Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment




Structural Biology Unit, XB20 Drug Design, Department of Pharmacy, University of Groningen, 9700AD Groningen, The Netherlands
*
Author to whom correspondence should be addressed.
Crystals 2020, 10(9), 725; https://doi.org/10.3390/cryst10090725
Submission received: 16 June 2020
/
Revised: 2 August 2020
/
Accepted: 12 August 2020
/
Published: 20 August 2020
(This article belongs to the Special Issue A 10 Years Journey: Chemical, Physical, and Biological Properties and Applications of Crystals)
Abstract
:The parallel advances of different scientific fields provide a contemporary scenario where collaboration is not a differential, but actually a requirement. In this context, crystallography has had a major contribution on the medical sciences, providing a “face” for targets of diseases that previously were known solely by name or sequence. Worldwide, cancer still leads the number of annual deaths, with 9.6 million associated deaths, with a major contribution from lung cancer and its 1.7 million deaths. Since the relationship between cancer and kinases was unraveled, these proteins have been extensively explored and became associated with drugs that later attained blockbuster status. Crystallographic structures of kinases related to lung cancer and their developed and marketed drugs provided insight on their conformation in the absence or presence of small molecules. Notwithstanding, these structures were also of service once the initially highly successful drugs started to lose their effectiveness in the emergence of mutations. This review focuses on a subclassification of lung cancer, non-small cell lung cancer (NSCLC), and major oncogenic driver mutations in kinases, and how crystallographic structures can be used, not only to provide awareness of the function and inhibition of these mutations, but also how these structures can be used in further computational studies aiming at addressing these novel mutations in the field of personalized medicine.
Keywords:
cancer; NSCLC; mutation; kinase; EGFR; KRAS; ALK; BRAF; personalized medicine; molecular modeling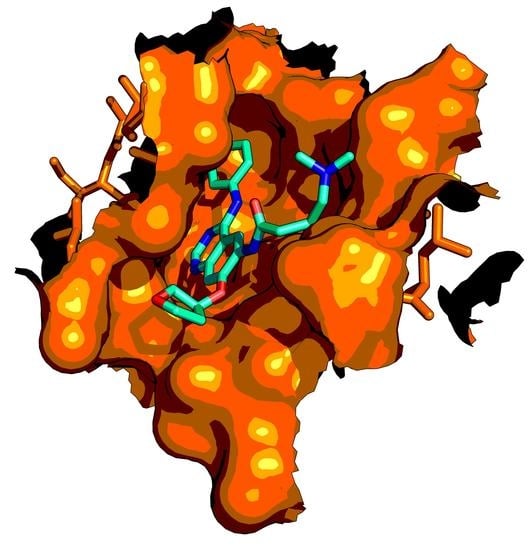
1. Introduction
Cancer is a general term to define a myriad of medical conditions that can affect different tissues in the body. A common characteristic is the abnormal growth of a cell that later develops the ability to spread to other tissues, in a process known as metastasis [1]. Worldwide, it is the second major cause of death, responsible for one in six deaths. Within cancer-related mortality, lung cancer is responsible for more than one million deaths annually—populating the top of the list of deadliest cancer types, a situation that is likely to increase [2]. Lung cancer can be subdivided into Small Cell Lung Cancer (SCLC) and Non-Small Cell Lung Cancer (NSCLC), with the latter being diagnosed in around 85% of lung cancer patients [3]. Advances in molecular techniques unveiled details of genes acting as drivers in NSCLC, identifying a family of proteins responsible for controlling key features of cell development: the kinase family [4].
Genetic events, such as gene amplification, activating mutations, and fusions or rearrangement were found in druggable kinases, including the Epidermal Growth Factor Receptor (EGFR) (~30%), Anaplastic Lymphoma Kinase (ALK)(~10%), and rapidly accelerated fibrosarcoma isoform B (BRAF) (~1.7%), among others (>1%) [5,6]. Despite the Kirsten rat sarcoma viral oncogene homolog (KRAS) often being described as a major oncotarget for lung cancer with the highest incidence (40%) in either smokers or non-smokers, this protein was often overlooked due to its “undruggability”. However, new results show the potential for a new class of KRAS targeting small molecules [7].
The first tyrosine kinase structure was published in 1991, in which researchers from the University of California, San Diego, solved the structure of the cyclic adenosine monophosphate dependent protein kinase (PKA). This structure opened the doors for what would be one of the most explored fields during the new millennium, the pharmacological assessment of kinase proteins [8]. During the early 2000s, the boom in the field brought to the clinic numerous kinase inhibitors, with imatinib, a bcr-ABL inhibitor used in the treatment of leukemia, at the vanguard [9]. With the NSCLC kinome unraveled, there was an emerging need for characterization of these targets, not solely to understand their molecular mechanisms but also for rational drug development.
In parallel with the advances in the medical sciences and structural biology techniques, another related field made huge strides over the years, namely molecular modeling (MM). MM comprises theoretic and computational methods for representing, mimicking, and manipulating molecules: from the small (water) to larger structures, such as cellular membranes [10]. In the context of this review, the highlight is the study of biomacromolecules, such as proteins, due to their pharmacological relevance in diseases. Molecular modeling is capable of analyzing atoms, the smallest unit of a molecule, in a molecular manner, or descend further into quantum chemistry [11]. In drug design, the mechanist approach is the most common due to its ability to describe a diversity of molecules, from water to oligomeric protein complexes, at the level of atoms, bonds, and angles, at high accuracy, in a relatively short time [12]. Despite significant developments within the field of MM, this review will focus on homology modeling, molecular docking, molecular dynamics, and free energy calculations, all of which are techniques within the field of MM.
Homology modeling (HM) is a bioinformatic tool commonly used to obtain the three-dimensional structures of proteins that are so far unresolved experimentally. Unlike the experimental elucidation of protein structures that might be delayed for difficulties with protein expression and further crystallization, HM is based on using proteins with a high level of homology as a template for the desired target. Like any technique, homology modeling has limitations, in this case, it is necessary to have a high-quality template, optimally another protein from the same family, with a high level of sequence homology (>40%) [13]. HM is also limited in the prediction of highly flexible motifs such as loops or tails. Common consensus classifies homology models as low-resolution structures [14]. In addition to contributing to providing structural models of novel structures, HM has also been extensively used to generate three-dimensional models of mutants in the absence of experimental structural information [15].
With a target structure in hand, it is possible to search for potential binding pockets, clefts, or cavities in the structure that would provide anchoring points for other molecules. Tools such as FTmap, AnchorQuery, and PocketQuery are a few of the hundreds that are used to map and recognize druggable clefts on proteins [16,17,18]. A more traditional manner of targeting macromolecules is to study the site in which native ligands bind and aim to design a competitive molecule. Molecular docking is a useful tool for probing libraries of molecules or chemical fragments in a desired binding pocket [19]. The prototype molecule conformation is accessed through the exploration of a large conformational space representing various potential binding modes and the prediction of the interaction energy associated with each of the predicted conformations. These steps are performed in a cyclic process by analyzing each ligand conformation with a specific scoring function, thereby converging to a minimum energy solution. During the conformational search step, the ligand undergoes modification on its torsional (dihedral), translational, and rotational degrees of freedom. This method explores the energy landscape of the conformational space and converges into the most likely binding mode with the minimum energy [19]. However, molecular docking is not limited to protein-small molecules studies, it is also a useful tool to explore protein-protein and DNA-ligand interactions [20,21].
Although homology modeling and molecular docking are useful tools to generate and assess proteins lacking experimental structure and their correlation with molecules of interest, they lack an appropriate assessment of protein dynamics. To address this characteristic, Molecular Dynamics (MD) simulations are an option. MD is based on the computational simulation of molecules, taking into account the physical motion of atoms. In this methodology, each atom of the whole protein structure has its position and velocity determined through Newton’s equations of motion. The first step is to attribute a specific profile to each atom to mimic the temperature and pressure of a physiologic environment. Secondly, by computation of the forces acting on each atom of the whole complex, it is possible to obtain the position and velocity of these atoms at a certain moment. This cycle is repeated through a determined period, specific for each experiment. The final result is the trajectory and progression in time of the receptor, in either the presence or absence of a ligand [22]. As the mass of every atom in the system is known, only the force is required to be calculated to obtain the acceleration. In MD, this force is determined by Force Fields such as AMBER, CHARMM, NOVA, and YASARA [23]. These force fields determine the force acting on the system using molecular interaction potentials, which can be established with the use of quantum chemistry calculations or experimental data aiming to compute how each type of interaction contributed to the global function, and thus to the whole system [23].
Force Field equations take intramolecular forces into consideration, such as the bond between atoms (often considered as “springs”), angles between bonds and dihedrals, as well as intermolecular forces, such as Van der Waals and electrostatic interactions. In this methodology, solvent layers are also computed, and include water molecules. Despite still being limited by computational requirements, MD simulation have massively improved, in both quality and computational system requirements, becoming an invaluable tool in modern science [24].
This review will focus on how structural biology provides insight into targets for NSCLC treatment and their potential interaction with available drugs to address the emergence of resistance mutations in the clinical setting. We will also summarize the use of these structures in computational studies to provide personalized medicine on demand.
2. Kinases: A Structural Overview
The involvement of kinase proteins in key regulatory aspects of cell biology is powered by their ability to modulate other proteins through a phosphorylation reaction, where the γ-phosphate group of Adenosine Triphosphate (ATP) molecules are transferred to selected amino acids of a substrate. Phosphorylation is the most common protein modification in signal transmission, mostly due to its reversible nature by dephosphorylation performed by phosphatases [25].
The nature of the phosphorylated residue guides the classification of kinase proteins into serine/threonine kinases or tyrosine kinases. However, a small group of proteins is able to target both threonine and tyrosine amino acids (dual specificity kinases) and are exemplified by the Dual Specificity Mitogen-Activated Protein Kinase 1(MEK1) and 2 (MEK2) [25,26]. The structure of the kinase domain can be generically depicted as a bilobal structure with a larger C-terminal lobe, presenting several conserved α-helices and β-strands, connected through a hinge to a smaller N-terminal lobe, a composition of a five-stranded antiparallel β-sheet (β1–β5) and a roving α-helix (αC-helix). In Figure 1, the crystal structure of the Hepatocyte Growth Factor Receptor (HGFR or c-MET) in the presence of ATP is used to exemplify the general kinase domain folding (Protein Data Bank (PDB): ID 3DKC).
The N-terminal presents a conserved glycine-rich (GxGxΦG) loop, occurring between the β1- and β2-strands, responsible for positioning the β- and γ-phosphate groups from the ATP molecule for catalysis. The glycine-rich loop is also known as the G-loop or P-loop, with the latter being the most common in kinase-related literature. The β1- and β2 strands also harbor the adenine moiety of ATP, contributing to its stabilization [27]. Within the N-terminal lobe, a characteristic interaction is often observed involving a conserved lysine from β3-strand and a glutamate residue occurring in the αC-helix, this salt bridge is a precondition for the active state. The presence or absence of such interaction is based on the positioning of the roving αC-helix. In the kinases, in this review, once the αC-helix is orientated toward the ATP binding pocket and the salt bridge is present it can be classified as αC-in. The outward positioning of the helix and absence of the lysine-glutamate interaction is known as αC-out [28]. The rotation of the αC-helix is often used in MD studies to classify the resultant structures into either active or inactive [29].
Although the αC-helix positioning and salt-bridge presence are necessary for the full activation of the protein, they are not sufficient. The C-terminal lobe contains a flexible segment, the activation segment, or A-loop, a motif often initiated by a conserved sequence of Asp-Phe-Gly (DFG) whose rotation has major effects on ATP binding pocket occupancy. Once the kinase is in an active conformation, the side chain of the aspartate residue occupies the ATP binding pocket (DFG-in), thereby coordinating a magnesium ion, and the activation segment is presented in an uncoiled, extended conformation [30,31].
Once the crystal structure of inactive c-SRC tyrosine kinase was elucidated, a different DFG conformation was observed [32]. The aspartate side chain was flipped 180° when compared to the active conformation, and swapping positions with the direct neighbor residue, phenylalanine, leading to a DFG-out conformation. Once the DFG-out state is formed, an allosteric pocket is formed contiguous to the ATP binding area. Guarding the new pocket there is a “gatekeeper” residue, a hotspot for resistance mutation against inhibitors in multiple kinases that will be addressed later in this review [28,31].
The kinases EGFR, ALK, and BRAF follow the aforementioned description with specific sequences for mentioned motif represented on the highlights in Figure 1. The highlighted structural motifs featured in Figure 1 are dynamic and have been extensively used not only for kinase state identification but also for the design of inhibitors. Due to their contribution for the proteins’ plasticity, they are commonly analyzed in computation studies for insights in behavior of a protein of interest.
3. Epidermal Growth Factor Receptor (EGFR)
The Epidermal Growth Factor Receptor (EGFR), also known as erbB1 or HER1 (UniProt ID: P00533), is a member of the erbB family of receptor tyrosine kinases (RTKs). Structurally, members of the erbB family (erbB 1-4) consist of an extracellular domain (ectodomain), a membrane-spanning domain, and an intracellular domain. The extracellular domain of the erbB family members is a target for a variety of molecules including most notably Epidermal Growth Factor (EGF), epiregulin (EPR), neuregulin (NRG family), and the transforming growth factor-α (TGF-α) [33,34]. In the absence of extracellular stimulus, all four receptors are found inactive in the cellular membrane, forming homo- or heterodimers upon ligand binding [35].
The intracellular region is further divided into a tyrosine kinase domain (residues 696–976), and a C-terminal regulatory region, residues 977–1210. The tyrosine kinase domain is responsible for the catalytic activity—conversion of an ATP molecule into ADP through cleavage of the bond between the γ and β phosphate groups and the release of a phosphate [33].
The erbB family, in a typical cellular setting, is responsible for translating the external stimulus from ligands into intracellular signaling as depicted in Figure 2. Upon activation of a tyrosine kinase receptor, such as erbB family members, the complex Grb2/SOS is recruited for further binding to inactive Ras proteins, promoting release of GDP and binding of GTP [36]. The activation of downstream RAS/RAF/MEK/ERK and AKT pathways is associated with cell growth and cell survival, as depicted in Figure 1 [37]. Consequently, the aberrant activation of these family members is often linked with a variety of human cancers, most notably NSCLC [38].
EGFR mutations are one of the major causes of NSCLC formation and progression and appear more frequently in never or light smokers, women, and East Asian NSCLC patients [39]. In a healthy cell, the absence of extracellular stimuli drives EGFR monomers into an auto-inhibitory, tethered conformation in which the dimerization arm is buried [40,41]. Activating ligands bind bivalently on EGFR and trigger a large conformational change in the extracellular domain, in which the dimerization arm, a β hairpin-like motif, becomes exposed to the aqueous environment and is thus able to dimerize [42]. Recent reports show the presence of a mixed population of inactive monomers and homodimers in the cellular membrane. In the inactive dimer, the monomers adopt a symmetric configuration and are kept together through interactions of the intracellular domain, the transmembrane domain, and the C-terminal tail of the extracellular domain. In response to ligand binding, a conformational change leads to a rotation of the transmembrane and intracellular domains of the receptor, yielding an active, asymmetric dimer [43].
Elucidation of EGFR kinase domain structure is in compliance with the canonical structure previous mentioned, as seen in Figure 1, with representations of the α-helix rich C-terminal lobe and an N-terminal lobe containing a five stranded β-sheet, its P-loop and the mobile αC-helix [44]. As shown in Figure 3, the plasticity of EGFR can be measured between its active and inactive conformation by comparison of the aforementioned structures with a clear coiled conformation of the activation segment in the inactive form against an elongated loop in the active EGFR, as example [45,46,47].
In analyzing the binding of ATP and its analogues to EGFR, the aromatic N1 nitrogen atom serves as a hydrogen bond acceptor for the backbone amino group from M793 while the N6 amino group serves as a hydrogen bond donor for Q791 (PDB: ID 2GS6) [45,48]. Combination of MD simulations with a Molecular Mechanics Generalized Born Surface Area (MMGBSA) method was employed to elucidate the specific structural elements that stabilize ATP binding in both active and inactive conformations. MMGBSA allows for assessment of binding free energy of protein-ligand complexes with a modest computational input but with proven contributions towards a higher quality evaluation of small ligands binding to biomacromolecules [49]. In the EGFR kinase domain, ATP binding is stabilized by the formation of hydrogen bonds and salt bridges between the negatively charged phosphate group and residues K745, R841, and D855, in both the active and inactive states throughout the simulations. Furthermore, in the inactive state, an ionic bond is formed between the negatively charged phosphate and Mg2+, which is correctly orientated through D855 and N842 [50,51].
The crystal structure of the N-terminal kinase domain of EGFR resembles that of other RTKs such as the Insulin Receptor Kinase (IRK) and the Fibroblast Growth Factor Receptor Kinase (FGFRK) [52]. However, the C-lobe kinase domain adopts a prototypical structure and activation mechanism, not previously observed in other RTKs. Typically, RTKs require phosphorylation on a conserved tyrosine residue located on the activation loop, which causes the conformational shift of the activation segment. This conformation is compatible with the active receptor and permits ATP binding [50]. However, crystallographic studies of the kinase domain of EGFR showed that the A-loop often adopts an active conformation and does not require phosphorylation of the conserved Y869 residue on the activation loop to become activated [53]. Consistent with this, a mutagenesis study provided evidence that Y869F substitution had no adverse effect on kinase activity [54].
In addition to the conformational changes required for EGFR activation, a dimerization process is also necessary. As previously mentioned, inactive EGFR is often found as symmetric dimers while activation is characterized by an asymmetric complex of two monomeric units. In the case of an EGFR-EGFR dimer, one of the units acts as an active partner contributing with its N-terminal while the other is the passive partner, contributing with its C-terminal. The dimer interface is governed by hydrophobic interactions involving residues L704, I706, L760, L782, and V786 from the active partner, and I941, Y944, M945, V948 and M952 from receiver kinase unit contributing with its C-lobe. The asymmetric dimer formation results in the transphosphorylation between the kinase domains and their activation [45].
Early attempts in crystallization of the monomeric inactive EGFR TK domain were challenging due to the spontaneous dimerization of EGFR at high concentrations. Wood et al. elucidated the first inactive structure in the presence of EGFR inhibitor lapatinib [55]. Lapatinib is an ATP competitive drug with a 4-anilinoquinazoline scaffold designed to interact with the hinge motif. Lapatinib explores the allosteric pocket guarded by the gatekeeper residue with an extended moiety using the hydrophobicity of phenolic rings substituted with chlorine and fluorine atoms to form Van der Waals interactions as depicted in Figure 4 (PDB: ID 1XKK) [56]. The inactive conformation closely resembles the structures of inactive Src and Cyclin-Dependent Kinase (CDK) proteins, mainly due to the positioning of the αC-helix [55].
A more elegant methodology has been employed to study the inactive TK domain in which the ATP nucleotide analog AMPPNP is coupled with the incorporation of the V948R point mutation (PDB: ID 2GS7). This mutation takes place in the dimer interface and disrupts the hydrophobic interactions between the active and the receiver units by introducing the polar residue arginine. Consequently, the mutation restricts the dimerization of erbB members and allows for studies of monomeric wild-type (WT) enzyme [45].
The first crystallographic structure obtained in the active state was WT EGFR in the apo form (PDB: ID 1M14). In the active EGFR TK domain conformation, the αC-helix becomes highly structured, adopting an αC-in conformation allowing the formation of the catalytically important salt bridge (E762- K745). Furthermore, the A-loop is positioned away from the active site, allowing substrate access to the catalytic site [52].
The increased availability of EGFR structures contributed to more in-depth computational studies to elucidate the dynamics of EGFR, both as a monomer and as a member of the activated dimer. A set of 25 EGFR crystallographic structures were used to build an ensemble structure that would be further submitted to molecular dynamic simulations and analyzed as standalone kinase domains or as active/passive components of an EGFR homodimer. This extensive study showed that asymmetric dimerization might not only stabilize the active conformation, thus organizing the ATP binding site, but also affect regions distal from the dimerization interface such as the A-loop. It is relevant to highlight concerns about the duration of the simulations, here consisting of 100 ns, which are potentially not enough to accurately model the intrinsically disordered intermediate between the active and inactive conformations. Songtawee et al. show that MD simulations can sample different experimentally obtained conformations by comparing the output of MD simulations with the aforementioned structures, the high level of similarity between MD outputs and crystallization data strengthen the reliability of computational methods such as molecular dynamics to explore kinase dynamics [58].
In the catalog of somatic mutations in cancer (COSMIC) database more than 594 types of mutations have been registered in the EGFR TK domain, of which the majority (93%) are located in exons 18,19,20 and 21. The most common class I activating mutations are deletions in exon 19, spanning through residues 746–750, accounting for approximately 44% of all EGFR mutations [59,60]. Class II mutations comprise of activating, and resistance-acquiring point mutations, and classical variants are exemplified by the activating L858R (39.8%), the resistance-acquiring T790M, and the rare sensitizing mutation G719A/C/D/S (3%). Class III mutations include exon 20 in-frame insertions and duplications [60,61,62]. Table 1 summarizes EGFR mutations of clinical relevance and their reported resistance or responsiveness to approved drugs.
The two most commonly found activating mutations, L858R and exon19 deletions account for over 90% of all sensitizing mutations and thus have been termed “classical” activating mutations [88]. L858 is located towards the N-terminus of the activation segment, directly neighboring the DFG motif. When in the inactive conformation the L858 is part of a hydrophobic cluster composed of hydrophobic and/or aromatic residues, F723, L747, M766, and L788, responsible for stabilizing the coiled stated of the activation segment. Structures of L858R mutants are available with a variety of ligands, from ATP analogs (PDB: ID 2EB3) to natural products (PDB: ID 2ITU). The multiple ligand complexes all share the same active conformation with an uncoiled A-loop and αC helix-in conformation [44,65].
The L858R point mutation is characterized by a 10 to 100-fold increase in the affinity for TKIs [75,89]. A structural comparison of WT/AMPPNP (PDB: ID 3VJO) with L858R/AMPPNP (PDB: ID 2EB3) identified that the side chain of F723 protrudes outwards towards the active site and interacts with R748. This enlarges the active site cleft, which allows faster release of ATP, while also making the active site more attainable for TKIs [65].
Further information on the L858R mutation was provided by Ding et al. through long duration (500ns or more) atomistic MD simulations, and experimental data. The MD studies provided evidence on the process of switching from the inactive state into an active conformation. Simulations involving bound gefitinib, (also an amino-4quinazole EGFR inhibitor) to either the WT or L858R show higher binding energy to the active than to the inactive conformation. This data explains why inhibitors such as gefitinib are more successful in the presence of activating mutations than in gene amplifications in the treatment of EGFR positive NSCLC patients. Since L858R drives the kinase domain into an active conformation, disturbing the equilibrium between active and inactive, it contributes to drug binding by providing a more accessible (open) conformation for the drug [90].
Deletions on exon19, and more specifically the common Δ746ELREA750 (Del19) take place in the N-terminal loop, in between the β3-strand and the αC-helix. MD studies performed by Tamirat et al. on the inactive and active form of the Del19 mutant found that the active state is favored due to stabilization of the E762-K745 salt bridge. This results from a decrease in the β3-αC loop flexibility, which stabilizes the αC-helix in the active αC-in conformation. Furthermore, the Δ746ELREA750 deletion causes an inwards conformational shift of the αC-helix, which disrupts the hydrophobic cluster in between the αC-helix, thereby promoting the activation of the TK domain [86].
Similarly, to L858R point-mutants, Del19 EGFR mutants display a decreased affinity for ATP binding (KM) and a lower Ki for first-generation TKIs such as erlotinib [66,86]. However, exon 19 deletions and consequent residue insertions display large heterogeneity and thus exhibit differential drug sensitivity [85,91]. Unfortunately, so far, there is no structure available for any of the exon19 deletions.
Upon identification of the aforementioned mutations as major biomarkers of NSCL cancer, there has been a keen interest in the development of drugs able to inhibit the enhanced kinase activity provided by these mutations. Efforts culminated with the development of first-generation inhibitors gefitinib and erlotinib, both reversible ATP competitive inhibitors sharing a 4-anilinoquinalzoline scaffold [92].
As pictured in Figure 4, both drugs are capable of interacting with M793 similarly to the adenosine ring of ATP, through a hydrogen bond with the residue backbone. The presence of the 3-chloro-4-fluoro aniline allows gefitinib to explore the allosteric hydrophobic pocket through interactions with L788, and T790. The methoxy moiety is within Van der Waals contact of G796 (PDB: ID 4WKQ). The 6-propyl morpholine ring on gefitinib extends into an area exposed to the solvent and was implemented to improve pharmacokinetic properties [93].
