRef] [PubMed]
- Bjoere, A.; Bostroem, J.; Davidsson, O.; Emtenaes, H.; Gran, H.; Iliefski, T.; Kajanus, J.; Olsson, R.; Sandberg, L.; Strandlund, G.I. Isondoline Derivatives for the Treatment of Arrhythmias. International Patent WO2008008022, 17 January 2008. [Google Scholar]
- Conn, E.L.; Hepworth, D.; Qi, Y.; Rocke, B.N.; Ruggeri, R.B.; Zhang, Y. 2-Phenyl Benzoylamides. International Patent WO 2011/145022 A1, 24 November 2011. [Google Scholar]
- Baldwin, J.J.; Claremon, D.A.; Tice, C.M.; Cacatian, S.; Dillard, L.H.; Ishchenko, A.V.; Yuan, J.; Xu, Z.; Mcgeehan, G.; Zhao, W. Renin Inhibitors. International Patent WO2008156816, 27 March 2008. [Google Scholar]
- Yang, G.; Shen, C.; Zhang, W. An asymmetric aerobic aza-Wacker-type cyclization: Synthesis of isoindolinones bearing tetrasubstituted carbon stereocenters. Angew. Chem. Int. Ed. 2012, 51, 9141–9145. [Google Scholar] [CrossRef] [PubMed]
- Scorzelli, F.; Di Mola, A.; De Piano, F.; Tedesco, C.; Palombi, L.; Filosa, R.; Waser, M.; Massa, A. A systematic study on the use of different organocatalytic activation modes for asymmetric conjugated addition reactions of isoindolinones. Tetrahedron 2017, 73, 819. [Google Scholar] [CrossRef]
- Wu, X.; Wang, B.; Zhou, Y.; Li, H. Propargyl Alcohols as One-Carbon Synthons: Redox-Neutral Rhodium(III)-Catalyzed C–H Bond Activation for the Synthesis of Isoindolinones Bearing a Quaternary Carbon. Org. Lett. 2017, 19, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xuan, P.; Lin, J.; Jiao, M. Copper(I)-catalyzed synthesis of 3,3-disubstituted isoindolin-1-ones from enamides via cascade radical addition and cyclization. Tetrahedron Lett. 2018, 59, 3636–3641. [Google Scholar] [CrossRef]
- Li, T.; Zhou, C.; Yan, X.; Wang, J. Solvent-Dependent Asymmetric Synthesis of Alkynyl and Monofluoroalkenyl Isoindolinones by CpRhIII -Catalyzed C-H Activation. Angew. Chem. Int. Ed. 2018, 57, 4048–4052. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guo, J.; Fang, D.; Huang, Y.; Wang, Q.; Bu, Z. A Brønsted Acid-Catalyzed Michael Addition/Cyclization Sequence for the Diastereoselective Assembly of Chroman-Bridged Polycyclic Isoindolinones. Adv. Synth. Catal. 2019, 361, 456–461. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y.; Sukhanova, A.A.; Zlotin, S.G. Stereoselective reactions of nitro compounds in the synthesis of natural compound analogs and active pharmaceutical ingredients. Tetrahedron 2016, 72, 6191–6281. [Google Scholar] [CrossRef]
- Novacek, J.; Waser, M. Syntheses and Applications of (Thio)Urea-Containing Chiral Quaternary Ammonium Salt Catalysts. Eur. J. Org. Chem. 2014, 2014, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Tiffner, M.; Novacek, J.; Busillo, A.; Gratzer, K.; Massa, A.; Waser, M. Design of chiral urea-quaternary ammonium salt hybrid catalysts for asymmetric reactions of glycine Schiff bases. RSC Adv. 2015, 5, 78941–78949. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Wang, C.; Liebich, J.X. Organocatalytic asymmetric aza-Michael additions. Chem. Eur. J. 2009, 15, 11058–11076. [Google Scholar] [CrossRef] [PubMed]
- Engman, M.; Cheruku, P.; Kaukoranta, P.; Bergquist, J.; Völker, S.F.; Andersson, P.G. Highly Selective Iridium-Catalyzed Asymmetric Hydrogenation of Trifluoromethyl Olefins: A New Route to Trifluoromethyl-Bearing Stereocenters. Adv. Synth. Catal. 2009, 351, 375–378. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R (eq) | Solvent | catalyst | Base (eq) | T (°C) | Time(h) | Yield 3 (%) a |
|---|---|---|---|---|---|---|---|
| 1 | Hexyl, (3 eq) | Toluene | Pd(PPh3)4 | Cs2CO3 (5 eq) | 100 | 2.5 | 3a, 20% |
| 2 | Hexyl, (5 eq) | Toluene | Pd(PPh3)4 | Cs2CO3 (5 eq) | 100 | 2.5 | 3a, 42% |
| 3 | Hexyl, (5 eq) | Toluene | Pd(PPh3)4 | Cs2CO3 (5 eq) | 100 | 18 | 3a, 16% |
| 4 | Hexyl, (5 eq) | Toluene | Pd(PPh3)4 | Cs2CO3 (3 eq) | 100 | 2.5 | 3a, 26% |
| 5 | Hexyl, (5 eq) | THF | PdCl2(PPh3)2 | K3PO4 (5 eq) | 60 | 24 | 3a, 42% |
| 6 | Hexyl, (5 eq) | THF | Pd(PPh3)4 | Cs2CO3 (5 eq) | 60 | 24 | 3a, 16% |
| 7 | Methyl, (5 eq) | Toluene | Pd(PPh3)4 | Cs2CO3 (5 eq) | 100 | 24 | 3b, 35% |
| 8 | Ethyl, (5 eq) | Toluene | Pd(PPh3)4 | Cs2CO3 (5 eq) | 100 | 24 | 3c, 36% |

