Effect of Cerium Precursor in the Synthesis of Ce-MCM-41 and in the Efficiency for Liquid-Phase Oxidation of Benzyl Alcohol


1
Group of New Materials for Sustainable Chemical Catalysis, Institute of Chemistry, Campus Darcy Ribeiro, University of Brasília, Brasília-DF 70910-900, Brazil
2
Laboratory of Materials and Fuels, Institute of Chemistry, Campus Darcy Ribeiro, University of Brasília, Brasília-DF 70910-900, Brazil
*
Authors to whom correspondence should be addressed.
Catalysts 2019, 9(4), 377; https://doi.org/10.3390/catal9040377
Submission received: 14 March 2019
/
Revised: 4 April 2019
/
Accepted: 17 April 2019
/
Published: 23 April 2019
(This article belongs to the Special Issue Catalysis for Global Development. Contributions around the Iberoamerican Federation of Catalysis)
Abstract
:Understanding the effects of synthetic parameters in the catalytic activity of heterogeneous catalysts is of utmost importance when aiming for optimal reaction conditions. Hence, we disclose in this work the synthesis and characterization of cerium-modified MCM-41 materials. In addition, it was observed for the first time, differences in catalytic activity when using different cerium synthetic precursors: CeCl3·7H2O and Ce(NO3)3·6H2O (Ce-MCM-Cl and Ce-MCM-NO3, respectively). A mechanism for cerium incorporation in MCM-41 was proposed, where [Ce(OH)3] species were hydrogen bonded to silicate anions, forming framework Ce-O-Si bonds during condensation and, consequently, causing distortion of the typical hexagonal mesophase. It was also observed that Ce(OH)3 formed aggregated layers with template assemblies during synthesis, resulting in non-framework CeO2 species on the MCM-41 surface after calcination. These CeO2 species were preferentially formed for Ce-MCM-NO3 and were attributed to the nitrate ions’ strong binding to template molecules. In the solvent free liquid-phase oxidation of benzyl alcohol (BzOH), Ce-MCM-Cl achieved better BzOH conversions and benzaldehyde (BzD) yields, while Ce-MCM-NO3 offered increased BzD selectivity. The catalysts’ reusability was also studied over three catalytic runs, where Ce-MCM-NO3 was more resistant than Ce-MCM-Cl towards deactivation. The observed catalytic behavior shows the importance of metal precursors in the obtainment of materials with desirable final properties.
1. Introduction
The need for starting materials in industrial processes makes fine chemistry one of the most relevant sectors in the modern industry, having an expressive economic impact in our society. By definition, fine chemicals are economically relevant products manufactured in a limited content (less than 5000 tons per year) with high added-value associated to its production process and end use [1]. In Brazil, more than 90% of the income from this sector belongs to the production of important starting materials for pharmaceutical, agricultural, and livestock industries [2]. According to data collected by the Brazilian Association of Chemical Industries (ABIQUIM), the cited industries ended 2017 with a net billing of approximately US$25.7 billion altogether, which corresponds to 21.4% of the national chemical industry total income [3]. It is noteworthy that the sustainability of industrial actions must be a major motive to investigate alternative routes for established chemical processes. This concern arises from the main disadvantage of fine chemistry: The generation of about 5 to 50 kg of waste per kg of product due to the use of homogeneous catalysis, stoichiometric reagents, and harsh reaction conditions [4,5].
Benzaldehyde (BzD) is an essential fine chemical material for several industrial sectors, being used as a flavoring agent, as a denaturant, as a fragrance, and as a precursor to the manufacture of important products, such as, cinnamaldehyde, mandelic acid, and malachite green [6]. According to an economic report from Transparent Market Research [7], the global revenue of the benzaldehyde market surpassed US$234 million in 2014 and the demand of approximately 80 Mt is expected to increase 3.06% until 2023. Since the last century, benzaldehyde is produced following two main industrial processes [6,8]: (i) The alkali hydrolysis of benzal chloride; and (ii) the aerobic oxidation of toluene. Both reaction pathways present environmental and procedural drawbacks: High basicity of effluents and a large amount of residues in the alkali hydrolysis; and high pressure and temperature at the expense of low conversions in the aerobic oxidation [6,8]. Thus, the development of safer, more efficient, and environmentally friendly technologies to improve the production of benzaldehyde is of great interest to the fine chemical industry. The liquid-phase oxidation of benzyl alcohol (BzOH) is an interesting alternative due to its intrinsic benefits: (i) Generation of low levels of effluents with little or no negative environmental effects [9]; (ii) use of milder conditions than the cited industrial processes, with temperatures and partial pressures (for oxidants, like molecular oxygen) not higher than 150 °C and 3 atm, respectively [9,10]; and (iii) the protective effect of BzOH towards over-oxidation of benzaldehyde to benzoic acid (HBz), mainly due to the low dissociation energy of the methylene group C-H bond [11]. Hence, the study of this catalytic oxidation process offers good opportunities for the renewal of the knowledge in catalytic oxidative processes, and in the preparation and use of heterogeneous catalysts as well.
The application of numerous heterogeneous catalysts for both gas- and liquid-phase oxidation of BzOH has been reported in the literature [12,13,14,15,16] and microstructural/physicochemical aspects of these materials are decisive in their efficient application. For example, Della Pina and co-workers [12] reported the gas-phase oxidation of BzOH by air using 1 wt.% of gold and/or copper supported on silica as catalysts. The monometallic materials exhibited lower conversion (75% for Au/SiO2) or selectivity (78% for Cu/SiO2) when compared to the bimetallic combination. Among these, the material Au-Cu/SiO2 with an Au/Cu weight ratio = 4 was the best catalyst, showing 98% of BzOH conversion and >99% of selectivity towards BzD at 313 °C. Similarly, Enache and co-workers [13] used Au-Pd/TiO2 catalysts in the liquid-phase oxidation of BzOH under O2 constant pressure. The material containing 2.5 wt.% of each metal showed good performance under mild conditions (pO2 = 0.2 MPa and T = 100 °C) when compared to their monometallic analogs, exhibiting 75% of BzOH conversion and 92% of selectivity towards BzD after 8 h. Even though the good redox activity of metal nanoparticles in the activation of oxygenated species to form peroxo/hydroperoxo intermediates via hydride transfer and dehydrogenative pathways is well known [17], the combination with other metallic nanoparticles seeks the enhancement of the catalyst conversion, selectivity, and durability. Jia and co-workers [14] modified ZSM-5 zeolites with sodium hydroxide to investigate their activity in the liquid-phase oxidation of BzOH with hydrogen peroxide (H2O2/BzOH ratio = 1.3) at reflux conditions. All obtained materials presented a higher Lewis acidity and external surface area after the alkali-treatment. The best catalyst was obtained after 0.5 h of the latter process, showing 53% of BzOH conversion and 86% of selectivity towards BzD. Both Lewis and Brønsted acid-base sites can play important roles in the efficiency of this process by performing a dehydrogenative step in the methylene carbon that allows BzOH oxidation [14,18]. Chen and co-workers [16] synthesized a mesoporous hybrid material based on the interaction between the heteropolyacid H3PW12O40 and N″-(3-amino-2-hydroxypropyl)-N,N,N′,N′-tetramethylguanidinium chloride (TMGHACl), an ionic liquid. The catalyst, [TMGHA]2.4H0.6PW12O40, was tested in the water-mediated liquid-phase oxidation of BzOH with H2O2 to test the reaction viability in a triphasic system. The catalyst gave 98% of BzOH conversion and 94% of selectivity towards BzD at 90 °C after 6 h. The interaction between the heteropolyanion PW12O403− in the catalyst and hydrogen peroxide forms {PO4[WO(O2)2]4}3− species that are active in oxidation reactions [19,20]. Also, the catalyst was efficient in a great variety of pure and mixed solvents due to the polar groups of the ionic liquid, providing a range of triphasic systems that allow better catalyst recovery and benzaldehyde separation.
MCM-41 (mobile composition of matter) is one of the most studied mesoporous silica-based molecular sieves [21]. These materials have desirable properties for heterogeneous catalysis due to: (i) Long-range ordering of cylindrical mesopores in an hexagonal array; (ii) specific surface areas higher than 700 m2 g−1; (iii) tunable pore volumes between 0.7 and 1.2 cm3 g−1; and (iv) pore diameter between 2.5 and 4.5 nm. Such properties allow a high dispersion of catalytic species, a better accessibility to these active sites, and increase the mass diffusion of reactants than those observed for microporous materials [22]. This singular structure is possible because the synthesis procedure employs long-chain alkyl-ammonium surfactants that arrange themselves in rod-like micelles in water [23]. Such supramolecular arrangements act as structural directing agents and define the manner that silicate oligomers will interact with the cationic head group before crystallizing in the final material [24]. Therefore, disturbances in this molecular assembly might lead to microstructural changes in MCM-41 solids, affecting their final properties as well [25,26].
In the context of benzyl alcohol oxidation, studies have shown the potential of MCM-41 as support for active catalysts, including heteropolyacids [27,28], metal complexes [29,30], and metallic nanoparticles [31]. Few reports in the literature have dedicated attention to the catalytic activity of metal-incorporated MCM-41 (i.e., isomorphically substituted) in this subject [32,33], although examples towards the oxidation of other substrates have been demonstrated over the years [34,35]. Specifically, cerium-incorporated MCM-41 (Ce-MCM-41) has been explored towards oxidation reactions with positive results [36,37], mainly because of the active Ce4+/Ce3+ redox pair that allows oxygen mobility in the structure of these materials. This feature refers to the ability of oxide-based Ce4+ materials to generate and relinquish oxygen vacancies, easily generating reactive sites for the activation of oxygenated species that can intermediate redox chemical processes. The insertion of cerium ions in the inorganic framework of MCM-41 could propitiate the formation of distorted coordinative environments not found on bulk materials and enhance the redox activity inherent to the element. So far, only Pal and co-workers [32] has explored this characteristic in the liquid-phase oxidation of BzOH. The material obtained by the researchers, named HSCS-20, presented 28.1 wt.% of the cerium content and showed 53.5% of BzOH conversion and 100% of selectivity towards BzD at room temperature for 24 h using tert-butyl hydroperoxide (TBHP) as the oxidant. Even though this result encourages the investigation of mesoporous cerium-silica catalysts in oxidation reactions, synthetic aspects involving these materials must be taken into account. For example, little has been done to unravel the effect that cerium precursors have in the catalytic activity of Ce-MCM-41 in any oxidation reaction whatsoever.
This work studies the preparation of Ce-MCM-41 materials by using different metal precursors, CeCl3·7H2O and Ce(NO3)3·6H2O (generating the catalysts, Ce-MCM-Cl and Ce-MCM-NO3, respectively). The structural, morphological, and textural differences between the two materials were demonstrated through characterization techniques (XRD, XRF, FTIR, UV-Vis, TG/DTG, DSC, N2 physisorption, solid state NMR, TEM, and Raman) and correlated with disturbances in the assembly of surfactant molecules and the stability of cerium species in the synthesis medium. In addition, the catalytic activity of the materials was evaluated in the liquid-phase oxidation of BzOH with TBHP.
2. Results and Discussion
2.1. Catalysts’ Characterization
The results described below are related to the calcined materials, except for the Raman spectroscopy and thermal analysis also done for the as-synthesized materials. Also, the Ce3+ ions used as a precursor in the synthesis procedure were oxidized to Ce4+ during the two step calcination to remove the template [38] (see Materials and Methods).
2.1.1. X-ray Fluorescence (XRF) and X-ray Diffraction (XRD)
The XRF elemental analysis of cerium-incorporated samples is compiled in Table 1. As can be seen, both materials have a similar cerium content, 5.89 and 5.24 wt.% for Ce-MCM-Cl and Ce-MCM-NO3, respectively. In both cases, the Ce content is lower than the expected value, which led to Si/Ce molar ratios higher than the one used in the synthesis gel (Si/Ce = 25), i.e., Si/Ce = 31 for Ce-MCM-Cl and Si/Ce = 36 for Ce-MCM-NO3. This result is not uncommon since the incorporation of heteroatom ions in silica matrices is strongly dependent on the synthetic procedure and precursors used [39,40,41,42].
Haller and Cesteros [39] studied the effects of several synthesis parameters during the preparation of Al-MCM-41 materials with Si/Al = 24, including different aluminum and silicon sources. Some samples showed large deviations from the desired Si/Al ratio, reaching values between 27.48 and 37.61, an error of 15% and 57%, respectively. Also, not all heteroatom ions are incorporated into the framework of silica materials, even when the silicon-to-metal molar ratio obtained in the solid is the same as the one used in the synthesis gel [43,44]. For example, Matsumoto and co-workers [43] studied Al-MCM-41 with different Si/Al ratios by solid state 27Al MAS-NMR and observed a signal at 53 ppm, assigned to tetrahedrally coordinated framework aluminum, and another at 0 ppm, associated with octahedrally coordinated non-framework aluminum.
The slightly higher Si/Ce ratio obtained for Ce-MCM-NO3, when compared to Ce-MCM-Cl, suggests that cerium(III) precursors with different anions might influence the outcome in the silica-based molecular sieves’ synthesis. Indeed, XRD patterns of the calcined Ce-MCM-41 materials in the low angle region (Figure 1) showed not only differences towards Si-MCM-41, but also among themselves. Si-MCM-41 displayed reflections at 2.30, 3.95, and 4.55°, corresponding to structural planes 100, 110, and 200 of the MCM-41 hexagonal mesophase (planar space group p6m) [45]. For the prepared Ce-MCM-41, the 100 reflection was noticeably shifted towards lower 2θ values and the 110 and 200 reflections overlap with each other, forming a broad signal between 3 and 5°. The first indicates an expansion of the unit cell due to cerium incorporation in the MCM-41 framework, which is expected since the ionic radii of the Ce4+ ions (87 pm for 6-coordination [46]) is higher than Si4+ (40 pm for 6-coordination [46]). The second observation is characteristic of hexagonal disordered mesoporous materials [47] and has been observed in the literature for Ce-MCM-41 [48,49,50,51]. This loss of mesophase ordering can be attributed to the partial substitution of Si4+ for Ce4+ ions due to changes in the Si-O-Si bond angles that lead to the emergence of local structural defects and bond strain [37,47]. However, metal precursor salts in the synthesis gel could cause changes in the assembly of CTA+ molecules, leading to less organized materials when compared to Si-MCM-41. As Barkam and co-workers [52] demonstrated, even the use of different anions for the same metal precursor can extensively affect the physicochemical properties of particles in solution [52].
Since MCM-41 is a molecular sieve with a bidimensional structure, the calculation of the hexagonal unit cell parameter (a) could be determined by using Equation (1) (see Materials and Methods). Unfortunately, the 100 reflection cannot be observed in the XRD pattern for the cerium-incorporated materials (Figure 1), as a consequence of the experimental limitations (i.e., the signal of the beam source was only obtainable starting at 2θ = 2°). As mentioned before, the 110 and 200 reflections could be distinguished, but are overlapped. If the sole 200 reflection could be observed in the modified materials, d100 could be calculated by a relationship between the interplanar spacing of the 200 and 100 reflections (Equation (2), see Materials and Methods). Therefore, deconvolution methods [53] were applied in the overlapped signal to gain information towards the 2θ position of both the 110 and 200 reflections to find d100 and, consequently, the unit cell, a, for the modified materials (see Figure 2).
The 110 and 200 reflections were found in 3.86 and 4.42° for Ce-MCM-Cl and in 3.56 and 4.10° for Ce-MCM-NO3, respectively. The values obtained for d100 and a using the information of the 200 deconvoluted reflections are compiled in Table 1. For Si-MCM-41, the d100 value calculated from the 200 reflection (3.88 nm) was close to the one observed experimentally (3.84 nm), validating our data obtained from Equations (1) and (2). As can be observed in Table 1, the values of a calculated for the cerium materials confirmed the unit cell expansion mentioned before and this effect was more expressive towards Ce-MCM-NO3.
Differences between both cerium-modified materials could also be observed at the high angle region (Figure S1 in Supplementary Material). While the Ce-MCM-Cl pattern only exhibited the usual broad signal around 25° common to amorphous silica in the MCM-41 walls, Ce-MCM-NO3 presented three reflections around 28, 47, and 57° that suggests the formation of CeO2 (reflections 110, 220, and 311 according to JCPDS 34-0394, respectively [54]). The surfacing of non-framework species is generally associated with the synthetic procedure [37,55] and the influence of a particular metal precursor is little discussed in the literature. However, the absence of peaks from segregated CeO2 in the particles of the Ce-MCM-Cl pattern does not mean there are not any; they might be highly dispersed on the MCM-41 surface. Nonetheless, the XRD analysis demonstrated the clear presence of CeO2 in one of the materials.
2.1.2. Spectroscopy Studies (FTIR, UV-Vis, Raman, and 29Si MAS-NMR)
To investigate the origin of non-framework CeO2 species, as well as the consequences of using different cerium precursors, Raman spectroscopy was performed in both as-synthesized and calcined MCM-41 materials. The Raman spectra of as-synthesized materials can give information on how an assembly of surfactants is affected by other ionic species in the synthetic medium. Raman signals of organic molecules are dominant in this situation and the occluded CTA+ cations can be in the same arrangement from the synthetic medium, counter-balancing the negative charges from deprotonated silanol groups. Also, the use of CTA+ as a probe to access information towards microstructural characteristics from the framework of as-synthesized MCM-41 has been little explored in the literature [51,56,57], encouraging the use of conformational-sensible spectroscopic techniques, such as Raman spectroscopy, in the unraveling of microstructural changes.