Compellingly, gefitinib has also been identified to bind in a second conformation to the L858R mutant. The second conformation exhibits a 180° rotation of the aniline ring, which allows the chloride substituent to interact with the sidechain of R855 via a halogen bond through the coordination of a water molecule. In both cases, the ether group extends outwards from the ATP binding pocket towards the aqueous environment [44].
In the active EGFR conformation, the erlotinib anilinoquinazoline ring is stabilized by seven hydrophobic (L718, A743, L788, L792, P794, and L844), three polar (T790, Q791, and T854) and three charged residues (E762, K45, and D855), while the solvent-exposed substituents interact with F795 and G796 (PDB: ID 1M17) [94]. In the inactive state, erlotinib is stabilized by the seven hydrophobic interactions stabilized in the active state and by an extra hydrophobic interaction from V726. Furthermore, it is stabilized by the same three polar residues and by three charged residues (K745, D800, and D855) [95].
Initial studies suggested that both drugs recognize the active conformation of EGFR. However, computational approaches indicated that erlotinib can bind to both active and inactive, conformations [96]. Crystallographic studies were able to co-crystallize the inactive TK domain in complex with erlotinib (PDB: ID 4HJO) providing further crystallographic evidence that erlotinib can bind to both states [96].
Erlotinib and gefitinib both showed greater potency for L858R EGFR against WT EGFR. Notably, gefitinib binds 20-fold more strongly to EGFR L858R mutant against WT EGFR and thus, preferentially inhibit L858R positive cancer cells, leading to their consequent apoptosis and cancer remission while sparing healthy cells [44].
Afatinib, a second-generation approved in 2013, is an irreversible ATP competitive anilinoquinazoline, which harbors an acrylamide reactive group as shown in Figure 5. Crystallographic studies by the Solca laboratory on the WT EGFR in complex with afatinib, (PDB 4G5J) showed a hydrogen bond between M793 and its core quinazoline ring but, most importantly, the electron density map identified a covalent bond formed between C797 and the acrylamide group in the active state of the kinase [77].
G719X (where X can be alanine, aspartic acid, cysteine, or serine) is a rare sensitizing point mutation on exon 18 accounting for approximately 3% of all EGFR TK domain mutations, with G719S (PDB 2EB2) being the most common variant [65,97]. G719 is located on the P-loop connecting strands β1 and β2, contributing to the stabilization of ATP phosphate groups. Furthermore, G719 is part of the hydrophobic cluster found during the inactive conformation creating a helical turn that generates a steric hindrance that helps position the αC-helix from the active site in a αC-out conformation. Deviation from glycine residues is not tolerated in the inactive state due to the disruption of the hydrophobic cluster thus favoring the active state. The structure of the G719S mutant in complex with AMP-PNP (PDB: ID 2ITN), gefitinib (PDB: ID 2ITO), and a staurosporine analog (PDB: ID 2ITQ) are available. Unlike L858R and del19 mutations, the G719X mutation does not promote receptor dimerization but rather influences intrinsic structural components favoring receptor activation [66].
The presence of a serine residue at position 719, shows to be sensitive to gefitinib with an inhibitory concentration (IC50) of 0.18 µM against 1.04 µM found for the WT EGFR. However, the presence of a secondary mutation at position 790 (T790M) increases the IC50 by 10-fold (IC50 = 1.86 µM). Interestingly, analysis of the constant of dissociation (Kd) for the double mutant (Kd = 5.6 nM) shows a tighter binding of gefitinib when compared to either the single mutant (Kd = 31.9 nM) or the WT (Kd 14.2 nM), indicating that double mutant diminished affinity for gefitinib is not due to reduced drug binding. However, a plausible explanation is raised by the Kinect studies, which show a ratio between the kinase activity (kcat) and the Michaelis–Menten constant (Km) comparable to the WT, indicating that the mutation T790M restores the nucleotide binding ability [65].
Treatment of L858R, Del19, and G719X with first- and second-generation TKIs show improved overall survival when compared to classical chemotherapy [69,98]. However, after a median of nine to thirteen months, EGFR positive NSCLC treated with first- or second-generation typically acquire the resistance mutation T790M [79,83]. The T790M resistance mutation is analogous to the imatinib-resistant bcr-ABL fusion harboring the T315I mutation and it accounts for more than 50% of all EGFR TKI- resistant mutations [99].
T790M is referred to as the gatekeeper mutation and it is located at the back of the ATP-binding site [67]. Just like L858R, the T790M mutation stabilizes the active conformation of the TK domain, but via a different mechanism as shown by free energy studies. M790 is part of a hydrophobic cluster formed in the back of the ATP binding site of the N-lobe and interacts with M766, located on the αC-helix. This hydrophobic interaction further extends towards the F856 and the catalytically relevant DFG motif [45,100]. Once the methionine replaces the threonine residue, there is a stabilization of αC-in conformation. Thermodynamic integration (IT) analysis is a theoretical method able to correlate free energy divergence between two given states of a system even in different spatial coordinates arising due to long simulation durations [101]. Park and colleagues’ combination of IT with MD simulations showed that the T790M stabilizes isoenergetically the active (αC-in) and intermediate disordered form of active apo EGFR, while disfavoring the inactive. However, it does not repress αC-intrinsic disorder and, consequently, is not believed to favor dimerization. Combination of MD simulation with MMGBSA in both active and inactive, in presence of the T790M alone or in presence of L858R shows that only erlotinib binding energy is decreased by the gatekeeper mutation while lapatinib is not affected [102].
The formation of resistance to first and second-generation TKIs by acquiring T790M is believed to emerge due to changes in the stability of ATP and drug binding [103]. It has recently been proposed that T790M resistance is a consequence of the restoration of ATP sensitivity similar to that of WT EGFR [50,104]. Comparisons of the L858R mutant with the WT EGFR showed that the variant amplifies the conformational landscape of the kinase domain. Interestingly, the co-existence of L858R with T790M as a double mutant presents a conformational landscape similar to the WT EGFR. Such a similar conformational profile is associated with the restored ATP affinity of the double mutant, being comparable to the WT [50].
Resistance also stems from steric hindrance clashes from the replacement of threonine by methionine within the ATP binding pocket, which contributes to the reduced binding of reversible TKIs [67]. However, more recent studies show that both gefitinib and erlotinib retain low nanomolar binding affinity towards the T790M mutant, proving that drug binding is still possible despite being limited [100]. In a scientific setting, limited binding affinity might still correlate with response to a drug yet, clinical assessment of these results might lead to a decision to withdraw the drug since the advantage of targeting the mutated kinase rather than its wild-type specie is lost. The therapeutic window for oncology drugs is a major point in medical decision-making [105].
Following the observation that first-generation drugs retain binding affinity for the T790M mutant free-energy studies found that gefitinib binding to the T790M and L858R are more energetically favorable than binding to WT EGFR, in a range of -12 and -15 kcal/mol, respectively. In addition, MD studies have shown that gefitinib binding alters the confirmation of the αC-helix whilst the activation loop maintains an active conformation. Overall, it has been observed that the T790M mutation does not ablate gefitinib binding as experimentally demonstrated by Gajiwala et al. through in vitro phosphorylation analysis and of T790M in the presence of L858R and in crystal structures (PDB: IDs 3UG2, 4I22) [103,104].
Emergence of T790M can also follow the primary G719X activating mutation, leading to a double mutant with synergistic interactions that stabilize the active conformation. Analysis of the gefitinib-double mutant complex (PDB: ID 3UG2) showed that gefitinib binds to the double mutant similarly to that of the WT EGFR [65]. Intriguingly, gefitinib binds the double mutant 6-fold stronger (Kd = 5.6nM) than the single G719S mutant (Kd =31.9 nM) providing evidence that binding is still possible and it is not prohibited via steric hindrance. However, the G719S does become 10-fold less sensitive to gefitinib when acquiring the T790M resistance mutation. When in the presence of AMPPNP (PDB: ID 3VJN), the methionine in position 790 forms a more stable structure with AMPPNP when compared to the WT. An important observation for future drug development is that the double mutant also decreases the size of the hydrophobic cleft formed between L718 and G796, and therefore future drug prototypes that aim to treat the T790M mutant should avoid this cleft [65].
Another piece of evidence proving that steric hindrance does not ablate drug binding is that afatinib has been co-crystallized in the active receptor conformation in presence of the T790M mutation (PDB: ID 4G5P) [77]. In vitro kinase assays identified that afatinib has 100-fold higher potency against the L858R/T790M double mutant when compared to gefitinib [77]. However, the concentrations necessary to bring an inhibitory effect to the T790M point-mutant might not be attained in the clinic under standard dosing regimens [106].
Due to the need to find a viable treatment option for the emergence of the resistant T790M mutation, third-generation TKIs have been developed. Osimertinib, which molecular structure is disclosed in Figure 5, is an irreversible EGFR inhibitor comprising a 2,4-diarylaminopyrimidine scaffold utilized for the L858R/T790M or exon19deletion/T790M mutants and shows a 200-fold preference for the double mutants over the WT EGFR [107]. A combination of the information provided by the crystal structure of osimertinib with WT EGFR (PDB: ID 4ZAU) and computational tools helped to elucidate the binding mode of osimertinib to the inactive state of the TK domain.
Yosatmadia et al. modeled osimertinib binding using the previously known crystal structure of T790M EGFR in complex with dacomitinib (PDB: ID 4I24) [103,107]. The L858R, T790M, and L858R/T790M mutations do not directly contribute to osimertinib binding but do favorably alter the TK domain conformation and dynamics that enhance drug binding [107]. Osimertinib engages in a hydrogen bond with the M793 backbone while Van der Waals interactions contribute to the drug orientation within the pocket. Specifically, the phenyl ring sits in a hydrophobic sandwich between L718 and G796, and the methyl-indole moiety is within Van der Waals distance from G719, F723, and V725. The interactions from the indole ring, especially with the aromatic side chain of F723, are believed to be responsible for the positioning of the P-loop towards the ATP binding pocket. As an irreversible ligand, osimertinib is capable of the formation of a covalent bond with C797 [107].
Unfortunately, similar to the emergence of first-generation resistance mutations, acquired resistance develops in response to osimertinib and afatinib treatment [108]. The most common resistance following T790M is C797S. The replacement of C797 with serine ablates the covalent binding ability of these irreversible drugs and thus confers resistance in approximately 15–25% of patients treated with osimertinib [74,109].
Uchibori et al. were able to identify a treatment option for a C797S/T790M/activating mutation triple mutant by running computational simulations and structure-activity relationship analyses that yielded brigatinib, a dual EGFR/ALK TKI, as a therapeutic agent [110]. Brigatinib binding to the C797/T790M/activating mutation EGFR ATP-binding site resembles that of EML4-ALK (PDB: ID 6MX8). Kinase studies identified that the inhibitor is more potent against Del19/T790M/C797S than in L858R activating mutations. When screened against different cell lines presenting the triple mutants, brigatinib was the only drug to inhibit EGFR phosphorylation and its downstream signaling. Effect of brigatinib on the triple mutant is improved once in combination with cetuximab, an anti-EGFR antibody [110].
L718 mutations significantly increase the IC50 value to osimertinib, with L718Q conferring the greatest resistance potential [64]. As previously mentioned, L718 is located on the P-loop in the proximity of the ATP-binding site and is important for the correct coordination of osimertinib during the covalent bond formation with C797. Substitution of leucine with glutamine sterically inhibits osimertinib binding due to the introduction of the larger, charged side chain, which decreases local hydrophobicity at the point of contact of osimertinib and spatially restricts its binding [64].
The L718Q mutant, in combination with L858R, confers resistance to gefitinib in either presence or absence of the T790M [64]. The L718Q mutation also confers gefitinib resistance to C797S positive NSCLC. Following the substitution of leucine with glutamic acid, the local hydrophobicity is disrupted, which impairs gefitinib binding [64]. Interestingly, although the L718Q point-mutant confers resistance to osimertinib and gefitinib, a patient with advanced metastatic NSCLC harboring the L858R/L718Q double mutant was successfully treated with afatinib, indicating a furan moiety might be suitable in the presence of an L718 mutation [81].
L792F/Y/H mutations constitute approximately 1.5% of resistance to osimertinib, with the L792H conferring the most remarkable resistance [64]. Structural and mutagenesis analysis of the complex of WT EGFR/osimertinib (PDB: ID 4ZAU) showed that replacement of L792 with the aforementioned amino acids sterically inhibits the binding of osimertinib to the ATP binding cleft thus disrupting the correct orientation of the inhibitor and its pharmacological action [111,112].
The G796C/D/R mutation had previously been identified to hamper the potency of erlotinib without further mechanistic disclosures [63]. The G796D mutation has also been identified to confer resistance to osimertinib due to the steric hindrance effect imposed by the replacement of glycine to aspartic acid thus, impairing the formation of the previously mentioned hydrophobic sandwich [71].
A novel G724S point mutation, identified in a set of NSCLC patients and linked with resistance to osimertinib was studied using computational modeling [113]. Interestingly, the G724S only confers osimertinib resistance to exon19del and not L858R mutants [82,113]. Resistance arises through the complementary action of exon19del, that reduces β3-αC loop flexibility, and the G724S point mutation, which lead to the destabilization of the αC-in conformation [82].
EGFR mutations are clearly rising faster than drug development can follow as seen by emerging clinical resistance to osimertinib and an associated poor prognostic for patients. Repurposing of already approved inhibitors can be of use as an accelerated methodology for clinicians as demonstrated for allopurinol and methotrexate, both initially developed to treat cancer but later repurposed for gout and rheumatoid arthritis [114]. The process of repurposing, despite being faster that following the pathway of developing a new molecular entity, remains hindered by the multitude of drugs to be assessed against a myriad of diseases, indicating a clear need for improvement on its methodological approach, opening a venue for application of in silico high throughput screening [115].
Computational tools, as the ones previously described, can be used for a rapid assessment of novel mutations as shown by Kemper et al. with the triple mutation exon19del/T790M/P794L. A team of clinicians and structural biologists were faced with the emergence of a novel mutation, from a proline to a leucine at position 794, in addition to an exon 19 deletion and T790M. Despite the presence of the T790M, the patient was responding poorly to osimertinib. Through docking studies, the Molecular Tumor Board compared the drugs osimertinib and afatinib, providing insight in how afatinib could retain binding affinity, thus being a suitable therapeutic option [87]. This is an example that drug repurposing associated with change of therapeutic indication of a medication, may also be used to reconsider drugs that were previously discarded.
4. Anaplastic Lymphoma Kinase (ALK)
ALK is a RTK that is subject to aberrations in 4–5% of NSCLC cases (UniProt: ID Q9UM73) [116]. The mutations cause ALK to become an essential growth driver of the tumor. This renders NSCLCs as part of the ALKoma entity [117]. The ALK protein was first discovered in 1994 as a fusion protein in a Non-Hodgkin’s Lymphoma subtype and has since been classified as a member of the insulin receptor tyrosine kinase (IRK) superfamily [118,119,120]. Subsequently, an increasing number of genetic aberrations have been identified in ALK with many of these mutations being caused by chromosomal rearrangements often driving to the hyperactivation of ALK. This leads to the overstimulation of downstream pathways that are involved in cell survival, differentiation, and apoptosis, resulting in oncogenesis [121].
There is an increasing number of treatments available for ALK-positive NSCLC in the form of ALK specific TKIs. However, most of them are met with drug resistance mutations after prolonged treatment. Computational studies can aid in the understanding of conformational changes due to aberrations in the ALK protein. This would be a key step towards personalized medicine for ALK-positive NSCLC patients by filling the gap brought by novel mutations and their response to available treatment.
Unlike many of the IRK subfamily members, the normal physiological function of ALK is yet to be fully elucidated. There is evidence that ALK plays a role in growth and fetal development of both the central nervous system (CNS) and peripheral nervous system (PNS) [119,120,122,123,124,125,126,127,128,129]. Furthermore, evidence indicates that constitutively active mutated ALK proteins induce neuronal growth and differentiation [130,131].
Like the physiological role of ALK, the downstream signaling pathways of the protein remain to be fully deciphered. A membrane-bound receptor, such as ALK, receives and transfers extracellular signals by activating intracellular signaling pathways. Upon ligand binding, the wild-type ALK receptor homodimerizes and activates via trans-autophosphorylation of tyrosine residues within the kinase activation loop. Docking sites for downstream Src homology 2 (SH2) and phosphotyrosine-binding domain (PTB)-containing effector and adaptor proteins are located within the cytoplasmic domain [132].
Ligands that activate ALK are under tremendous debate. Some scientists show that the growth factors pleiotrophin and midkine induce ALK activation, whereas other refute this or propose that the small secreted, FAM150 peptides generate ALK activation [132]. What does have a consensus is that upon ligand binding, ALK can induce a multitude of pathways. These include the JAK/STAT, PI3K/AKT/mTOR, SOS/RAS/MEK/ERK1/2, and PLC-γ/DAG/PKC pathway as depicted in Figure 2 [133,134,135]. Cell growth and proliferation are induced via the PLC-γ/DAG/PKC and SOS/RAS/ERK1/2 pathways, while cell survival is directed through JAK/STAT and PI3K/AKT/mTOR. The diverse pathogenic signaling profile of ALK is caused by the different aberrations within the protein and the large number of pathways ALK can induce [133,135,136].
ALK consists of 1620 amino acids in its native full-length single-chain receptor form. Multiple subdomains encompass the 1030-residue extracellular region, including the LDL-A domain (low-density lipoprotein class A domain), a MAM (meprin, A5, mu) domain, and a glycine-rich region [119,120]. The cytoplasmic domain contains 563 residues and includes the catalytic kinase domain. ALK was first identified in a chimeric protein, where the catalytic kinase domain of ALK was fused with the extracellular region of nucleophosmin (NPM) [118]. NPM mediates constitutive dimerization of the protein, thereby inducing constant activation via trans-autophosphorylation of the chimeric protein [117]. Currently, nearly 30 fusion partners with ALK have been identified of which the most common in NSCLC is the echinoderm microtubule-associated protein-like 4 (EML4)-ALK gene fusion [137].
EML4-ALK is the main rearrangement form in ALK-positive NSCLC, with 3–7% of all cases [138,139]. EML4-ALK positive NSCLC patients share clinical characteristics with patients who harbor activating mutations in EGFR, both groups are non- or light smokers and manifest adenocarcinoma histology [88,116,117,140]. Notably, the EML4-ALK fusion gene and mutations in EGFR or Kirsten rat sarcoma virus (KRAS) are mutually exclusive, albeit with rare exceptions [141,142,143].
The protein resulting from the fusion gene consists of the amino-terminal portion of EML4-fused to the intracellular region, including the catalytic domain, of ALK. The chimeric protein is constitutively active, which is caused by the coiled-coil trimerization domain of the EML4 portion [144]. This results in a transforming ability in a manner dependent on the associated upregulation of RTK activity [117].
Furthermore, there are not only variations within the EML4-ALK fusion oncogene, but rearrangements of the ALK gene with a different partner have also been reported, such as kinesin family member 5B (KIF5B)-ALK [145], Huntington-interacting protein 1 (HIP1)-ALK [146], translocated promoter region (TPR)-ALK [147], baculoviral inhibition of apoptosis protein repeat-containing 6 (BIRC6)-ALK [148], and many more [149]. Most of these fusion proteins rely on ALK catalytic activity. Furthermore, recent evidence suggests that the different fusion partners influence kinase activity, transforming ability, protein stability, and notably ALK TKI drug sensitivity [150]. The development of a wide range of ALK specific inhibitors is therefore imperative.
Understanding the structure and mechanism of the ALK catalytic domain aids in the development of ALK specific TKIs, on the road towards personalized medicine. An important step in understanding the unique substrate specificity of ALK was the release of its x-ray crystal structure in 2010 by Lee et al. (PDB: ID 3L9P) [132]. It was to be expected that ALK’s catalytic domain is similar to other IRK family members as there is a high degree of structural and sequence conservation amongst this kinase family; 45% sequence identity and 62% sequence conservation over 280-290 residues between ALK and IRK/IGF1RK. Indeed, ALK has the canonical kinase domain architecture and topology, with the ATP-binding site residing at the interlobar cleft. As with EGFR and BRAF, the cleft is formed by a smaller N-terminal lobe (N-lobe) and a larger C-terminal lobe (C-lobe) connected through the hinge region, as portrayed in Figure 6 [132].
Although ALK is believed to follow the same switch between active and inactive, Lee et al. determined the structure of ALK in an intermediate conformation that, despite having the αC-in, characteristic of an active conformation, the A-loop is not fully extended as previously described for similar kinases [132]. Accelerated MD (aMD) is an enhancement from the classical molecular dynamics (MD), on this method the energy barriers between two different states of a system are reduced, thus improving the conformational sampling to be analyzed, boosting free energy calculations [151]. Analyzing an apo WT ALK system with aMD suggests a smooth switch from the αC-helix from the in into the out state, as would be expected in a cellular setting [132,152]. The juxtamembrane segment at the N-terminus, residues 1096-1103 (YCFAGKTS), contains a β-turn motif. This β-motif packs against the distal end of the αC-helix, on the opposite side of the β-sheet. This β-turn motif is not observed in the structures of IGF1RL or IRK and seems to be unique to ALK although its contribution to the general structure is yet to be disclosed [132].
The most solvent-exposed A-loop tyrosine residue, Y1282 is positioned below R1284. The other tyrosine residue, Y1283, is flipped towards the active site and sandwiched between hydrophobic residues of the proximal, M1273 of αAl, and distal, M1290/L1291, ends of the A-loop [132]. Y1278, critical for ALK transforming activity, forms a hydrogen bond with the backbone amide nitrogen of C1097 of the N-terminal β-turn motif. Y1278 is separated from the second and third tyrosine in the ALK A-loop sequence by the RAS (Arg-Ala-Ser) motif [132]. This RAS sequence motif has been predicted to be a distinguishing A-loop feature, contributing to ALK A-loop autophosphorylation efficiency and the preference for Y1278. The RAS motif is not present in IGF1RK/IRK, instead, they have the ETD (Glu-Thr-Asp) motif [153].
The ALK catalytic domain is intrinsically autoinhibited due to the aforementioned β-turn motif in the N-lobe [132]. The hydrogen atom of the C1097 amide backbone forms a hydrogen bond with the hydroxy group of Y1278, which prohibits the phosphorylation of the latter residue [154]. The A-loop also plays a role in the autoinhibition of ALK. A short, two helical turned α-helix, named αAL (residues 1272-1279; GMARDIYR), is found at the proximal portion of the A-loop immediately following the phenylalanine of the DFG-motif. αAL is packed orthogonally below the αC-helix. This prevents ALK from relaxing to its active conformation [154].
The previously mentioned accelerated molecular dynamics study supported the findings of Lee et al. that wild-type ALK resides in an autoinhibitory state. It was published that the A-loop is not fully extended and, thereby, blocks the space for peptide binding. Furthermore, the A-loop blocks the ATP-binding site when deviating inward. It thereby adopts a closed conformation that is similar to other inactive tyrosine kinases [152]. IRK family members form a pseudosubstrate with their A-loop in the inactive and unphosphorylated form. Furthermore, the phosphor-acceptor site (P-site) of the substrate peptide binding region is bound in a cis-auto-inhibitory fashion to the second A-loop tyrosine residue [132]. However, during the simulation ALK adopts a DFG-in conformation, where D1270 points inward, allowing for more space in the ATP binding site, an indication of an active conformation [154]. Furthermore, computational studies indicate that the more active A-loop conformation does not display the pseudosubstrate cis-inhibitory pose as it does in other IRK family members. This means that dormant wild-type ALK resides in a unique partially inactive tyrosine kinase conformation as it lacks the negative regulatory structural elements consistent with a fully inactive kinase conformation [132,152].
The x-ray crystal structure in combination with the aMD simulation demonstrates that the hydrogen bond network is essential in the construction of the conformational framework [132,152]. Amongst the polar face of αAL, basic residues along with K1285 draw the proximal portion of the A-loop downward toward the lower and inner aspect of the αC-helix, stabilizing this relative position in the partial inactive conformation of ALK. D1163 of the αC-helix forms two hydrogen bonds with the positively charged residues of R1275 and R1279 on the face of αAL [132,152]. These hydrogen bonds are considered the key factors in the stabilization of the αC-in formation in the partially inactive state [152]. As for the semi-closed A-loop conformation in this state, Q1159 of the αC-helix and Y1283, form a hydrogen bond that results in a sharp U-turn at the end of αAL. As a result, the A-loop cannot be fully extended or adopt a fully closed conformation. The electrostatic and hydrophobic interactions within the protein further cement the partially inactive conformation. The hydrophobic stem consisting of M1290, L1291, and P1292, is stabilized by the hydrophobic cluster of M1296, F1301, and L1339 in combination with the C1288 and L1291 hydrogen bond [152].