| Entry | Catalyst (mol%) | Base (1 eq) | Time (Days) | Yield (%) a | ee (%) b |
|---|---|---|---|---|---|
| 1 | A (20) | K2CO3 | 3d | 99 | 6 |
| 2 | B (20) | -- | 7d | Traces | -- |
| 3 | C1 (20) | K2CO3 | 3d | 99 | 41 |
| 4 | D (20) | -- | 7d | Traces | -- |
| 5 | E1 (20) | K2CO3 | 7d | Traces | -- |
| 6 | E2 (20) | K2CO3 | 7d | Traces | -- |

| Entry | Catalyst (10 mol%) | Time (Days) | Yield (%) a | ee (%) b |
|---|---|---|---|---|
| 1 c | C1 | 3d | 99 | 41 |
| 2 | C1 | 3d | 99 (42) d | 45 (87) d |
| 3 | C2 | 3d | 95 | 39 |
| 4 | C3 | 3d | 93 | 38 |
| 5 | C4 | 7d | 15 | 20 |
| 6 | C5 | 3d | 95 | 40 |
| 7 | C6 | 7d | 99 | 7 |
| 8 | C7 | 7d | 50 | 39 |
| 9 e | C1 | 3d | 99 | 45 |
| 10 f,g | C1 | 3d | 99 | 43 |

| Entry | X | R | Time (Days) | Yield (8) (%) a | ee (%) b |
|---|---|---|---|---|---|
| 1 | H | Me | 3d | 99 (8a) | 45 |
| 2 | Cl | Me | 2d | 99 (8b) | 30 |
| 3 | Br | Me | 2d | 99 (8c) | 26 |
| 4 |  | Me | 1d | 99 (8d) | 4 |
| 5 | NO2 | Me | 2d | 95 (8e) | 4 |
| 6 | H | Et | 3d | 99 (8f) | 36 |
| 7 | H | Hexyl | 3d | 99 (8g) | 36 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, F.; Di Mola, A.; Palombi, L.; Tiffner, M.; Waser, M.; Massa, A. Synthesis and Organocatalytic Asymmetric Nitro-aldol Initiated Cascade Reactions of 2-Acylbenzonitriles Leading to 3,3-Disubstituted Isoindolinones. Catalysts 2019, 9, 327. https://doi.org/10.3390/catal9040327
Romano F, Di Mola A, Palombi L, Tiffner M, Waser M, Massa A. Synthesis and Organocatalytic Asymmetric Nitro-aldol Initiated Cascade Reactions of 2-Acylbenzonitriles Leading to 3,3-Disubstituted Isoindolinones. Catalysts. 2019; 9(4):327. https://doi.org/10.3390/catal9040327
Chicago/Turabian StyleRomano, Fabio, Antonia Di Mola, Laura Palombi, Maximilian Tiffner, Mario Waser, and Antonio Massa. 2019. "Synthesis and Organocatalytic Asymmetric Nitro-aldol Initiated Cascade Reactions of 2-Acylbenzonitriles Leading to 3,3-Disubstituted Isoindolinones" Catalysts 9, no. 4: 327. https://doi.org/10.3390/catal9040327