Figure 3 displays the spectra of the as-synthesized materials and, for means of comparison, the spectra of template molecules with two different molecular orderings were acquired: A 23 wt.% aqueous solution of cetyltrimethylammonium bromide (CTAB) and solid CTAB (Figure S2 in Supplementary Material). The concentration of 23 wt.% was chosen because it is in the range of the concentration where surfactant molecules form an hexagonal liquid-crystal phase in solution [58]. The comparison between the spectra of the as-synthesized materials and the two template standards clearly shows that CTA+ cations are organized as cylindrical micelles inside the pores of as-synthesized materials, as it is expected based on the mechanism for MCM-41 crystallization [59].
The assignment of each vibrational mode observable in the spectra of the as-synthesized materials and template standards is summarized in Table S1 (see Supplementary Material). For the solid CTAB, two of the most important Raman bands that contain information towards molecular configuration are those in 1061 and 1126 cm−1, attributed to C-C symmetric and asymmetric stretching, respectively [57,60,61]. These are characteristic of extended hydrocarbon chains with all-trans ordered structures in which alternate carbon atoms move in the opposite direction. Other important vibrational modes at 2848/2880 and 2941/2958 cm−1 were assigned to the C-H symmetric/asymmetric stretching of CH2 and CH3, respectively. These bands are the strongest in the Raman spectra and highly sensitive to molecular conformation [57,62]. In comparison with solid CTAB, the Raman spectra of CTAB solution and occluded CTA+ in as-synthesized materials showed less and broader signals, as well as other relevant differences. First, a new signal appeared at 1076 cm−1 for both as-synthesized materials and CTAB solution. This signal, which was not observed for solid CTAB, was assigned to the C-C stretching mode of hydrocarbon chains in the gauche configuration and is expected in more disordered systems [57,60]. Second, common signals found for solid CTAB were absent or became overlapped for CTA+ molecules in as-synthesized materials or in solution, especially those at 1175 and 1464 cm−1, associated to the dynamic motion of molecules in supramolecular assemblies [57]. This result indicates that occluded CTA+ molecules in as-synthesized materials act as it were in a liquid-like state [57]. Finally, intensity and shift changes in conformational-sensitive peaks between 2850 and 3000 cm−1 were also observed for CTA+ occluded and in solution. The asymmetric methylene C-H mode around 2885 cm−1 is less intense than in the solid CTAB and the signals assigned to methyl C-H modes were considerably displaced, both evidence of higher statistical chances for attaining the gauche configuration, i.e., a more disordered state [57,61,62]. This was even more noticeable for the broad band around 2920 to 2928 cm−1 for CTA+ occluded and in solution. This band is a composite of two overlapping modes, one from the polymethylene chain (usually around 2920 cm−l) and the other from the methyl group (at 2941 cm−1 for solid CTAB), making it sensitive to conformational disorder of the long methylene chain, since the former contributes more than the latter [61,62].
As mentioned before, changes in the intensity of C-H and C-C stretching modes can reveal effects towards disordering since liquid-like states permit an increase in the number of molecules that can attain the gauche conformation. In the literature, it was demonstrated that the ratio between the intensities of certain long alkyl chain Raman bands can reflect their conformational state [61,62,63]. Therefore, we employed such parameters to understand the influence of different cerium precursors in the assembly of the template molecules. Two of the most employed ratios are those between the C-C gauche stretching around 1080 cm−1 and the C-C asymmetric stretching around 1120 cm−1 [57,62]; and the C-H stretchings around 2920 and 2850 cm−1 [61,63]. Higher values for these parameters are related to an increase of feasibility to attain the gauche conformation and, consequently, disordered the template assembly more in the synthetic medium that was retained in MCM-41 pores. Both ratios for the as-synthesized materials were calculated and are compiled in Table 2.
The I2920/I2850 ratio demonstrates small changes in its value when compared to the I1080/I1120 ratio, which indicates that the latter is a more sensible parameter towards conformational disordering than the former [62]. Nonetheless, the values presented for both ratios allowed the materials to be organized in increasing disorder of template assemblies: A-Si-MCM-41 < A-Ce-MCM-NO3 < A-Ce-MCM-Cl. Thus, it was possible to perceive that: (i) The presence of cerium species altered the supramolecular arrangement of CTA+ cations; (ii) the disordering was more prominent when utilizing cerium(III) chloride as the precursor; and (iii) the degree of disordering obtained for A-Ce-MCM-NO3 was close to A-Si-MCM-41. To explain these observations, it is worth recalling the chemistry of cerium species in high pH aqueous mediums [64,65,66] and the synthetic procedure used in this work to prepare Ce-MCM-41 materials (see Materials and Methods). Under basic conditions, the hydrolysis of Ce(III) generates [Ce(OH)y](3−y) species, where y goes from 0 to 3. During our synthesis, the dropwise addition of cerium(III) precursor turned the clear solution into a dark slurry before it became a light brown/purple solid. This could be addressed by assuming that [Ce(OH)]2+, [Ce(OH)2]+, and [Ce(OH)3] formation occurred progressively and continuously [65]. In addition, the oxidation of Ce3+ to Ce4+ is not thermodynamically favored under the conditions used in this work (i.e., non-aerated solution and pH = 11.2) [64] and [Ce(OH)3] can be regarded as the main cerium compound in solution [67].
Based on the above discussion and the well-known self-assembled systems used for synthesizing MCM-41 [59], this work proposes the following mechanism for cerium incorporation: (i) CTA+ cations are counterbalanced by basic silicate anions; (ii) the latter interacts through hydrogen bonds with [Ce(OH)3] species; and (iii) the gradual condensation under basic conditions leads to silicate oligomers containing Si-O-Si and Ce-O-Si bonds. The higher disorder detected by Raman for occluded CTA+ indicates that cerium incorporation into silica causes different electrostatic interactions between CTA+ cations and the anionic surface. Since both cerium(III) precursors are highly soluble salts with similar stability constants [68], the differences observed between Ce-MCM-Cl and Ce-MCM-NO3 by the techniques used in this work must be related to the different anions. Berr and co-workers [69] studied the effect of anions on alkyltrimethylammonium micelles and observed that micelle aggregation increases in the following order: OH− << Cl− < CH3SO4− < Br− < NO3−. For that reason, nitrate anions induce longer rods of CTA+ cations in solution than chloride [70]. In addition, the molar ratio used in the synthesis of Ce-MCM-41 materials presented a concentration of nitrate or chloride anions close to CTAB, exhibiting 1TEOS:0.04Ce:0.12A:0.152CTAB:2.8NH4OH:141.2H2O, where A = NO3− or Cl−. Since NO3− ions have a higher ionic exchange constant on CTAB micelles than Cl− [71], they are expected to present a stronger binding to CTA+ rods. For Ce-MCM-NO3, nitrate ions bonded to CTA+ could also be hydrogen bonded to [Ce(OH)3]. This layer of concentrated cerium(III) species will aggregate during calcination to form non-framework CeO2 species on the surface of the MCM-41 structure. The higher disordering level observed for Ce-MCM-Cl can be associated to a higher degree of framework Ce-O-Si due to the lower binding between chloride ions and CTA+ molecules. Thus, the physicochemical differences among both cerium modified samples may arise fundamentally from an anion effect in the disturbance of CTA+ molecule assemblies, especially the formation of different cerium species in the calcined materials.
Further effects in the formation of cerium species can be perceived in the calcined materials. Figure 4 displays the Raman spectra of the calcined materials between 650 and 150 cm−1. In general, they all present signals common to the amorphous nature of the silica present in MCM-41. At the studied range, the Si-MCM-41 spectrum presents a signal at 483 cm−1, typical of the stretching mode of Si-O bonds for fourfold cyclosiloxane groups [72,73,74]. For Ce-MCM-Cl, a weak peak appears aside the cyclosiloxane group mode at 460 cm−1 and this signal becomes dominant over the 483 cm−1 mode in the spectrum of Ce-MCM-NO3. This feature is commonly attributed in the literature to the symmetric stretching of Ce-O bonds of the fluorite crystal structure of CeO2 [75] and was assigned as the F2g mode of CeO2. The spectrum of crystalline CeO2 was obtained for means of comparison (Figure S3 in Supplementary Material) and the F2g mode presents itself as a strong signal centered in 463 cm−1. In the case of the modified materials, the presence of this extra signal in both samples indicates the existence of non-framework cerium and that Ce-MCM-NO3 possess a higher content of these species due to a much stronger intensity for this vibrational mode, which is in agreement with the XRD data presented earlier. A similar observation in the F2g mode appearance was made by Zhan and co-workers [48] for Ce-MCM-48 materials, where the mode increased in intensity with the increase of cerium content. However, such a difference between two cerium-modified MCM-41 materials with similar Si/Ce ratios as it is presented in this work has not been observed in the literature so far.
Correlating with the information extracted for the as-synthesized materials, it is possible to ascertain the mechanism of crystallization proposed earlier. Thus, the disordering observed in the assembly of CTA+ molecules of Ce-MCM-Cl was driven by isomorphic substitution. As for Ce-MCM-NO3, part of the cerium added in the synthetic medium does not react with silicate oligomers and precipitates to form non-framework [Ce(OH)3], which converts completely to CeO2 after calcination. Such a hypothesis is scarcely pointed out to explain the difference in the reactivity of cerium precursors in the synthesis of oxides, but there is an extensive literature regarding the differences in the use of precursors that leads to singular microstructural properties of these materials [52,76,77]. These works clearly suggest that the anions in cerium precursors have major roles in defining the properties of the final cerium-based materials more than just the mere electrostatic counterparts in the synthetic medium.
Figure 5 displays the Fourier transform infrared spectroscopy (FTIR) spectra for all calcined MCM-41 materials.
The vibrational modes between 1400 and 400 cm−1 are typical of Si-O-Si bonds [78]. In the case of Si-MCM-41: (i) Asymmetric stretching of Si-O-Si bonds at 1232 and 1084 cm−1; (ii) symmetric stretching of Si-OH bonds at 964 cm−1; (iii) symmetric stretching of Si-O-Si at 802 cm−1; and (iv) the Si-O-Si bending mode at 459 cm−1. Changes in the wavenumber of these modes are frequently associated to isomorphic substitution of heteroelements in the framework of silica-based materials, because the average length of T-O bonds (T corresponds to any heteroatom) in the walls of the MCM-41 is increased with the insertion of a larger atom than silicon [37,49,51]. However, the shifts observed for the cerium-incorporated materials were too small to make any assumption of isomorphic substitution. In addition, no vibrational modes attributed to Ce-O bonds, between 1600 and 900 cm−1 and below 700 cm−1, were evidenced. These observations (i.e., small shifts and absence of Ce-O bonds) could be respectively attributed to the low concentration of framework Ce4+ ions and non-framework CeO2 when compared to the amount of silica in MCM-41.
Diffuse reflectance UV-Vis (DRS UV-Vis) spectra for the calcined materials are displayed in Figure 6A. When compared to Si-MCM-41, cerium-incorporated materials presented stronger absorption in the ultraviolet range and a red shift of the absorption edge due to O2−→Ce4+ charge transfer [32]. These experimental observations are proof of cerium incorporation in the materials, as already described by Pal and co-workers [32]. Moreover, the greater intensity of the Ce-MCM-Cl spectrum can be due to its slightly higher cerium content (see Table 1).
When compared to a standard sample of CeO2 (Figure S4 in Supplementary Material), it is possible to gain information towards the coordination environment of the heteroatom, since the charge transfer transition is symmetry-sensible [37,51,79]. The CeO2 spectrum presented more intense bands at 380 and 250 nm, owing to Ce4+ ions in octahedral/polymeric and tetrahedral coordination environments, respectively. The octahedral/polymeric environment is the main coordination type for cerium in the CeO2 fluorite crystal structure. Its non-stoichiometric lattice [80] gives the dioxide a high oxygen mobility and absorptions near or above 380 nm is strong evidence of non-framework species. The spectra of cerium-incorporated materials in Figure 6A showed that UV-absorption finishes around 310 nm for Ce-MCM-Cl with a steep absorption edge until 450 nm, while for Ce-MCM-NO3, it ends at 355 nm with a less steep edge until 500 nm. These results are evidence that the heteroatoms in Ce-MCM-Cl have more tetrahedral coordination sites than Ce-MCM-NO3, which absorbs more radiation in the visible range of the spectrum due to non-framework species [37,51,79].
The different positions found for the absorption edge in Ce-MCM-41 samples can be used to obtain important information towards the heteroatom by determining the optical band gap. The latter was calculated by Equation (3) (see Materials and Methods) using n = ½, due to direct allowed transitions, and Davis-Mott plots with suitable linear ranges were obtained (R2 > 0.99). CeO2 has an optical band gap of 3.02 eV, which is near to the values described in the literature for the bulk material [81]. Ce-MCM-Cl and Ce-MCM-NO3 showed band gap values of 3.50 and 3.33 eV, respectively, which were lower than Si-MCM-41 (4.97 eV). As studied by Casas-Orozco and co-workers [73], higher band gap values of metal-incorporated MCM-41 than the metal oxide counterpart indicates high dispersion of the heteroatom in the siliceous framework. Also, the lower band gap value observed for Ce-MCM-NO3 could be associated with the higher content of non-framework species identified by XRD and Raman, resulting in less dispersed CeO2 particles.
29Si MAS-NMR spectra of the calcined materials are available in Figure 7. As can be seen, the signals obtained for all samples were broad and overlapped in the range between −80 and −120 ppm. This interval is typical for the Qn-groups with a general formula of Si(OSi)n(OH)4−n [82]: (i) Geminal silanols or Q2 groups (resonating around −90 ppm); (ii) terminal silanols or Q3 groups (resonating around −100 ppm); and (iii) siloxane or Q4 groups (resonating around −110 ppm). The overlapping effect is typical in systems based on amorphous silica due to the wide distribution of Si-O-Si bond angles and Si-O bond lengths [82]. It was possible to perceive that, in the case of Ce-MCM-Cl, there was a slight diminishment in the signal of Q3 groups, indicating a decrease in its content. This type of change was even more noticeable for Ce-MCM-NO3, where all defect-related environments (Q2 and Q3) were significantly reduced when compared to Si-MCM-41. Such qualitative observations indicate that the cerium incorporation is modifying the condensation of Si-O-Si bonds in the structure of these materials. To access more information towards the latter, deconvolution methods were applied and are denoted in Figure 7.
No additional peaks to those mentioned earlier were found after applying the deconvolution methods. Therefore, the traditional parameters associated to the condensation of Si-O-Si bonds were applied to further analyze the changes perceived for cerium modified materials. Among them, the percentage of each siliceous chemical environment was calculated based on the area of each peak and they were used to determine Q4/(Q2 + Q3) ratios and the molar percentage of silanol groups (Si molar%) using Equation (4) (see Materials and Methods). Higher values for the mentioned ratio and lower ones for Si molar% indicates more condensed Si-O-Si bonds in the inorganic framework. These quantitative data were compiled in Table 3.
From the values displayed in Table 3, it was possible to infer that: (i) For Ce-MCM-Cl, even though the content of terminal silanols (Q3) decreases, the siloxane group (Q4) does not increase significantly when compared to geminal silanols (Q2), suggesting that even if the condensation of Si-O-Si bonds slightly increases, the percentage of silanol groups (Si molar%) remains relatively constant if Q2 increases; and (ii) for Ce-MCM-NO3, the modification decreases drastically the content of silanol groups, indicating that the non-framework cerium species might be located in the mesopores’ wall or external surface, but Raman data suggests the former. Once again, there is little contribution from the literature to explain effectively the reason for differences in 29Si MAS NMR arising solely with varying the counterion in cerium precursor. Even though this is a scarce topic, it is well known that such effects in siliceous molecular sieves are dependent more in the synthetic procedure than in any other aspect and the fact that cerium incorporation can lead to a more or less condensed degree of chemical bonds has already been reported [32,51]. Thus, the MAS-NMR data is in good agreement with the observations made earlier due to the crystallization mechanism proposed: (i) The smaller aggregation effect of chloride anions on CTA+ assemblies and the formation of framework Ce-O-Si bonds led to the emergence of silanol defects (Q2); and (ii) the precipitation of Ce(OH)3 and conversion to non-framework CeO2 resulted in a coating effect in the MCM-41 surface, leading to the decrease of Q2.
2.1.3. Morphological (TEM and HRTEM), Textural (Surface Area and Pore Volume), and Thermal Analyses (TG/DTG/DSC)
The morphology of the calcined MCM-41 materials was accessed through low-resolution transmission electron microscopy (TEM). Representative images are selected and displayed in Figure 8.