As previously discussed, the apo ALK structure rarely adopts an active conformation and the dormant protein resides in a partially inactive state. Upon binding of ATP, aMD simulations show that the structure adopts another energetically favorable conformation comparable to an obtained structure (PDB: ID 3LCT). This new conformation adopts an active-like state. Generally, ATP is used by protein kinases to phosphorylate their specific substrate with their phosphate groups [152]. However, recently ATP has also been reported as an allosteric modulator [155,156,157,158]. This active-like state of ATP bound ALK is also the result of such allosteric effects [152].
Whereas the apo kinase domains mostly transition between the αC-in and αC-out conformation, ATP bound ALK has a more compact active site as the triphosphate moiety positions the αC-helix towards the active site [152]. The triphosphate moiety engages in electrostatic interactions with K1150 thereby anchoring the side chain of said residue to E1167, protecting the stability of the salt bridge partner. Furthermore, a hydrogen bond network is formed between ATP, the charged R1275, and D1063, pulling the αC-helix even more toward the active site [152].
A MD study also shows that ATP binding induces conformational rearrangements in the A-loop of ALK [152]. Upon binding of ATP, the sharp U-turn at the end of αAL is disrupted. This allows for the full extension of the A-loop and creates space for peptide substrate binding. This is considered a determinant step in kinase activation [159]. Due to the hydrogen bond formed between ATP and R1275, αAL moves nearly 4 Å forward [152]. This leads to the disruption of the hydrogen bond of D1276 and R1284 and the sharp U-turn is no longer formed. Additionally, C1288 and K1285 form a hydrogen bond stabilizing the distal A-loop residues in a position that no longer blocks the substrate-binding site. Upon ATP binding, the hydrophobic region, consisting of M1290, L1291, and P1292, moves away from the active site, and a new hydrophobic core is formed as M1273 packs against P1292 and V1293. This new hydrophobic core resides closer to the active site than the original hydrophobic core and is considered to support the open A-loop conformation [152].
To combat ALK-positive lung cancer, small-molecule inhibitors have been developed over the past decade. Like EGFR, the drugs are designed specifically to stop the pro-survival signaling provided by ALK mutants. To date, the Food and Drug Administration (FDA) has approved five TKIs for the treatment of ALK-positive NSCLC [116].
Crizotinib was the first ATP-competitive small-molecule ALK inhibitor to be FDA-approved for lung cancer in 2014. Treatment with crizotinib resulted in longer progression-free survival at higher response rates and substantially reduced symptoms compared to standard-second line chemotherapy [160]. The compound is a type I inhibitor, as such it binds in the typical fashion by binding to the active protein kinase formation (PDB: ID 2XP2). Crizotinib inhibits ALK by occupying the front ATP binding pocket in the αC-in/DFG-in conformation, forming two hydrogen bonds with E1197 and M1199, as depicted in Figure 7 [133]. Hydrophobic interactions between L1256 and crizotinib stabilize the 2-aminopyridine core, the 3-benzyloxy group, and anchors the scaffold in an L-shape [161].
Unfortunately, drug resistance quickly emerged after prolonged treatment with the first-generation inhibitor crizotinib as outlined in Table 2 [162]. To overcome these resistances, multiple generations of drugs were developed. The resistances are frequently induced by single point mutations, either within or surrounding the catalytic ALK pocket (such as the L1196M gatekeeper mutation). These mutations alter the structure of the binding pocket, preventing ALK specific inhibitors from binding in their normal “bioactive” pose. X-ray crystallography and computational simulations allow us to examine the effect of mutations on the ALK-TKI complex and thereby design and develop new TKIs [163].
Ceritinib, a second-generation ALK inhibitor, was discovered through high-throughput screening by Novartis [175]. Ceritinib is almost 10-fold more potent than crizotinib against wild-type ALK and proved highly effective against L1196M, S1206Y, G1269A, and I1171T EML4-ALK mutants [162]. Like crizotinib, ceritinib is a type I inhibitor and binds to the active ALK conformation. As portrayed in Figure 8, ceritinib forms two hydrogen bonds with M1199 located at the hinge region (PDB: ID 4MKC). Furthermore, a salt bridge is formed between the piperidine ring and the side chain of E1210. The isopropoxy group of the compound forms favorable interactions with R1120, E1132, and the L1198–A1200-G1201–G1202 hinge segment [133,189].
Another type I second-generation inhibitor, alectinib, was approved in 2015. This compound is more effective than crizotinib in untreated ALK-positive lung cancer and has an IC50 of 1.9 nM against native ALK [190,191]. Moreover, alectinib may inhibit both native ALK (Ki = 0.83 nM) and the L1196M ALK mutation (Ki = 1.56 nM) [190]. Furthermore, alectinib is more effective against tumors with brain metastasis, due to higher penetration across the blood-brain barrier (BBB) [192]. The structure of ALK co-crystallized with alectinib (PDB: ID 3AOX) indicates that the backbone nitrogen of M1199, like other inhibitors, forms a hydrogen bond with the carbonyl oxygen of the benzo[b]carbazole moiety. Furthermore, the tetracyclic benzo[b]carbazolone of alectinib adopts a planar conformation as observed in its co-crystalized structure with ALK. This allows for the formation of a hydrogen bond network via solute or water molecules to K1150, E1269, E1270, and R1253 [190].
Brigatinib is an inhibitor of native EML4-ALK, with an IC50 of 14 nM whereas crizotinib, ceritinib, and alectinib all display a lower potency. The potency of brigatinib is substantially larger than that of crizotinib, ceritinib, and alectinib in 17 EML4-ALK variants. Brigatinib is also 2.2- to 77-fold more potent than crizotinib (IC50 1.1–170 nM) against all mutants, except L1198F. Furthermore, brigatinib is also 3.2- to 40-fold more potent than ceritinib against five mutants; L1152R/P, C1156Y, L1198F, and G1269A. Finally, brigatinib was substantially more potent than alectinib (3.2- to 54-fold) against the six mutants L1152R, I1171N, V1180L, L1196M, G1202R, and G1269A. Against any of the 17 mutants tested, neither ceritinib nor alectinib had more than two-fold greater potency than brigatinib [193]. As such, the drug was approved by the FDA in 2017 as a treatment option for ALK-positive NSCLC patients [121].
Brigatinib has also been approved for EGFR-positive NSCLC, classifying it as a dual inhibitor. The compound undergoes multiple interactions with the target; the methoxy group interacts with L1198, the C5-chlorine atom with L1196, and the unique dimethyl phosphine oxide (DMPO) moiety with the DFG motif (PDB: ID 6MX8), as illustrated in Figure 8 [194]. Moreover, the hydrogen bond between the protein and the DMPO moiety stabilizes the compound in a U-shaped conformation.
To date, all FDA approved ALK TKIs contain an ATP-adenine equivalent kinase hinge binder and an extra motif extending to the solvent area. This renders the compounds sensitive towards solvent-front mutations, e.g., G1202R. In 2018 a new type of compound, a third-generation ALK inhibitor was approved by the FDA. This compound, lorlatinib, contains an amido-linked 12-membered macrocycle [195]. Johnson et al. compared the apo ALK structure with the ALK/crizotinib co-crystallized structure and used crizotinib as the base to design the compound. As illustrated in Figure 7, the macrocycle of lorlatinib is precisely anchored in the adenine binding site (PDB: ID 4CLI) [195]. Furthermore, two stable hydrogen bonds are formed with M1199 and one with E1197 through the N3, N17, and N24 of lorlatinib [196]. Moreover, van der Waals interactions are established between the compound and R1270 [133]. Hydrophobic interactions between lorlatinib and the residues L1122, A1148, L1196, A1200, G1202, and L1256 are conserved between crizotinib and lorlatinib. However, the interactions between G1123, V1130, F1198, and G1269, and the drug are unique to the ALK-lorlatinib interaction [196].
Unfortunately, resistance mutations in ALK-positive NSCLC against lorlatinib has already been predicted [197] and reported as highlighted on Table 2. Notably, the L1198F substitution induces resistance against lorlatinib, but it resensitizes cells for crizotinib [176].
The inevitability of resistance mutations leads the scientific community to develop a wider range of ALK specific TKIs. One such TKI in development is ensartinib (X-396), which is seen as a promising candidate as its potency values are ten-fold greater than crizotinib, with an IC50 of 22 nM [198]. The drug is effective against multiple ALK mutants, including G1269A and C1156Y and it is currently undergoing Phase II clinical trials [199,200,201,202].
Entrectinib (PDB: ID 5FTO) is a promising potent drug against ALK with an IC50 of 2 nM [203]. It was approved in 2019 for the treatment of ROS1-positive NSCLC patients, but still awaits approval in ALK-positive treatment [204]. In phase I trials it is well tolerated and, like ensartinib, seems to have antitumor activity in the central nervous system [205]. Molecular docking and molecular dynamic studies have examined the behavior of the drug in the ALK catalytic domain [206]. This provides early knowledge on the ALK-TKI relationship, and whether inhibitory activity is sufficient for clinical use, findings that might be further used in the study of activating and resistant mutations [207].
Computational studies are heavily reliant on x-ray crystallography. The protein structures that these studies unravel allow for the creation of 3D models of mutants with unknown structure When an x-ray crystal structure is not available, homology modeling via computational means provides an accurate structure that still allows for the analysis of certain ALK mutations, as was the case for Okamoto et al. [206].
The L1196M mutation has been dubbed the gatekeeper mutation and is analogous to the T790M EGFR resistance mutation. L1196 controls access of inhibitors to a hydrophobic pocket within the active site. Substituting L1196 with bulky side chain residues leads to the resistance of ATP-competitive ALK inhibitors, which is considered a common mechanism of resistance in tyrosine kinases [208]. Studies not only revealed that the mutation causes steric hindrance but also how the mutation affects the structure, shifting the αC-helix, thereby increasing its affinity for ATP [163,209].
Extensive in silico analyses have led to the understanding of the resistance mechanism of L1196M against crizotinib [163,209]. The results indicate that L1196M causes flexibility in the P-loop and A-loop, thereby causing fluctuations in the conformation of the protein. This increased flexibility of the P-loop, including its GxGxΦG motif, increases the binding affinity for ATP [210]. A-loop flexibility alters the hydrophobic interaction of the protein with crizotinib, leading to poor binding of the inhibitor [163]. Furthermore, studies indicate that the L1196M mutation causes a secondary structural modification in the A-loop region, which reduces the stability of the αAL motif (residues 1272-1278). Therefore, the preferentially autophosphorylated Y1278 is destabilized by the L1196M mutation. The van der Waals interactions that Y1278 undergoes with Y1096 and M1166, located within the hydrophobic face of the C-helix, are thereby compromised. Changes in van der Waals interactions as caused by L1196M have been identified as a major component of the binding energy difference between all wild-type and mutated complexes [209,211]. In silico experiments suggest that the L1196M mutation decreases the total binding energy of crizotinib. This occurs through a decrease in the non-bonded contribution and increases in the entropy upon binding of the ligand. The main cause of resistance to other drugs is associated with a change in the overall binding energy [163].
In summary, the L1196M mutation introduces conformational flexibility that leads to a shift in the αC-helix that increases the binding affinity for ATP while simultaneously decreasing the binding affinity for the ALK inhibitor [209,210]. Many other computational studies have uncovered different resistance mechanisms of ALK mutations, such as F1174V, L1198F, and the frequent G1202R [196,212,213,214]. For example, molecular dynamics and binding energy calculations have shown that the C1156Y mutation disrupts the van der Waals interactions and electrostatic interactions between the mutated ALK and crizotinib [215]. The mutation alters the conformations of the P-loop, β-sheet, and αC-helix, causing the displacement of crizotinib.
Similarly, molecular dynamics together with free binding energy calculations revealed that the F1174C mutation disrupts the aromatic-aromatic network that is formed amongst residues F1098, F1174, F1245, and F1271 [216]. This allosterically affects the dynamics of the P-loop, as it leads to its upward movement, away from the ATP-binding site. The mutation causes weak binding of ALK to ceritinib as hydrophobic interactions, are disrupted by the mutation, causing drug resistance. Computational studies are not limited to single ALK point mutations. Double mutated ALK catalytic domains have also been subjected to virtual molecular studies, such as the L1196M/G1269A [217] or the L1198F/G1202R double mutant, where the effects of the individual mutations as well as the double mutant effects were considered [218].
The computational studies may not only clarify the mechanism of resistance of mutations but also indicate the compatibility of certain drugs with the mutated ALK protein. In the case of L1198F, computational studies showed that the mutation alters the conformation of the ATP binding pocket - which is unfavorable for lorlatinib binding. This conformational change allows for the resensitization of the protein to crizotinib, with a new hydrogen bond being formed between the fluorine atom and K1150. MMGBSA analysis of the simulations indicate an ameliorated binding of crizotinib to the L1198F single or double mutant when compared to the WT ALK [176,218]. The divergence for experimental and theoretical data of the G1202R mutant is elucidated once the apo structure of the mutant is analyzed through MD simulations. The resistance is suggested to be due a hydrogen bond involving R1202 and the backbone of L1122, hindering the access of drugs to the binding pocket. This blockage is absent in the double mutant L1198F/G1202R due to the F1198 interaction with E11132, dislocating R1202 [196].
Recently, Shaw et al. reported a case in which they used virtual molecular studies to aid in the treatment regimen of the patient [176]. A patient was diagnosed with ALK-positive NSCLC and treated accordingly with crizotinib. However, after prolonged treatment, resistance mutations arose and the patient was treated with ceritinib after acquiring the C1156Y mutation. After entering a clinical trial, in which the patient received lorlatinib, the patient acquired a second mutation; C1156Y/L1198F. Interestingly, the patient was resensitized to crizotinib as observed for the single mutant L1198F [176].
Besides, Shaw et al. set out to understand the mechanism of the patient’s mutated ALK protein and included a structural study. The computational study proposed that the C1156Y substitution increased the kinase activity by shifting the P-loop even though the C1156 is positioned 13 Å from the inhibitor binding site. The novel resistance mutation, L1198F, resides near the ATP-binding site. This mutation forces the rigid macrocyclic inhibitor lorlatinib to rotate away from the introduced phenylalanine. This disrupts the hinge-binding interaction with the inhibitor and introduces strain into the kinase, reducing the binding energy compared to wild-type ALK. Interestingly, the newly introduced phenylalanine does not clash with crizotinib. Instead, it moves slightly closer to the inhibitor. In the double mutation, the computational study revealed that the enhanced binding caused by L1198F offsets the increased kinase activity introduced by C1156Y, resulting in crizotinib sensitization [176].
This case is a prime example of computational biology in the setting of diagnosis and personalized medicine by its effort to evaluate their own theoretical data with different source of experimental output. Structure-based study led to the understanding of the mechanism of the double mutant with a novel resistance mutation. Upon understanding the structure and mechanism, an informed clinical decision could be made and the treatment with crizotinib was restarted. The patient had a “rapid and dramatic clinical improvement” [176].
5. Rapidly Accelerated Fibrosarcoma Homologue B (BRAF)
The RAF (rapidly accelerated fibrosarcoma) protein family members, ARAF, BRAF, and CRAF are one of the core protein kinases in the mitogen-activated protein kinase (MAPK) pathway represented in Figure 2 [219]. The MAPK cascade plays a major role in cellular signal transduction, and it is activated by different extracellular ligands like growth factors, cytokines, and hormones. This activation, based on specific cellular conditions mediates cell growth, survival, and differentiation [220,221]. RAS (rat sarcoma viral oncogene) a membrane-associated protein, when loaded with GTP, induces dimerization and activates RAF proteins. The activation of RAF kinases leads to phosphorylation of mitogen-activated protein (MEK) which in turn phosphorylate ERK (extracellular signal-regulated kinase) downstream the pathway [221,222]. Despite being an ATPase, such as EGFR and ALK, BRAF is classified as serine-threonine kinase and does not contact the extracellular environment directly, instead depending on TKRs and RAS binding for its activation. RAF activity is tightly regulated and its malfunction is often linked to cancer [223].
The three isoforms share conserved regions; CR1 and CR2 in the N-terminus and CR3, which encodes the kinase domain in the C-terminus [224]. CR1 is composed of a RAS-GTP binding domain (RBD) and cysteine-rich domain, which binds with two zinc ions [225]. In the autoinhibitory state, the cysteine-rich domain also functions as an auto inhibitor of the kinase domain [226]. The CR2 region is rich in serine and threonine residues and contains a domain for binding of 14-3-3 protein [225].
Confined between CR1 and CR3, the CR2 function as a flexible hinge between the two regions [227]. The CR3 region besides, the kinase domain, presents a binding site for 14-3-3 protein a class of helical regulatory molecules. The N-terminus of CR3 contains a glycine-rich motif, responsible for the stabilization of ATP binding that also contributes to the stabilization of the inactive conformation. The substrate-binding pocket is found in the C-terminal end of CR3 and has a catalytic loop that promotes the smooth transfer of phosphate group from ATP to the enzyme substrate [228,229]. Despite all three isoforms’ contribution to physiological processes, the focus of this review will be on the BRAF isoform due to its contribution to NSCLC (UniProt: ID P15056) [230].
Structurally, BRAF presents two lobes connected by a hinge region, as mentioned for EGFR and ALK. Regulatory components such as the P-loop (G464-G469), the movable αC-helix (T491-R506) and, the dynamic A-loop, and its DFG (D594/F595/G596) motif are also present, as depicted in Figure 1 and Figure 9 [231,232].
ATP binding to BRAF follows a similar pattern as described for the previous kinases, with the adenosine ring anchored through hydrogen bonds to the backbone of Q530 and C532 in the hinge region. In addition to stabilization by the P-loop and the DFG, the non-transferable phosphate groups are in contact with K483 and E501, with the positive charge of K484 bending towards the ATP α and β phosphate groups, and being stabilized by E501 in the absence of a nucleotide [233].
In quiescent cells, RAF proteins are localized in the cytosol and are devoid of subcellular localization motifs [234]. They exist in an autoinhibited manner in which the N-terminus interacts with the C-terminus and represses kinase activity [222]. Dimerization of the kinase is believed to be an important step in RAF activation and, binding of RAS-GTP to RBD activates the dimerization of RAF members leading to the formation of homo- or hetero monomers [235,236].
In its quiescent state, besides existing in the autoinhibited state, BRAF forms a complex with MEK. RAF and MEK form a dimer interface by primarily using the C-lobe of both kinases including the activation segment.. Characterization of the BRAF-MEK1 complex (PDB: ID 4MNE and 6PP9) provides insights into the dynamics of the protein-protein interaction and casts light on the inactive complex in the presence of the inhibitory 14-3-3 protein [237,238]. Upon RAS-GTP mediated pathway activation, BRAF bounded to MEK will dimerize with CRAF or another BRAF monomer to form a transient active heterotetrameric RAF-MEK complex, as shown in Figure 2, resulting in a phosphorylated MEK. Phosphorylation of MEK destabilizes the heterotetrameric complex leading to dissociation of MEK and allowing MEK to phosphorylate the kinase domain of ERK [237].
Studies conducted to examine RAF dimerization have shown that all RAF proteins are capable of forming homo or heterodimers [236,239]. However, growth factor stimulus predominantly induces BRAF/CRAF heterodimerization, with a low level of BRAF/ARAF and little to no CRAF/ARAF heterodimers observed [239]. Moreover, BRAF/CRAF heterodimers have a higher kinase activity when compared to their respective monomer or homodimers [235]. The process of dimerization involving the members of the RAF family relies on the αC-helix of both units of the dimer packing against each other. Therefore, mutations on the intermolecular interface are linked to the disruption of RAF dimers. As an example, the mutation of a conserved arginine on both BRAF (R509) and CRAF (R401) into a histidine residue results in disruption of the dimerization process [239,240]. The introduction of this mutation is useful when seeking crystallization of the monomer BRAF instead of its dimeric state (PDB: ID 4RZV) [241].
In the inactive kinase conformation, the activation segment is packed and the αC- helix is in the out conformation, disrupting the salt bridge between the conserved lysine of the β3 strands and the glutamate of αC- helix. Activation is usually triggered by phosphorylation and leads the disruption of the inactive conformation, leading the activation segment to take a position needed for catalysis, the DFG in state, and αC-helix to turn to “in” to form a catalytic salt bridge [242].
RAF paralogs are subject to complex regulation and their activity depends on the phosphorylation of different regions, binding of inhibitory proteins, and intramolecular autoinhibitory interactions between the different domains of the protein. The detail of their regulation also varies based on the regions conserved in each isoform [221,222]. The activation segment that is found at the center of the catalytic domain plays a key role in the regulation, and phosphorylation of residues positioned at the activation segment transforms the inactive kinase orientation into the active kinase conformation [243]. ARAF and CRAF require additional phosphorylation within the N region of the kinase domain whereas BRAF does not need this phosphorylation [221,244]. This difference in their activation implies that those isoforms can be regulated independently [244].
Although all RAF isomers are important in normal physiology, BRAF has higher basal kinase activity. Moreover, it is by far the most mutated RAF kinase in different types of malignancies [229,245]. BRAF mutations have been identified in different types of malignancies such as thyroid cancer, colorectal cancer, and hairy cell leukemia. When mutated, it is a major therapeutic target in melanoma and interestingly also in NSCLC [246]. Mutations in BRAF are mostly found in the activation segment (A-loop) near position 600 or the glycine-rich phosphate-binding loop (P-loop) with rare cases in flanking regions [245].
While all BRAF mutations constitutively hyperactivate the MAPK pathway, they have a different mechanism of activation. Based on their effect on kinase activity, RAS independency, and dimerization state, BRAF mutations can be categorized into three distinct groups [227]: class 1 BRAF mutations act as RAS independent active monomers whereas, class 2 BRAF mutations are active dimers. Class 3 mutations are RAS dependent and possess low kinase activity [227]. As mentioned above a significant number of oncogenic BRAF mutations occur in the conserved region of the kinase domain specifically in the P-loop and the DGF motif, destabilizing the inactive conformation leading to auto-activation of BRAF independent of upstream signaling [244].
Class 2 mutants have an intermediate to high kinase activity, whereas class 3 mutants have an impaired kinase activity [234]. Class 2 mutations predominately reside in the activation segment or P-loop, whereas Class 3 mutants are located in the P-loop, catalytic loop, and DFG motif [227]. Class 2 mutants that are found in the activation segment block the interaction with the P-loop, which is important for the auto-inhibition of BRAF leading to increased intrinsic kinase activity. Class 1 and 2 mutated kinases act independently of their upstream activator RAS GTPase for signaling growth and proliferation in cancer cells [229].
In contrast, class 3 mutants are RAS dependent and these mutants possess low kinase activity compared to the wild-type BRAF, or they lack kinase activity [227]. However, those non-oncogenic BRAF mutations, which have a lower kinase activity stimulate ERK activity by disfavoring BRAF-MEK complex formation and potentiate BRAF-CRAF dimerization. Those mutants trigger the dimerization of WT BRAF and CRAF and their stimulation of the MAPK pathway is dependent on WT CRAF [237,244]
BRAF V600E/D/K/R mutations are categorized as class 1 mutations and are the most frequently identified BRAF mutations in cancer. These mutations lead to a 500-fold increase in kinase activity compared to the wild-type protein [244]. The WT BRAF kinase domain structure shows that the aliphatic side chain of V600 contacts the phenyl ring of residue F468 found in the P-loop. Substitution of valine with a larger and hydrophilic amino acid—such as glutamic or aspartic acid, lysine, or arginine—is presumed to disrupt the interaction that maintains the DFG motif in the inactive conformation, holding the activation segment in an active position [244]. Additionally, mutation at position 600 (V600X) could also guide the activation segment into the active conformation, resulting in increased BRAF activity [244]. This conformational change is similar to the one formed during dimerization, which explains why BRAF V600E is active as a monomer and does not depend on dimerization for kinase activity and MEK hyperactivation [222,228].
Although BRAF V600E mutants are functional as monomers, an in vitro study by Cope et al. revealed that BRAF V600E mutants have increased oligomeric formation, indicating that V600E contributes to a higher potential of BRAF for dimerization. In the same study, MD simulations showed that the BRAF V600E mutant preferentially adopts a conformation in which the activation segment is fully uncoiled and the αC-helix takes the αC-in position. As a result, αC-in is associated with BRAF dimerization, providing evidence on how BRAF V600E shows an amplified dimerization capacity. However, the introduction of R509H severely disrupts the BRAF dimers, but does not abolish kinase activity of BRAF V600E. This implies that the V600E mutation could influence BRAF kinase activity by playing two functions, serving as an active monomer and also increasing dimerization potential [247].
