
Review of First Principles Simulations of STO/BTO, STO/PTO, and SZO/PZO (001) Heterostructures



Abstract
1. Introduction
2. Computational Methods and Materials
3. Ab Initio Calculation Results
3.1. Ab Initio B3PW-Computed ABO3 Perovskite Bulk Properties
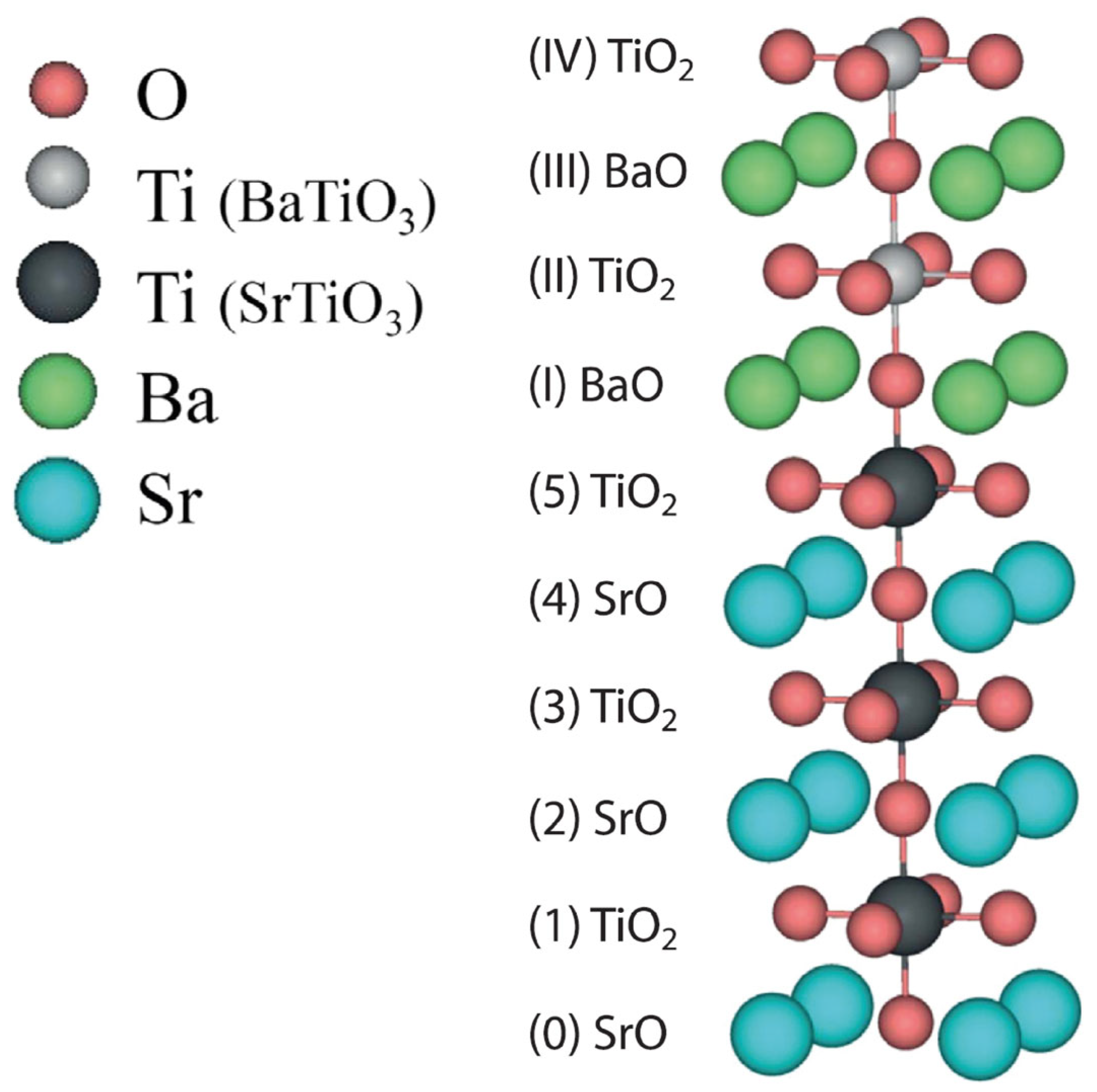
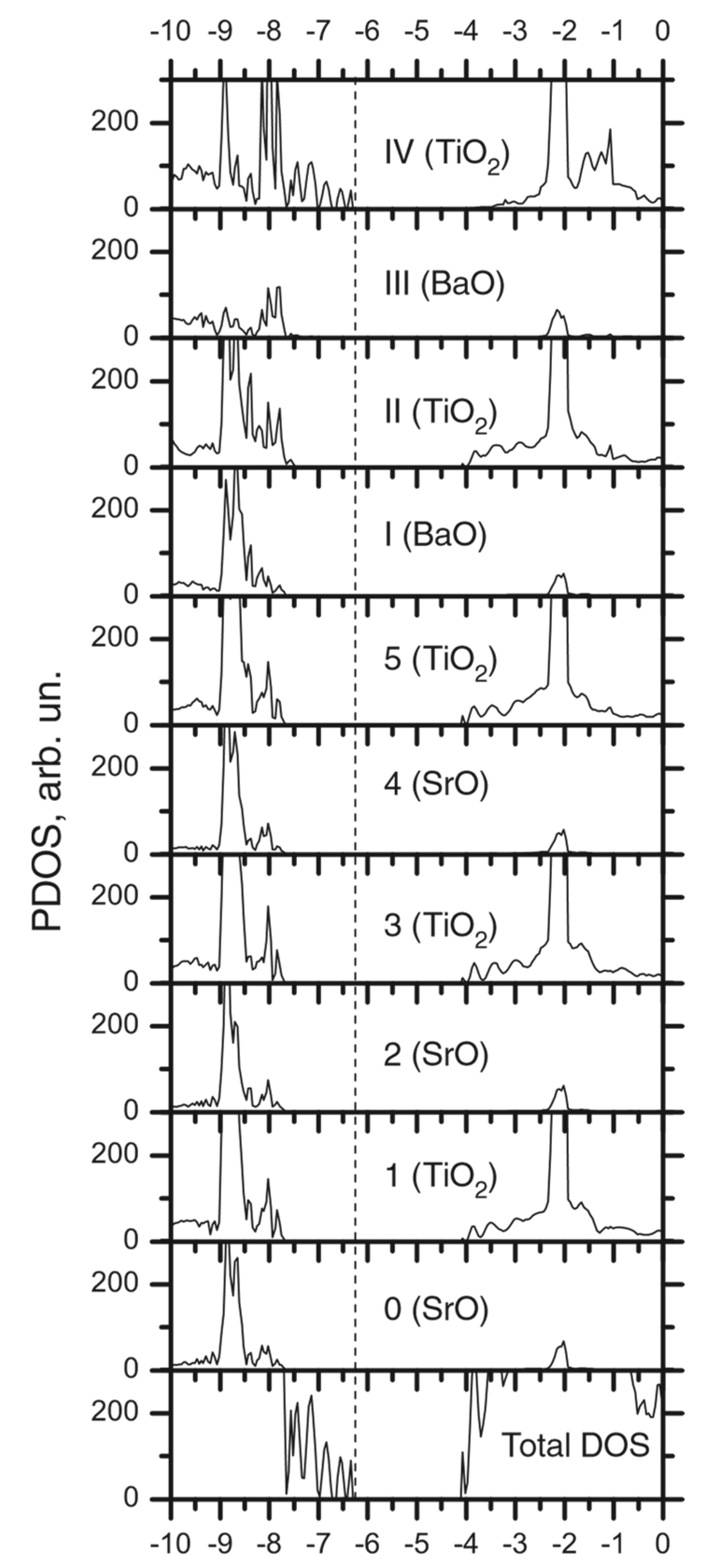
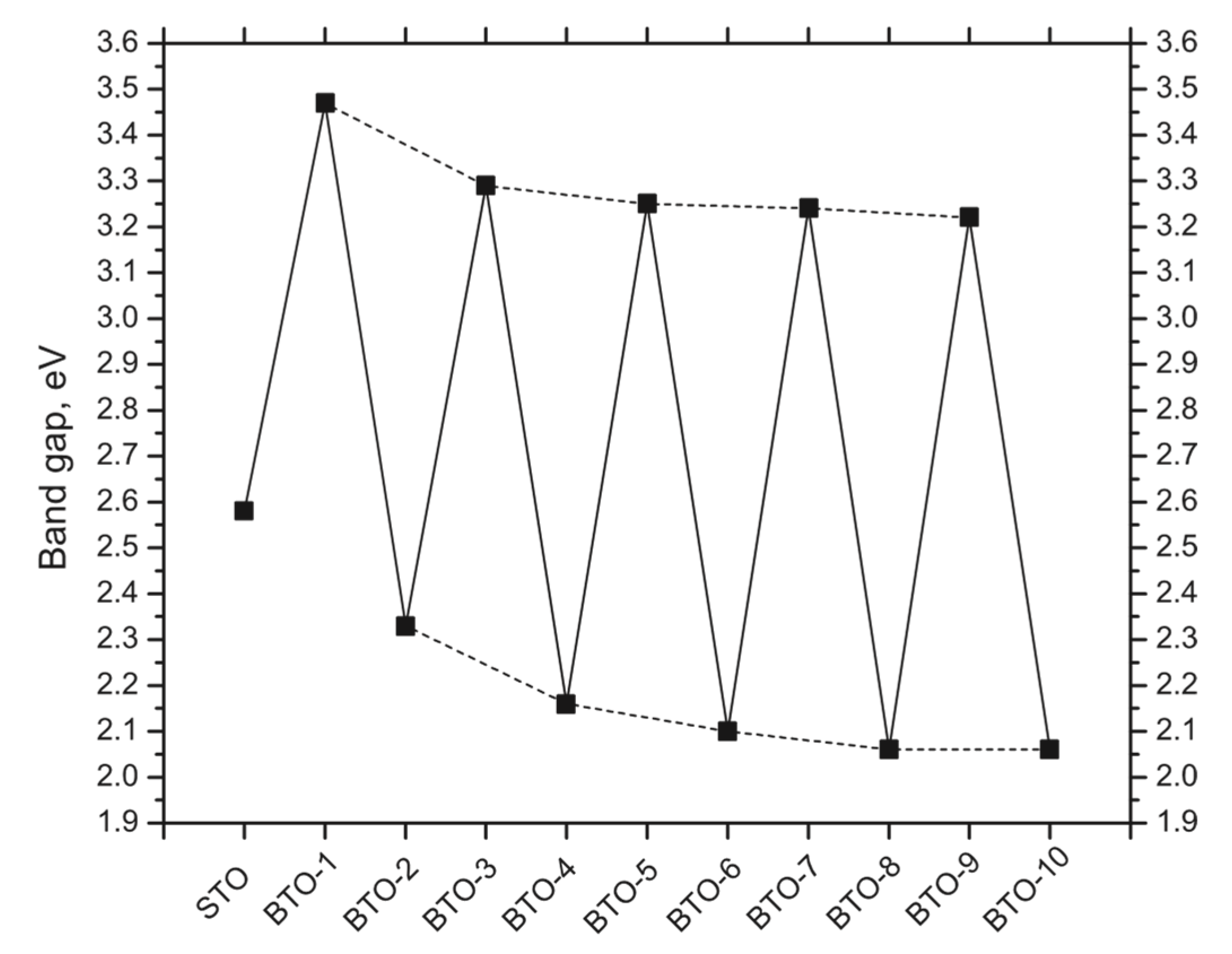