For Si-MCM-41, perfectly spherical nanoparticles were obtained (mean size of 61.8 ± 15.9 nm). There is little aggregation between the particles, possibly a synthetic consequence of the rapid formation of ethanol as a side product during TEOS hydrolysis [83]. On the other hand, the modified materials present smaller and spherical nanoparticles, with a mean size of around 34.2 ± 14.5 nm and 38.7 ± 9.1 nm for Ce-MCM-Cl and Ce-MCM-NO3, respectively. Also, the nanoparticles have a more aggregated aspect when compared with Si-MCM-41, exhibiting more irregular shapes. These observations are possibly an evidence of a faster nucleation process, a consequence of the presence of cerium precursors in the synthetic medium. This supports the loss of long-range ordering observed previously by XRD, as well as the disordering effect of the supramolecular assembly of CTA+ cations evidenced by Raman spectroscopy.
To further study the modified materials, high-resolution transmission electron microscopy (HRTEM) was used to access the formation of CeO2 species on Ce-MCM-41 materials and representative images are displayed in Figure 9. As one can see, some of the irregular entities observed in TEM for both materials showed dark nanorods on their surface that were not observable on Si-MCM-41. High magnifications of these nanorods (Figure S5 in Supplementary Material) displayed lattices fringes around 0.33 nm, consistent with the interplanar distance characteristic to 110 XRD reflection of CeO2, thus confirming their identity. For Ce-MCM-Cl, these nanorods displayed lengths between 22.0 and 70.0 nm and widths between 1.75 and 2.50 nm, while for Ce-MCM-NO3, these entities were bigger and thicker, with lengths between 100 and 250 nm and widths between 5.0 and 6.7 nm. The differences observed in their measures were consistent with the crystallization mechanism proposed for non-framework cerium species, where longer and thicker rods were expected for Ce-MCM-NO3 due to the aggregation effect of micelles attributed to the nitrate anion. Also, the formation of these nanorods was consistent with the mechanism proposed for non-framework cerium species. The formation of ceria nanorods is widely studied in the literature [84,85] and includes surfactant-assisted synthetic procedures using CTAB. However, these methodologies are normally performed under hydrothermal conditions with high temperatures (80–160 °C), longer time periods (8–96 h), and low Ce/CTAB molar ratios (0.33–0.66) [84,85]. The results observed in this work suggest that nanorods can also form under milder conditions (30–35 °C and 4 h) and high Ce/CTAB molar ratios (3.8).
The textural characterization of the calcined MCM-41 materials was performed by N2 physisorption using t-plot curves obtained from analytical functions for each portion of the P/P0 domain (see Equations (5)–(8) in Materials and Methods) [86]. The initial part of the curve was used to calculate the total specific surface area (Stotal) of the materials using the slope of the linear fit going through the origin (red line in Figure 10a). On the other hand, the final part of the curve was used to calculate the external specific surface area (Sext), indicated in Figure 10a by the blue line.
Figure 10b exhibits the N2 adsorption/desorption isotherms obtained for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO3, respectively. According to the IUPAC classification [87], all curves were type IV isotherms, presenting a characteristic hysteresis loop associated with capillary condensation in mesopores. This hysteresis was type H1 according to the IUPAC classification [87] and could be associated with a narrow and uniform pore size distribution. This result implies that the MCM-41 pore system in cerium-incorporated materials was preserved and that the long-range disorder was responsible for the loss of the mesophase observed by XRD. This is not uncommon and even in highly disordered materials, such as titanosilicate ETS-10, short-range ordering could not be affected by long-range disordering [88].
The initial part of the isotherm, before capillary condensation, is associated to monolayer-multilayer adsorption on both mesopores and external surface, and consequently, was used to calculate Stotal, as described above [86]. Similarly, in the final part of the isotherm, after capillary condensation, adsorption occurs only on the external surface and, for that reason, was used to determine Sext. The capillary condensation is identified by an increase of adsorbed N2 at the relative pressure range of P/P0 = 0.30 to 0.45, due to the filling of mesopores.
The data obtained by N2 physisorption experiments (Table 4) showed a reduction of Stotal, mesoporous surface area (Smeso), and mesopore volume (Vmeso) for the cerium-incorporated samples, which could be attributed to the loss of long-range order. The sharp increase observed in all isotherms at the relative pressure range of P/P0 = 0.9 to 1.0 is related to the presence of interparticle porosity [32] and its effect can be observed in a higher value of the total pore volume (Vtotal) than Vmeso. For that reason, the average pore diameter (PD) was calculated using the penultimate point from the adsorption isotherm (see Materials and Methods) to avoid contributions from interparticle porosities, since the materials showed different particle sizes. The values obtained for PD were almost identical, which is in agreement with the narrow and uniform pore size distribution expected for the type H1 hysteresis loop observed for all materials and discussed above. However, the sample of Ce-MCM-NO3 showed a small increase that could be attributed to its different mechanism of pore formation, possibly due to the coating effect of non-framework CeO2. Additionally, PD values were used to determine the wall thickness (WT) of MCM-41 materials, evidencing that cerium incorporation affects this parameter due to framework cerium and non-framework CeO2. In the case of Ce-MCM-Cl, the increase in WT was consistent with the incorporation of Ce in the inorganic framework, causing an expansion of the unit cell (a) and, ultimately, of the pore walls. However, in the case of Ce-MCM-NO3, not only isomorphic substitution could cause such an increase, but also the existence of non-framework CeO2 and the higher value of WT corroborates the possible coating effect mentioned previously, which can make the pore walls appear thicker. These results are in accordance with the higher value of a found for Ce-MCM-NO3 (4.97 nm) when compared to Ce-MCM-Cl (4.61 nm), explaining the cell expansion observed by XRD. Finally, the higher Sext value obtained for cerium-incorporated materials are linked to their smaller particle size when compared to Si-MCM-41, as observed by TEM.
Thermal analyses were performed for the as-synthesized and calcined MCM-41 materials to further analyze the consequences of texture changes towards microstructural aspects. The thermogravimetry (TG), differential thermogravimetry (DTG), and differential scanning calorimetry (DSC) curves for the as-synthesized materials are displayed in Figure 11, Figure 12 and Figure 13. The values of mass loss, temperature range, and DTG maximum were collected in Table S2 (see Supplementary Material).
For A-Si-MCM-41, five thermal events were observed and are consistent with previously reported events for similar as-synthesized MCM-41 materials [89]: (i) Evaporation of physically adsorbed water (endothermic, from room temperature to 129 °C); (ii) initial decomposition of occluded CTA+ molecules through Hofmann elimination (endothermic, from 129 to 270 °C), generating triethylamine and alkenes; (iii) further fragmentation of carbon chains that resisted Hofmann elimination (exothermic; from 270 to 322 °C); (iv) oxidation of reminiscent organic residues from previous thermal events, like coke (exothermic; from 322 to 482 °C); and (v) condensation of silanol groups (with almost undetectable DTG and DSC maximums, from 482 to 1000 °C).
The comparison between the modified as-synthesized materials and A-Si-MCM-41 showed differences in the DTG and DSC curves, even though the TG curves display similar profiles and CTA+/SiO2 ratios. The different thermal behavior for both cerium incorporated materials, especially after the Hofmann elimination, revealed that: (i) In the temperature range associated to CTA+ decomposition, one extra endothermic event occurs at 304 °C for A-Ce-MCM-Cl and 309 °C for A-Ce-MCM-NO3; (ii) these extra events were overlapped with the first oxidation process, now shifted in the range of 315 to 360 °C; (iii) increase in the temperature of the DTG maximum for the final oxidation process, occurring around 70 °C higher than for A-Si-MCM-41; and (iv) increase in mass loss for the final oxidation processes, 5.59% for A-Si-MCM-41 and above 8.50% for both A-Ce-MCM-41 samples. Overall, this evidence demonstrates clearly that template molecules in the modified materials are thermally more resistant than in the as-synthesized Si-MCM-41. The extra thermal event might be due to an extra Hofmann elimination of molecules that survived the first decomposition process, delaying both exothermic events. Such phenomena can be either related to an increase of the interaction stability between CTA+ and the inorganic framework or to diffusion limitations of products from thermal processes, since there was a decrease in Vmeso after cerium incorporation. Similar observations have been made by Khalil [55] and de Souza and co-workers [90], especially as further evidence of isomorphic substitution of heteroelements in the inorganic framework.
The TG curves of the calcined MCM-41 materials (Figure 14) and the mass loss associated to all processes (Table S3) are displayed in the Supplementary Material. These curves provided significant information to differentiate the materials among themselves, where a difference in total mass loss from room temperature to 1000 °C was observed in all solids. The two thermal events were associated to the evaporation of adsorbed water (endothermic event from room temperature to temperatures around 115 to 150 °C) and the condensation of silanol groups (water evaporation until 1000 °C). The major mass loss was associated to the initial evaporation of water: 4.58%, 14.23%, and 12.29% for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO3, respectively. Also, this event ends in higher temperatures for the cerium-modified samples than for Si-MCM-41 (usually above 130 °C). Altogether, these observations indicate that cerium incorporation increases the surface polarity of the materials, somewhat expected since cerium is less electronegative than silicon. Also, the mass loss from silanol condensation was different for the three materials: 1.70%, 3.50%, and 2.67% for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO3, respectively. Thus, it is noticeable that the isomorphic substitution leads to less thermally stable materials, highlighting the fact that Ce-MCM-Cl is less stable than Ce-MCM-NO3, evidence of its higher degree of silicon substitution in the framework. This is expected once the insertion of heteroelements causes bond angle tension and loss of the crystalline mesophase as indicated by XRD data, which is in accordance with the disordering effect proposed for the crystallization of the materials and corroborated previously with other characterization techniques.
2.2. Catalytic Tests
The liquid-phase oxidation of benzyl alcohol (BzOH) was used to further analyze the consequences of cerium precursors in the generation of catalytic active species. In all reaction analyses, the only products detected were benzaldehyde (BzD) and benzoic acid (HBz), both oxidation products. The other possible product, benzyl benzoate, was not observed. The reaction parameters were varied to achieve an optimal condition (see Materials and Methods for reaction details), where the maximum BzD yield was used as a reference. The results of all catalytic essays were compiled in Table 5 and Table 6 for Ce-MCM-Cl and Ce-MCM-NO3, respectively.
The first parameter studied was the type of oxidant: Hydrogen peroxide (H2O2, 30 wt.%); tert-butyl hydroperoxide (TBHP, 70 wt.%); and di-tert-butyl peroxide (DTBP), runs 1 to 3. As can be perceived in Table 5 and Table 6, the only peroxide that produced significant values of BzOH conversion (C, %) and BzD yield (Y, %) was TBHP. Other studies involving the use of cerium-modified molecular sieves in different oxidation reactions suggest the use of H2O2 as a milder oxidant [91,92]. However, the catalysts’ behavior in this work were consistent with the reactive conditions reported by Pal and co-workers [32], which performed liquid-phase BzOH oxidation with cerium-modified mesoporous materials. The values of Y and TON with TBHP as the oxidant were different for both cerium materials: 22.0% and 74.0 mol molcat−1 for Ce-MCM-Cl; 17.5% and 49.5 mol molcat−1 for Ce-MCM-NO3, respectively. Thus, in these conditions, Ce-MCM-Cl presented better Y and was more active than Ce-MCM-NO3. This result was consistent with previous literature indicating that non-framework CeO2 species (observed mostly in Ce-MCM-NO3) are less active and selective as catalytic sites [48,51].
To further optimize the system, the use of solvents was tested to observe if the changes in hydrophobicity/hydrophilicity of the medium can affect positively the reaction towards better BzD yields. Acetonitrile (CH3CN) and isopropanol (i-PrOH) were selected to observe this influence (runs 4 and 5, respectively). For both cerium materials, the use of CH3CN and i-PrOH resulted in less efficient conditions towards BzD yield and catalyst activity, even though BzD selectivity improved significantly (up to 99%). The lowering of C was the main issue, and this parameter was more affected using i-PrOH, possibly due to adsorptive competition of the protic solvent molecules [93].
The next parameter studied was the BzOH:TBHP molar ratio, since reactions so far were done in stoichiometric quantities. Changes in the chemical equilibrium can possibly affect the catalytic output and two conditions were tested: Excess of alcohol (2:1, run 6) and excess of TBHP (1:2, run 7). For Ce-MCM-Cl and Ce-MCM-NO3, excess of alcohol decreased C, 25.8% and 19.7%, and TON, 56.8 and 48.8 mol molcat−1, respectively. Even though there was a better peroxide efficiency in the molar ratio of 2:1, lower Y values were obtained (19.4% and 15.9% for Ce-MCM-Cl and Ce-MCM-NO3). With the excess of peroxide, the main improvement was in the catalytic efficiency (TON), exhibiting 76.1 mol molcat−1 for Ce-MCM-Cl and 68.4 mol molcat−1 for Ce-MCM-NO3. However, the little improvements observed in Y for both materials do not justify such an increase in the oxidant amount. Thus, the following essays were done in stoichiometric proportions of alcohol:oxidant.
Next, the effect of temperature was studied, increasing it from 70 to 80 and 90 °C (runs 8 and 9). With increasing temperature, significant enhancements in C were obtained (up to 60.2% and 52.5% for Ce-MCM-Cl and Ce-MCM-NO3, respectively), but with concomitant decreases in S (close to 41% for both materials at 90 °C). In addition, improvements in Y and TON were significant, especially at 80 °C (26.4% and 119.4 mol molcat−1 for Ce-MCM-Cl, and 21.7% and 95.1 mol molcat−1 for Ce-MCM-NO3). At higher temperatures, a better activation of oxidants and generation of reactive species were obtained, but it was only beneficial until a certain point, since a loss of selectivity was visible at 90 °C for both materials when compared to 80 °C. Therefore, the following essays were performed at 80 °C.
The final parameter evaluated was the catalyst amount, 5 and 15 wt.%, runs 10 and 11, respectively. In the case of Ce-MCM-Cl, both changes led to less efficient catalytic situations in terms of C and Y, where decreases of up to 20.0% for the latter and around 40.0% of the former were perceived when compared to run 8 (10 wt.%). This can be associated to a more exacerbated decomposition of TBHP in the case of 15 wt.% and a less efficient activation of the oxidant at 5 wt.%, achieving the optimal mass and active sites’ amount around 10 wt.% for this catalyst. Surprisingly, a different trend was observed for Ce-MCM-NO3, where the catalyst amount of 5 wt.% showed a negligible decrease of C and a considerable increase of S (33.7% and 64.3%, respectively). This combination resulted in the same Y and better catalytic performance (TON = 160.3 mol molcat−1) than from run 8 (10 wt.%), while the use of 15 wt.% catalyst increased C to 63.7% with concomitant decrease in S to 31.9%, resulting in a decrease in Y (20.3%) and catalytic performance (TON = 103.5 mol molcat−1). This can be associated to the mass transfer in Ce-MCM-NO3 mesopores, while 5 wt.% offered an optimal condition of catalytic site usage and diffusion of reactants and products; 10 or 15 wt.% seems to lead to conditions where over-oxidation was favored due to an excess of active sites. These results are consistent with the fact that mainly non-framework CeO2 (less selective) is present in the material’s constitution as mentioned before. Thus, the amount of catalyst plays a significant role in attaining better Y, but also in differentiating the applicability of both active materials in this study. Also, for comparison means, Si-MCM-41 was tested in the optimal conditions found for Ce-MCM-Cl and Ce-MCM-NO3 (run 12), and negligible values of C and Y (less than 1%) were obtained, proving that the activity of MCM-41 materials is due to the presence of cerium species.
In order to evaluate the reusability of the catalysts, the solids from the optimized conditions were recovered and studied for further cycles, see Materials and Methods. After each cycle, the cerium content and coordination state were analyzed by XRF and Raman to understand possible changes in the catalytic performance. The recycled results are compiled in Figure 15.
As can be perceived from Figure 15, BzD Y of both catalysts maintains a stable state after three catalytic cycles, but the conversion of both materials decreases. For Ce-MCM-Cl, the conversion decreases from 54.3% to 43.9% in the second cycle and 39.3% in the third cycle. Meanwhile, the conversion of Ce-MCM-NO3 decreases from 33.4% to 27.3% in the second cycle and 25.0% in the third cycle. Also, according to XRF analyses of the recovered solids, the amount of cerium in Ce-MCM-Cl diminishes from 5.89% to 5.77% in the last cycle, while for Ce-MCM-NO3, the diminishment goes from 5.24% to 5.16%. Thus, it is possible to conclude from the previous data that: (i) Ce-MCM-Cl deactivates in a higher rate than Ce-MCM-NO3; and (ii) the main reason for deactivation can be associated to microstructural changes in the recovered materials, since cerium leaching was negligible.