Over 40 mutations have been identified in BRAF. However, the substitution of a valine residue by glutamine acid at position 600 (V600E) is responsible for 92% of BRAF mutation in different types of cancer [245]. This mutation is located proximal to the DFG motif and introduces a negatively charged side chain in the previously hydrophobic position. The introduction of the negatively charged side chain of glutamic acid has a comparable effect on the activation loop as the phosphate group of ATP on neighboring residues T599 and S602, classifying this variant as phosphomimetic. Similarly, to the L858R variant of EGFR, the V600E mutation is believed to favor the active conformation by increasing the energy threshold required to transition from the active into the inactive conformation - locking the kinase in a constitutively active conformation. This loss of inhibition increases the basal kinase activity, creating an oncogenic mutation [248].
Identification of BRAF gene alteration in different cancers and a better understanding of BRAF three-dimensional structure has enabled the development and design of small-molecule inhibitors that specifically deactivate BRAF kinase activity [229,249]. The first-generation RAF inhibitors were ATP-competitive inhibitors, of which sorafenib was the first to be approved. Sorafenib has a multi-kinase profile and acts on wild-type and V600E BRAF, CRAF, VEGFR, PDGFR, c-KIT, and FLT3 [229,249].
Sorafenib was first designed as a CRAF inhibitor, targeting the inactive conformation of RAF proteins [244,250]. Sorafenib was shown to be inefficient in the treatment of melanoma as a monotherapy or with combination with chemotherapy [229], but was found to be effective in renal cell and hepatocellular carcinoma without evidence of efficiency on the NSCLC treatment [251,252]. The co-crystal structure of sorafenib with WT BRAF (PDB: ID 1UWH) demonstrated that the drug locks the kinase domain in the inactive conformation by inducing a DFG-out conformation and decreasing activation loop mobility. The DFG-out locked conformation is stabilized by the interaction of sorafenib’s urea carbonyl moiety acting as a hydrogen bond acceptor for D594 backbone carbonyl, with further sterically hindrance created by the trifluoromethyl ring. The pyridyl rings mimic the adenosine ring interaction with W531, F583, F595—part of the hinge region, catalytic loop, and DFG motif, respectively. Residue F595 in the DFG motif also interacts with the central benzene ring of sorafenib as demonstrated on Figure 10. The other end of the inhibitor contains a lipophilic trifluoromethyl phenyl ring and is buried in the hydrophobic pocket that is constructed between the αC and αE helices and the N-terminal region of the catalytic loop and DFG motif [244]. The same binding mode is observed in the presence of the V600E mutation with the DFG-in conformation conserved (PDB: ID 1UWJ) [244].
Second-generation inhibitors were designed to specifically inhibit V600E. Through structure-based drug design principles, vemurafenib (PLX4032) was developed and approved by the FDA for the treatment of advanced-stage melanoma [253]. Dabrafenib (GSK2118436), another mutant selective BRAF inhibitor likewise gained approval in 2013 [254]. Encorafenib (LGX818), also an ATP competitive second-generation RAF inhibitor, has a higher potency than vemurafenib and dabrafenib due to its significant slower off-rate from BRAF V600E and it is currently in phase II clinical trial in combination with binimetinib, a MEK inhibitor (clinical trial identifier: NCT03915951). Moreover, in vivo and in vitro data showed that it is inactive against BRAF wild-type tumors [255,256].
Studies conducted to analyze the binding mode of BRAF inhibitors have shown that different sulfonamide-based inhibitors, like vemurafenib, bind to the off state conformation of BRAF [257]. Interaction of the sulfonamide moiety with residues at the N-terminus of the DFG region leads to the binding of the alkyl chain to an “RAF-selective pocket”, which is unique to the RAF family. This binding is critical and drives selective inhibition of oncogenic mutations [258]. This unique pocket contains residues L505, L514, F516, and F595, and interaction between the functionalized sulfonamide moiety with the L505 side chain stabilizes the αC helix in an outward position [257].
RAF inhibitors are also classified by the conformation of the regulatory αC-helix and DFG motif resulting from drug binding. In this classification, inhibitors can be type I (αC-in/DFG-in), type I 1/2 (αC-out/DFG-in), or type II inhibitors (αC-in/DFG-out) [241,259,260]. BRAF first- and second-generation inhibitors belong mostly to type I and I 1/2, with vemurafenib and dabrafenib belonging to the type I 1/2 class inhibitors. Type II inhibitors target the inactive kinase domain αC-in/ DFG-out kinase state, whereas type I 1/2 kinase inhibitors bind to another inactive conformation, αC-out/DFG-in which blocks the formation of catalytical competent enzyme form. Type II and Type I 1/2 kinase inhibitors are more selective since those molecules target a particular inactive conformation that could be structurally unique for BRAF [261]. The crystal structure of the different inhibitors with the BRAF kinase domain has been solved including vemurafenib (PDB: ID 3OG7, 4RZV), sorafenib (PDB: ID 1UWH, 1UWJ) and, dabrafenib (PDB: ID 4XV2, 5CSW) [244,258,262].
The crystal structure of type II inhibitor sorafenib with WT BRAF and V600E BRAF has shown that type II inhibitors bind strongly to both monomers and exhibit a long inhibition period, and may be relatively weak dimerization inducers. Sorafenib binds to both of the protomers and lead to the dimerization of BRAF [244]. The recent discovery of LY3009120 (PDB: ID 5C9C) led to a better understanding of “paradoxical” BRAF inhibitor activation. LY3009120 is a type II pan-RAF dimer inhibitor of three RAF isoforms and has a minimal activation pathway, due to its effective binding with both protomers of the dimer [263]. Vemurafenib, a typical type I 1/2 inhibitor binds only to one of the monomers of the dimer and stabilizes the αC-out conformation. This position induces negative allosteric changes to the αC helix of the other monomer, resulting in a conformation which is resistant to the binding of vemurafenib and activation of the heterodimer BRAF-CRAF (leading to the term “paradoxical”). This mechanism of vemurafenib is best visualized in the BRAF-vemurafenib co-crystal structure (PDB: ID 3OG7) [258].
Dabrafenib is categorized as a type I 1/2 inhibitor and binds to both protomers with identical DFG-in/αC-helix-out conformation (PDB: ID 4XV2, 5CSW) [262,264]. The potential of type I 1/2 inhibitor to induce dimerization could be correlated with the extent of their paradoxical activation. Likewise, whereas dabrafenib is a relatively strong promoter of dimerization and paradoxical activation, vemurafenib may weakly induces dimerization and shows a decreased paradoxical activation in biological assays [262,264,265]. Responses of dabrafenib, vemurafenib, and encorafenib to BRAF mutations are summarized in Table 3.
MD simulations, combined with cell assays of paradox inducers (vemurafenib and PLX4720) and paradox breakers (PLX8394 and PLX7904) indicated that a subtle structural difference could result in a profound conformational change and the overall dynamics of BRAF dimer complex [266]. Arora et al. showed that paradox inducers could increase the movement of both the αC-helix and the A-loop region, whereas paradox breakers promote a relatively stable αC-helix and activation segment.
Analysis of hydrogen bonds throughout MD simulation identified that paradoxical inducers present a significantly higher number of hydrogen bonds with the gatekeeper residue T529 when compared to the paradox breakers, providing an explicit discrimination between paradox inducers and breakers [266].
As observed for EGFR and ALK, gatekeeper mutations are a common mechanism of resistance as they do not hamper ATP binding but preclude interactions with the hydrophobic pocket composed of the αC-helix and A-loop - a common pocket for drug development. This residue is also considered an essential structural feature that could influence kinase activation [208]. Arguably, the addition of the sulfonylurea group in paradox breakers blocks the interaction with the gatekeeper residue T529, inducing an overall conformation change in the paradox blocker complex, and promoting an open conformation. In contrast, the sulfonamide group of the paradox inducer promotes a closed conformation facilitating BRAF dimerization [266], which can lead to paradoxical activation of the MAPK pathway in the presence of ATP competitive BRAF inhibitors [240,260].
The mutational landscape of BRAF despite being limited when compared to EGFR and ALK for NSCLC patients remains a topic of interest due to its complex signaling contribution and possibility of future role in lung cancer [229].
6. Kirsten Rat Sarcoma Viral Oncogene Homologue (KRAS)
The rat sarcoma viral oncogene (RAS) family includes Kirsten rat sarcoma viral oncogene homolog (KRAS), neuroblastoma rat sarcoma viral oncogene homolog (NRAS) and Harvey rat sarcoma viral oncogene homolog (HRAS). The RAS family is a membrane-bound GTPase family with a pivotal role in cell homeostasis. RAS is responsible for the conversion of a plethora of extracellular signals from multiple TKRs (i.e., EGFR, insulin receptors) and G protein-coupled receptors (GPCRs) into intracellular signals, through the activation of canonical cascades such as PI3K/AKT/mTOR, and RAF/MEK/ERK. Therefore, this family is associated with a multitude of typical cellular functions, from proliferation and survival to motility and gene transcription [281].
RAS exists in two conformations; an inactive conformation with a GDP molecule bound and active conformation with GTP. The association of GTP with RAS leads to conformational changes that oblige downstream effectors to bind and be further activated by one of the three RAS isoforms. [282].
A slow intrinsic hydrolysis ratio is not compatible with the dynamic role of RAS on cell survival. A combination of RAS-associated proteins, such as GTPase activating proteins (GAPS) and Guanine nucleotide exchange factors (GEFs) are responsible for strict amplification and control of RAS activity. GAPs are associated with an increase of RAS hydrolysis, increasing GTPase activity. Once GDP molecules are generated, GEF assists RAS to eject GDP allowing for a new GTP molecule to bind [283].
Despite being rather small proteins (~190 amino acids), RAS family members have well-defined regions associated with either their function (binding site, switch I, switch II, effector binding site) or regulation (GEF binding site and GAP binding site). GTP binds to the nucleotide-binding region where it is converted into GDP. Upon GTP hydrolysis, the switch I (Q25-Y40) and switch II (D57-G75) regions change their conformation allowing GAP and GEF binding. The changes also allow the binding of effector proteins while in the active conformation [283].
RAS signaling is structurally linked to conformational changes in the GTP-bound (active) state. In the presence of GTP, RAS proteins present a closed conformation of two structural elements (switch I and switch II), allowing the binding of effectors such as RAF kinases and PI3K. The positively charged P-loop stabilizes the negatively charged phosphate group, thereby maintaining the GDP bound conformation. The P loop is conserved throughout the family as a conserved GxxxxGKS/T motif. A conserved lysine stabilizes the negative charges of the β and γ phosphate groups, and the remaining negative charges are stabilized by the bidentate interaction with a magnesium ion. Another conserved motif, NKxD, stabilizes the guanine base [284,285].
Structurally, the RAS family follows the canonical α,β-fold with six β-strand and five α-helices like other nucleotide binding proteins as shown in Figure 11 [286]. The variability of conformations, especially of the switch II region, was analyzed through the lens of computational biology. Interestingly, the output of MD simulations shows that there is not only a change of flexibility in the three isoforms but also a change in their degree of freedom upon binding of different nucleotide states. Kapoor and Travesset also uncovered a transient new pocket in KRAS, neighboring the nucleotide-binding site. The proximity of this new transient pocket to the effector domain and the membrane-binding domain provides an insight into a possible targetable site for the abrogation of the nucleotide exchange [287].
Probe-based Molecular Dynamics (pMD) differ from the classical method by using probe molecules in conjunction with usual solvents (i.e., water) [288,289]. This method is comparable with fragment-based crystallography and NMR spectroscopy [290,291]. By applying the principles of pMD to the RAS family, the initial binding pockets were analyzed dynamically indicating that these are better formed upon protein relaxation [292,293]. Besides four allosteric pockets, pMD also highlighted reactive surfaces that might be involved with protein-protein or protein-membrane interactions [294].
Due to its association with different cancer types, KRAS has gained the spotlight [4], with its role in tumor survival evident from KRAS-driven mouse models [295]. Despite its critical role in pancreatic and colorectal cancer, the KRAS mutational landscape has also been in focus in lung cancer with related prognostic and predictive power [296]. Due to its relevance for oncogenic addiction, multiple attempts to target the RAS family have been described. However, they failed, either due to moderate efficacy or lack of selectivity leading to toxic effects [292,297,298,299,300,301,302].
Oncogenic mutations in KRAS were observed to cluster around the nucleotide-binding site in positions 12, 13, and 61 (UniProt: ID P01116). In the Western countries, approximately 20–35% of lung adenocarcinomas contain activating mutations in KRAS [303,304,305]. The most common KRAS mutation in lung cancer is the G12C substitution (PDB: ID 4LDJ), along with G12D (PDB: ID 5US4) and G12V (PDB: ID 6GOE) [304,306].
Recent advances provided experimentally determined structures for less common mutants such as G13C (PDB: ID 6OB3), D33E located on switch I (PDB: ID 6BP1) and A59G (PD: ID 6ASA) [307,308]. Q61 is located on the switch II region, and related to effector binding. In KRAS, the P-loop is adjacent to both switch I and II leading to the hypothesis that these mutations, as well as variations on codon 61, could affect KRAS binding to effector partners [287].
As previously mentioned, wild-type KRAS exists in two states. A molecular dynamics study indicates that hotspot mutations influence the inactive-to-active conformational transition [309]. The G12C mutant still exists in two, active and inactive, states compared to WT. Both the G12C and the G12D mutants alter the dynamics of KRAS, shifting the protein such that the GTP-binding pocket now resides in a more open conformation compared to wild-type KRAS-GDP. These open conformations increase the solvent-accessible surface area (SASA) by almost 23% and 14% respectively [309].
Of note is that the increased SASA of the G12D substitution culminated in a more prominent open conformation than mutation G13D. The G12D mutation also causes increased atomic fluctuations at the P-loop and switch II regions) [310]. Interestingly, the open conformation is not limited to the common mutations in the P-loop. The rare switch mutations D33E (switch I) (PDB: ID 6BP1) and A59G (switch II) (PDB: ID6ASA) also adopt an open switch I conformation in silico [308]. The dynamic and open conformation of the mutated KRAS is suggested to hamper nucleotide exchange [311].
Hunter and colleagues showed that all three residues positions are linked to a decreased affinity between RAS and RAF although with different impacts [312]. The impact of mutations was studied in the context of intrinsic GTP conversion, with the mutation G12C presenting minimal impact in the hydrolysis rate, while the presence of alanine or arginine residues in the same position led to around 80%-fold decrease and aspartic acid and valine showing an intermediate effect. A considerable decrease of hydrolysis constants was observed for position 61, where mutation Q61H/L has similar profiles to G12D/V. Hotspot position G13 also presented decreased hydrolysis when mutation G13D was present. A similar outcome is found when in the presence of GAPs [312].
The common mutation G12C impairs the arrangement necessary for GAP-mediated hydrolysis by disturbing the arrangements of the catalytic residues Q61 of KRAS and the arginine finger of GAP in the active site for catalysis. The mutation also disrupts the formation of a hydrogen bond between the side chain NE2 atom of Q61 and the γ-phosphate of GTP [309].
Similarly, the side chain of alanine in the G12A substitution causes a clash with Q61. In addition, there is a clash between the side chain of T32 and the side chain of R789 of GAP caused by the G12A substitution. The formation of a hydrogen bond involving the backbone of A12 with the hydroxyl group of the T32 side chain in the switch I stabilize the γ-phosphate of GTP in its pre-catalytic form. This restricts the phosphates from stretching and rotating their bonds, increasing the difficulty of reaching the transition state, explaining the reduced intrinsic GTP-hydrolysis [313].
Other position 12 mutations, such as G12D, cause the OE1 atom of Q61 to move away from the γ-phosphate of GTP, preventing hydrogen to be extracted from a catalytic water molecule [309,314]. Instead, the side chain of G12D takes the place of Q61, thereby impairing the charge distribution in the active site for intrinsic hydrolysis. Furthermore, position 12 of the P-loop is less flexible than Q61 of switch II, thereby sterically interfering with the GAP arginine finger, supported through a computational study [311,314].
Interestingly, it is not the common G12C, but rather the G13D substitution that induces rapid nucleotide exchange kinetics compared to other KRAS mutants. The kinetics of nucleotide exchange between wild-type KRAS and mutant KRAS are the same, except for KRAS G13D. The G13D substituted KRAS has a GDP exchange rate that is 13.5 times faster than the wild-type KRAS. Furthermore, the GTP exchange rate of KRAS G13D is nine times faster than WT KRAS. The G13D mutation decreases the intrinsic and GAP-mediated GTPase activity of KRAS, thereby decreasing the auto-inactive signal propagation of KRAS. Furthermore, the substitution causes a 2.4-fold decrease in the affinity for RAF-RBD, an effector protein of KRAS [312].
Mutations at position 13 alter the side-chain conformation of Q61 in such a manner that it can no longer interact with a catalytic water. Furthermore, G13D substitution prevents any direct interaction between Q61 and GTP [309]. However, the kinetic abnormalities induced by the G13D mutation can be explained by changes in the electrostatic charge distribution of the active site [312]. The crystal structure shows that the aspartate side chain of G13D is positioned above the α-phosphate. The carboxyl group of the residue points towards the carbon-5 of the sugar group of the GDP ribose. As the G13D mutation resides in the strongly positive P-loop region above the negatively charged phosphate groups, it disturbs the local electrostatic binding pocket. The aspartic acid introduces a strongly negative charge into the phosphate-binding pocket, increasing binding difficulty. Of note is that the neighboring G12D induces much less of a disruption to the electrostatic density of the GTP-binding pocket [312].
KRAS has been considered problematic in the past, as the high in vivo concentration of GTP and GDP in combination with the high affinity of KRAS for these nucleotides has been proven troublesome for the direct targeting of the protein [312]. However, data collated over the years suggest that direct targeting of the mutated KRAS proteins is possible. For example, the data collated on the structure and difference in conformational states was used to develop a specific, high-affinity non-covalent inhibitor for G12D KRAS over the wild-type protein [315]. As familiarity with mutated KRAS increased, some have used the mutations as a means for the development of small-molecule inhibitors. Lim, Hunter, and colleagues, developed a GDP analog with an electrophilic warhead that covalently binds to the mutant cysteine in the G12C KRAS [316,317].
Another challenge in the KRAS drug development process is the lack of a sufficiently large and deep hydrophobic pocket for small-molecule binding, aside from the precarious nucleotide-binding pocket. There have been numerous efforts in the identification and targeting of shallow sites on RAS protein, which have led to the ambitious effort of Welsch et al. to develop a compound that is capable of simultaneously inhibiting HRAS, NRAS, and KRAS (also known as pan-RAS) [298]. However, these allosteric binding sites may also be used in a mutant-specific manner [318]. Ostrem et al. show that the binding of G12C specific inhibitors in an allosteric pocket disrupts both switches I and II, thereby undermining the KRAS preference to favor GDP over GTP and impairing binding to effector proteins [319]. The combination of protein-protein interaction assays has also been proven fruitful in the development of Q61H KRAS specific inhibitors, again using an allosteric binding site [320].
The most promising development is the compound AMG-510. Upon the discovery of a surface groove that may be occupied by aromatic rings due to an alternative conformation of H95, AMG-510 emerged as a top candidate after an optimization campaign. Preclinical data show that AMG-510 selectively targets G12C KRAS tumors and cause durable regression as monotherapy. The compound may not only be applied individually but also works synergistically with cytotoxic and targeted agents. Furthermore, there are indications that AMG-510 also synergizes with immunotherapy. Moreover, the combination of AMG-510 with immunotherapy seems to result in an adaptive immune response that can recognize and eradicate related non-G12C KRAS tumors [321].
Elucidating the structures, mechanisms, and kinetics of KRAS mutants is essential in the drug development and personalized medicine process, as seen in the aforementioned tyrosine kinases, computational biology will aid in the KRAS drug development process, with molecular dynamics studies allowing for the understanding of the mutant structures and their mechanisms. Using techniques such as docking, possible compounds may be virtually assessed. Gupta and colleagues have developed a pipeline that involves molecular dynamics, high-throughput ensemble docking, and biophysical and cell assays that yield novel lead compounds [322]. Similarly, this pipeline may be applied in the future as a means of personalized medicine if novel mutations emerge. The KRAS mutant of individual patients, even those with a novel or double mutation, can be evaluated through molecular dynamics, and in combination with docking their treatment options may be ranked according to viability. This allows for fast and accurate diagnostics and development of treatment regimens tailored to an individual patient.
7. Molecular Modeling as a Supporting Tool for Personalized Medicine: The Future Is Knocking on the Door
Despite medical advances, the prognosis of advanced NSCLC remains gruesome, especially for those with a late diagnosis. In the case of metastasis, surgery can no longer be performed and these patients might not be suitable for chemoradiation approaches. In those cases, genetic profiling of these patients is performed searching for driver mutations in EGFR, ALK, BRAF, RAS, RET, and ROS that can undergo treatment with small molecules that would inhibit the abnormal kinase activity. While there are many kinase inhibitors in the market, physicians are still struggling to address the overall survival of these patients, which ranges between 7 to 14 months [323,324]
The lingering time between emergence/identification of resistance and treatment availability is not only due to the long process of drug development and approval but also due to the complexity of cancer—which can result from anomalies in a multitude of pathways within the same tumor cell population. Additionally, there is a clear gap between the medical and the research community, which delays the classification of mutations once they emerge and is thereby detrimental for patient care to address this issue [325].
An alternative from the traditional pursuit of novel molecular entities to target novel mutations is the repurposing of already approved drugs, as described for ALK single mutant L1198F and crizotinib and EGFR triple mutant exon19del/T790M/P794L and afatinib[87,176]. These cases show how computational studies combined with experimental data can not only grant possible treatments but also provide insight into the mechanism of resistance. Another remarkable example of combining theoretical and experimental methodologies is the potential to predict future BRAF resistance mutations by combining previous knowledge on EGFR and ALK hotspot positions [278,280].
Throughout this review, computational methods contributions were highlighted in the study of proteins and small molecules dynamics when experimental data was limited or absent. A highly noteworthy contribution of MD simulations is depicted in uncovering a transient pocket on KRAS, yielding a molecule for a target previously considered undruggable [321].
Experimental data might vary upon different techniques used or even the settings for the same experiment, as reproducibility grows as a scientific concern that is addressed with sizable initiatives as established by the Brazilian government or the European Union (Findability, Accessibility, Interoperability, and Reusability-FAIR project), as examples [326,327]. Computational studies are not different; a simulation for the same system using different force fields, temperature and pressure controls, and cluster sizes can provide different outputs. Docking is also susceptible to such phenomena, since there are hundreds of commercial and non-commercial software available, and their reckoning in the weight of types of bonds between protein-ligands might differ [328]. Following the steps set for the FAIR project, a sustainable approach for computational assessment of proteins and related mutations can no longer be postponed due to its remarkable contribution to the biomedical field.
In an attempt to close the gap between medical teams and scientists, an increasingly common approach is establishing tumor boards at cancer treatment centers, gathering not only health care providers, but also experts in scientific fields such as molecular biology. The main goal is to provide a personalized approach to patients who do not fit into the conventional treatment regiments, either in the presence of novel mutations or limited response to an established regiment [329].
Although tumor boards initially rely solely on previous patient reports, an innovative approach is the use of structural data obtained either from experimental or theoretical sources. As described by Koopman et al., structural biologists are able to provide computational data regarding novel mutations in a timeframe to contribute to patient care [87,330,331]. Ultimately, the combination of experimental and theoretical structural biology, when applied judiciously, is shown to be an advantageous supporting tool for the process of decision-making in personalized medicine and, hopefully, it can be included in standard care in the not-too-distant future.