According to Timofeeva and coworkers [92], changes in the Ce coordination environment can occur after oxidation processes due to etching effects of peroxides in the surface of cerium-incorporated silica materials, where framework sites (more active) can turn to non-framework species (less active). In our study, the Raman spectra of the recovered materials (Figure 16) proved such a point, where an increase in the F2g mode intensity of CeO2 after each cycle confirmed the change in the cerium coordination state towards non-framework species. It was also noticeable in the Ce-MCM-Cl spectrum that the intensity of this band doubles itself after the second cycle, while for Ce-MCM-NO3, it happens to a lesser degree. Such an observation is consistent with the steadier loss in catalytic performance observed in Ce-MCM-NO3, indicating that this catalyst is more resistant towards deactivation. This effect is possibly a consequence of the CeO2 coating mentioned earlier in this work, since pore walls were thicker for Ce-MCM-NO3 (3.64 nm) than for Ce-MCM-Cl (3.43 nm), making the former less susceptible towards possible etching effects from TBHP. The protective effect described is not usually highlighted in the literature for metal-incorporated MCM-41 materials and shows how relevant the choice of metal precursors is to attain not only active materials, but more stable ones towards unfavorable processes inherent of catalytic applications.
Overall, the best conditions for the BzOH liquid-phase oxidation were the ones in run 8 for Ce-MCM-Cl (TBHP, molar ratio of 1:1, 10 wt.% of catalyst, and 80 °C), while the conditions of run 10 were best for Ce-MCM-NO3 (TBHP, molar ratio of 1:1, 5 wt.% of catalyst, and 80 °C). In situations where better BzOH conversions, BzD yields, and heteroelement incorporation are aimed for, cerium(III) chloride precursor affords the ideal material. In situations where BzD selectivity and catalytic endurance is the objective, cerium(III) nitrate precursor might be the best choice of synthetic precursor.
3. Materials and Methods
3.1. Materials
Cetyltrimethylammonium bromide (CTAB, Sigma-Aldrich, >99.9%, Darmstadt, Germany), ammonium hydroxide (NH4OH, Sigma-Aldrich, 28–30 wt.% aqueous solution, Darmstadt, Germany), ethyl orthosilicate (TEOS, Sigma-Aldrich, 98%, Darmstadt, Germany), silver nitrate (AgNO3, Honeywell Fluka, ≥99.5%, Seelze, Germany), cerium chloride heptahydrate (CeCl3·7H2O, Sigma-Aldrich, >98%, Darmstadt, Germany), cerium nitrate hexahydrate (Ce(NO3)3·6H2O, Vetec, 99%, Duque de Caxias, RJ, Brazil), benzyl alcohol (BzOH, Tedia®, >99%, Fairfield, OH, USA), tert-butyl hydroperoxide (TBHP, Sigma-Aldrich, 70 wt.% aqueous solution, Darmstadt, Germany), hydrogen peroxide (H2O2, Vetec, 30 wt.% aqueous solution, Duque de Caxias, RJ, Brazil), di-tert-butyl peroxide (DTBP, Sigma-Aldrich, 98%, Darmstadt, Germany), deuterated chloroform (CDCl3, Sigma-Aldrich, ≥ 99.8% D atom and 0.03% v/v of tetramethylsilane, TMS, Darmstadt, Germany), acetonitrile (CH3CN, J.T.Baker, 99.99%, Phillipsburg, NJ, USA), and isopropanol (i-PrOH, Cromoline, 99.5%, Diadema, SP, Brazil) were used as supplied.
3.2. Synthesis of MCM-41 Materials
The catalysts were synthesized following an adaptation of the precipitation method proposed by Grün and co-workers [94]. In the synthesis of Si-MCM-41, 55 mL of deionized water and 1.24 g of CTAB were added in a round bottom flask. The mixture was stirred at 400 rpm and heated at 30 to 35 °C until homogenization. Then, 4.1 mL of concentrated NH4OH and 5 mL of TEOS were added, the latter being dropwise with a Pasteur pipette. The synthetic mixture contained the following molar ratio of the reactants: 1TEOS:0.152CTAB:2.8NH4OH:141.2H2O. After 4 h of reaction, the resulting white solid was filtered and washed with distilled water until it was free of halide anions (tested with a 1 mol L−1 aqueous solution of AgNO3). The material was dried at room temperature for 3 days and then calcined in two steps: 300 °C for 3 h and 550 °C for 3 h. Ce-MCM-41 samples were prepared by adding, simultaneously, 5 mL of an aqueous solution of CeCl3·7H2O or Ce(NO3)3·6H2O and TEOS in a dropwise manner. The synthetic mixture contained a Si/Ce molar ratio = 25, generating the materials of Ce-MCM-Cl and Ce-MCM-NO3, respectively. The resulting light brown/purple solids were filtered, washed, dried, and calcined under the same conditions as Si-MCM-41. After the calcination procedure, both cerium-incorporated materials showed a light-yellow appearance. The as-synthesized materials (obtained after the drying step) were designated as A-Si-MCM-41, A-Ce-MCM-Cl, and A-Ce-MCM-NO3.
3.3. Characterization of the Catalysts
X-ray fluorescence (XRF) analyses were made under vacuum on a Shimadzu EDX-720 (Tokyo, Japan) equipped with a rhodium tube as the X-ray source.
X-ray diffraction (XRD) patterns were obtained on a Rigaku Ultima IV diffractometer (Tokyo, Japan) with CuKα radiation (λ = 0.15409 nm) at 1° min−1, 2θ range between 2 and 80°, and a step of 0.01°. For peak deconvolution, Gaussian functions were fitted with an exponential decay baseline using the Peak Analyzer tool in OriginPro 2019. The calculation of the cell lattice parameter (a) was made using crystallographic knowledge of hexagonal crystal lattices and Bragg’s law [95] according to Equations (1) and (2):
where d100 and d200 are interplanar spaces of the 100 and 200 reflections, respectively.
a = (2/31/2)d100
d100 = 2d200
Fourier transform infrared spectroscopy (FTIR) spectra were obtained on a Varian 640-IR (Palo Alto, CA, USA) using a 4 cm−1 resolution, 32 scans, and 1 wt.% of sample in KBr pellets.
The Raman spectroscopy measurements were performed on an In-ViaTM Raman system from Renishaw (Wotton-under-Edge, Glos., England) equipped with a thermoelectric cooled CCD detector and a 1200 lines/groove diffraction grating. A Leica microscope (Wetzlar, Germany) with a 50× objective lens was used to focus the radiation of the excitation laser on the sample and to collect the scattered photons. The wavenumbers were calibrated using the Raman phonon of a silicon wafer at 520.5 cm−1. Each spectrum was the accumulation of 10 scans with a 20 s integration time at 632.8 nm (HeNe laser). All Raman spectra were recorded at a laser power of 20 mW and the nominal spectra resolution was 6 cm−1. A 1 mm optical path quartz cuvette was used to perform Raman measurements of the solutions. All Raman measurements were performed at room temperature.
Diffuse reflectance UV-Vis spectroscopy (UV-Vis-DRS) spectra were obtained on a Varian Cary 5000 UV-Vis-NIR spectrophotometer (Palo Alto, CA, USA) equipped with a Harrick diffuse reflectance accessory and using BaSO4 as a background standard. The optical band gap was calculated using the Davis-Mott method [96], in which a plot is done according to Equation (3). After obtaining the Davis-Mott plot, an extrapolation to zero of the linear portion where the curvature of the graph first changes gives the optical band gap value:
where α is the absorption coefficient, replaced in this work by the Kubelka-Munk function, F(R); Eg is the optical band gap; hυ is energy; A is a proportionality constant; and 1/n is a coefficient dependent on the type of transition responsible for the absorption edge observed.
[αhυ]1/n = A(Eg − hυ)
Nuclear magnetic resonance (NMR) experiments were conducted in the solid state at 14 T with a Bruker AscendTM 600 (Kontich, AN, Belgium) equipped with a 4 mm magic-angle spinning (MAS) probe and zirconia rotors. 29Si MAS-NMR spectra (119.237 MHz) were obtained with spinning rate at 10 kHz, pulse length of 4.25 μs (π/2), 512 scans, and a recycle delay of 20 s. The spectra were referenced to TMS (δ = 0.0 ppm). The molar percentage of silanol groups (Si molar%) was calculated using Equation (4) [97]:
where Q2, Q3, and Q4 correspond to the relative proportions in the percentage of geminal silanols, terminal silanols, and siloxane groups, respectively. Peak areas were obtained after deconvolution by fitting the signals to Voigt peaks in the range between −110 and −90 ppm.
Si molar% = [(2Q2 + Q3)/(Q2 + Q3 + Q4)]100
Thermogravimetry (TG), derivative thermogravimetry (DTG), and differential scanning calorimetry (DSC) curves were obtained on a TA Instruments SDT Q600 (New Castle, DE, USA) from room temperature to 1000 °C (20 °C min−1), using synthetic air (99.999%) as the purge gas (flow rate of 60 mL min−1). The analysis was performed on alumina crucibles and the external standard employed for calibration was sapphire.
N2 physisorption measurements were performed at −196 °C on a Quantachrome Instruments NOVA 4200e apparatus (Boynton Beach, FL, USA). All samples were outgassed at 200 °C under vacuum for 24 h prior to the analysis. Surface area and porosity data were obtained from N2 adsorption/desorption isotherms. Total specific surface area (Stotal), external specific surface area (Sext), and mesopore volume (Vmeso) were determined using a t-plot curve obtained from analytical functions for each portion of the P/P0 domain (see Equations (5)–(8) and Figure 10a) [86]. Mesoporous surface area (Smeso) was calculated by subtracting Sext from Stotal. Total pore volume (Vtotal) was calculated using the penultimate point from the adsorption isotherm. Average pore diameter (PD) was calculated according to Equation (9). Wall thickness (WT) was calculated according to Equation (10):
where A = 0.5675647, a = 0.199735, B = 0.4116168, b = 2.00834, C = 0.1423566, D = 0.1078, c = 0.4888, E = 0.08309076, F = 0.02995, e = 0.369, G = 1.268066, g = 1.931, H = 0.76934, and h = 51.09.
t (nm) = A(P/P0)a + B(P/P0)b for 0.03 ≤ P/P0 < 0.25
t (nm) = {C/[D − log(P/P0)]}c for 0.25 ≤ P/P0 < 0.60
t (nm) = {E/[F − log(P/P0)]}e for 0.60 ≤ P/P0 < 0.90
t (nm) = G(P/P0)g + H(P/P0)h for 0.90 ≤ P/P0
PD (nm) = 4Vtotal/Stotal
WT (nm) = a − PD
Low-resolution transmission electron microscopy (TEM) images were obtained on a JEOL-1011 microscope (Tokyo, Japan) at 120 kV. Before analysis, each sample was dispersed in ethanol (0.1 mg mL−1) through sonication. Subsequently, 5 µL of this suspension was deposited to a carbon-coated copper grid and dried overnight at room temperature. The mean particle size was determined using the software, ImageJ (version 1.52a), counting at least 100 particles for each sample.
High-resolution transmission electron microscopy (HRTEM) images were obtained on a JEOL JEM-2100 microscope (Tokyo, Japan) at 200 kV under vacuum and N2 purge. Before analysis, each sample was dispersed in ethanol and subsequently deposited to a carbon-coated copper grid and dried at room temperature.
3.4. Liquid-Phase Oxidation of Benzyl Alcohol
In a typical essay, the desired catalyst amount (5, 10, or 15 wt.% of BzOH) was transferred to a 10 mL glass reactor and dried for 30 min at 70 °C. Then, 240 μL of BzOH and the required amount of TBHP were added for reactions with 1:1, 1:2, and 2:1 (alcohol:oxidant) molar ratios. The glass reactor was closed with a cover and subjected to magnetic stirring and heating at the desired temperature (70, 80, or 90 °C) for 24 h in an aluminum dry bath block/hot plate stirrer system. In addition, H2O2 and DTBP were also studied as oxidant agents, while acetonitrile and isopropanol were tested as solvents. After the reaction, the system was cooled at room temperature, centrifuged at 2500 rpm, and an aliquot removed for analysis by 1H NMR (see the schematic representation of these procedures in Figure 17). For the reusability studies, the catalysts used in the first cycle of the optimized conditions were washed with ethanol twice, dried at room temperature, and calcined at 300 °C for 2 h in order to eliminate the residual organic content.
The reaction analysis was performed by NMR on a Bruker Magneto Oxford YH300 using CDCl3 as the solvent and TMS as internal standard for spectra referencing (δ = 0.0 ppm). 1H NMR spectra (300 MHz) were obtained with a spinning rate at 20 Hz, pulse length of 4.90 μs (π/4), 16 scans, and a delay recycle of 1.36 s. Identification of the substrate and main products was performed by comparison with known standards. The BzOH conversion (C, %), BzD selectivity (S, %), BzD yield (Y, %), and turnover number (TON, mol molcat−1) were determined using Equations (11)–(14), respectively:
where ABzOH, ABzD, and AHBz are the integrated areas of the methylene singlet at 4.69 ppm, the aldehyde hydrogen singlet at 10.02 ppm, and the deshielded aromatic double multiplet centered at 8.10 ppm, respectively; n = moles of BzOH converted; and ncat = moles of Ce in the catalyst.
C, % = [(2ABzD + AHBz)/(2ABzD + AHBz + ABzOH)]100
S, % = [2ABzD/(2ABzD +AHBz)]100
Y, % = (CS)/100
TON, mol molcat−1 = n/ncat
4. Conclusions
For the first time, the effect of different cerium(III) precursors was reported in the synthesis of Ce-MCM-41 materials. Cerium incorporation into the MCM-41 framework led to long-range disordered materials, but the results obtained by several characterization techniques showed the influence of different anions in the microstructural properties and catalytic activity of Ce-MCM-Cl and Ce-MCM-NO3. A mechanism for cerium incorporation in the MCM-41 structure was proposed, where [Ce(OH)3] species were hydrogen bonded to silicate anions, forming framework Ce-O-Si bonds during condensation. It was also observed that the synthesis of Ce-MCM-NO3 follows a different path when compared to Ce-MCM-Cl. The former induced Ce(OH)3 monomers to aggregate, forming non-framework CeO2 during calcination on the MCM-41 surface, while the latter preferentially formed framework Ce-O-Si bonds. The preferential formation of CeO2 species on Ce-MCM-NO3 was attributed to the nitrate ions’ strong binding to template molecules. The prepared materials were tested in the solvent free oxidation of benzyl alcohol to produce benzaldehyde, an important fine chemical compound for several industrial sectors, and the reaction parameters were optimized for each catalyst. Overall, Ce-MCM-Cl achieved better BzOH conversions and benzaldehyde (BzD) yields, while Ce-MCM-NO3 offered increased BzD selectivity. The catalysts’ reusability was also studied over three cycles. Ce-MCM-Cl and Ce-MCM-NO3 showed a reduction in conversion, but increase in selectivity, resulting in negligible changes in BzD yield values. Also, it was observed that the different crystallization mechanism differentiates both catalysts’ endurance towards deactivation, where Ce-MCM-NO3 was more resistant than Ce-MCM-Cl. This effect was attributed to the CeO2 coating on the Ce-MCM-NO3 surface, making the latter less prone to possible etching effects from TBHP. Thus, the manuscript shows the importance of the selection of suitable heteroelement precursors in the synthesis of materials. Even more, there is scarce literature regarding differences in the same metal-incorporated silicas that can achieve different conditions for liquid-phase reactions, an aspect very important when seeking the best choices for the synthetic preparation of catalysts with the desired activity/selectivity in industrial processes.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/9/4/377/s1, Figure S1: XRD patterns of Ce-MCM-Cl and Ce-MCM-NO3 materials at high angle region, Figure S2. Raman spectra of CTA+ systems between 400–1700 cm−1 (A) and 2800–3000 cm−1 (B), Figure S3. Raman spectrum of crystalline CeO2, Figure S4. Absorption spectra (A) and Davis-Mott plot (B) for of CeO2, Figure S5. High magnification in HRTEM images highlighting CeO2 110 interplanar spacing for: Ce-MCM-Cl (A) and Ce-MCM-NO3 (B), Table S1. Raman band assignments for CTA+ cations in as-synthesized MCM-41 materials and template standards, Table S2. TGA results for the as-synthesized MCM-41 materials, Table S3. TGA results for the calcined MCM-41 materials.
Author Contributions
Conceptualization, formal analysis, methodology, project administration, visualization, writing—Review and editing, C.M.A. and J.L.M.; investigation, C.M.A. and K.V.d.O.; writing—Original draft preparation, C.M.A., K.V.d.O. and J.L.M.; validation, C.M.A.; funding acquisition, resources, supervision, J.L.M.
Funding
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico—Brasil (CNPq), grant number 308061/2015-2; Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF)—Brasil, grant number 0193001347/2016; and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES), finance code 001.
Acknowledgments
The authors would like to thank Financiadora de Inovação e Pesquisa—Brasil (Finep), Instituto de Química da Universidade de Brasília—Brasil, Ministério da Ciência, Tecnologia e Inovação—Brasil (MCTI), Linconl Araújo Teixeira and Sandra Maria da Luz (Faculdade Gama da Universidade de Brasília—Brasil) for TG/DTG/DSC measurements, Rafael Brasil Figueras (Instituto de Geociências da Universidade de Brasília—Brasil) for XRD measurements, Tatiane Oliveira dos Santos (Laboratório Multiusuário de Alta Resolução da Universidade Federal do Goiás—Brasil) for high resolution TEM measurements, Ingrid Gracielle Martins da Silva (Laboratório de Microscopia Eletrônica e Microanálise do Instituto de Ciências Biológicas da Universidade de Brasília—Brasil) for low resolution TEM measurements, Maryene Alves Camargo for theoretical discussion and Claudia Cristina Gatto for the support during this manuscripts writing.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Ciriminna, R.; Pagliaro, M. Green chemistry in the fine chemicals and pharmaceutical industries. Org. Process Res. Dev. 2013, 17, 1479–1484. [Google Scholar] [CrossRef]
- de Oliveira, N.B. Inovação e produção na química fina. Quim. Nova 2005, 28, S79–S85. [Google Scholar] [CrossRef]
- O desempenho da indústria quimica em 2017. Available online: http://www.webcitation.org/75xqkk7e9 (accessed on 5 February 2019).
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E Factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Brühne, F.; Wright, E. Benzaldehyde. In Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Elvers, B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Volume 5, pp. 223–235. [Google Scholar]
- Benzaldehyde Market for Aroma Chemicals, Pharmaceuticals, Agriculture, Coatings and Other End-Users—Global Industry Analysis, Size, Share, Growth, Trends and Forecast 2015–2023. Available online: http://www.webcitation.org/75xscDvAV (accessed on 5 February 2019).
- Shearon, W.H.; Hall, H.E.; Stevens, J.E. Fine chemicals from coal. Ind. Eng. Chem. 1949, 41, 1812–1820. [Google Scholar] [CrossRef]
- Sheldon, R.A. Recent advances in green catalytic oxidations of alcohols in aqueous media. Catal. Today 2015, 247, 4–13. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Ribeiro, A.P.C.; Alegria, E.C.B.A.; Martins, N.M.R.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Catalytic oxidation of alcohols: Recent advances. Adv. Organomet. Chem. 2015, 63, 91–174. [Google Scholar]
- Sankar, M.; Nowicka, E.; Carter, E.; Murphy, D.M.; Knight, D.W.; Bethell, D.; Hutchings, G.J. The benzaldehyde oxidation paradox explained by the interception of peroxy radical by benzyl alcohol. Nat. Commun. 2014, 5, 3332–3337. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Rossi, M. Highly selective oxidation of benzyl alcohol to benzaldehyde catalyzed by bimetallic gold–copper catalyst. J. Catal. 2008, 260, 384–386. [Google Scholar] [CrossRef]