Author Contributions
Conceptualization, J.F.V. and M.R.V.; writing—original draft preparation, J.F.V., S.C.M., M.Z.A., S.E.; writing—review and editing, J.F.V. and M.R.G.; supervision, M.R.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to acknowledge Santos Adel-Sayed for graphic support on the figures of this manuscript, and Rick Oerlemans for helpful comments on the final version.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the result.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.D. Lung Cancer Pathology. Clin. Chest Med. 2020, 41, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [Green Version]
- Brustugun, O.T.; Khattak, A.M.; Trømborg, A.K.; Beigi, M.; Beiske, K.; Lund-Iversen, M.; Helland, Å. BRAF-mutations in non-small cell lung cancer. Lung Cancer 2014, 84, 36–38. [Google Scholar] [CrossRef]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014. [Google Scholar] [CrossRef]
- Pimentel, A.S.; Guimarães, C.R.W.; Miller, Y. Molecular Modeling: Advancements and Applications. J. Chem. 2013, 2013, 1–2. [Google Scholar] [CrossRef]
- Smondyrev, A.M.; Voth, G.A. Molecular dynamics simulation of proton transport tnrougn tne influenza A virus M2 channel. Biophys. J. 2002, 83, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z. Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. PocketQuery: Protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res. 2012, 40, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Dömling, A.; Camacho, C.J. AnchorQuery: Rapid online virtual screening for small-molecule protein-protein interaction inhibitors. Protein Sci. 2018, 27, 229–232. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Geng, C.; Narasimhan, S.; Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J. Information-driven, ensemble flexible peptide docking using HADDOCK. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1561, pp. 109–138. [Google Scholar]
- Ricci, C.G.; Netz, P.A. Docking Studies on DNA-Ligand Interactions: Building and Application of a Protocol To Identify the Binding Mode. J. Chem. Inf. Model. 2009, 49, 1925–1935. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defelipe, L.A.; Arcon, J.P.; Modenutti, C.P.; Marti, M.A.; Turjanski, A.G.; Barril, X. Solvents to fragments to drugs: MD applications in drug design. Molecules 2018, 23, 3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Madhusudan; Trafny, E.A.; Xuong, N.-H.; Adams, J.A.; Eyck, L.F.T.; Taylor, S.S.; Sowadski, J.M. cAMP-dependent protein kinase: Crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 2008, 3, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Kuriyan, J. The Conformational Plasticity of Protein Kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- D’Abramo, M.; Besker, N.; Chillemi, G.; Grottesi, A. Modeling conformational transitions in kinases by molecular dynamics simulations: Achievements, difficulties, and open challenges. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Möbitz, H. The ABC of protein kinase conformations. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1555–1566. [Google Scholar] [CrossRef]
- Meharena, H.S.; Chang, P.; Keshwani, M.M.; Oruganty, K.; Nene, A.K.; Kannan, N.; Taylor, S.S.; Kornev, A.P. Deciphering the Structural Basis of Eukaryotic Protein Kinase Regulation. PLoS Biol. 2013, 11, e1001680. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Doshi, A.; Lei, M.; Eck, M.J.; Harrison, S.C. Crystal Structures of c-Src Reveal Features of Its Autoinhibitory Mechanism. Mol. Cell 1999, 3, 629–638. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Kamata, N.; Takechi, M.; Mitani, Y.; Ohta, K.; Fujimoto, S.; Hiraoka, M.; Higashikawa, K.; Shigeishi, H. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol. Rep. 2008, 19, 1557–1564. [Google Scholar] [CrossRef]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gureasko, J.; Galush, W.J.; Boykevisch, S.; Sondermann, H.; Bar-Sagi, D.; Groves, J.T.; Kuriyan, J. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat. Struct. Mol. Biol. 2008, 15, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.; Gee, J.M.; Harper, M. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.; Lee, S.-H.; Choi, Y.-L.; Han, J. Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6,595 lung cancers. Oncotarget 2016, 7, 23874–23884. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.P.; Berger, M.B.; Lin, C.-C.; Schlessinger, J.; Lemmon, M.A.; Ferguson, K.M. Epidermal Growth Factor Receptor Dimerization and Activation Require Ligand-Induced Conformational Changes in the Dimer Interface. Mol. Cell. Biol. 2005, 25, 7734–7742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.-H.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights into Differential Inhibitor Sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic Control in the EGF Receptor and Its Connection to General Kinase Regulatory Mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Arkhipov, A.; Kim, E.T.; Pan, A.C.; Shaw, D.E. Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 7270–7275. [Google Scholar] [CrossRef] [Green Version]
- George Priya Doss, C.; Rajith, B.; Chakraborty, C.; NagaSundaram, N.; Ali, S.K.; Zhu, H. Structural signature of the G719S-T790M double mutation in the EGFR kinase domain and its response to inhibitors. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Saldaña-Rivera, L.; Bello, M.; Méndez-Luna, D. Structural insight into the binding mechanism of ATP to EGFR and L858R, and T790M and L858R/T790 mutants. J. Biomol. Struct. Dyn. 2019, 37, 4671–4684. [Google Scholar] [CrossRef]
- Keyloun, K.R.; Reid, M.C.; Choi, R.; Song, Y.; Fox, A.M.W.; Hillesland, H.K.; Zhang, Z.; Vidadala, R.; Merritt, E.A.; Lau, A.O.T.; et al. The gatekeeper residue and beyond: Homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 2014, 141, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, N.; Tojo, A.; Hino, M.; Yazaki, Y.; Shibuya, M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 1992, 186, 768–774. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.H.; Dolder, C.R. Lapatinib: A Novel Dual Tyrosine Kinase Inhibitor with Activity in Solid Tumors. Ann. Pharmacother. 2006, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio Visualization; Dassault Systemes BIOVIA: San Diego, CA, USA, 2016.
- Songtawee, N.; Bevan, D.R.; Choowongkomon, K. Molecular dynamics of the asymmetric dimers of EGFR: Simulations on the active and inactive conformations of the kinase domain. J. Mol. Graph. Model. 2015, 58, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Yang, M.; Liang, N.; Li, S. Determining EGFR-TKI sensitivity of G719X and other uncommon EGFR mutations in non-small cell lung cancer: Perplexity and solution. Oncol. Rep. 2017, 37, 1347–1358. [Google Scholar] [CrossRef] [Green Version]
- Noronha, V.; Choughule, A.; Patil, V.M.; Joshi, A.; Kumar, R.; Philip, D.S.J.; Banavali, S.; Dutt, A.; Prabhash, K. Epidermal growth factor receptor exon 20 mutation in lung cancer: Types, incidence, clinical features and impact on treatment. Onco. Targets. Ther. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Avizienyte, E.; Ward, R.A.; Garner, A.P. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem. J. 2008, 415, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non–Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, S.; Kukimoto-Niino, M.; Parker, L.; Handa, N.; Terada, T.; Fujimoto, T.; Terazawa, Y.; Wakiyama, M.; Sato, M.; Sano, S.; et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 2013, 32, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and Clinical Relevance of the Epidermal Growth Factor Receptor in Human Cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta-Proteins Proteomics 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassunke, J.; Müller, F.; Keul, M.; Michels, S.; Dammert, M.A.; Schmitt, A.; Plenker, D.; Lategahn, J.; Heydt, C.; Brägelmann, J.; et al. Overcoming EGFRG724S-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat. Commun. 2018, 9, 4655. [Google Scholar] [CrossRef]
- Takeda, M.; Nakagawa, K. First- and Second-Generation EGFR-TKIs Are All Replaced to Osimertinib in Chemo-Naive EGFR Mutation-Positive Non-Small Cell Lung Cancer? Int. J. Mol. Sci. 2019, 20, 146. [Google Scholar] [CrossRef] [Green Version]
- El Meskini, R.; Iacovelli, A.; Kulaga, A.; Gumprecht, M.; Spencer, M.; Ileva, L.; Simmons, A.; Weaver Ohler, Z. Abstract A162: Continuous treatment with rociletinib in EGFR transgenic mice results in acquired resistance to both rociletinib and osimertinib and intra-“patient” tumor heterogeneity. In EGFR/Her2; American Association for Cancer Research: Philadelphia, PA, USA, 2018; p. A162. [Google Scholar]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Rangachari, D.; To, C.; Shpilsky, J.E.; VanderLaan, P.A.; Kobayashi, S.S.; Mushajiang, M.; Lau, C.J.; Paweletz, C.P.; Oxnard, G.R.; Jänne, P.A.; et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J. Thorac. Oncol. 2019, 14, 1995–2002. [Google Scholar] [CrossRef]
- Niederst, M.J.J.; Hu, H.; Mulvey, H.E.E.; Lockerman, E.L.L.; Garcia, A.R.R.; Piotrowska, Z.; Sequist, L.V.V.; Engelman, J.A.A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, K.D.; Garton, A.J.; Romero, M.S.; Kahler, J.; Thomson, S.; Ross, S.; Park, F.; Haley, J.D.; Gibson, N.; Sliwkowski, M.X. Kinetic Analysis of Epidermal Growth Factor Receptor Somatic Mutant Proteins Shows Increased Sensitivity to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Erlotinib. Cancer Res. 2006, 66, 8163–8171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, N.; Azuma, K.; Sakai, K.; Hattori, S.; Kawahara, A.; Ishii, H.; Tokito, T.; Kinoshita, T.; Yamada, K.; Nishio, K.; et al. Association of EGFR Exon 19 Deletion and EGFR-TKI Treatment Duration with Frequency of T790M Mutation in EGFR-Mutant Lung Cancer Patients. Sci. Rep. 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, A.; Husain, H. First-Line Treatment in EGFR Mutant Non-Small Cell Lung Cancer: Is There a Best Option? Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Ou, Q.; Wu, X.; Wang, X.; Shao, Y.W.; Ying, J. Acquired EGFR L718V mutation mediates resistance to osimertinib in non-small cell lung cancer but retains sensitivity to afatinib. Lung Cancer 2018, 118, 1–5. [Google Scholar] [CrossRef]
- Liu, J.; Jin, B.; Su, H.; Qu, X.; Liu, Y. Afatinib helped overcome subsequent resistance to osimertinib in a patient with NSCLC having leptomeningeal metastasis baring acquired EGFR L718Q mutation: A case report. BMC Cancer 2019, 19, 702. [Google Scholar] [CrossRef]
- Brown, B.P.; Zhang, Y.-K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target resistance to the mutant-selective EGFR inhibitor osimertinib can develop in an allele specific manner dependent on the original EGFR activating mutation. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Fujino, T.; Nishino, M.; Koga, T.; Chiba, M.; Sesumi, Y.; Ohara, S.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. EGFR T790M and C797S Mutations as Mechanisms of Acquired Resistance to Dacomitinib. J. Thorac. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokudome, N.; Koh, Y.; Akamatsu, H.; Fujimoto, D.; Okamoto, I.; Nakagawa, K.; Hida, T.; Imamura, F.; Morita, S.; Yamamoto, N. Differential significance of molecular subtypes which were classified into EGFR exon 19 deletion on the first line afatinib monotherapy. BMC Cancer 2020, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Tamirat, M.Z.; Koivu, M.; Elenius, K.; Johnson, M.S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE 2019, 14, e0222814. [Google Scholar] [CrossRef] [PubMed]
- van Kempen, L.C.; Wang, H.; Aguirre, M.L.; Spatz, A.; Kasymjanova, G.; Vilacha, J.F.; Groves, M.R.; Agulnik, J.; Small, D. Afatinib in Osimertinib-Resistant EGFR ex19del/T790M/P794L Mutated NSCLC. J. Thorac. Oncol. 2018, 13, e161–e163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.B. Kinase inhibitor-responsive genotypes in EGFR mutated lung adenocarcinomas: Moving past common point mutations or indels into uncommon kinase domain duplications and rearrangements. Transl. Lung Cancer Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Liu, X.; Song, X.; Yao, J. Chemotherapy Drug Response to the L858R-induced Conformational Change of EGFR Activation Loop in Lung Cancer. Mol. Inform. 2016, 35, 529–537. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Cheng, G.; Song, Z.; Chen, D. Clinical efficacy of first-generation EGFR-TKIs in patients with advanced non-small-cell lung cancer harboring EGFR exon 20 mutations. Oncol. Targets. Ther. 2016, 9, 4181–4186. [Google Scholar] [CrossRef] [Green Version]
- Barker, A.J.; Gibson, K.H.; Grundy, W.; Godfrey, A.A.; Barlow, J.J.; Healy, M.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Scarlett, L.; et al. Studies leading to the identification of ZD1839 (iressaTM): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. [Google Scholar] [CrossRef]
- Yasuda, H.; Park, E.; Yun, C.-H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.-L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci. Transl. Med. 2014, 5, 225er1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, M. Binding mechanism of kinase inhibitors to EGFR and T790M, L858R and L858R/T790M mutants through structural and energetic analysis. Int. J. Biol. Macromol. 2018, 118, 1948–1962. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and Biological Features Associated With Epidermal Growth Factor Receptor Gene Mutations in Lung Cancers. JNCI J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Z.; Hao, X.; Hu, X.; Wang, H.; Wang, Y.; Ying, J. Clinical characteristics and response to tyrosine kinase inhibitors of patients with non-small cell lung cancer harboring uncommon epidermal growth factor receptor mutations. Chin. J. Cancer Res. 2017, 29, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Skaggs, B.J.; Gorre, M.E.; Ryvkin, A.; Burgess, M.R.; Xie, Y.; Han, Y.; Komisopoulou, E.; Brown, L.M.; Loo, J.A.; Landaw, E.M.; et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 19466–19471. [Google Scholar] [CrossRef] [Green Version]
- Sos, M.L.; Rode, H.B.; Heynck, S.; Peifer, M.; Fischer, F.; Klüter, S.; Pawar, V.G.; Reuter, C.; Heuckmann, J.M.; Weiss, J.; et al. Chemogenomic Profiling Provides Insights into the Limited Activity of Irreversible EGFR Inhibitors in Tumor Cells Expressing the T790M EGFR Resistance Mutation. Cancer Res. 2010, 70, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Kästner, J.; Senn, H.M.; Thiel, S.; Otte, N.; Thiel, W. QM/MM Free-Energy Perturbation Compared to Thermodynamic Integration and Umbrella Sampling: Application to an Enzymatic Reaction. J. Chem. Theory Comput. 2006, 2, 452–461. [Google Scholar] [CrossRef]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef] [Green Version]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the Aberrant Activity of Mutant EGFR Kinase Domain and Drug Recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wykosky, J.; Fenton, T.; Furnari, F.; Cavenee, W.K. Therapeutic targeting of epidermal growth factor receptor in human cancer: Successes and limitations. Chin. J. Cancer 2011, 30, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Masood, A.; Kancha, R.K.; Subramanian, J. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer harboring uncommon EGFR mutations: Focus on afatinib. Semin. Oncol. 2019, 46, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; Liguori, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; D’Aveni, A.; Marabello, G.; Altavilla, G.; Rosell, R. Osimertinib in the treatment of non-small-cell lung cancer: Design, development and place in therapy. Lung Cancer Targets Ther. 2017, 8, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liam, C.-K. The role of osimertinib in epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer. J. Thorac. Dis. 2019, 11, S448–S452. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhou, F.; Shen, W.; Jiang, T.; Wu, X.; Tong, X.; Shao, Y.W.; Qin, S.; Zhou, C. Novel Mutations on EGFR Leu792 Potentially Correlate to Acquired Resistance to Osimertinib in Advanced NSCLC. J. Thorac. Oncol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Klempner, S.J.; Mehta, P.; Schrock, A.B.; Ali, S.M.; Ou, S.-H.I. Cis-oriented solvent-front EGFR G796S mutation in tissue and ctDNA in a patient progressing on osimertinib: A case report and review of the literature. Lung Cancer 2017, 8, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Oztan, A.; Fischer, S.; Schrock, A.B.; Erlich, R.L.; Lovly, C.M.; Stephens, P.J.; Ross, J.S.; Miller, V.; Ali, S.M.; Ou, S.H.I.; et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 2017, 111. [Google Scholar] [CrossRef]
- Yella, J.; Yaddanapudi, S.; Wang, Y.; Jegga, A. Changing Trends in Computational Drug Repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, A.; Szabo, C. Inventing new therapies without reinventing the wheel: The power of drug repurposing. Br. J. Pharmacol. 2018, 175, 165–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, P.L.; John, T.; Dobrovic, A.; Mitchell, P. Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin. Epidemiol. 2014, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mano, H. ALKoma: A cancer subtype with a shared target. Cancer Discov. 2012, 2, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.; Kirstein, M.; Valentine, M.; Dittmer, K.; Shapiro, D.; Saltman, D.; Look, A. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Cancer Sci. 2018, 109, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Vernersson, E.; Khoo, N.K.S.; Henriksson, M.L.; Roos, G.; Palmer, R.H.; Hallberg, B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr. Patterns 2006, 6, 448–461. [Google Scholar] [CrossRef]
- Pulford, K.; Lamant, L.; Morris, S.W.; Butler, L.H.; Wood, K.M.; Stroud, D.; Delsol, G.; Mason, D.Y. Detection of Anaplastic Lymphoma Kinase (ALK) and Nucleolar Protein Nucleophosmin (NPM)-ALK Proteins in Normal and Neoplastic Cells With the Monoclonal Antibody ALK1. Blood 1997, 89, 1394–1404. [Google Scholar] [CrossRef]
- Yao, S.; Cheng, M.; Zhang, Q.; Wasik, M.; Kelsh, R.; Winkler, C. Anaplastic Lymphoma Kinase Is Required for Neurogenesis in the Developing Central Nervous System of Zebrafish. PLoS ONE 2013, 8, e63757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbough, J.; Broadie, K. Anterograde Jelly belly ligand to Alk receptor signaling at developing synapses is regulated by Mind the gap. Development 2010, 137, 3523–3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klincumhom, N.; Tharasanit, T.; Thongkittidilok, C.; Tiptanavattana, N.; Rungarunlert, S.; Dinnyés, A.; Techakumphu, M. Selective TGF-β1/ALK inhibitor improves neuronal differentiation of mouse embryonic stem cells. Neurosci. Lett. 2014, 578, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Delisle, L.; Pierre-Eugène, C.; Bloch-Gallego, E.; Birling, M.-C.; Duband, J.-L.; Durand, E.; Bourgeois, T.; Matrot, B.; Sorg, T.; Huerre, M.; et al. Hyperactivation of Alk induces neonatal lethality in knock-in AlkF1178L mice. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, R.; Lwigale, P. Expression of the heparin-binding growth factors Midkine and pleiotrophin during ocular development. Gene Expr. Patterns 2019, 32, 28–37. [Google Scholar] [CrossRef]
- Motegi, A. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J. Cell Sci. 2004, 117, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Souttou, B.; Carvalho, N.B.-D.; Raulais, D.; Vigny, M. Activation of Anaplastic Lymphoma Kinase Receptor Tyrosine Kinase Induces Neuronal Differentiation through the Mitogen-activated Protein Kinase Pathway. J. Biol. Chem. 2001, 276, 9526–9531. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Jia, Y.; Li, N.; Sun, X.; Ng, K.; Ambing, E.; Gao, M.-Y.; Hua, S.; Chen, C.; Kim, S.; et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem. J. 2010, 430, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Mol. Case Stud. 2017, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Long, X.; Song, Y.; Chen, P.; Li, S.; Yang, H.; Wu, P.; Wang, Y.; Bing, Z.; Cao, Z.; et al. Distribution of ALK Fusion Variants and Correlation with Clinical Outcomes in Chinese Patients with Non-Small Cell Lung Cancer Treated with Crizotinib. Target. Oncol. 2019, 14, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.-S.; Leung, E.L.-H.; So, K.K.-T.; Tam, I.Y.-S.; Sihoe, A.D.-L.; Cheng, L.-C.; Ho, K.-K.; Au, J.S.-K.; Chung, L.-P.; Pik Wong, M. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009, 115, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; et al. EML4-ALK Fusion Gene and Efficacy of an ALK Kinase Inhibitor in Lung Cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Varghese, A.M.; Ou, S.-H.I.; Kabraji, S.; Awad, M.M.; Katayama, R.; Pawlak, A.; Mino-Kenudson, M.; Yeap, B.Y.; Riely, G.J.; et al. ALK Rearrangements Are Mutually Exclusive with Mutations in EGFR or KRAS: An Analysis of 1,683 Patients with Non-Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4273–4281. [Google Scholar] [CrossRef] [Green Version]
- Tiseo, M.; Gelsomino, F.; Boggiani, D.; Bortesi, B.; Bartolotti, M.; Bozzetti, C.; Sammarelli, G.; Thai, E.; Ardizzoni, A. EGFR and EML4-ALK gene mutations in NSCLC: A case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer 2011, 71, 241–243. [Google Scholar] [CrossRef]
- Campos-Gomez, S.; Lara-Guerra, H.; Routbort, M.J.; Lu, X.; Simon, G.R. Lung adenocarcinoma with Concurrent KRAS Mutation and ALK Rearrangement Responding to Crizotinib: Case Report. Int. J. Biol. Markers 2015, 30, 254–257. [Google Scholar] [CrossRef]
- Bayliss, R.; Choi, J.; Fennell, D.A.; Fry, A.M.; Richards, M.W. Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell. Mol. Life Sci. 2016, 73, 1209–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a Novel Fusion Oncokinase Identified by an Immunohistochemistry-based Diagnostic System for ALK-positive Lung Cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.-H.I.; Klempner, S.J.; Greenbowe, J.R.; Azada, M.; Schrock, A.B.; Ali, S.M.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Identification of a Novel HIP1-ALK Fusion Variant in Non–Small-Cell Lung Cancer (NSCLC) and Discovery of ALK I1171 (I1171N/S) Mutations in Two ALK-Rearranged NSCLC Patients with Resistance to Alectinib. J. Thorac. Oncol. 2014, 9, 1821–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.-L.; Lira, M.E.; Hong, M.; Kim, R.N.; Choi, S.-J.; Song, J.-Y.; Pandy, K.; Mann, D.L.; Stahl, J.A.; Peckham, H.E.; et al. A Novel Fusion of TPR and ALK in Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Jiang, P.; Xu, F.; Zhang, W.; Guo, L.; Wu, J.; Zeng, Y.; Jiao, Y.; Ying, J. BIRC6-ALK, a Novel Fusion Gene in ALK Break-Apart FISH-Negative Lung Adenocarcinoma, Responds to Crizotinib. J. Thorac. Oncol. 2015, 10, e37–e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef]
- Childress, M.A.; Himmelberg, S.M.; Chen, H.; Deng, W.; Davies, M.A.; Lovly, C.M. ALK Fusion Partners Impact Response to ALK Inhibition: Differential Effects on Sensitivity, Cellular Phenotypes, and Biochemical Properties. Mol. Cancer Res. 2018, 16, 1724–1736. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Zhang, Y.; Li, Y.; Liu, Q.; Chu, H.; Zhang, D.; Li, G. Integrating Multiple Accelerated Molecular Dynamics To Improve Accuracy of Free Energy Calculations. J. Chem. Theory Comput. 2018, 14, 1216–1227. [Google Scholar] [CrossRef]
- He, M.-Y.; Li, W.-K.; Zheng, Q.-C.; Zhang, H.-X. Conformational Transition of Key Structural Features Involved in Activation of ALK Induced by Two Neuroblastoma Mutations and ATP Binding: Insight from Accelerated Molecular Dynamics Simulations. ACS Chem. Neurosci. 2018, 9, 1783–1792. [Google Scholar] [CrossRef]
- Donella-Deana, A.; Marin, O.; Cesaro, L.; Gunby, R.H.; Ferrarese, A.; Coluccia, A.M.L.; Tartari, C.J.; Mologni, L.; Scapozza, L.; Gambacorti-Passerini, C.; et al. Unique Substrate Specificity of Anaplastic Lymphoma Kinase (ALK): Development of Phosphoacceptor Peptides for the Assay of ALK Activity. Biochemistry 2005, 44, 8533–8542. [Google Scholar] [CrossRef]
- Jiang, C.-H.; Huang, C.-X.; Chen, Y.-J.; Chuang, Y.-C.; Huang, B.-Y.; Yang, C.-N. Molecular Modeling for Structural Insights Concerning the Activation Mechanisms of F1174L and R1275Q Mutations on Anaplastic Lymphoma Kinase. Molecules 2018, 23, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, M.; Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Li, S.; Zhang, J. Harnessing Allostery: A Novel Approach to Drug Discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Huang, W.; Wang, Q.; Shen, Q.; Li, S.; Nussinov, R.; Zhang, J. The Structural Basis of ATP as an Allosteric Modulator. PLoS Comput. Biol. 2014, 10, e1003831. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in Advanced ALK -Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.-Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK Inhibitor Ceritinib Overcomes Crizotinib Resistance in Non-Small Cell Lung Cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Kay, M.; Dehghanian, F. Exploring the crizotinib resistance mechanism of NSCLC with the L1196M mutation using molecular dynamics simulation. J. Mol. Model. 2017, 23, 323. [Google Scholar] [CrossRef]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a Novel ALK G1123S Mutation in a Patient with ALK-rearranged Non–small-cell Lung Cancer Exhibiting Resistance to Ceritinib. J. Thorac. Oncol. 2015, 10, e55–e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Niu, X.; Chang, L.; Chen, R.; Ou, S.-H.I.; Lu, S. Next generation sequencing reveals a novel ALK G1128A mutation resistant to crizotinib in an ALK-Rearranged NSCLC patient. Lung Cancer 2018, 123, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.; Dô, P.; Richard, N.; Dubos, C.; Michels, J.J.; Bonneau, J.; Gervais, R. Identification of I1171N resistance mutation in ALK-positive non-small-cell lung cancer tumor sample and circulating tumor DNA. Lung Cancer 2016, 99, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.-H.I.; Greenbowe, J.; Khan, Z.U.; Azada, M.C.; Ross, J.S.; Stevens, P.J.; Ali, S.M.; Miller, V.A.; Gitlitz, B. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer 2015, 88, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.; Milliken, J.C.; Azada, M.C.; Miller, V.A.; Ali, S.M.; Klempner, S.J. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer 2016, 91, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two Novel ALK Mutations Mediate Acquired Resistance to the Next-Generation ALK Inhibitor Alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK Mutations in Lung Cancer That Confer Resistance to ALK Inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK -rearranged lung cancer. Cancer Discov. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.-W.; Lee, M.-S.; Lee, S.E.; Song, J.-Y.; Shin, H.-T.; Kim, Y.J.; Oh, D.Y.; Jung, K.; Sung, M.; Kim, M.; et al. Molecular breakdown: A comprehensive view of anaplastic lymphoma kinase ( ALK )-rearranged non-small cell lung cancer. J. Pathol. 2017, 243, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.Y.; Friboulet, L.; Kodack, D.P.; Engstrom, L.D.; Li, Q.; West, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell 2015, 28, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro- N 2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)- N 4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,-4. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.-L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. ALK Mutations Conferring Differential Resistance to Structurally Diverse ALK Inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezquita, L.; Planchard, D. The role of brigatinib in crizotinib-resistant non-small cell lung cancer. Cancer Manag. Res. 2018, 10, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; Pao, W. Escaping ALK Inhibition: Mechanisms of and Strategies to Overcome Resistance. Sci. Transl. Med. 2012, 4, 120ps2. [Google Scholar] [CrossRef]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [Green Version]