- Enache, D.I.; Edwards, J.K.; Landon, P.; Solsona-Espriu, B.; Carley, A.F.; Herzing, A.A.; Watanabe, M.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Solvent-free oxidation of primary alcohols to aldehydes using Au-Pd/TiO2 catalysts. Science 2006, 311, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Jia, A.; Lou, L.-L.; Zhang, C.; Zhang, Y.; Liu, S. Selective oxidation of benzyl alcohol to benzaldehyde with hydrogen peroxide over alkali-treated ZSM-5 zeolite catalysts. J. Mol. Catal. A Chem. 2009, 306, 123–129. [Google Scholar] [CrossRef]
- Bansal, V.K.; Thankachan, P.P.; Prasad, R. Oxidation of benzyl alcohol and styrene using H2O2 catalyzed by tetraazamacrocycle complexes of Cu(II) and Ni(II) encapsulated in zeolite-Y. Appl. Catal. A Gen. 2010, 381, 8–17. [Google Scholar] [CrossRef]
- Chen, G.; Zhou, Y.; Long, Z.; Wang, X.; Li, J.; Wang, J. Mesoporous polyoxometalate-based ionic hybrid as a triphasic catalyst for oxidation of benzyl alcohol with H2O2 on water. ACS Appl. Mater. Interfaces 2014, 6, 4438–4446. [Google Scholar] [CrossRef] [PubMed]
- Della Pina, C.; Falletta, E.; Prati, L.; Rossi, M. Selective oxidation using gold. Chem. Soc. Rev. 2008, 37, 2077–2095. [Google Scholar] [CrossRef]
- Corma, A.; García, H. Lewis Acids as Catalysts in Oxidation Reactions: From Homogeneous to Heterogeneous Systems. Chem. Rev. 2002, 102, 3837–3892. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Yamawaki, K.; Ura, T.; Yamada, H.; Yoshida, T.; Ogawa, M. Hydrogen peroxide oxidation catalyzed by heteropoly acids combined with cetylpyridinium chloride. Epoxidation of olefins and allylic alcohols, ketonization of alcohols and diols, and oxidative cleavage of 1,2-diols and olefins. J. Org. Chem. 1988, 53, 3587–3593. [Google Scholar] [CrossRef]
- Venturello, C.; D’Aloisio, R.; Bart, J.C.J.; Ricci, M. A New peroxotungsten heteropoly anion with special oxidizing properties: Synthesis and structure of tetrahexylammonium tetra(diperoxotungsto)phosphate(3-). J. Mol. Catal. 1985, 32, 107–110. [Google Scholar] [CrossRef]
- Kresge, C.T.; Roth, W.J. The discovery of mesoporous molecular sieves from the twenty year perspective. Chem. Soc. Rev. 2013, 42, 3663–3670. [Google Scholar] [CrossRef]
- Alothman, A.Z. A review: Fundamental aspects of silicate mesoporous materials. Materials 2012, 5, 2874–2902. [Google Scholar] [CrossRef]
- Linares, N.; Silvestre-Albero, A.M.; Serrano, E.; Silvestre-Albero, J.; García-Martínez, J. Mesoporous materials for clean energy technologies. Chem. Soc. Rev. 2014, 43, 7681–7717. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-P.; Mou, C.-Y. Salt effect in post-synthesis hydrothermal treatment of MCM-41. Microporous Mesoporous Mater. 2002, 55, 69–80. [Google Scholar] [CrossRef]
- Lin, H.-P.; Kao, C.-P.; Mou, C.-Y. Counterion and alcohol effect in the formation of mesoporous silica. Microporous Mesoporous Mater. 2001, 48, 135–141. [Google Scholar] [CrossRef]
- Leontidis, E. Hofmeister anion effects on surfactant self-assembly and the formation of mesoporous solids. Curr. Opin. Colloid Interface Sci. 2002, 7, 81–91. [Google Scholar] [CrossRef]
- Singh, S.; Patel, A. Environmentally benign oxidations of alkenes and alcohols to corresponding aldehydes over anchored phosphotungstates: Effect of supports as well as oxidants. Catal. Lett. 2016, 146, 1059–1072. [Google Scholar] [CrossRef]
- Patel, A.; Singh, S. Undecatungstophosphate anchored to MCM-41: An ecofriendly and efficient bifunctional solid catalyst for non-solvent liquid-phase oxidation as well as esterification of benzyl alcohol. Microporous Mesoporous Mater. 2014, 195, 240–249. [Google Scholar] [CrossRef]
- Wang, X.; Wu, G.; Li, J.; Zhao, N.; Wei, W.; Sun, Y. Selective oxidation of benzyl alcohol catalyzed by Cr(salen) complexes immobilized on MCM-41. J. Mol. Catal. A Chem. 2007, 276, 86–94. [Google Scholar] [CrossRef]
- Hamza, A.; Srinivas, D. Selective oxidation of benzyl alcohol over copper phthalocyanine immobilized on MCM-41. Catal. Lett. 2009, 128, 434–442. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, V.P.; Vishwanathan, V.; Chary, K.V.R. Synthesis, characterization, and reactivity of Au/MCM-41 catalysts prepared by homogeneous deposition–precipitation (HDP) method for vapor phase oxidation of benzyl alcohol. Mater. Res. Bull. 2015, 61, 105–112. [Google Scholar] [CrossRef]
- Pal, N.; Cho, E.-B.; Kim, D. Synthesis of ordered mesoporous silica/ceria–silica composites and their high catalytic performance for solvent-free oxidation of benzyl alcohol at room temperature. RSC Adv. 2014, 4, 9213–9222. [Google Scholar] [CrossRef]
- Cánepa, A.L.; Elías, V.R.; Vaschetti, V.M.; Sabre, E.V.; Eimer, G.A.; Casuscelli, S.G. Selective oxidation of benzyl alcohol through eco-friendly processes using mesoporous V-MCM-41, Fe-MCM-41 and Co-MCM-41 materials. Appl. Catal. A Gen. 2017, 545, 72–78. [Google Scholar] [CrossRef]
- Dang, T.T.H.; Seeburg, D.; Radnik, J.; Kreyenschulte, C.; Atia, H.; Vu, T.T.H.; Wohlrab, S. Influence of V-sources on the catalytic performance of VMCM-41 in the selective oxidation of methane to formaldehyde. Catal. Commun. 2018, 103, 56–59. [Google Scholar] [CrossRef]
- Corma, A.; Navarro, M.T.; Nemeth, L.; Renz, M. Sn-MCM-41—A heterogeneous selective catalyst for the Baeyer–Villiger oxidation with hydrogen peroxide. Chem. Commun. 2001, 2190–2191. [Google Scholar] [CrossRef]
- Yao, W.; Chen, Y.; Min, L.; Fang, H.; Yan, Z.; Wang, H.; Wang, J. Liquid oxidation of cyclohexane to cyclohexanol over cerium-doped MCM-41. J. Mol. Catal. A Chem. 2006, 246, 162–166. [Google Scholar] [CrossRef]
- González Vargas, O.A.; de los Reyes Heredia, J.A.; Montesinos Castellanos, A.; Chen, L.F.; Wang, J.A. Cerium incorporating into MCM-41 mesoporous materials for CO oxidation. Mater. Chem. Phys. 2013, 139, 125–133. [Google Scholar] [CrossRef]
- Ghesti, G.F.; de Macedo, J.L.; Parente, V.C.; Dias, J.A.; Dias, S.C. Synthesis, characterization and reactivity of Lewis acid/surfactant cerium trisdodecylsulfate catalyst for transesterification and esterification reactions. Appl. Catal. A Gen. 2009, 355, 139–147. [Google Scholar] [CrossRef]
- Cesteros, Y.; Haller, G.L. Several factors affecting Al-MCM-41 synthesis. Microporous Mesoporous Mater. 2001, 43, 171–179. [Google Scholar] [CrossRef]
- Ghiaci, M.; Seyedeyn-Azad, F.; Kia, R. Fast and efficient synthesis of ZSM-5 in a broad range of SiO2/Al2O3 without using seeding gel. Mater. Res. Bull. 2004, 39, 1257–1264. [Google Scholar] [CrossRef]
- Zhao, D.; Nie, C.; Zhou, Y.; Xia, S.; Huang, L.; Li, Q. Comparison of disordered mesoporous aluminosilicates with highly ordered Al-MCM-41 on stability, acidity and catalytic activity. Catal. Today 2001, 68, 11–20. [Google Scholar] [CrossRef]
- Reddy, K.M.; Song, C. Effect of Al sources on the synthesis and acidic characteristics of mesoporous aluminosilicates of MCM-41 type. In Studies in Surface Science and Catalysis; Bonneviot, L., Béland, F., Danumah, C., Giasson, S., Kaliaguine, S., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 117, pp. 291–299. [Google Scholar]
- Matsumoto, A.; Chen, H.; Tsutsumi, K.; Grün, M.; Unger, K. Novel route in the synthesis of MCM-41 containing framework aluminum and its characterization. Microporous Mesoporous Mater. 1999, 32, 55–62. [Google Scholar] [CrossRef]
- Lin, W.; Cai, Q.; Pang, W.; Yue, Y.; Zou, B. New mineralization agents for the synthesis of MCM-41. Microporous Mesoporous Mater. 1999, 33, 187–196. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef] [Green Version]
- El Haskouri, J.; Morales, J.M.; Ortiz de Zárate, D.; Fernández, L.; Latorre, J.; Guillem, C.; Beltrán, A.; Beltrán, D.; Amorós, P. Nanoparticulated silicas with bimodal porosity: Chemical control of the pore sizes. Inorg. Chem. 2008, 47, 8267–8277. [Google Scholar] [CrossRef]
- Zhan, W.; Lu, G.; Guo, Y.; Guo, Y.; Wang, Y.; Wang, Y.; Zhang, Z.; Liu, X. Synthesis of cerium-doped MCM-48 molecular sieves and its catalytic performance for selective oxidation of cyclohexane. J. Rare Earths 2008, 26, 515–522. [Google Scholar] [CrossRef]
- de Souza, L.K.C.; Pardauil, J.J.R.; Zamian, J.R.; da Rocha Filho, G.N.; Barrado, C.M.; Angélica, R.S.; da Costa, C.E.F. Rapid synthesis and characterization of CeMCM-41. Powder Technol. 2012, 229, 1–6. [Google Scholar] [CrossRef]
- Bing, J.; Li, L.; Lan, B.; Liao, G.; Zeng, J.; Zhang, Q.; Li, X. Synthesis of cerium-doped MCM-41 for ozonation of p-chlorobenzoic acid in aqueous solution. Appl. Catal. B 2012, 115–116, 16–24. [Google Scholar] [CrossRef]
- Laha, S.C.; Mukherjee, P.; Sainkar, S.R.; Kumar, R. Cerium containing MCM-41-type mesoporous materials and their acidic and redox catalytic properties. J. Catal. 2002, 207, 213–223. [Google Scholar] [CrossRef]
- Barkam, S.; Ortiz, J.; Saraf, S.; Eliason, N.; McCormack, R.; Das, S.; Gupta, A.; Neal, C.; Petrovici, A.; Hanson, C.; et al. Modulating the catalytic activity of cerium oxide nanoparticles with the anion of the precursor salt. J. Phys. Chem. C 2017, 121, 20039–20050. [Google Scholar] [CrossRef]
- Mihailova, B.; Valtchev, V.; Mintova, S.; Faust, A.C.; Petkov, N.; Bein, T. Interlayer stacking disorder in zeolite beta family: A Raman spectroscopic study. Phys. Chem. Chem. Phys. 2005, 7, 2756–2763. [Google Scholar] [CrossRef]
- Zhou, L.; Li, X.; Yao, Z.; Chen, Z.; Hong, M.; Zhu, R.; Liang, Y.; Zhao, J. Transition-metal doped ceria microspheres with nanoporous structures for CO oxidation. Sci. Rep. 2016, 6, 23900. [Google Scholar] [CrossRef]
- Khalil, K.M.S. Cerium modified MCM-41 nanocomposite materials via a nonhydrothermal direct method at room temperature. J. Colloid Interface Sci. 2007, 315, 562–568. [Google Scholar] [CrossRef]
- Kolodziejski, W.; Corma, A.; Navarro, M.-T.; Pérez-Pariente, J. Solid-state NMR study of ordered mesoporous aluminosilicate MCM-41 synthesized on a liquid-crystal template. Solid State Nucl. Magn. Reson. 1993, 2, 253–259. [Google Scholar] [CrossRef]
- Chao, K.J.; Wu, C.N.; Chang, H.; Lee, L.J.; Hu, S.-f. Incorporation of vanadium in mesoporous MCM-41 and microporous AFI zeolites. J. Phys. Chem. B 1997, 101, 6341–6349. [Google Scholar] [CrossRef]
- Hill, P.J.; Shrestha, K.L.; Ishihara, S.; Ji, Q.; Ariga, K. Self-assembly: From amphiphiles to chromophores and beyond. Molecules 2014, 19, 8589–8609. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Burkett, S.L.; Li, H.-X.; Davis, M.E. Studies on mesoporous materials II. Synthesis mechanism of MCM-41. Microporous Mater. 1993, 2, 27–34. [Google Scholar] [CrossRef]
- Dendramis, A.L.; Schwinn, E.W.; Sperline, R.P. A surface-enhanced Raman scattering study of CTAB adsorption on copper. Surf. Sci. 1983, 134, 675–688. [Google Scholar] [CrossRef]
- Snyder, R.G.; Strauss, H.L.; Elliger, C.A. Carbon-hydrogen stretching modes and the structure of n-alkyl chains. 1. Long, disordered chains. J. Phys. Chem. 1982, 86, 5145–5150. [Google Scholar] [CrossRef]
- Orendorff, C.J.; Ducey, M.W.; Pemberton, J.E. Quantitative correlation of Raman spectral indicators in determining conformational order in alkyl chains. J. Phys. Chem. A 2002, 106, 6991–6998. [Google Scholar] [CrossRef]
- Jamieson Lauren, E.; Li, A.; Faulds, K.; Graham, D. Ratiometric analysis using Raman spectroscopy as a powerful predictor of structural properties of fatty acids. R. Soc. Open Sci. 2018, 5, 181483. [Google Scholar] [CrossRef]
- Terribile, D.; Trovarelli, A.; Llorca, J.; de Leitenburg, C.; Dolcetti, G. The synthesis and characterization of mesoporous high-surface area ceria prepared using a hybrid organic/inorganic route. J. Catal. 1998, 178, 299–308. [Google Scholar] [CrossRef]
- Hayes, S.A.; Yu, P.; O’Keefe, T.J.; O’Keefe, M.J.; Stoffer, J.O. The phase stability of cerium species in aqueous systems: I. E-pH diagram for the Ce-HClO4–H2O system. J. Electrochem. Soc. 2002, 149, C623–C630. [Google Scholar] [CrossRef]
- Bumajdad, A.; Eastoe, J.; Mathew, A. Cerium oxide nanoparticles prepared in self-assembled systems. Adv. Colloid Interface Sci. 2009, 147–148, 56–66. [Google Scholar] [CrossRef]
- Bouchaud, B.; Balmain, J.; Bonnet, G.; Pedraza, F. pH-distribution of cerium species in aqueous systems. J. Rare Earths 2012, 30, 559–562. [Google Scholar] [CrossRef]
- Peppard, D.F.; Mason, G.W.; Hucher, I. Stability constants of certain lanthanide(III) and actinide(III) chloride and nitrate complexes. J. Inorg. Nucl. Chem. 1962, 24, 881–888. [Google Scholar] [CrossRef]
- Berr, S.; Jones, R.R.M.; Johnson, J.S. Effect of counterion on the size and charge of alkyltrimethylammonium halide micelles as a function of chain length and concentration as determined by small-angle neutron scattering. J. Phys. Chem. 1992, 96, 5611–5614. [Google Scholar] [CrossRef]
- Ye, F.; Vallhov, H.; Qin, J.; Daskalaki, E.; Sugunan, A.; Toprak, M.S.; Fornara, A.; Gabrielsson, S.; Scheynius, A.; Muhammed, M. Synthesis of high aspect ratio gold nanorods and their effects on human antigen presenting dendritic cells. Int. J. Nanotechnol. 2011, 8, 22. [Google Scholar] [CrossRef]
- Gamboa, C.; Sepulveda, L.; Soto, R. Free energies of transfer of anions from water to cationic micelles from ionic exchange measurements. J. Phys. Chem. 1981, 85, 1429–1434. [Google Scholar] [CrossRef]
- Xiong, G.; Li, C.; Li, H.; Xin, Q.; Feng, Z. Direct spectroscopic evidence for vanadium species in V-MCM-41 molecular sieve characterized by UV resonance Raman spectroscopy. Chem. Commun. 2000, 677–678. [Google Scholar] [CrossRef] [Green Version]
- Casas-Orozco, D.; Alarcón, E.; Carrero, C.A.; Venegas, J.M.; McDermott, W.; Klosterman, E.; Hermans, I.; Villa, A.-L. Influence of tin loading and pore size of Sn/MCM-41 catalysts on the synthesis of nopol. Ind. Eng. Chem. Res. 2017, 56, 6590–6598. [Google Scholar] [CrossRef]
- Li, C. Identifying the isolated transition metal ions/oxides in molecular sieves and on oxide supports by UV resonance Raman spectroscopy. J. Catal. 2003, 216, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chen, F.; Lu, J.-Q.; Luo, M.-F. Study of defect sites in Ce1–xMxO2−δ (x = 0.2) solid solutions using Raman spectroscopy. J. Phys. Chem. A 2011, 115, 7972–7977. [Google Scholar] [CrossRef]
- He, H.; Yang, P.; Li, J.; Shi, R.; Chen, L.; Zhang, A.; Zhu, Y. Controllable synthesis, characterization, and CO oxidation activity of CeO2 nanostructures with various morphologies. Ceram. Int. 2016, 42, 7810–7818. [Google Scholar] [CrossRef]
- Soren, S.; Bessoi, M.; Parhi, P. A rapid microwave initiated polyol synthesis of cerium oxide nanoparticle using different cerium precursors. Ceram. Int. 2015, 41, 8114–8118. [Google Scholar] [CrossRef]
- Ghiaci, M.; Abbaspur, A.; Kia, R.; Belver, C.; Trujillano, R.; Rives, V.; Vicente, M.A. Vapor-phase alkylation of toluene by benzyl alcohol on H3PO4-modified MCM-41 mesoporous silicas. Catal. Commun. 2007, 8, 49–56. [Google Scholar] [CrossRef]
- Fu, Y.; Zhan, W.; Guo, Y.; Wang, Y.; Liu, X.; Guo, Y.; Wang, Y.; Lu, G. Effect of surface functionalization of cerium-doped MCM-48 on its catalytic performance for liquid-phase free-solvent oxidation of cyclohexane with molecular oxygen. Microporous Mesoporous Mater. 2015, 214, 101–107. [Google Scholar] [CrossRef]
- Mullins, D.R. The surface chemistry of cerium oxide. Surf. Sci. Rep. 2015, 70, 42–85. [Google Scholar] [CrossRef] [Green Version]
- Zdravković, J.; Simović, B.; Golubović, A.; Poleti, D.; Veljković, I.; Šćepanović, M.; Branković, G. Comparative study of CeO2 nanopowders obtained by the hydrothermal method from various precursors. Ceram. Int. 2015, 41, 1970–1979. [Google Scholar] [CrossRef]
- Zhao, X.S.; Lu, G.Q.; Whittaker, A.K.; Millar, G.J.; Zhu, H.Y. Comprehensive study of surface chemistry of MCM-41 using 29Si CP/MAS NMR, FTIR, pyridine-TPD, and TGA. J. Phys. Chem. B 1997, 101, 6525–6531. [Google Scholar] [CrossRef]
- Varache, M.; Bezverkhyy, I.; Saviot, L.; Bouyer, F.; Baras, F.; Bouyer, F. Optimization of MCM-41 type silica nanoparticles for biological applications: Control of size and absence of aggregation and cell cytotoxicity. J. Non-Cryst. Solids 2015, 408, 87–97. [Google Scholar] [CrossRef]
- Vantomme, A.; Yuan, Z.-Y.; Du, G.; Su, B.-L. Surfactant-Assisted Large-Scale Preparation of Crystalline CeO2 Nanorods. Langmuir 2005, 21, 1132–1135. [Google Scholar] [CrossRef]
- Pan, C.; Zhang, D.; Shi, L. CTAB assisted hydrothermal synthesis, controlled conversion and CO oxidation properties of CeO2 nanoplates, nanotubes, and nanorods. J. Solid State Chem. 2008, 181, 1298–1306. [Google Scholar] [CrossRef]
- Galarneau, A.; Villemot, F.; Rodriguez, J.; Fajula, F.; Coasne, B. Validity of the t-plot method to assess microporosity in hierarchical micro/mesoporous materials. Langmuir 2014, 30, 13266–13274. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquérol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl. Chem. 1985, 57, 17. [Google Scholar] [CrossRef]
- Anderson, M.W. Solid-state NMR as a probe of porous catalysts and catalytic processes. Top. Catal. 1996, 3, 195–220. [Google Scholar] [CrossRef]
- Kleitz, F.; Schmidt, W.; Schüth, F. Evolution of mesoporous materials during the calcination process: Structural and chemical behavior. Microporous Mesoporous Mater. 2001, 44–45, 95–109. [Google Scholar] [CrossRef]
- de Souza, L.K.C.; Pardauil, J.J.R.; Zamian, J.R.; da Rocha Filho, G.N.; da Costa, C.E.F. Influence of the incorporated metal on template removal from MCM-41 type mesoporous materials. J. Therm. Anal. Calorim. 2011, 106, 355–361. [Google Scholar] [CrossRef]
- Selvaraj, M.; Park, D.W.; Ha, C.S. Well ordered two-dimensional mesoporous CeSBA-15 synthesized with improved hydrothermal stability and catalytic activity. Microporous Mesoporous Mater. 2011, 138, 94–101. [Google Scholar] [CrossRef]
- Timofeeva, M.N.; Jhung, S.H.; Hwang, Y.K.; Kim, D.K.; Panchenko, V.N.; Melgunov, M.S.; Chesalov, Y.A.; Chang, J.S. Ce-silica mesoporous SBA-15-type materials for oxidative catalysis: Synthesis, characterization, and catalytic application. Appl. Catal. A Gen. 2007, 317, 1–10. [Google Scholar] [CrossRef]
- Wang, Z.; Balkus, K.J. Liquid phase propylene oxidation with tert-butyl hydroperoxide over titanium containing wrinkled mesoporous silica. Catal. Commun. 2017, 96, 15–18. [Google Scholar] [CrossRef]
- Grün, M.; Unger, K.K.; Matsumoto, A.; Tsutsumi, K. Novel pathways for the preparation of mesoporous MCM-41 materials: Control of porosity and morphology. Microporous Mesoporous Mater. 1999, 27, 207–216. [Google Scholar] [CrossRef]
- Cullity, B.D. Elements of X-ray Diffraction, 2nd ed.; Addison-Wesley Publishing Company, Inc.: Reading, PS, USA, 1967; pp. 81–349. [Google Scholar]
- Davis, E.A.; Mott, N.F. Conduction in non-crystalline systems V. Conductivity, optical absorption and photoconductivity in amorphous semiconductors. Philos. Mag. 1970, 22, 0903–0922. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, N.; Koyano, K.A.; Tanaka, Y.; Nakata, S.; Hashimoto, K.; Tatsumi, T. Investigation of the factors influencing the structural stability of mesoporous silica molecular sieves. Microporous Mesoporous Mater. 2003, 59, 43–52. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns of the calcined MCM-41 materials at the low angle region.
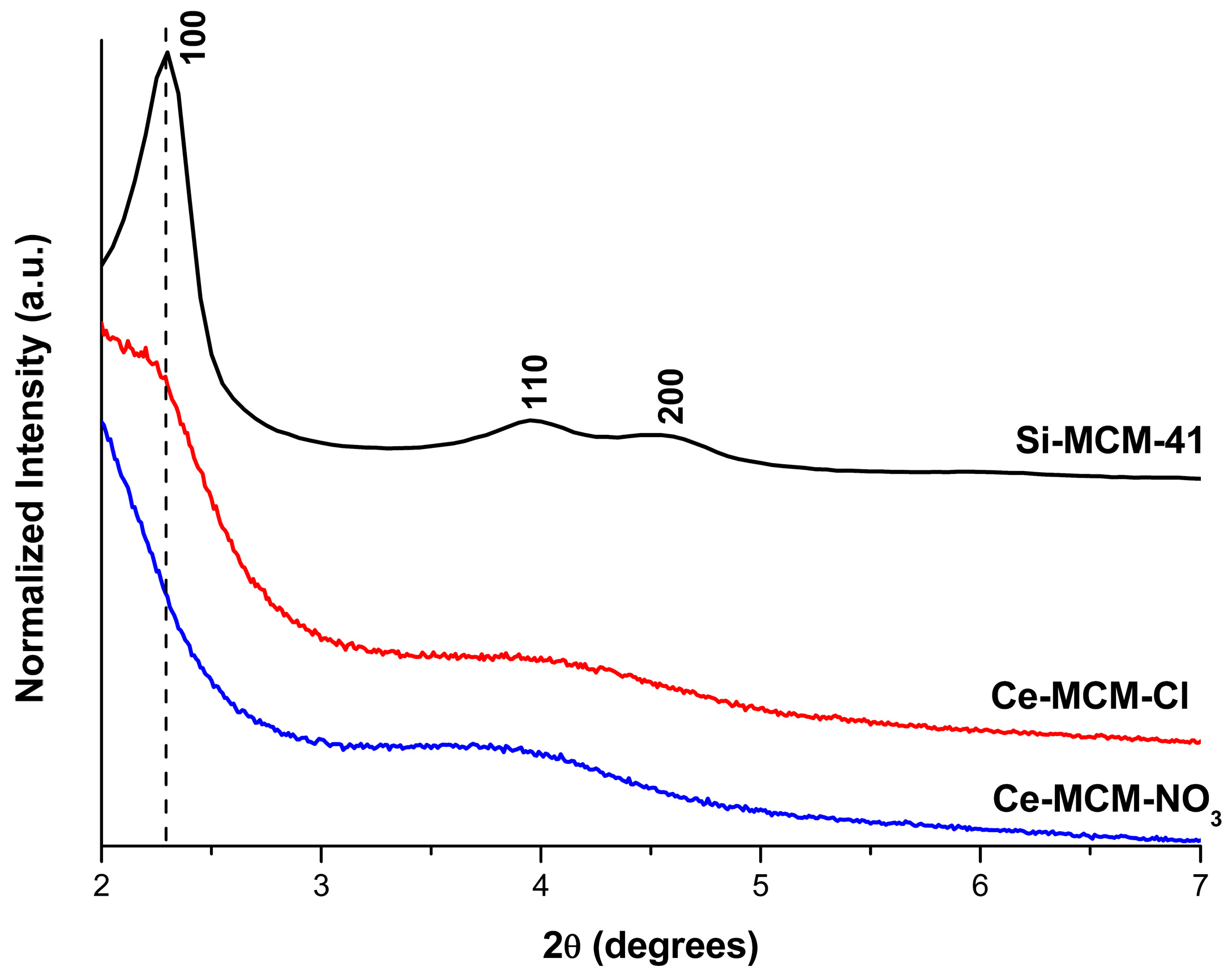
Figure 2.
Deconvolution of the overlapped XRD reflections and their 2θ positions for Ce-MCM-Cl and Ce-MCM-NO3 materials.
Figure 2.
Deconvolution of the overlapped XRD reflections and their 2θ positions for Ce-MCM-Cl and Ce-MCM-NO3 materials.

Figure 3.
Raman spectra of the as-synthesized materials between 400 and 1700 cm−1 (A) and 2800 and 3000 cm−1 (B). The signals used for the determination of the disorder parameters are indicated.
Figure 3.
Raman spectra of the as-synthesized materials between 400 and 1700 cm−1 (A) and 2800 and 3000 cm−1 (B). The signals used for the determination of the disorder parameters are indicated.

Figure 4.
Raman spectra of the calcined materials between 150 and 650 cm−1.

Figure 5.
FTIR spectra of the calcined MCM-41 materials.

Figure 6.
Absorption spectra (A) and Davis-Mott plots (B) for the calcined MCM-41 materials.
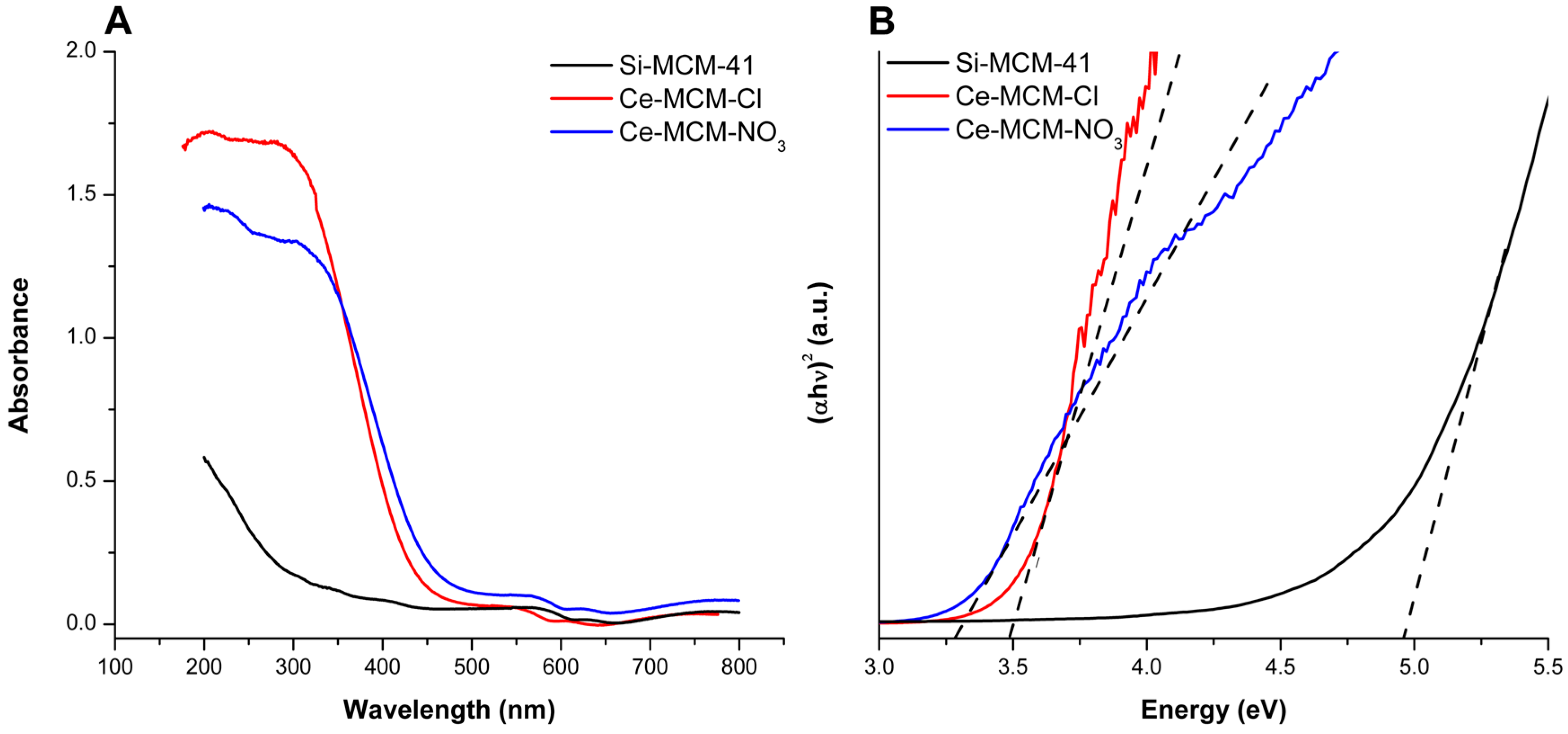
Figure 7.
29Si MAS-NMR spectra for the calcined MCM-41 materials, highlighting the position of each chemical environment detected by deconvolution.
Figure 7.
29Si MAS-NMR spectra for the calcined MCM-41 materials, highlighting the position of each chemical environment detected by deconvolution.

Figure 8.
TEM images for the calcined MCM-41 materials: Si-MCM-41 (A); Ce-MCM-Cl (B); and Ce-MCM-NO3 (C).
Figure 8.
TEM images for the calcined MCM-41 materials: Si-MCM-41 (A); Ce-MCM-Cl (B); and Ce-MCM-NO3 (C).

Figure 9.
HRTEM images for the calcined Ce-MCM-Cl (a) and Ce-MCM-NO3 (b) materials.

Figure 10.
Thickness or t-plot curve for Si-MCM-41 (a) where the blue line slope corresponds to Stotal, the red line slope corresponds to Sext, and the dashed black line intercept with the y-axis corresponds to Vmeso; N2 adsorption/desorption isotherm for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO3 (b) where filled circles correspond to adsorption and hollow circles corresponds to desorption.
Figure 10.
Thickness or t-plot curve for Si-MCM-41 (a) where the blue line slope corresponds to Stotal, the red line slope corresponds to Sext, and the dashed black line intercept with the y-axis corresponds to Vmeso; N2 adsorption/desorption isotherm for Si-MCM-41, Ce-MCM-Cl, and Ce-MCM-NO3 (b) where filled circles correspond to adsorption and hollow circles corresponds to desorption.

Figure 11.
Thermal analysis obtained under synthetic air for A-Si-MCM-41: TG and DTG curves (A); DTG and DSC curves (B). The temperatures indicated refers to the DTG maximums of observable peaks.
Figure 11.
Thermal analysis obtained under synthetic air for A-Si-MCM-41: TG and DTG curves (A); DTG and DSC curves (B). The temperatures indicated refers to the DTG maximums of observable peaks.

Figure 12.
Thermal analysis obtained under synthetic air for A-Ce-MCM-Cl: TG and DTG curves (A); DTG and DSC curves (B). The temperatures indicated refers to DTG maximums of observable peaks.
Figure 12.
Thermal analysis obtained under synthetic air for A-Ce-MCM-Cl: TG and DTG curves (A); DTG and DSC curves (B). The temperatures indicated refers to DTG maximums of observable peaks.

Figure 13.
Thermal analysis obtained under synthetic air for A-Ce-MCM-NO3: TG and DTG curves (A); DTG and DSC curves (B). The temperatures indicated refers to DTG maximums of observable peaks.
Figure 13.
Thermal analysis obtained under synthetic air for A-Ce-MCM-NO3: TG and DTG curves (A); DTG and DSC curves (B). The temperatures indicated refers to DTG maximums of observable peaks.

Figure 14.
TG curves for the calcined MCM-41 materials.
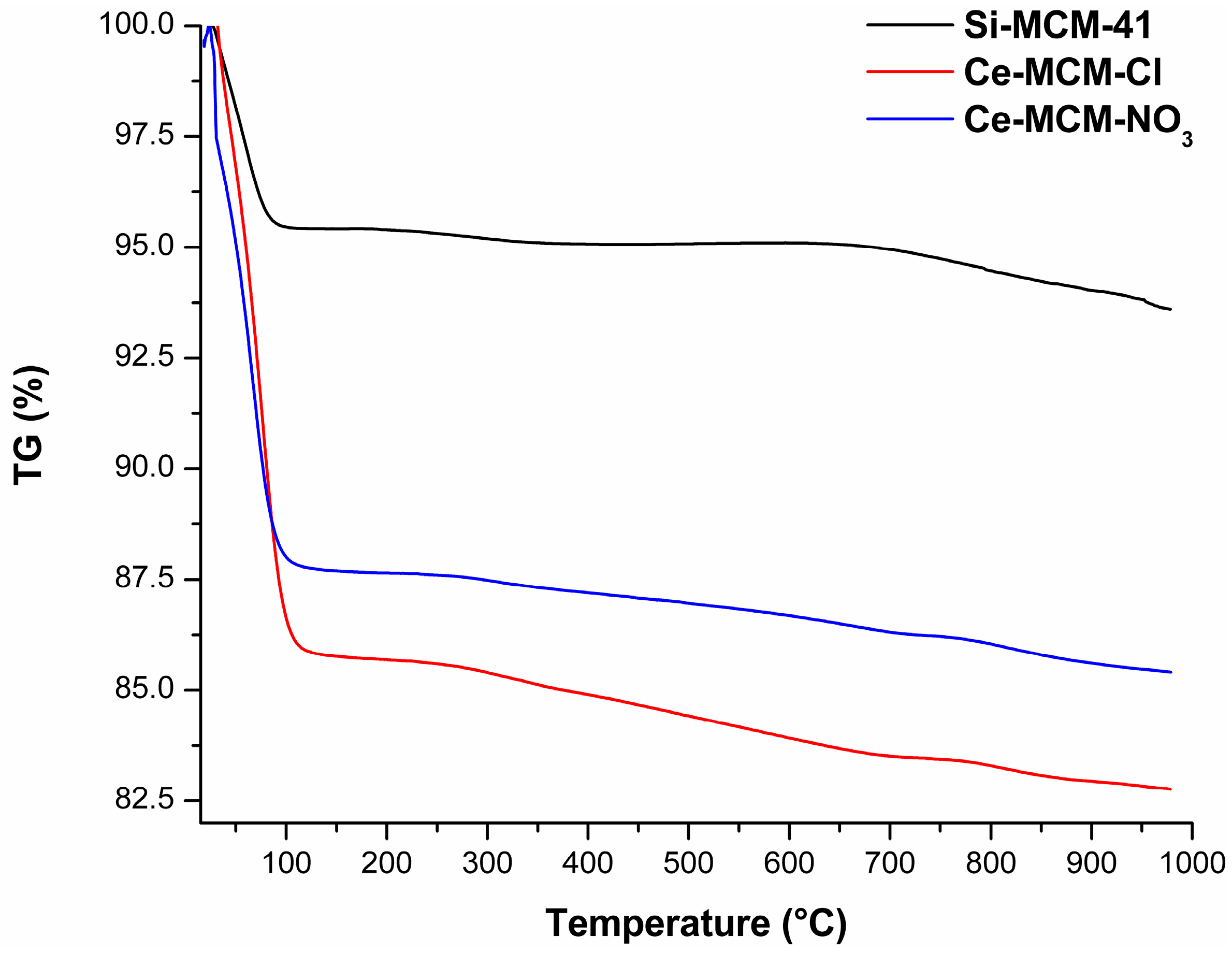
Figure 15.
Performance of recovered solids after three catalytic cycles: Ce-MCM-Cl (A) and Ce-MCM-NO3 (B).
Figure 15.
Performance of recovered solids after three catalytic cycles: Ce-MCM-Cl (A) and Ce-MCM-NO3 (B).

Figure 16.
Raman spectra of recovered solids: Ce-MCM-Cl (A) and Ce-MCM-NO3 (B).

Figure 17.
Schematic description for liquid-phase oxidation of BzOH and catalytic measurements.