- Kodityal, S.; Elvin, J.A.; Squillace, R.; Agarwal, N.; Miller, V.A.; Ali, S.M.; Klempner, S.J.; Ou, S.-H.I. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer 2016, 92, 19–21. [Google Scholar] [CrossRef]
- Michels, S.Y.F.; Scheel, A.H.; Wündisch, T.; Heuckmann, J.M.; Menon, R.; Puesken, M.; Kobe, C.; Pasternack, H.; Heydt, C.; Scheffler, M.; et al. ALK G1269A mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. NPJ Precis. Oncol. 2017, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.G.; Cortinovis, D.; Agustoni, F.; Arosio, G.; Villa, M.; Cordani, N.; Bidoli, P.; Bisson, W.H.; Pagni, F.; Piazza, R.; et al. A Compound L1196M/G1202R ALK Mutation in a Patient with ALK-Positive Lung Cancer with Acquired Resistance to Brigatinib Also Confers Primary Resistance to Lorlatinib. J. Thorac. Oncol. 2019, 14, e257–e259. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.-Y.; et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin. Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Seto, Y.; Okada, K.; Uematsu, S.; Uchibori, K.; Tsukahara, M.; Oh-hara, T.; Fujita, N.; Yanagitani, N.; Nishio, M.; et al. Overcoming resistance by ALK compound mutation (I1171S + G1269A) after sequential treatment of multiple ALK inhibitors in non-small cell lung cancer. Thorac. Cancer 2020, 11, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglivo, S.; Ricciuti, B.; Ludovini, V.; Metro, G.; Siggillino, A.; De Giglio, A.; Chiari, R. Dramatic Response to Lorlatinib in a Heavily Pretreated Lung Adenocarcinoma Patient Harboring G1202R Mutation and a Synchronous Novel R1192P ALK Point Mutation. J. Thorac. Oncol. 2018, 13, e145–e147. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; He, Z.; Xie, D.; Zheng, L.; Zhao, T.; Zhang, X.; Cheng, D. Molecular Mechanism Behind the Resistance of the G1202R-Mutated Anaplastic Lymphoma Kinase to the Approved Drug Ceritinib. J. Phys. Chem. B 2018, 122, 4680–4692. [Google Scholar] [CrossRef]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a Selective ALK Inhibitor Capable of Blocking the Resistant Gatekeeper Mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK -Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.-L.; Dinh, D.; et al. Discovery of (10 R )-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro- 2H -8,4-(metheno)pyrazolo[4,3- h ][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sun, R.; Wu, Y.; Song, M.; Li, J.; Yang, Q.; Chen, X.; Bao, J.; Zhao, Q. L1198F Mutation Resensitizes Crizotinib to ALK by Altering the Conformation of Inhibitor and ATP Binding Sites. Int. J. Mol. Sci. 2017, 18, 482. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Araki, M.; Sakashita, T.; Ma, B.; Kanada, R.; Yanagitani, N.; Horiike, A.; Koike, S.; Oh-hara, T.; Watanabe, K.; et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 2019, 41, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; Heuckmann, J.M.; de Stanchina, E.; Chen, H.; Thomas, R.K.; Liang, C.; Pao, W. Insights into ALK-Driven Cancers Revealed through Development of Novel ALK Tyrosine Kinase Inhibitors. Cancer Res. 2011, 71, 4920–4931. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, J.; Zhou, J.; Feng, J.; Zhuang, W.; Chen, J.; Zhao, J.; Zhong, W.; Zhao, Y.; Zhang, Y.; et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell lung cancer: A multicentre, phase 2 trial. Lancet Respir. Med. 2020, 8, 45–53. [Google Scholar] [CrossRef]
- Horn, L.; Infante, J.R.; Reckamp, K.L.; Blumenschein, G.R.; Leal, T.A.; Waqar, S.N.; Gitlitz, B.J.; Sanborn, R.E.; Whisenant, J.G.; Du, L.; et al. Ensartinib (X-396) in ALK-Positive Non–Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin. Cancer Res. 2018, 24, 2771–2779. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Zhang, X.; Yang, D.; Wang, H.; Si, X.; Zhang, L. Single-center study to determine the safety and efficacy of CT-707 in Chinese patients with advanced anaplastic lymphoma kinase-rearranged non-small-cell lung cancer. Thorac. Cancer 2020, 11, 1216–1223. [Google Scholar] [CrossRef]
- Horn, L.; Reckamp, K.L.; Patel, S.; Blumenschein, G.; Neal, J.W.; Gitlitz, B.; Waqar, S.; Oxnard, G.; Brzezniak, C.; Dukart, G.; et al. Abstract CT151: CNS activity of ensartinib in ALK-positive non-small cell lung cancer patients. In Clinical Trials; American Association for Cancer Research: Philadelphia, PA, USA, 2017; p. CT151. [Google Scholar]
- Menichincheri, M.; Ardini, E.; Magnaghi, P.; Avanzi, N.; Banfi, P.; Bossi, R.; Buffa, L.; Canevari, G.; Ceriani, L.; Colombo, M.; et al. Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J. Med. Chem. 2016, 59, 3392–3408. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pizzutilo, E.G.; Marrapese, G.; Tosi, F.; Cerea, G.; Siena, S. Entrectinib for the treatment of metastatic NSCLC: Safety and efficacy. Expert Rev. Anticancer Ther. 2020, 20, 333–341. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Kojima, H.; Saito, N.; Okabe, T.; Masuda, Y.; Furuya, T.; Nagano, T. Virtual screening and further development of novel ALK inhibitors. Bioorg. Med. Chem. 2011, 19, 3086–3095. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; Mcgrady, P.W.; et al. ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell 2014, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, M.; Seeliger, M.A.; Gray, N.S.; Kuriyan, J.; Daley, G.Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 2008, 15, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, J.; Zhu, W. Mutation L1196M-induced conformational changes and the drug resistant mechanism of anaplastic lymphoma kinase studied by free energy perturbation and umbrella sampling. Phys. Chem. Chem. Phys. 2017, 19, 30239–30248. [Google Scholar] [CrossRef]
- Ai, X.; Shen, S.; Shen, L.; Lu, S. An interaction map of small-molecule kinase inhibitors with anaplastic lymphoma kinase (ALK) mutants in ALK-positive non-small cell lung cancer. Biochimie 2015, 112, 111–120. [Google Scholar] [CrossRef]
- Leonis, G.; Steinbrecher, T.; Papadopoulos, M.G. A Contribution to the Drug Resistance Mechanism of Darunavir, Amprenavir, Indinavir, and Saquinavir Complexes with HIV-1 Protease Due to Flap Mutation I50V: A Systematic MM–PBSA and Thermodynamic Integration Study. J. Chem. Inf. Model. 2013, 53, 2141–2153. [Google Scholar] [CrossRef]
- Dehghanian, F.; Kay, M.; Vallian, S. F1174V mutation alters the ALK active conformation in response to Crizotinib in NSCLC: Insight from molecular simulations. J. Mol. Graph. Model. 2017, 75, 287–293. [Google Scholar] [CrossRef]
- Liu, C.; Chen, C. P1.03-012 Using Computational Modeling to Simulate Clinical Response of ALK Inhibitors to G1202R ALK and Possible Mechanisms of Resistance. J. Thorac. Oncol. 2017, 12, S1955. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Guo, W.; Du, B.; Huang, X.; Wu, R.; Yang, B.; Lin, X.; Wu, Y. Insight into resistance mechanism of anaplastic lymphoma kinase to alectinib and JH-VIII-157-02 caused by G1202R solvent front mutation. Drug Des. Devel. Ther. 2018, 12, 1183–1193. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-Y.; Ji, F.-Q. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem. Biophys. Res. Commun. 2012, 423, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Wang, X.; Zhang, T.; Jin, R.Z. Molecular dynamics simulations reveal the allosteric effect of F1174C resistance mutation to ceritinib in ALK-associated lung cancer. Comput. Biol. Chem. 2016, 65, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Nagasundaram, N.; Wilson Alphonse, C.R.; Samuel Gnana, P.V.; Rajaretinam, R.K. Molecular Dynamics Validation of Crizotinib Resistance to ALK Mutations (L1196M and G1269A) and Identification of Specific Inhibitors. J. Cell. Biochem. 2017, 118, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-C.; Huang, B.-Y.; Chang, H.-W.; Yang, C.-N. Molecular Modeling of ALK L1198F and/or G1202R Mutations to Determine Differential Crizotinib Sensitivity. Sci. Rep. 2019, 9, 11390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras / Raf / MEK / ERK signaling pathway and its role in the occurrence and development of HCC ( Review ). Oncol. Lett. 2016, 3045–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.; Kondo, M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 2011, 49, 248–268. [Google Scholar] [CrossRef]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [Green Version]
- Aramini, J.M.; Vorobiev, S.M.; Tuberty, L.M.; Janjua, H.; Campbell, E.T.; Seetharaman, J.; Su, M.; Huang, Y.J.; Acton, T.B.; Xiao, R.; et al. The RAS-Binding Domain of Human BRAF Protein Serine/Threonine Kinase Exhibits Allosteric Conformational Changes upon Binding HRAS. Structure 2015, 23, 1382–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, R.E.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.E.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural history and molecular characteristics of lung cancers harboring egfr exon 20 insertions. J. Thorac. Oncol. 2013, 8, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.S.; Keshwani, M.M.; Steichen, J.M.; Kornev, A.P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2517–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClendon, C.L.; Kornev, A.P.; Gilson, M.K.; Taylora, S.S. Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. USA 2014, 111, E4623–E4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Gademann, K.; Portmann, C.; Blom, J.F.; Zeder, M.; Jüttner, F. Multiple toxin production in the cyanobacterium Microcystis: Isolation of the toxic protease inhibitor cyanopeptolin 1020. J. Nat. Prod. 2010, 73, 980–984. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.K.; Slupsky, J.R.; Andreas Kalmes, H.; Rapp, U.R. Active ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61, 3595–3598. [Google Scholar] [PubMed]
- Haling, J.R.; Sudhamsu, J.; Yen, I.; Sideris, S.; Sandoval, W.; Phung, W.; Bravo, B.J.; Giannetti, A.M.; Peck, A.; Masselot, A.; et al. Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling. Cancer Cell 2014, 26, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Del Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. The importance of Raf dimerization in cell signaling. Small GTPases 2013, 4, 180. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, T.; Sahmi, M.; Lefrançois, M.; Sicheri, F.; Therrien, M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 2009, 461, 542–545. [Google Scholar] [CrossRef]
- Karoulia, Z.; Wu, Y.; Ahmed, T.A.; Xin, Q.; Bollard, J.; Krepler, C.; Wu, X.; Zhang, C.; Bollag, G.; Herlyn, M.; et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 2016, 30, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Endicott, J.A.; Noble, M.E.M.; Johnson, L.N. The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 2012, 81, 587–613. [Google Scholar] [CrossRef]
- Johnson, L.N.; Noble, M.E.M.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, N.; Novak, B.; Candelora, C.; Wong, K.; Cavallo, M.; Gunderwala, A.; Liu, Z.; Li, Y.; Wang, Z. Biochemical Characterization of Full-Length Oncogenic BRAFV600E together with Molecular Dynamics Simulations Provide Insight into the Activation and Inhibition Mechanisms of RAF Kinases. ChemBioChem 2019, 20, 2850–2861. [Google Scholar] [CrossRef] [PubMed]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Pan, Y.L. Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant colorectal cancer. Cancer Manag. Res. 2018, 10, 2289–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef] [Green Version]
- Lang, L. FDA Approves Sorafenib for Patients With Inoperable Liver Cancer. Gastroenterology 2008, 134, 379. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, D.D.; Li, N.; Poon, D.J.; Aardalen, K.; Kaufman, S.; Merritt, H.; Salangsang, F.; Lorenzana, E.; Li, A.; Ghoddusi, M.; et al. Abstract 3790: Preclinical profile of LGX818: A potent and selective RAF kinase inhibitor. Cancer Res. 2012, 72, 3790. [Google Scholar] [CrossRef]
- Thevakumaran, N.; Lavoie, H.; Critton, D.A.; Tebben, A.; Marinier, A.; Sicheri, F.; Therrien, M. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat. Struct. Mol. Biol. 2014, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Zhang, Y.; van Horn, R.D.; Yin, T.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, H.; Thevakumaran, N.; Gavory, G.; Li, J.J.; Padeganeh, A.; Guiral, S.; Duchaine, J.; Mao, D.Y.L.; Bouvier, M.; Sicheri, F.; et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat. Chem. Biol. 2013, 9, 428–436. [Google Scholar] [CrossRef]
- Tse, A.; Verkhivker, G.M. Exploring Molecular Mechanisms of Paradoxical Activation in the BRAF Kinase Dimers: Atomistic Simulations of Conformational Dynamics and Modeling of Allosteric Communication Networks and Signaling Pathways. PLoS ONE 2016, 11, e0166583. [Google Scholar] [CrossRef]
- Waizenegger, I.C.; Baum, A.; Steurer, S.; Stadtmüller, H.; Bader, G.; Schaaf, O.; Garin-Chesa, P.; Schlattl, A.; Schweifer, N.; Haslinger, C.; et al. A novel RAF kinase inhibitor with DFG-out-binding mode: High efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol. Cancer Ther. 2016, 15, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Di Michele, M.; Stes, E.; Vandermarliere, E.; Martens, L.; Gevaert, K.; Van Heerde, E.; Linders, J.T.M.; Brehmer, D.; Jacoby, E.; et al. Structural investigation of B-Raf paradox breaker and inducer inhibitors. J. Med. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Noeparast, A.; Verschelden, G.; Umelo, I.; de Brakeleer, S.; Decoste, L.; Teugels, E.; de Greve, J. Investigation of non-V600 BRAF mutations commonly found in NSCLC for their sensitivity to Dabrafenib or Trametinib. Ann. Oncol. 2015, 26, ii36. [Google Scholar] [CrossRef]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.J.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAFV600 and BRAFnonV600 mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Casadei Gardini, A.; Chiadini, E.; Faloppi, L.; Marisi, G.; Delmonte, A.; Scartozzi, M.; Loretelli, C.; Lucchesi, A.; Oboldi, D.; Dubini, A.; et al. Efficacy of sorafenib in BRAF-mutated nonsmall-cell lung cancer (NSCLC) and no response in synchronous BRAF wild typehepatocellular carcinoma: A case report. BMC Cancer 2016, 16. [Google Scholar] [CrossRef]
- Sereno, M.; Moreno, V.; Rubio, J.M.; Gómez-Raposo, C.; Sánchez, S.G.; Jusdado, R.H.; Falagan, S.; Tébar, F.Z.; Sáenz, E.C. A significant response to sorafenib in a woman with advanced lung adenocarcinoma and a BRAF non-V600 mutation. Anticancer. Drugs 2015, 26, 1004–1007. [Google Scholar] [CrossRef]
- Gautschi, O.; Peters, S.; Zoete, V.; Aebersold-Keller, F.; Strobel, K.; Schwizer, B.; Hirschmann, A.; Michielin, O.; Diebold, J. Lung adenocarcinoma with BRAF G469L mutation refractory to vemurafenib. Lung Cancer 2013, 82, 365–367. [Google Scholar] [CrossRef]
- Hallmeyer, S.; Gonzalez, R.; Lawson, D.H.; Cranmer, L.D.; Linette, G.P.; Puzanov, I.; Taback, B.; Cowey, C.L.; Ribas, A.; Daniels, G.A.; et al. Vemurafenib treatment for patients with locally advanced, unresectable stage IIIC or metastatic melanoma and activating exon 15 BRAF mutations other than V600E. Melanoma Res. 2017, 27, 585–590. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Gadaleta-Caldarola, G.; Divella, R.; Mazzocca, A.; Infusino, S.; Ferraro, E.; Filippelli, G.; Daniele, A.; Sabbà, C.; Abbate, I.; Brandi, M. Sorafenib: The gold standard therapy in advanced hepatocellular carcinoma and beyond. Futur. Oncol. 2015, 11, 2263–2266. [Google Scholar] [CrossRef]
- Rubinstein, J.C.; Sznol, M.; Pavlick, A.C.; Ariyan, S.; Cheng, E.; Bacchiocchi, A.; Kluger, H.M.; Narayan, D.; Halaban, R. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J. Transl. Med. 2010, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, O.; Clements, A.; Menzies, A.M.; O’Toole, S.; Kefford, R.F.; Long, G.V. BRAF inhibitor activity in V600R metastatic melanoma. Eur. J. Cancer 2013, 49, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Moiseyenko, F.V.; Egorenkov, V.V.; Kramchaninov, M.M.; Artemieva, E.V.; Aleksakhina, S.N.; Holmatov, M.M.; Moiseyenko, V.M.; Imyanitov, E.N. Lack of Response to Vemurafenib in Melanoma Carrying BRAF K601E Mutation. Case Rep. Oncol. 2019, 12, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Landrette, S.F.; Wang, T.; Evans, P.; Bacchiocchi, A.; Bjornson, R.; Cheng, E.; Stiegler, A.L.; Gathiaka, S.; Acevedo, O.; et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res. 2014, 27, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yao, Z.; Jonsson, P.; Allen, A.N.; Qin, A.C.R.; Uddin, S.; Dunkel, I.J.; Petriccione, M.; Manova, K.; Haque, S.; et al. A secondary mutation in BRAF confers resistance to RAF inhibition in a BRAFV600E-mutant brain tumor. Cancer Discov. 2018, 8, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.; Kirk, R.; Hayward, R.; Zambon, A.; Viros, A.; Cantarino, N.; Affolter, A.; Nourry, A.; Niculescu-Duvaz, D.; Springer, C.; et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med. 2010, 2, 35ra41. [Google Scholar] [CrossRef]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The ras superfamily of small GTPases: The unlocked secrets. Methods Mol. Biol. 2014, 1120, 1–18. [Google Scholar]
- Liao, J.; Shima, F.; Araki, M.; Ye, M.; Muraoka, S.; Sugimoto, T.; Kawamura, M.; Yamamoto, N.; Tamura, A.; Kataoka, T. Two conformational states of Ras GTPase exhibit differential GTP-binding kinetics. Biochem. Biophys. Res. Commun. 2008, 369, 327–332. [Google Scholar] [CrossRef]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: Role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Dreusicke, D.; Schulz, G.E. The glycine-rich loop of adenylate kinase forms a giant anion hole. FEBS Lett. 1986, 208, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Klockow, B.; Ahmadian, M.R.; Block, C.; Wittinghofer, A. Oncogenic insertional mutations in the P-loop of Ras are overactive in MAP kinase signaling. Oncogene 2000, 19, 5367–5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, A.; Travesset, A. Differential dynamics of RAS isoforms in GDP- and GTP-bound states. Proteins Struct. Funct. Bioinforma. 2015, 83, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Prakash, P.; Sayyed-Ahmad, A.; Gorfe, A.A. pMD-Membrane: A Method for Ligand Binding Site Identification in Membrane-Bound Proteins. PLOS Comput. Biol. 2015, 11, e1004469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayyed-Ahmad, A.; Gorfe, A.A. Mixed-Probe Simulation and Probe-Derived Surface Topography Map Analysis for Ligand Binding Site Identification. J. Chem. Theory Comput. 2017, 13, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.N.; Bellamacina, C.R.; Ding, X.; Jeffery, C.J.; Mattos, C.; Petsko, G.A.; Ringe, D. An experimental approach to mapping the binding surfaces of crystalline proteins. J. Phys. Chem. 1996, 100, 2605–2611. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMr-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Hocker, H.J.; Cho, K.J.; Chen, C.Y.K.; Rambahal, N.; Sagineedu, S.R.; Shaari, K.; Stanslas, J.; Hancock, J.F.; Gorfe, A.A. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc. Natl. Acad. Sci. USA 2013, 110, 10201–10206. [Google Scholar] [CrossRef] [Green Version]
- Rosnizeck, I.C.; Graf, T.; Spoerner, M.; Tränkle, J.; Filchtinski, D.; Herrmann, C.; Gremer, L.; Vetter, I.R.; Wittinghofer, A.; König, B.; et al. Stabilizing a weak binding state for effectors in the human ras protein by cyclen complexes. Angew. Chem. Int. Ed. 2010, 49, 3830–3833. [Google Scholar] [CrossRef]
- Prakash, P.; Hancock, J.F.; Gorfe, A.A. Binding hotspots on K-ras: Consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins Struct. Funct. Bioinforma. 2015, 83, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Alexander, R.E.; Maclennan, G.T.; Cummings, O.W.; Montironi, R.; Lopez-beltran, A.; Cramer, H.M.; Davidson, D.D. Molecular pathology of lung cancer: Key to personalized medicine. Mod. Pathol. 2012, 25, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Marín-Ramos, N.I.; Piñar, C.; Vázquez-Villa, H.; Martín-Fontecha, M.; González, Á.; Canales, Á.; Algar, S.; Mayo, P.P.; Jiménez-Barbero, J.; Gajate, C.; et al. Development of a Nucleotide Exchange Inhibitor That Impairs Ras Oncogenic Signaling. Chem. A Eur. J. 2017, 23, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Burke, J.P.; Phan, J.; Burns, M.C.; Olejniczak, E.T.; Waterson, A.G.; Lee, T.; Rossanese, O.W.; Fesik, S.W. Discovery of Small Molecules that Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chem. Int. Ed. 2012, 124, 6244–6247. [Google Scholar] [CrossRef] [Green Version]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [Green Version]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- El Osta, B.E.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and outcomes of patients (pts) with metastatic KRAS mutant lung adenocarcinomas: Lung Cancer Mutation Consortium (LCMC) database. J. Clin. Oncol. 2017, 35, 9021. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Domerg, C.; Hainaut, P.; Jänne, P.A.; Pignon, J.-P.; Graziano, S.; Douillard, J.-Y.; Brambilla, E.; Le Chevalier, T.; Seymour, L.; et al. Pooled Analysis of the Prognostic and Predictive Effects of KRAS Mutation Status and KRAS Mutation Subtype in Early-Stage Resected Non–Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J. Clin. Oncol. 2013, 31, 2173–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aredo, J.V.; Padda, S.K.; Kunder, C.A.; Han, S.S.; Neal, J.W.; Shrager, J.B.; Wakelee, H.A. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer 2019, 133, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Bera, A.K.; Gondi, S.; Westover, K.D. KRAS Switch Mutants D33E and A59G Crystallize in the State 1 Conformation. Biochemistry 2018, 57, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-C.; Er, T.-K.; Liu, Y.-Y.; Hwang, J.-K.; Barrio, M.J.; Rodrigo, M.; Garcia-Toro, E.; Herreros-Villanueva, M. Computational Analysis of KRAS Mutations: Implications for Different Effects on the KRAS p.G12D and p.G13D Mutations. PLoS ONE 2013, 8, e55793. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.A.; Volmar, A.Y.; Pavlopoulos, S.; Mattos, C. K-Ras Populates Conformational States Differently from Its Isoform H-Ras and Oncogenic Mutant K-RasG12D. Structure 2018, 26, 810–820.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Long, B.N.; Boris, G.H.; Chen, A.; Ni, S.; Kennedy, M.A. Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 970–984. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef] [Green Version]
- Kauke, M.J.; Traxlmayr, M.W.; Parker, J.A.; Kiefer, J.D.; Knihtila, R.; McGee, J.; Verdine, G.; Mattos, C.; Wittrup, K.D. An engineered protein antagonist of K-Ras/B-Raf interaction. Sci. Rep. 2017, 7, 5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Gurbani, D.; Ficarro, S.B.; Carrasco, M.A.; Lim, S.M.; Choi, H.G.; Xie, T.; Marto, J.A.; Chen, Z.; Gray, N.S.; et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 2014, 111, 8895–8900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic targeting of oncogenic K-ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. 2014, 53, 199–204. [Google Scholar] [CrossRef]
- Sun, Q.; Phan, J.; Friberg, A.R.; Camper, D.V.; Olejniczak, E.T.; Fesik, S.W. A method for the second-site screening of K-Ras in the presence of a covalently attached first-site ligand. J. Biomol. NMR 2014, 60, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Migoni, A.; Canning, P.; Quevedo, C.E.; Bataille, C.J.R.; Bery, N.; Miller, A.; Russell, A.J.; Phillips, S.E.V.; Carr, S.B.; Rabbitts, T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 2545–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Gupta, A.K.; Wang, X.; Pagba, C.V.; Prakash, P.; Sarkar-Banerjee, S.; Putkey, J.; Gorfe, A.A. Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem. Biol. Drug Des. 2019, cbdd.13519. [Google Scholar] [CrossRef]
- Putora, P.M.; Ess, S.; Panje, C.; Hundsberger, T.; van Leyen, K.; Plasswilm, L.; Früh, M. Prognostic significance of histology after resection of brain metastases and whole brain radiotherapy in non-small cell lung cancer (NSCLC). Clin. Exp. Metastasis 2015, 32, 143–149. [Google Scholar] [CrossRef]
- Su, S.F.; Hu, Y.X.; Ouyang, W.W.; Ma, Z.; Lu, B.; Li, Q.S.; Li, H.Q.; Wang, Z.Y.; Wang, Y. The survival outcomes and prognosis of stage IV non-small-cell lung cancer treated with thoracic three-dimensional radiotherapy combined with chemotherapy. Radiat. Oncol. 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Guise, J.-M.; Savitz, L.A.; Friedman, C.P. Mind the Gap: Putting Evidence into Practice in the Era of Learning Health Systems. J. Gen. Intern. Med. 2018, 33, 2237–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, O.B.; Neves, K.; Wasilewska-Sampaio, A.P.; Carneiro, C.F. The Brazilian Reproducibility Initiative. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, D.L.; van Herpen, C.M.L.; van Laarhoven, H.W.M.; Smit, E.F.; Groen, H.J.M.; Willems, S.M.; Nederlof, P.M.; Langenberg, M.H.G.; Cuppen, E.; Sleijfer, S.; et al. Molecular Tumor Boards: Current practice and future needs. Ann. Oncol. 2017, 28, 3070–3075. [Google Scholar] [CrossRef]
- Santos, J.F.V.M.R.; Hiddinga, B.I.; van Kempen, L.C.; ter Elst, A.; Hijmering, L.B.; Hiltermann, T.J.; Schuuring, E.; Groves, M.R.; Groen, H.J.; Wekken, A.J. van der Abstract 1398: Modeling of drug-protein interactions to support clinical decision making for therapy-resistant EGFR or ALK -positive non-small cell lung carcinoma. In Clinical Research (Excluding Clinical Trials); American Association for Cancer Research: Philadelphia, PA, USA, 2019; Volume 79, p. 1398. [Google Scholar]
- Koopman, B.; van der Wekken, A.J.; ter Elst, A.; Hiltermann, T.J.N.; Vilacha, J.F.; Groves, M.R.; van den Berg, A.; Hiddinga, B.I.; Hijmering-Kappelle, L.B.M.; Stigt, J.A.; et al. Relevance and Effectiveness of Molecular Tumor Board Recommendations for Patients With Non–Small-Cell Lung Cancer With Rare or Complex Mutational Profiles. JCO Precis. Oncol. 2020, 393–410. [Google Scholar] [CrossRef]
Figure 1.