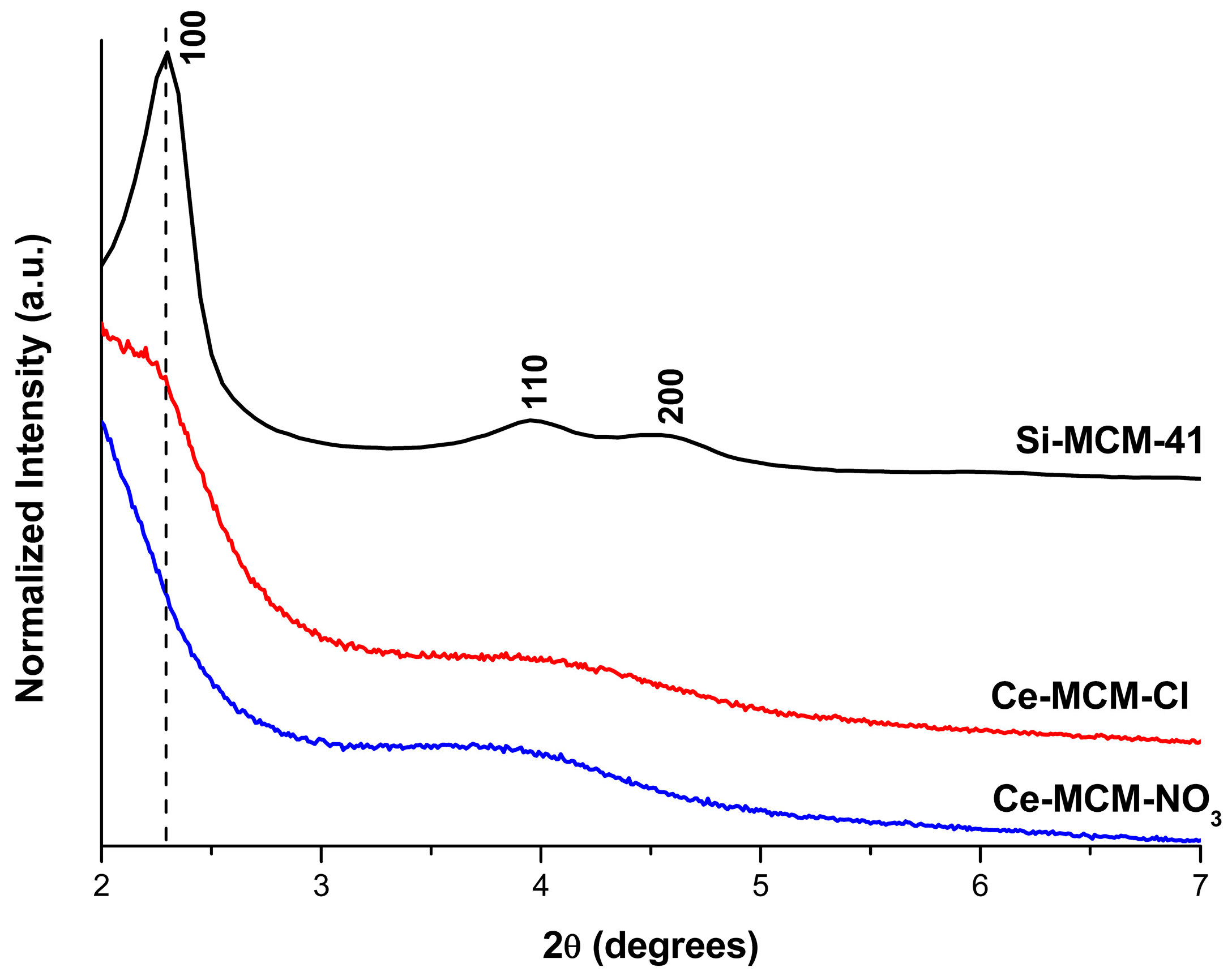{kind=link}
{kind=link}
{kind=link}
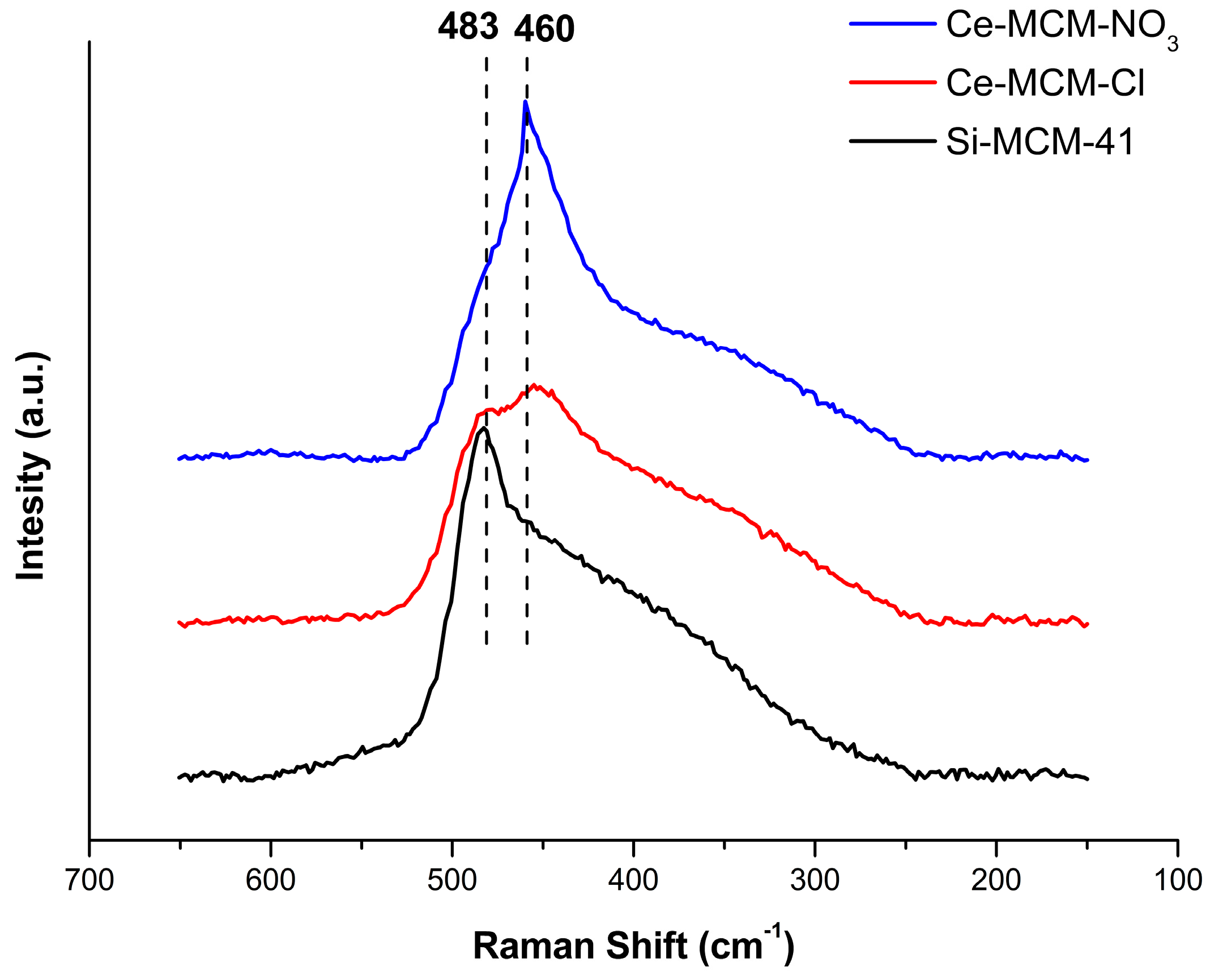{kind=link}
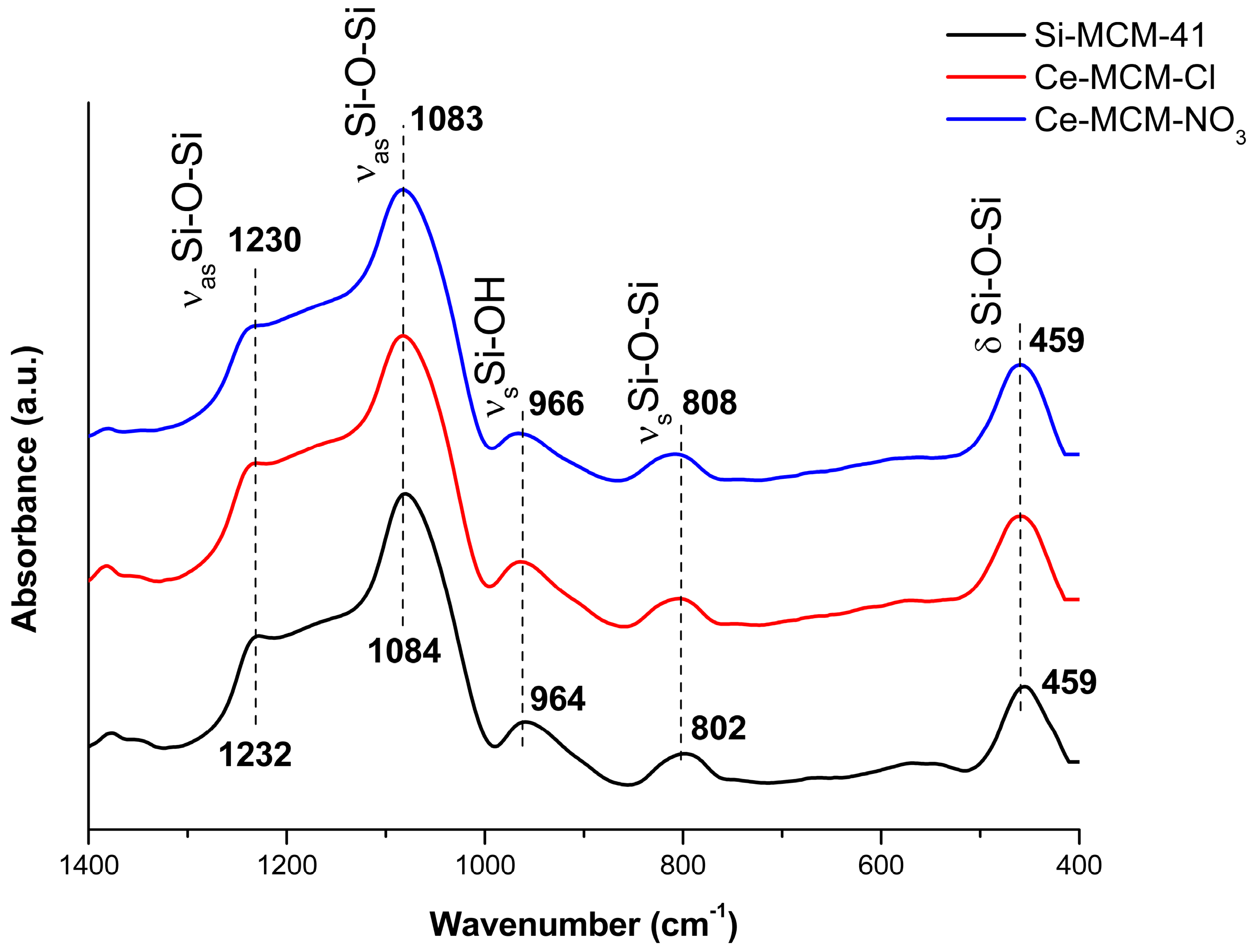{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
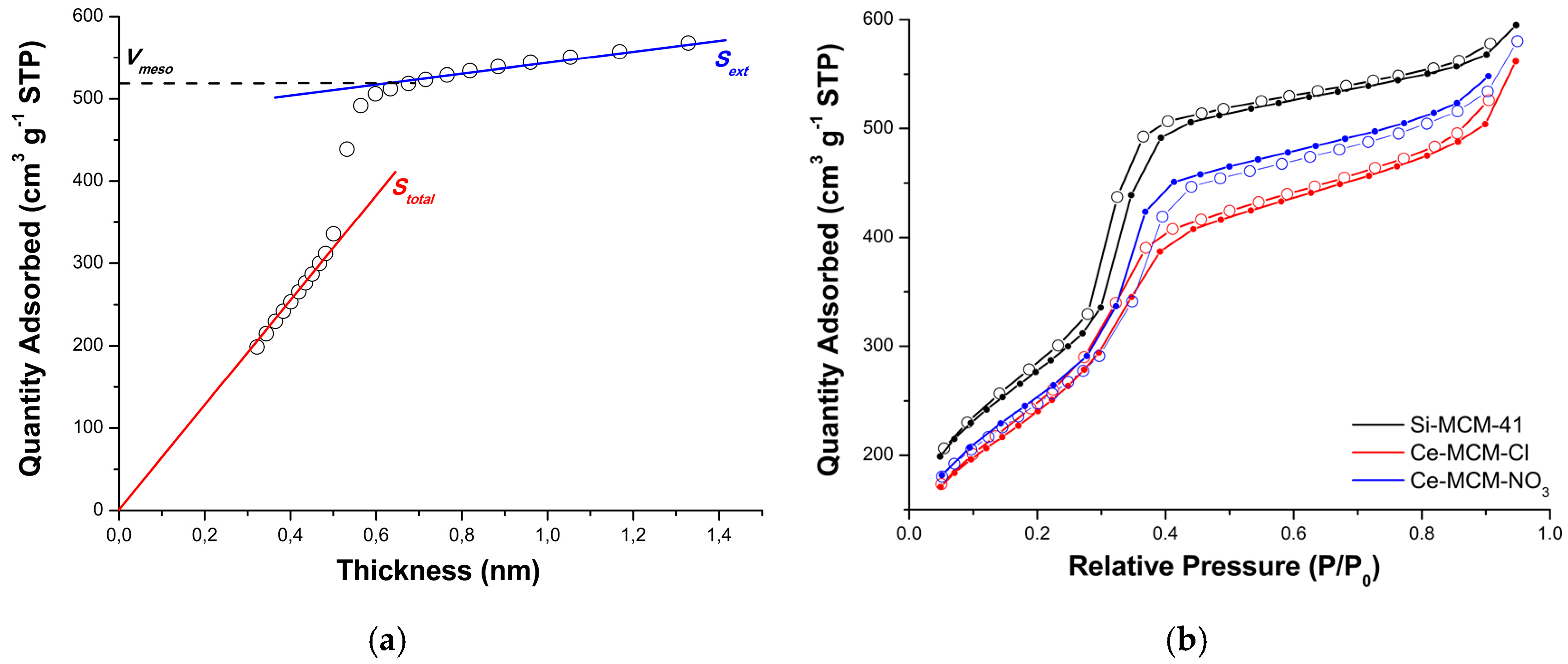{kind=link}
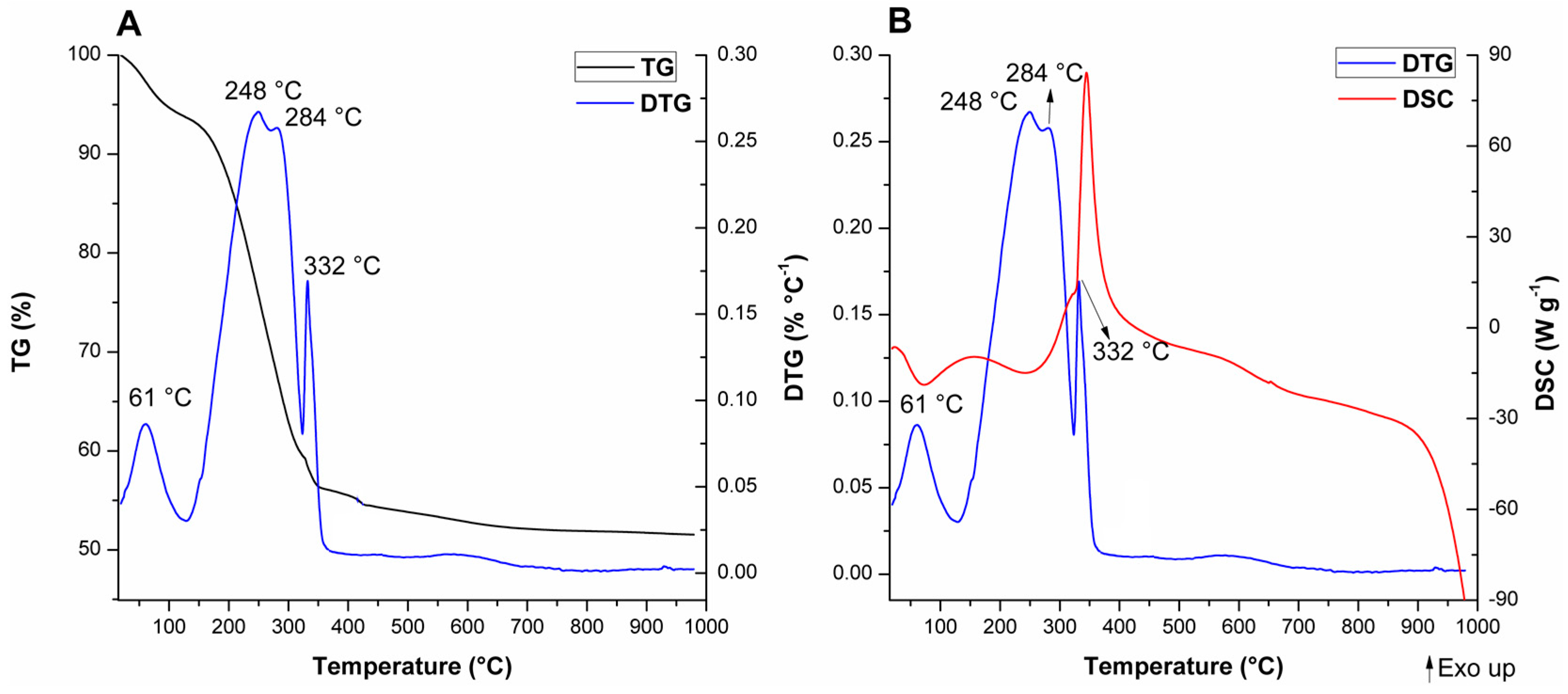{kind=link}
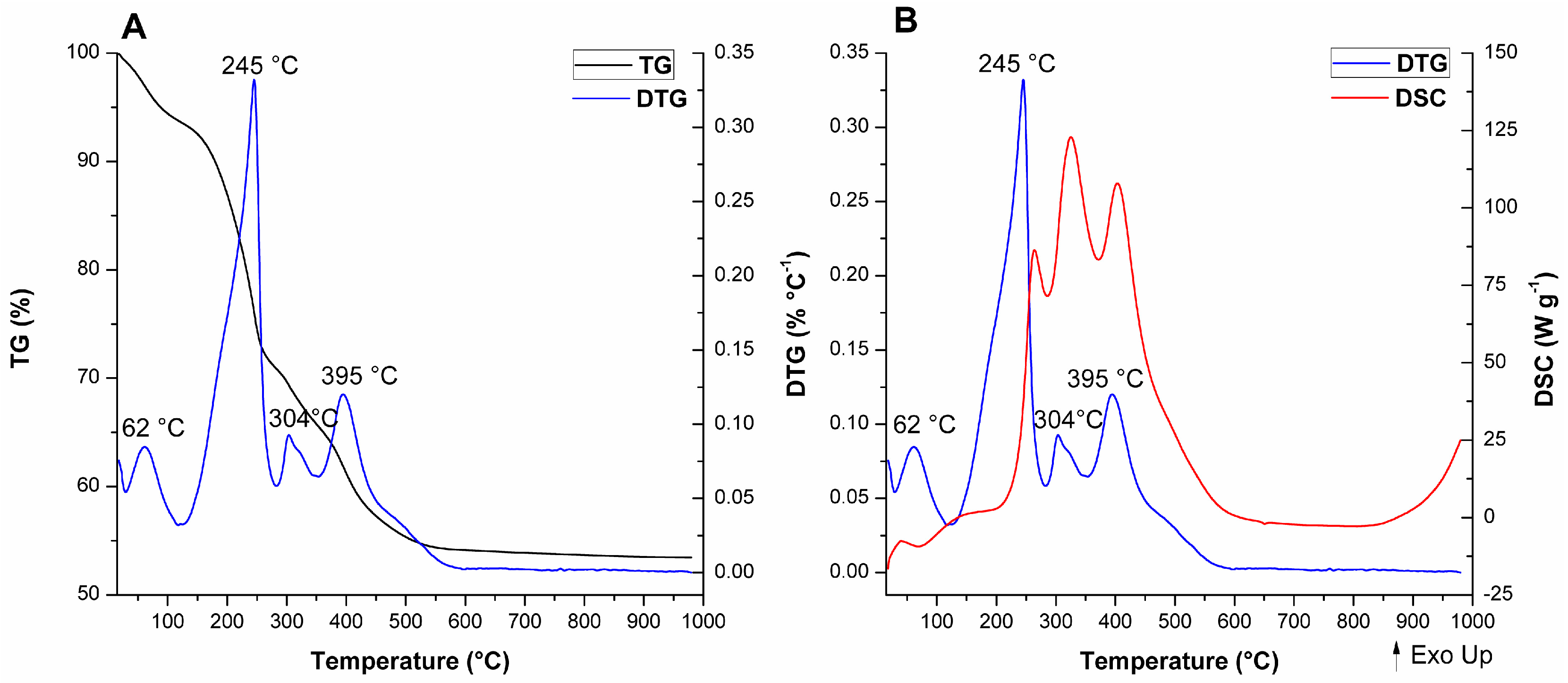{kind=link}
{kind=link}
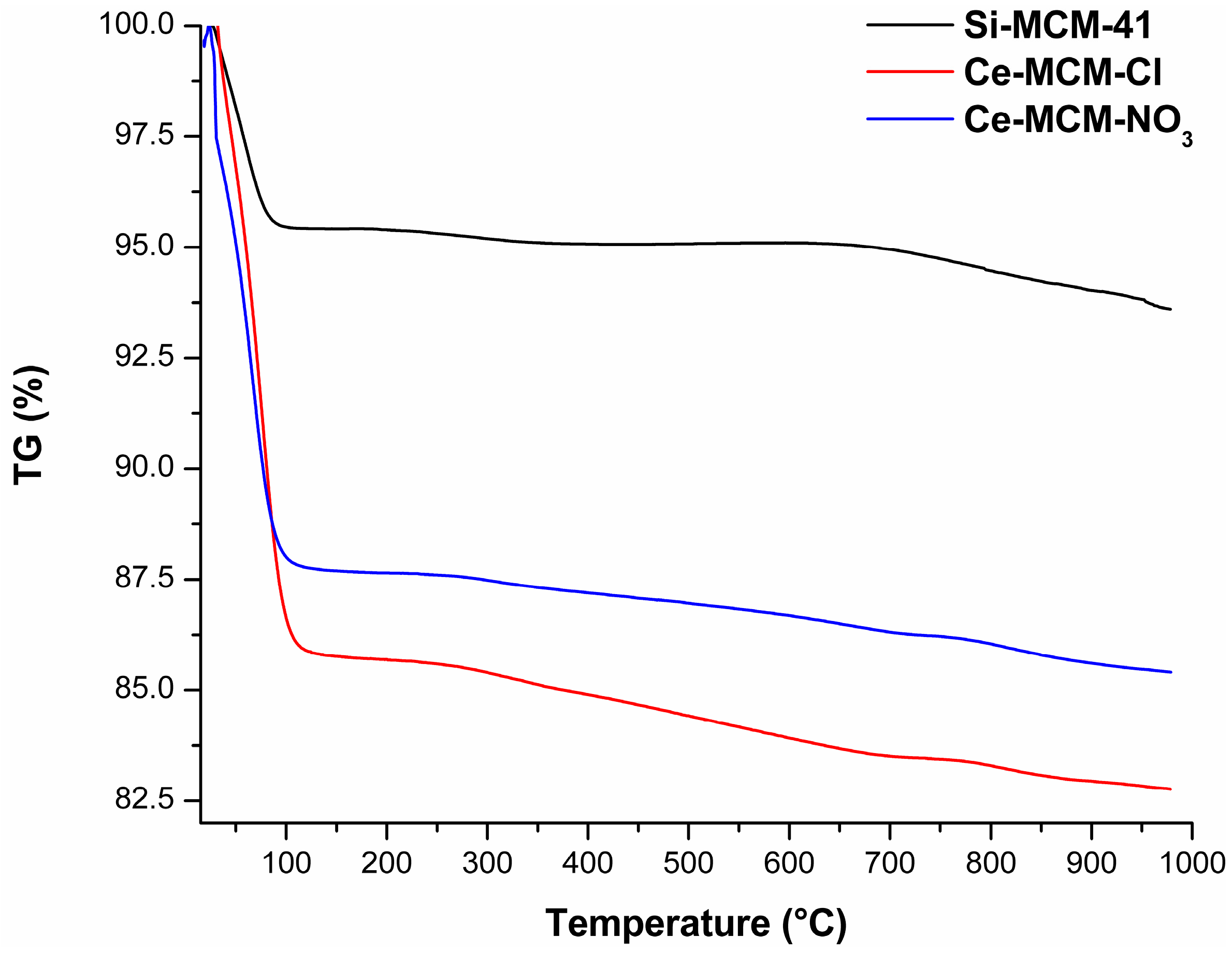{kind=link}
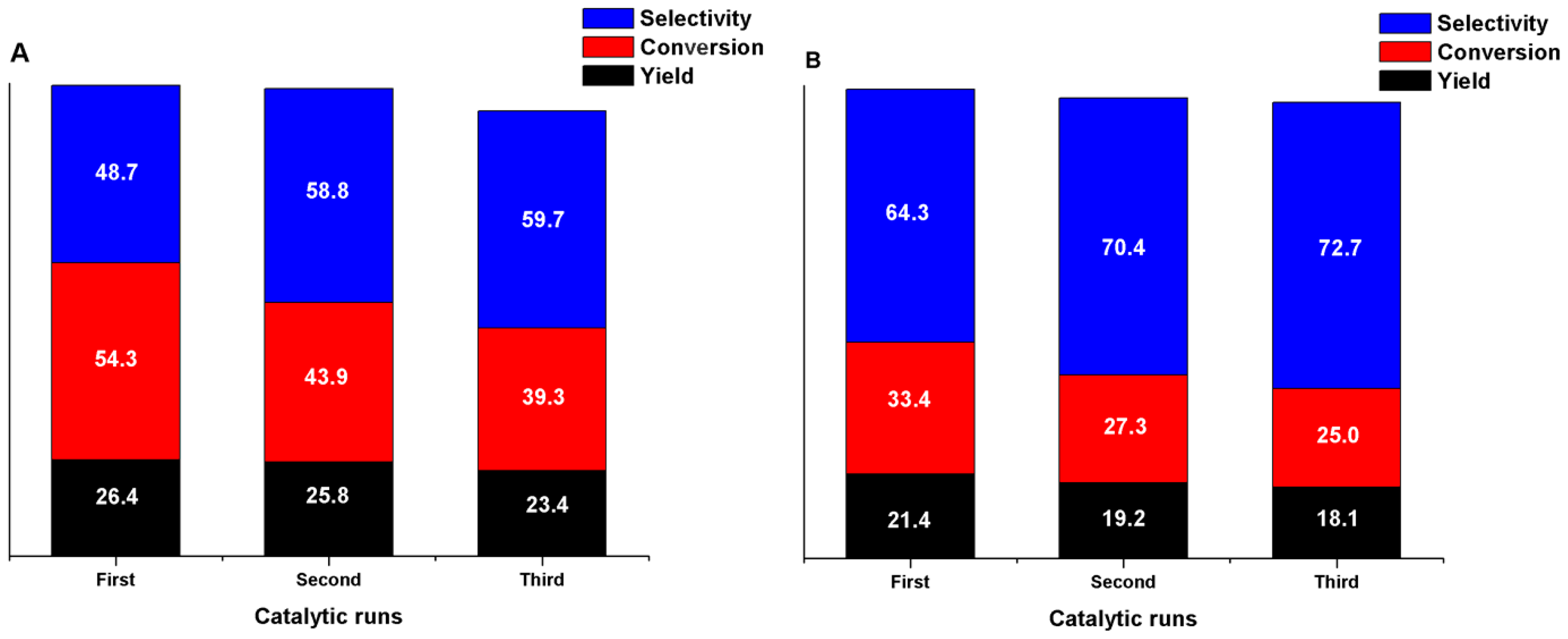{kind=link}
{kind=link}
{kind=link}
Table 1.
XRD and XRF data of calcined MCM-41 materials.
| Sample | d200 (nm) | d100 (nm) 1 | a (nm) 2 | Ce content (wt.%) | Si/Ce Molar Ratio |
|---|---|---|---|---|---|
| Si-MCM-41 | 1.94 | 3.88 | 4.48 | - | ∞ |
| Ce-MCM-Cl | 2.00 | 4.00 | 4.61 | 5.89 | 31 |
| Ce-MCM-NO3 | 2.15 | 4.30 | 4.97 | 5.24 | 36 |
1 Calculated from Equation (2). 2 Calculated from Equation (1).
Table 2.
Template assembly disorder for the as-synthesized MCM-41 materials.
| As-Synthesized Sample | I1080/I1120 1 | I2920/I2850 2 |
|---|---|---|
| A-Si-MCM-41 | 1.994 | 0.731 |
| A-Ce-MCM-Cl | 2.310 | 0.760 |
| A-Ce-MCM-NO3 | 2.024 | 0.747 |
1 Ratio between the intensities of bands around 1080 and 1120 cm−1. 2 Ratio between intensities of bands around 2920 and 2850 cm−1.
Table 3.
Content of Qn-groups and related parameters obtained from 29Si MAS-NMR spectra of the calcined MCM-41 materials.
Table 3.
Content of Qn-groups and related parameters obtained from 29Si MAS-NMR spectra of the calcined MCM-41 materials.
| Sample | Q2 (%) 1 | Q3 (%) 1 | Q4 (%) 1 | Q4/(Q2 + Q3) | Silanol (Si Molar%) 2 |
|---|---|---|---|---|---|
| Si-MCM-41 | 12.4 | 33.1 | 54.5 | 1.20 | 57.9 |
| Ce-MCM-Cl | 19.3 | 21.2 | 59.5 | 1.47 | 59.7 |
| Ce-MCM-NO3 | 0.4 | 25.6 | 74.0 | 2.85 | 26.4 |
1 Calculated from normalization of the area of each peak. 2 Calculated from Equation (4).
Table 4.
Textural properties for the calcined MCM-41 materials.
| Properties | Si-MCM-41 | Ce-MCM-Cl | Ce-MCM-NO3 |
|---|---|---|---|
| Stotal (m2 g−1) 1 | 1032.1 | 909.7 | 907.0 |
| Sext (m2 g−1) 2 | 158.1 | 267.6 | 252.4 |
| Smeso (m2 g−1) 3 | 874.0 | 642.1 | 654.6 |
| Vtotal (cm3 g−1) 4 | 0.92 | 0.87 | 0.90 |
| Vmeso (cm3 g−1) 5 | 0.80 | 0.68 | 0.71 |
| PD (nm) 6 | 3.40 | 3.43 | 3.64 |
| WT (nm) 7 | 1.08 | 1.18 | 1.33 |
1 Total specific surface area. 2 External specific surface area. 3 Mesoporous surface area. 4 Total pore volume. 5 Mesopore volume. 6 Average pore diameter. 7 Wall thickness.
Table 5.
Catalytic performance optimization of Ce-MCM-Cl for benzyl alcohol oxidation.
| Runs | Oxidant | BzOH:Oxidant Molar Ratio | T (°C) | Catalyst Amount (wt.%) | C (%) 1 | S (%) 2 | Y (%) 3 | TON4 |
|---|---|---|---|---|---|---|---|---|
| run 1 | H2O2 | 1:1 | 70 | 10 | 0.80 | 50.0 | 0.40 | 1.85 |
| run 2 | TBHP | 1:1 | 70 | 10 | 33.6 | 65.4 | 22.0 | 74.0 |
| run 3 | DTBP | 1:1 | 70 | 10 | 2.00 | 70.0 | 1.40 | 4.39 |
| run 4 5 | TBHP | 1:1 | 70 | 10 | 15.1 | 89.0 | 13.4 | 27.3 |