The sequences of the three kinases focused on within this review are represented with relevant residues highlighted. (A) Epidermal Growth Factor Receptor (EGFR) (UniProt ID: P00533), (B) Anaplastic Lymphoma Kinase (ALK) (UniProt: ID Q9UM73), and (C) Rapidly Accelerated Fibrosarcoma Homologue B (BRAF) sequences are represented with relevant motifs highlighted. (D) c-MET in complex with ATP (PDB: ID 3DKC) is used as a general representation of a kinase domain with the C-terminal lobe colored blue with hinge motif colored pink, N-terminal lobe is colored yellow with the P-loop residues backbone depicted as sticks and the regulatory αC-helix colored in orange. Nitrogen and oxygen atoms are colored blue and red, respectively. The ATP molecule is depicted with carbon atoms in green and phosphate atoms in orange, magnesium ion is depicted as a green sphere.
Figure 1.
The sequences of the three kinases focused on within this review are represented with relevant residues highlighted. (A) Epidermal Growth Factor Receptor (EGFR) (UniProt ID: P00533), (B) Anaplastic Lymphoma Kinase (ALK) (UniProt: ID Q9UM73), and (C) Rapidly Accelerated Fibrosarcoma Homologue B (BRAF) sequences are represented with relevant motifs highlighted. (D) c-MET in complex with ATP (PDB: ID 3DKC) is used as a general representation of a kinase domain with the C-terminal lobe colored blue with hinge motif colored pink, N-terminal lobe is colored yellow with the P-loop residues backbone depicted as sticks and the regulatory αC-helix colored in orange. Nitrogen and oxygen atoms are colored blue and red, respectively. The ATP molecule is depicted with carbon atoms in green and phosphate atoms in orange, magnesium ion is depicted as a green sphere.

Figure 2.
Summary of the main signaling pathways involving the erbB family, EML4-ALK fusion protein and RAF family. RAS family implication is also represented in either GDP- or GTP-bound states. These signaling pathways regulate and control important downstream cellular functions such as apoptosis, cell growth, survival, angiogenesis, and migration.
Figure 2.
Summary of the main signaling pathways involving the erbB family, EML4-ALK fusion protein and RAF family. RAS family implication is also represented in either GDP- or GTP-bound states. These signaling pathways regulate and control important downstream cellular functions such as apoptosis, cell growth, survival, angiogenesis, and migration.

Figure 3.
Schematic representation of (A) active EGFR kinase domain structure with erlotinib (PDB: ID 1M17). Mutation hotspots are indicated in red with T790 depicted as spheres and L858 as sticks. The most common sequence deletion in exon 19, E746-A750 is colored orange. Comparison of regulatory motifs (B) β1 and β2 strand along with the P-loop (residues G719-G724), (C) αC-helix (residues P741-A767), (D) Activation segment starting with DFG motif (residues D855/F856/G857) and ending with AxE motif (A882/L883/E884). Regulatory motifs are highlighted and aligned with the inactive conformation (PDB: ID 1XKK) in gray. (E) K745 from β3 strand engaging in a salt bridge with E762 in the αC-helix.
Figure 3.
Schematic representation of (A) active EGFR kinase domain structure with erlotinib (PDB: ID 1M17). Mutation hotspots are indicated in red with T790 depicted as spheres and L858 as sticks. The most common sequence deletion in exon 19, E746-A750 is colored orange. Comparison of regulatory motifs (B) β1 and β2 strand along with the P-loop (residues G719-G724), (C) αC-helix (residues P741-A767), (D) Activation segment starting with DFG motif (residues D855/F856/G857) and ending with AxE motif (A882/L883/E884). Regulatory motifs are highlighted and aligned with the inactive conformation (PDB: ID 1XKK) in gray. (E) K745 from β3 strand engaging in a salt bridge with E762 in the αC-helix.
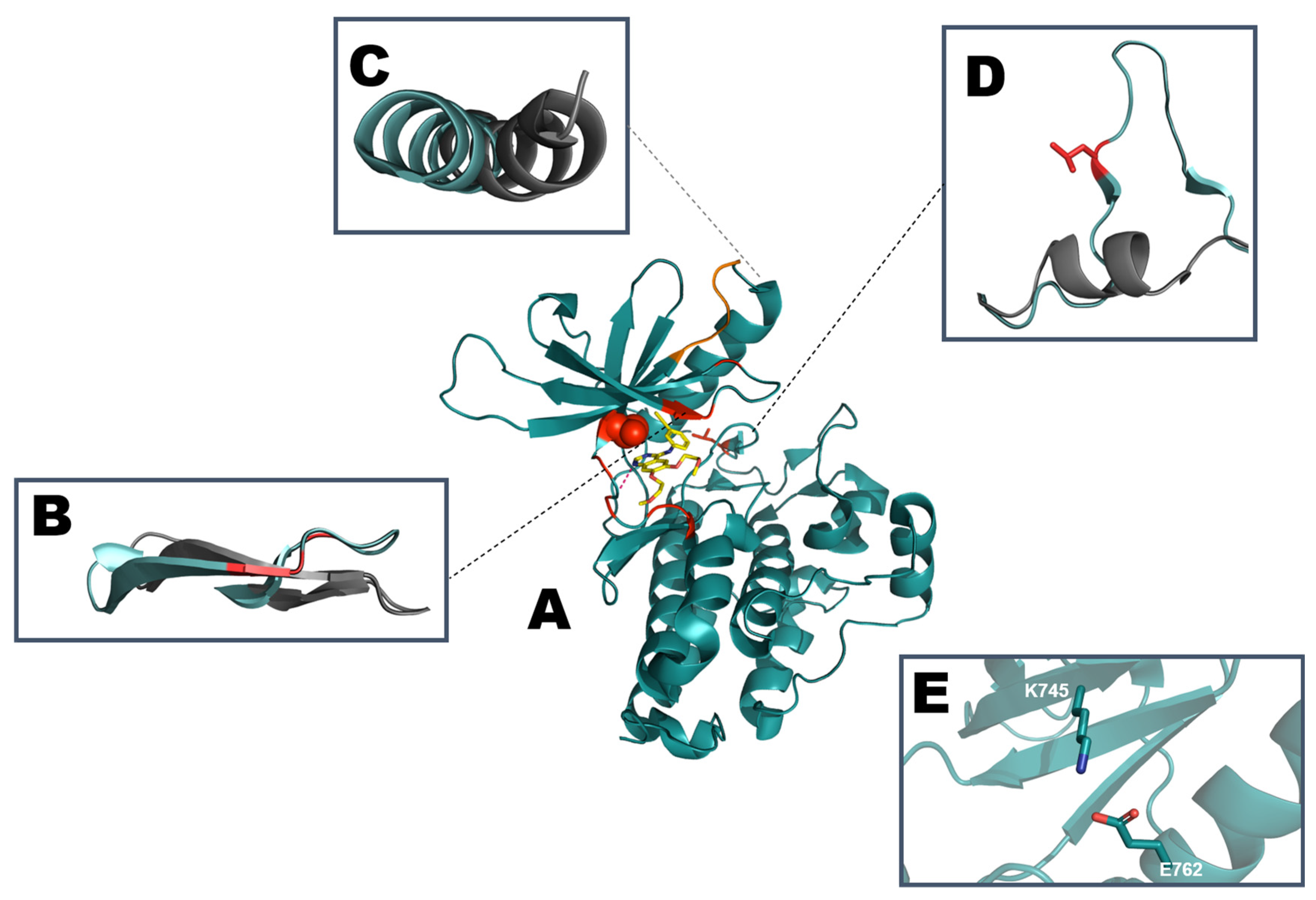
Figure 4.
Two-dimensional representation of lapatinib and first-generation inhibitors, gefitinib and erlotinib. The 4-aminoquinaoline scaffold, common to all drugs, engages in hydrogen bonds with hinge residue M793 while hydrophobic substituents contribute with van de Waals interactions with residues from the allosteric pocket, such as L788, V766, and L858. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon(grey), nitrogen (blue), oxygen (red), sulfur (beige), chlorine (green), and fluorine (light blue).
Figure 4.
Two-dimensional representation of lapatinib and first-generation inhibitors, gefitinib and erlotinib. The 4-aminoquinaoline scaffold, common to all drugs, engages in hydrogen bonds with hinge residue M793 while hydrophobic substituents contribute with van de Waals interactions with residues from the allosteric pocket, such as L788, V766, and L858. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon(grey), nitrogen (blue), oxygen (red), sulfur (beige), chlorine (green), and fluorine (light blue).

Figure 5.
Two-dimensional representation of second-generation inhibitors, afatinib and dacomitinib, and third-generation inhibitor, osimertinib. Although all three drugs follow a similar binding mode to first-generation inhibitors through hinge-binding scaffolds, osimertinib lacks interactions with the allosteric pocket. The proximity of the drugs’ warhead to C797, its covalent bond partner, is also depicted. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon (grey), nitrogen (blue), oxygen (red), chlorine (green), and fluorine (light blue).
Figure 5.
Two-dimensional representation of second-generation inhibitors, afatinib and dacomitinib, and third-generation inhibitor, osimertinib. Although all three drugs follow a similar binding mode to first-generation inhibitors through hinge-binding scaffolds, osimertinib lacks interactions with the allosteric pocket. The proximity of the drugs’ warhead to C797, its covalent bond partner, is also depicted. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon (grey), nitrogen (blue), oxygen (red), chlorine (green), and fluorine (light blue).
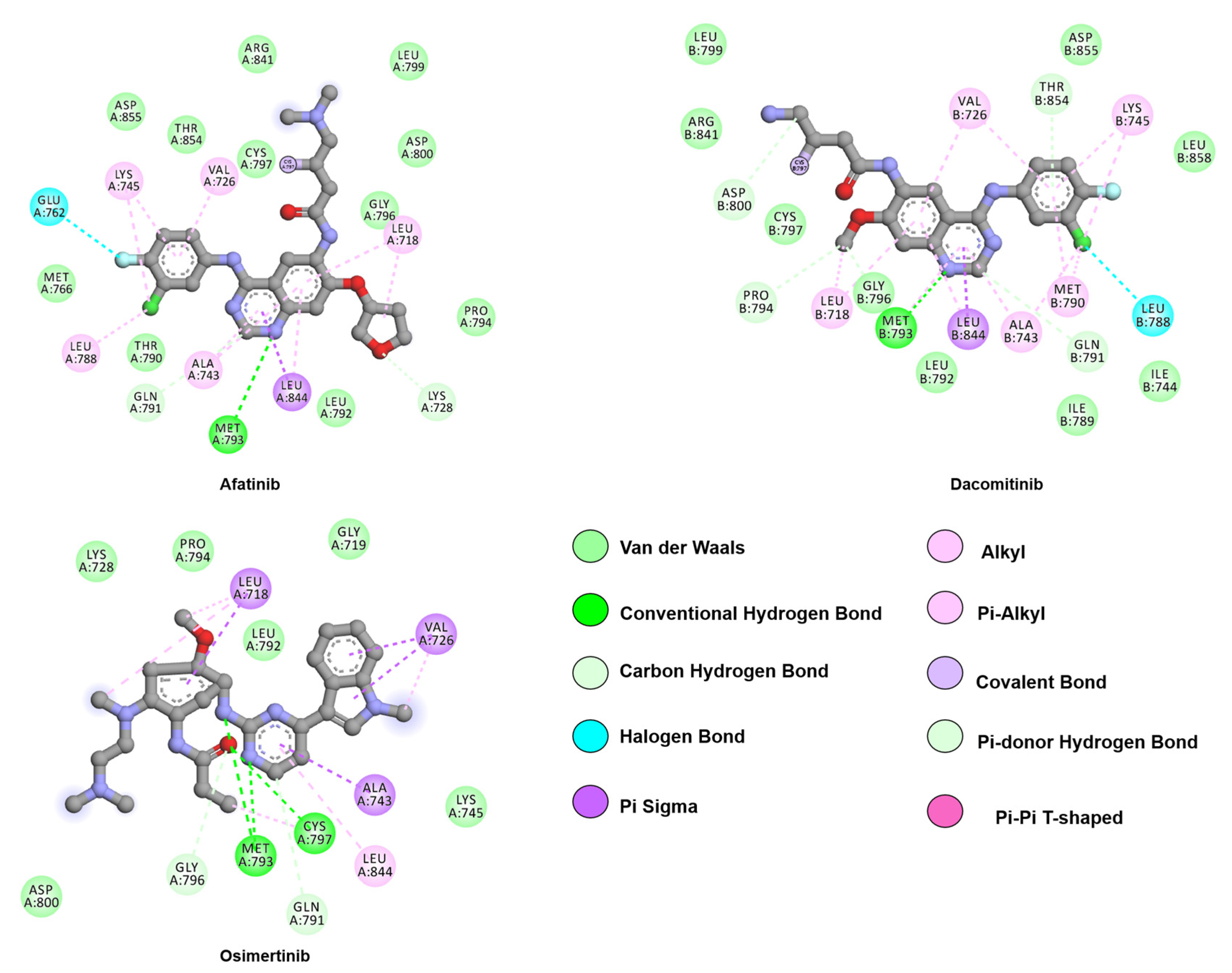
Figure 6.
Schematic representation of (A) partially active ALK kinase domain with crizotinib (PDB: ID 5FTO). Mutation hotspots are indicated in blue with L1196 depicted as spheres. Representation of the regulatory (B) β1 and β2 strands with P-loop (residues G1123-G1128), (C) unique YCFAGKTS motif (residues Y1096-S1103) in grey packed against the αC-helix (residues E1158-K1173), and (D) αAL motif (G1272-R1279) from the activation segment (residues D1270/F1271/G1272 to E1299) are highlighted and aligned with the inactive conformation in gray (PDB: ID 3L9P). (E) K1150 from β3 strand engaging in a salt bridge with E1167 in the αC-helix.
Figure 6.
Schematic representation of (A) partially active ALK kinase domain with crizotinib (PDB: ID 5FTO). Mutation hotspots are indicated in blue with L1196 depicted as spheres. Representation of the regulatory (B) β1 and β2 strands with P-loop (residues G1123-G1128), (C) unique YCFAGKTS motif (residues Y1096-S1103) in grey packed against the αC-helix (residues E1158-K1173), and (D) αAL motif (G1272-R1279) from the activation segment (residues D1270/F1271/G1272 to E1299) are highlighted and aligned with the inactive conformation in gray (PDB: ID 3L9P). (E) K1150 from β3 strand engaging in a salt bridge with E1167 in the αC-helix.
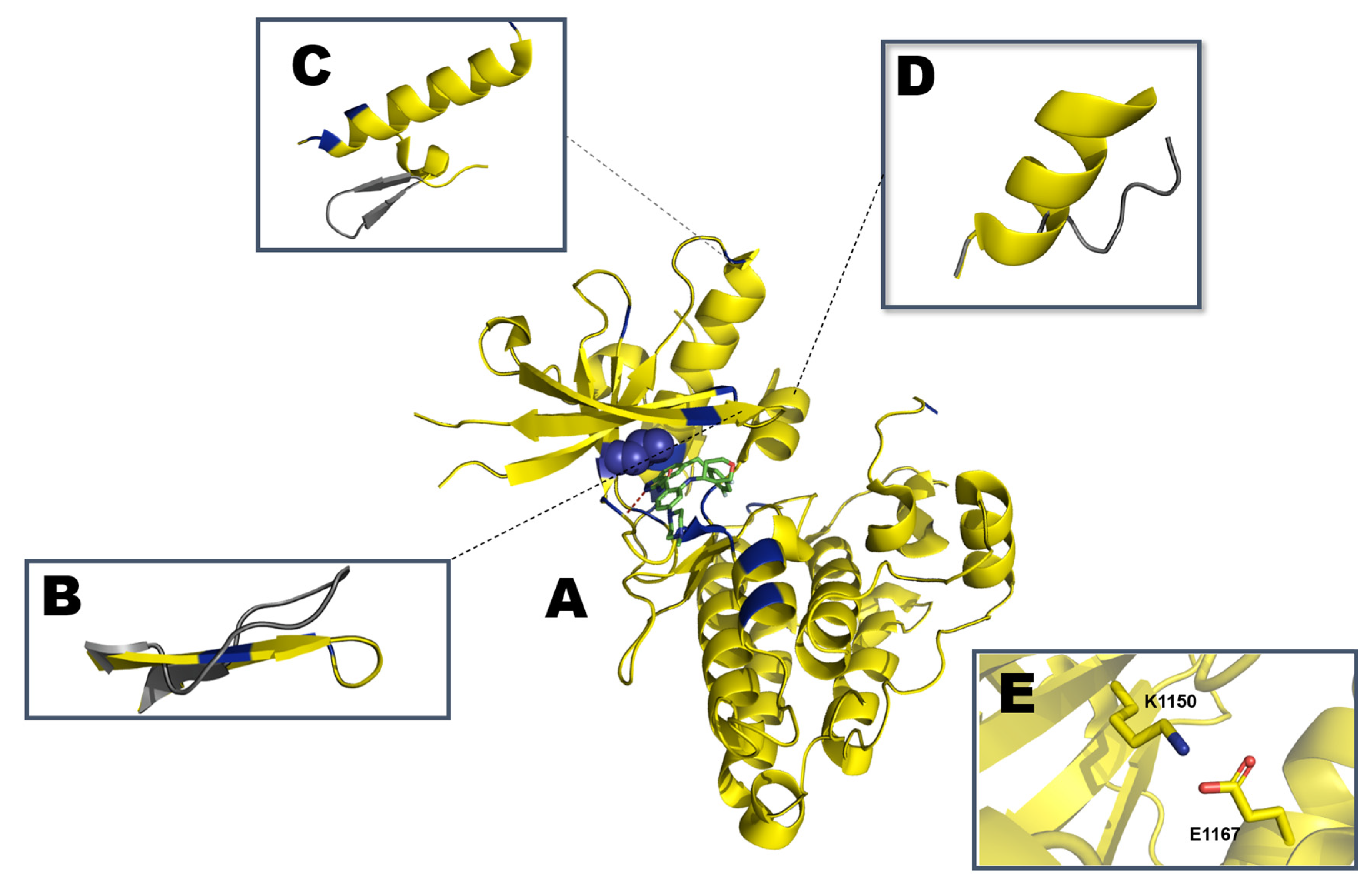
Figure 7.
Two-dimensional representation of first-generation inhibitors, crizotinib and entrectinib, and third-generation inhibitor, lorlatinib. Both generations interact with the hinge region through hydrogen bonds with E1197 and M1199. Crizotinib and entrectinib are within reach of G1202, while lorlatinib lacks this close approach. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon (grey), nitrogen (blue), oxygen (red), chlorine (green), and fluorine (light blue).
Figure 7.
Two-dimensional representation of first-generation inhibitors, crizotinib and entrectinib, and third-generation inhibitor, lorlatinib. Both generations interact with the hinge region through hydrogen bonds with E1197 and M1199. Crizotinib and entrectinib are within reach of G1202, while lorlatinib lacks this close approach. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon (grey), nitrogen (blue), oxygen (red), chlorine (green), and fluorine (light blue).
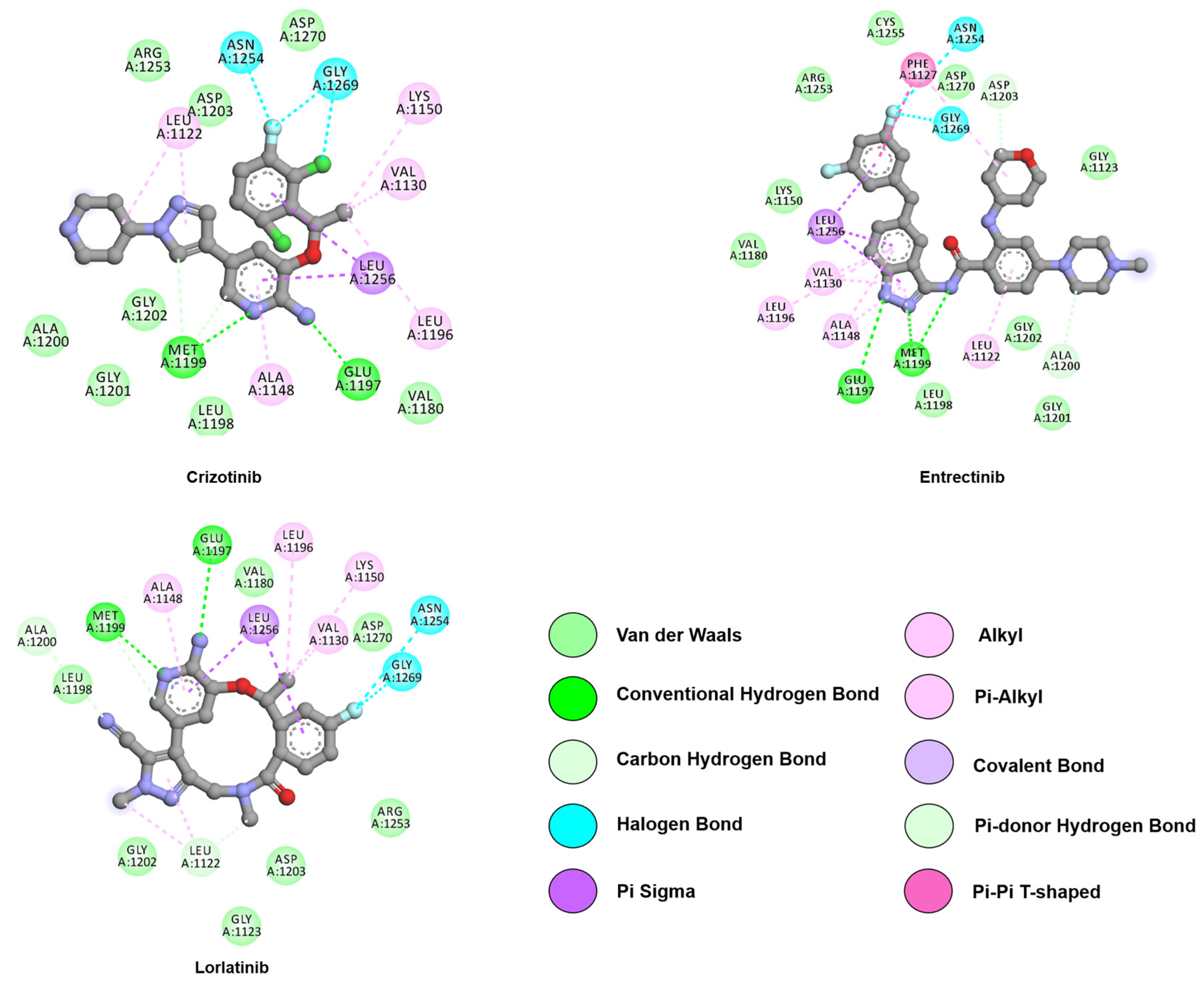
Figure 8.
Two-dimensional representation of second-generation inhibitors, alectinib, brigatinib, and ceritinib. All represented drugs interact with the hinge region through hydrogen bonds with M1199. Alectinib 6-dimethyl-5,6-dihydro-11H-benzo[b]carbazol-11-one scaffold is depicted as illustration of its constrained pose. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon (grey), nitrogen (blue), oxygen (red), phosphorus (orange), sulfur (beige), and chlorine (green).
Figure 8.
Two-dimensional representation of second-generation inhibitors, alectinib, brigatinib, and ceritinib. All represented drugs interact with the hinge region through hydrogen bonds with M1199. Alectinib 6-dimethyl-5,6-dihydro-11H-benzo[b]carbazol-11-one scaffold is depicted as illustration of its constrained pose. Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon (grey), nitrogen (blue), oxygen (red), phosphorus (orange), sulfur (beige), and chlorine (green).
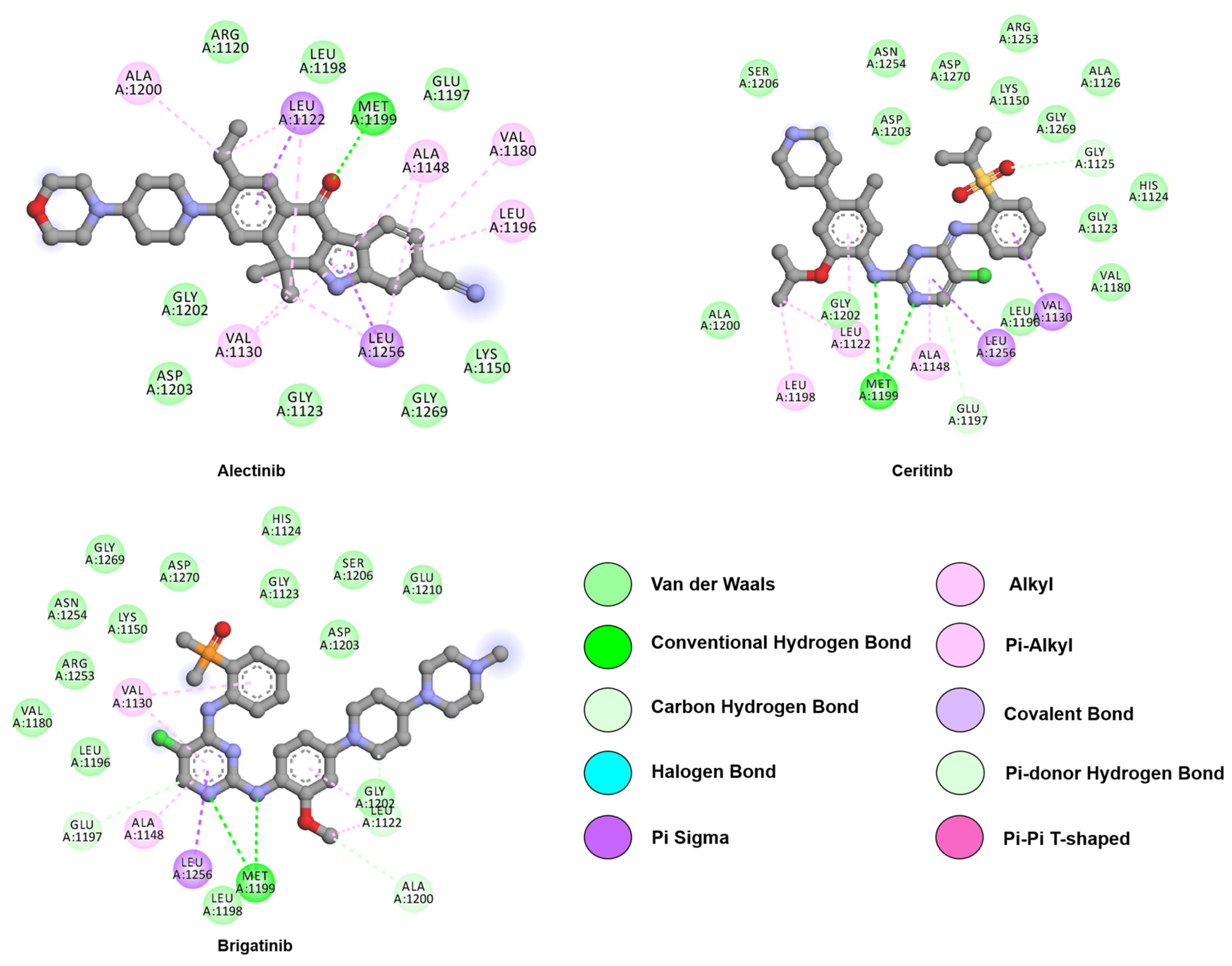
Figure 9.
Schematic representation of (A) the BRAF kinase domain with vemurafenib (PDB: ID 3OG7). Mutation hotspots are indicated in yellow with L529 depicted as spheres and V600 as sticks. Comparison of regulatory motifs (B) β1 and β2 strands containing the P-loop (G464-G469), (C) αC-helix (residues T591-R506), and (D) the activation segment starting with DFG (D594/F595/G596) and ending with AxE motif (A621/P622/E623). Regulatory motifs are highlighted and aligned with the inactive conformation (PDB: ID 4RZV) in gray. (E) The position of the αC-helix in the αC-out conformation hampers the interaction involving K483 and E501.
Figure 9.
Schematic representation of (A) the BRAF kinase domain with vemurafenib (PDB: ID 3OG7). Mutation hotspots are indicated in yellow with L529 depicted as spheres and V600 as sticks. Comparison of regulatory motifs (B) β1 and β2 strands containing the P-loop (G464-G469), (C) αC-helix (residues T591-R506), and (D) the activation segment starting with DFG (D594/F595/G596) and ending with AxE motif (A621/P622/E623). Regulatory motifs are highlighted and aligned with the inactive conformation (PDB: ID 4RZV) in gray. (E) The position of the αC-helix in the αC-out conformation hampers the interaction involving K483 and E501.
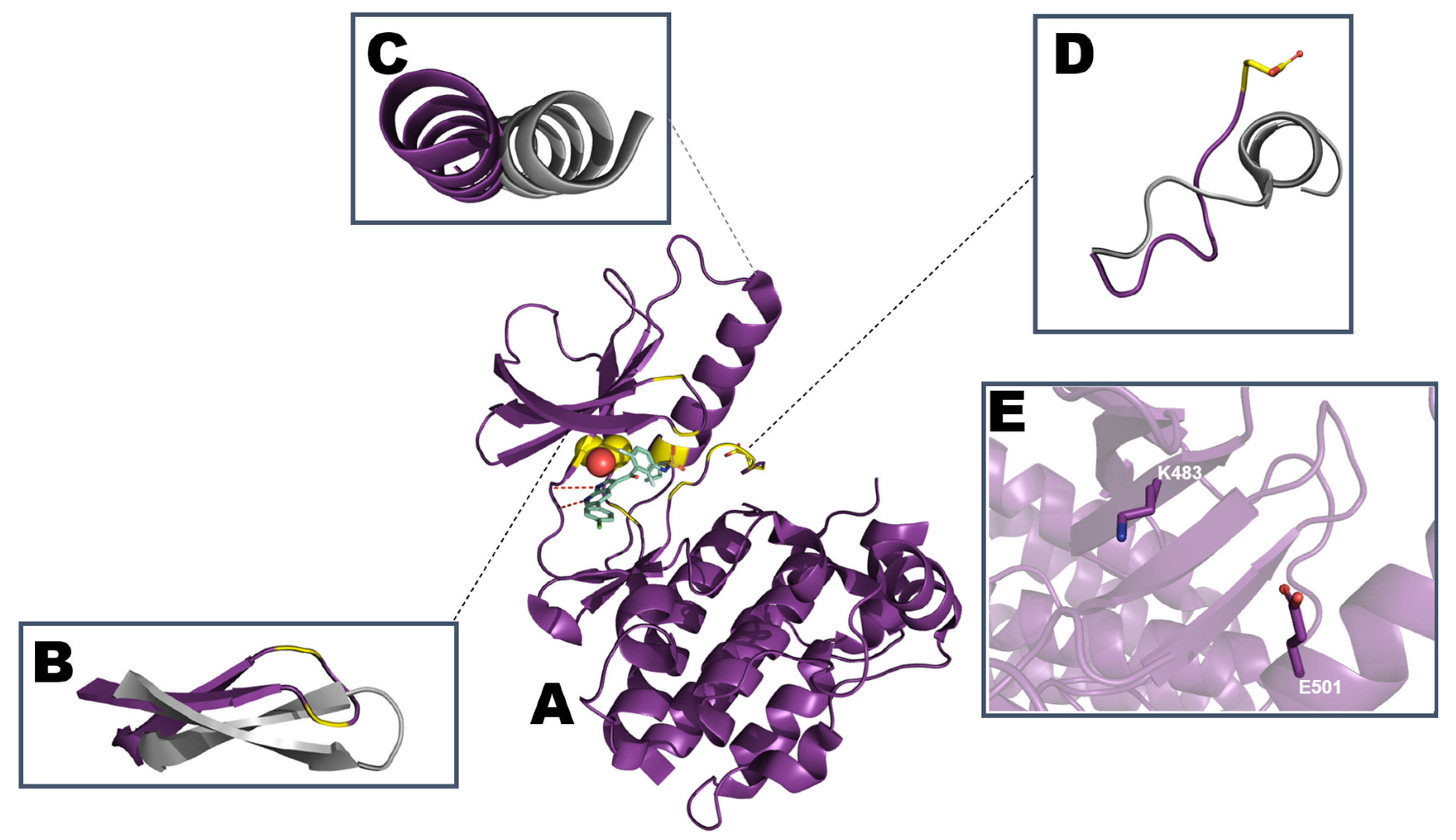
Figure 10.
Two-dimensional representation of BRAF inhibitors, sorafenib, dabrafenib, and vemurafenib. All represented drugs interact with the hinge region through hydrogen bonds with Q530 and C532. Vemurafenib and dabrafenib present a functionalized sulfonamide moiety providing an anchoring point with the allosteric pocket and the αC-helix (L505). Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon(grey), nitrogen (blue), oxygen (red), sulfur (beige), chlorine (green), and fluorine (light blue).
Figure 10.
Two-dimensional representation of BRAF inhibitors, sorafenib, dabrafenib, and vemurafenib. All represented drugs interact with the hinge region through hydrogen bonds with Q530 and C532. Vemurafenib and dabrafenib present a functionalized sulfonamide moiety providing an anchoring point with the allosteric pocket and the αC-helix (L505). Interactions were analyzed and plotted using Discovery Studio Visualizer [57]. Atoms color scheme: carbon(grey), nitrogen (blue), oxygen (red), sulfur (beige), chlorine (green), and fluorine (light blue).

Figure 11.
Schematic representation of Kirsten rat sarcoma viral oncogene homolog (KRAS) nucleotide binding domain in the presence of GTP (PDB: ID 5VPI). Mutation hotspots are indicated in pink with GTP binding mode highlighted with magnesium being depicted as a yellow sphere. The GTP molecule is depicted with carbon (pink), nitrogen (blue), oxygen (red,) and phosphorus (orange) atoms and represented as sticks.
Figure 11.
Schematic representation of Kirsten rat sarcoma viral oncogene homolog (KRAS) nucleotide binding domain in the presence of GTP (PDB: ID 5VPI). Mutation hotspots are indicated in pink with GTP binding mode highlighted with magnesium being depicted as a yellow sphere. The GTP molecule is depicted with carbon (pink), nitrogen (blue), oxygen (red,) and phosphorus (orange) atoms and represented as sticks.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of EGFR mutations and reported response to approved drugs.
| Mutations | 1st Generation | 2nd Generation | 3rd Generation | ||
|---|---|---|---|---|---|
| Erlotinib | Gefitinib | Afatinib | Dacomitinib | Osimertinib | |
| L718V/Q | Resistant [63] | Resistant [64] | N/A | N/A | Resistant [64] |
| G719A | N/A | N/A | Sensitive [59] | N/A | N/A |
| G719S | Sensitive [65] | Sensitive [44,65,66,67] | N/A | N/A | N/A |
| G724S | N/A | N/A | Sensitive [68] | N/A | Resistant [68] |
| T790M | Resistant [44,50] | Resistant [44] | N/A | N/A | Sensitive [59,69] |
| L792H/F/Y | N/A | Resistant [64] | N/A | N/A | Resistant [64] |
| G796C | Resistant [63] | N/A | N/A | N/A | Resistant [70] |
| G796D | Resistant [63] | N/A | N/A | N/A | Resistant [71] |
| G796R | Resistant [63] | N/A | N/A | N/A | Resistant [64] |
| G796S | N/A | N/A | N/A | N/A | Resistant [72] |
| C797S | Resistant [73] | Sensitive [64] | Resistant [63] | N/A | Resistant [74] |
| L858R | Sensitive [60,65,75,76] | Sensitive [60,65,75,76] | Sensitive [59,77] | Sensitive [78] | Sensitive [69,79] |
| L858R/L718V | N/A | N/A | Sensitive [80] | N/A | Resistant [80] |
| L858R/L718Q | N/A | Resistant [64] | Sensitive [81] | N/A | Resistant [64,81] |
| L858R/G724S | N/A | N/A | N/A | N/A | Resistant [82] |
| L858R/T790M | Resistant [50,59,60,65,77,78] | Resistant [59,60,65,67,77] | Inconclusive [77] | Resistant [78] | Sensitive [78,79,83] |
| L858R/C797S | Sensitive [78] | Sensitive [78] | Sensitive [78] | N/A | Resistant [84] |
| T790M/G719S | N/A | Resistant [48,65] | N/A | N/A | N/A |
| T790M/C797S | Resistant [63,83] | Resistant [83] | Resistant [83] | Resistant [83] | Resistant [59,83] |
| L858R/ T790M/ C797S | Resistant [78] | N/A | Resistant [78] | Resistant [78] | Resistant [78] |
| Exon19del* | Sensitive [66,75,76] | Sensitive [66] | Sensitive [85,86] | Sensitive [78] | Sensitive [64] |
| Exon19del*/ G724S | N/A | N/A | Sensitive [68] | N/A | Resistant [68] |
| Exon19del* / T790M | Resistant [65] | Resistant [65] | N/A | Resistant [74] | Sensitive [78] |
| Exon19del*/ C797S | N/A | Sensitive [74] | Sensitive [74] | N/A | Resistant [64,84] |
| L858R/T790M/ L718Q | Resistant [81] | Resistant [81] | Resistant [81] | Resistant [81] | Resistant [81] |
| Exon19del*/ T790M/ P794L | N/A | N/A | Sensitive [87] | N/A | Resistant [87] |
| Exon19del*/ T790M/ C797S | Resistant [74] | Resistant [74] | Resistant [74] | Resistant [74] | Resistant [74] |
* In this table, exon19del is the exon 19 deletion Δ746ELREA750.N/A: Not Available.
Table 2.
Summary of ALK mutations and reported response to approved drugs.
| Mutations | 1st Generation | 2nd Generation | 3rd Generation | |||
|---|---|---|---|---|---|---|
| Crizotinib | Entrectinib * | Alectinib | Brigatinib | Ceritinib | Lorlatinib | |
| G1123S | Resistant [164] | N/A | N/A | N/A | Resistant [164] | N/A |
| G1128A | Resistant [165] | N/A | N/A | N/A | N/A | N/A |
| I1171N | N/A | N/A | Resistant [146,166,167] | N/A | Sensitive [168] | Sensitive [167] |
| I1171S | N/A | N/A | Resistant [146,169] | N/A | N/A | N/A |
| I1171T | Resistant [170] | N/A | Resistant [170] | N/A | Sensitive [162] | N/A |
| C1156Y | Resistant [171] | N/A | N/A | N/A | N/A | N/A |
| F1174C | Resistant [162,170,172] | N/A | Sensitive [162,170,172] | N/A | Resistant [162,170,172] | N/A |
| F1174L | N/A | N/A | Sensitive [162,170,172] | N/A | Resistant [162,170,172] | N/A |
| F1174V | Resistant [169] | N/A | Sensitive [162,169,170,172] | N/A | Resistant [162,170,172] | N/A |
| V1180L | Resistant [170] | N/A | Resistant [170] | N/A | Sensitive [170] | N/A |
| R1192P | Resistant [173] | N/A | N/A | N/A | Resistant [173] | N/A |
| L1196M | N/A | N/A | N/A | Sensitive [174] | Sensitive [162,175] | Sensitive [174] |
| L1196Q | N/A | N/A | Sensitive [167] | N/A | N/A | N/A |
| L1198F | Sensitive [176] | N/A | N/A | N/A | N/A | Resistant [176] |
| L1198P | Resistant [177] | N/A | N/A | N/A | N/A | N/A |
| G1202R | Resistant [162,172] | N/A | Resistant [167,170,172] | Sensitive [178] | Resistant [162,172] | Sensitive [167,179] |
| D1203N | Resistant [177] | N/A | N/A | N/A | N/A | N/A |
| S1206Y | Resistant [180,181,182] | N/A | N/A | N/A | N/A | N/A |
| E1210K | N/A | N/A | N/A | N/A | N/A | Resistant [167] |
| F1245C | Resistant [183] | N/A | N/A | N/A | Sensitive [183] | N/A |
| G1269A | Resistant [184] | N/A | Resistant [180,181,182] | N/A | Sensitive [162,175] | Resistant [167] |
| A1280V | Resistant [173] | N/A | N/A | N/A | Resistant [173] | N/A |
| L1535Q | Resistant [173] | N/A | N/A | N/A | Resistant [173] | N/A |
| E1210K/D1203N | Resistant [172] | N/A | Resistant [172] | Resistant [172] | Resistant [172] | Sensitive [172] |
| C1156Y/L1198F | N/A | N/A | N/A | N/A | N/A | Resistant [167] |
| D1203N/E1210K | Resistant [172] | N/A | Resistant [172] | Resistant [172] | Resistant [172] | Sensitive [172] |
| D1203N/F1174C | Resistant [172] | N/A | Resistant [172] | Resistant [172] | Resistant [172] | Sensitive [172] |
| L1196M/G1202R | N/A | N/A | N/A | Resistant [185] | N/A | Resistant [185] |
| L1196M/D1203N | N/A | N/A | N/A | N/A | N/A | Resistant [186] |
| I1171S/G1269A | N/A | N/A | N/A | Sensitive [187] | Sensitive [187] | Resistant [187] |
| C1156Y/G169A | Resistant [186] | Resistant [186] | Resistant [186] | N/A | N/A | Sensitive [186] |
| F1174L/G1202R | N/A | N/A | N/A | N/A | N/A | Resistant [186] |
| G1202R/R1192P | Resistant [188] | N/A | N/A | Resistant [188] | N/A | Sensitive [188] |
* Entrectinib is considered a first-generation inhibitor for TRK fusions. N/A: Not Available.
Table 3.
Summary of BRAF mutations and reported response to approved drugs.
| Mutations | 1st Generation | 2nd Generation | ||
|---|---|---|---|---|
| Sorafenib | Dabrafenib | Encorafenib | Vemurafenib | |
| G466A | N/A | Sensitive [267] | N/A | Resistant [268] |
| G466V | N/A | Sensitive [267] | N/A | Resistant [268] |
| G469A | N/A | Sensitive [267] | N/A | Resistant [268] |
| G469V | Sensitive [269] | Sensitive [267] | N/A | Resistant [268] |
| G469R | Sensitive [270] | N/A | N/A | N/A |
| G469L | N/A | N/A | N/A | Resistant [271] |
| D594E | N/A | Sensitive [267] | N/A | N/A |
| D594G | N/A | N/A | N/A | Sensitive [272] |
| D594N | N/A | Sensitive [267] | N/A | N/A |
| G596C | N/A | Sensitive [267] | N/A | N/A |
| G596R | N/A | N/A | N/A | Resistant [268] |
| L597S | N/A | N/A | N/A | Sensitive [272] |
| V600D | N/A | Sensitive [273] | N/A | Resistant [268] |
| V600E | Inconclusive ** [269,274] | Sensitive [253] | N/A | Sensitive [253] |
| V600K | N/A | Sensitive [275] | N/A | Sensitive [275] |
| V600M | N/A | Sensitive [273] | N/A | N/A |
| V600R | N/A | Sensitive [276] | N/A | Sensitive [276] |
| K601E | N/A | N/A | N/A | Resistant [277] |
| V600E_L505H | N/A | N/A | N/A | Resistant [278] |
| V600E_L514V | N/A | Resistant [279] | N/A | Resistant [279] |
| V600E_T529I | Resistant [280] | N/A | N/A | N/A |
** Studies are divergent, there are reports of sorafenib having residual inhibitory activity against BRAF V600E in vitro but, responses in clinical settings vary. The type of cancer may play a role in drug efficacy. N/A—not available.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vilachã, J.F.; Mitchel, S.C.; Akele, M.Z.; Evans, S.; Groves, M.R. Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment. Crystals 2020, 10, 725. https://doi.org/10.3390/cryst10090725
AMA Style
Vilachã JF, Mitchel SC, Akele MZ, Evans S, Groves MR. Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment. Crystals. 2020; 10(9):725. https://doi.org/10.3390/cryst10090725
Chicago/Turabian StyleVilachã, Juliana F., Sarah C. Mitchel, Muluembet Z. Akele, Stephen Evans, and Matthew R. Groves. 2020. "Making NSCLC Crystal Clear: How Kinase Structures Revolutionized Lung Cancer Treatment" Crystals 10, no. 9: 725. https://doi.org/10.3390/cryst10090725
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.