| run 5 6 | TBHP | 1:1 | 70 | 10 | 7.00 | 99.0 | 6.93 | 15.4 |
| run 6 | TBHP | 2:1 | 70 | 10 | 25.8 | 74.9 | 19.4 | 56.8 |
| run 7 | TBHP | 1:2 | 70 | 10 | 34.6 | 63.5 | 22.0 | 76.1 |
| run 8 | TBHP | 1:1 | 80 | 10 | 54.3 | 48.7 | 26.4 | 119.4 |
| run 9 | TBHP | 1:1 | 90 | 10 | 60.2 | 41.0 | 24.7 | 132.3 |
| run 10 | TBHP | 1:1 | 80 | 5 | 35.9 | 57.1 | 20.5 | 151.8 |
| run 11 | TBHP | 1:1 | 80 | 15 | 43.0 | 50.0 | 21.5 | 62.2 |
| run 12 7 | TBHP | 1:1 | 80 | 10 | 0.50 | 100.0 | 0.50 | n.a. 8 |
1 Conversion, calculated from Equation (11). 2 BzD selectivity, calculated from Equation (12). 3 BzD yield, calculated from Equation (13). 4 Turnover number, calculated from Equation (14). 5 Acetonitrile as solvent. 6 Isopropanol as solvent. 7 Si-MCM-41 as catalyst. 8 Not available.
Table 6.
Catalytic performance optimization of Ce-MCM-NO3 for benzyl alcohol oxidation.
| Runs | Oxidant | BzOH:Oxidant Molar Ratio | T (°C) | Catalyst Amount (wt.%) | C (%) 1 | S (%) 2 | Y (%) 3 | TON4 |
|---|---|---|---|---|---|---|---|---|
| run 1 | H2O2 | 1:1 | 70 | 10 | 1.30 | 30.8 | 0.40 | 3.23 |
| run 2 | TBHP | 1:1 | 70 | 10 | 23.2 | 75.6 | 17.5 | 49.5 |
| run 3 | DTBP | 1:1 | 70 | 10 | 1.30 | 32.0 | 0.42 | 3.17 |
| run 4 5 | TBHP | 1:1 | 70 | 10 | 14.1 | 92.0 | 13.0 | 30.1 |
| run 5 6 | TBHP | 1:1 | 70 | 10 | 14.5 | 59.7 | 8.60 | 35.8 |
| run 6 | TBHP | 2:1 | 70 | 10 | 19.7 | 80.3 | 15.9 | 48.8 |
| run 7 | TBHP | 1:2 | 70 | 10 | 27.7 | 64.5 | 17.8 | 68.4 |
| run 8 | TBHP | 1:1 | 80 | 10 | 38.5 | 56.5 | 21.7 | 95.1 |
| run 9 | TBHP | 1:1 | 90 | 10 | 52.5 | 40.8 | 21.4 | 129.6 |
| run 10 | TBHP | 1:1 | 80 | 5 | 33.7 | 64.3 | 21.7 | 160.3 |
| run 11 | TBHP | 1:1 | 80 | 15 | 63.7 | 31.9 | 20.3 | 103.5 |
| run 12 7 | TBHP | 1:1 | 80 | 5 | 0.35 | 100.0 | 0.35 | n.a. 8 |
1 Conversion, calculated from Equation (11). 2 BzD selectivity, calculated from Equation (12). 3 BzD yield, calculated from Equation (13). 4 Turnover number, calculated from Equation (14). 5 Acetonitrile as solvent. 6 Isopropanol as solvent. 7 Si-MCM-41 as catalyst. 8 Not available.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aiube, C.M.; Oliveira, K.V.d.; Macedo, J.L.d. Effect of Cerium Precursor in the Synthesis of Ce-MCM-41 and in the Efficiency for Liquid-Phase Oxidation of Benzyl Alcohol. Catalysts 2019, 9, 377. https://doi.org/10.3390/catal9040377
AMA Style
Aiube CM, Oliveira KVd, Macedo JLd. Effect of Cerium Precursor in the Synthesis of Ce-MCM-41 and in the Efficiency for Liquid-Phase Oxidation of Benzyl Alcohol. Catalysts. 2019; 9(4):377. https://doi.org/10.3390/catal9040377
Chicago/Turabian StyleAiube, Carlos M., Karolyne V. de Oliveira, and Julio L. de Macedo. 2019. "Effect of Cerium Precursor in the Synthesis of Ce-MCM-41 and in the Efficiency for Liquid-Phase Oxidation of Benzyl Alcohol" Catalysts 9, no. 4: 377. https://doi.org/10.3390/catal9040377
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.