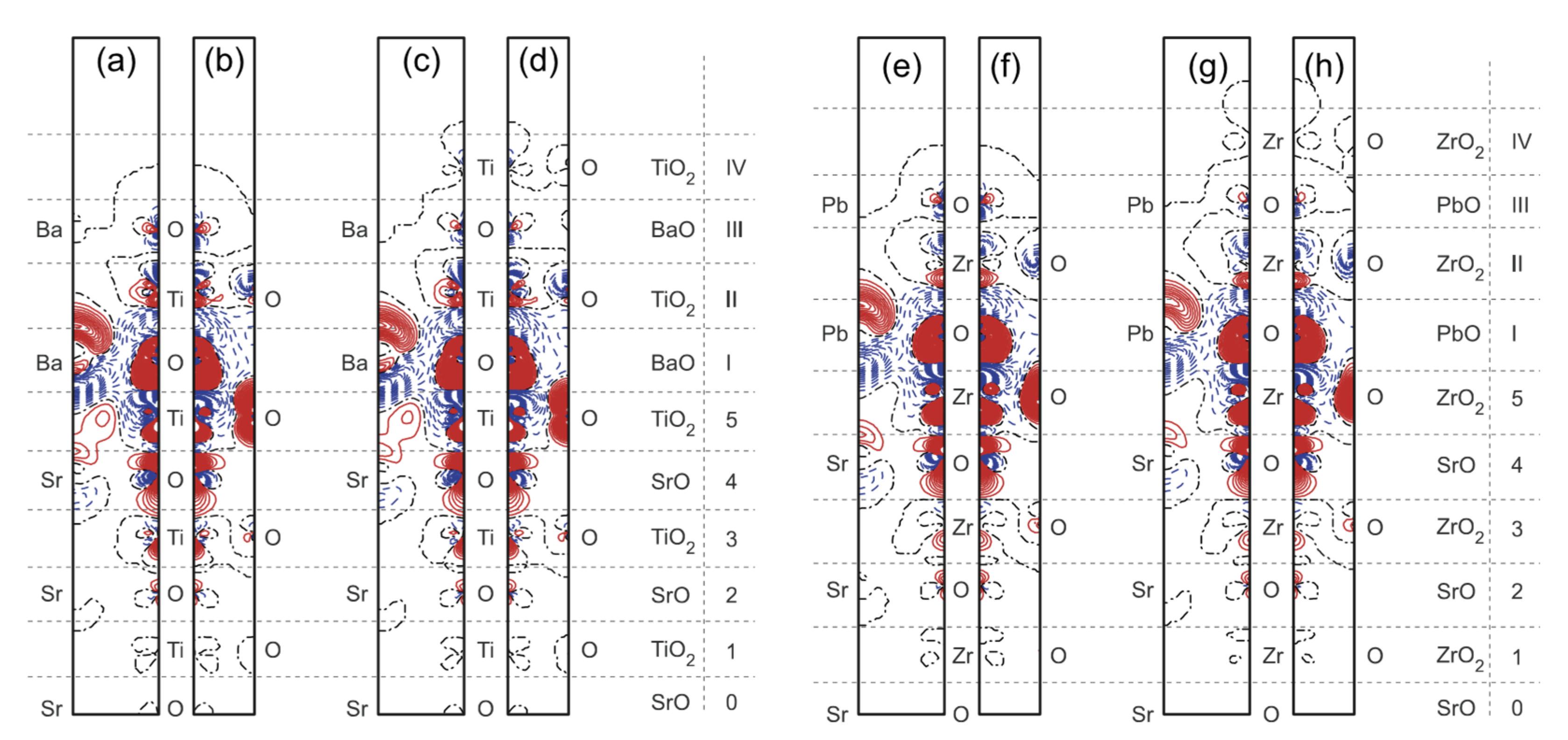
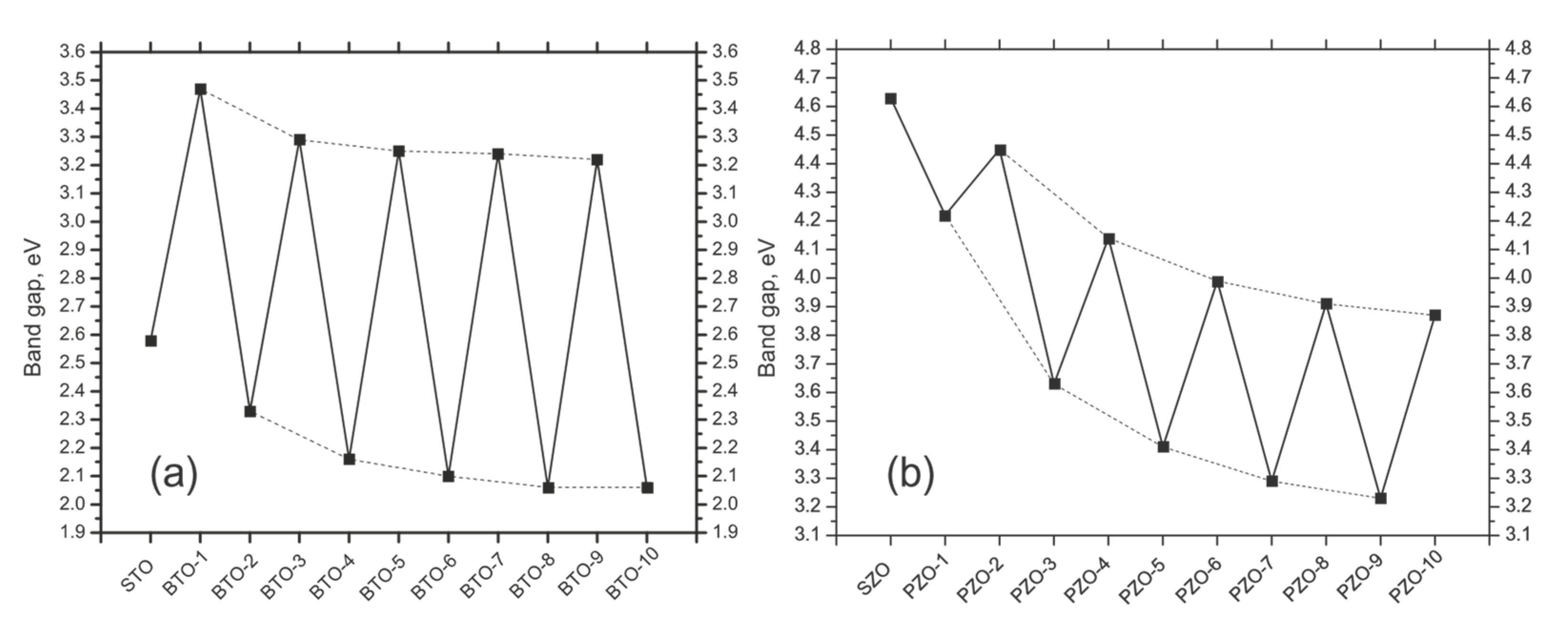
3.2. Ab Initio B3PW-Computed STO/BTO (001) Interfaces
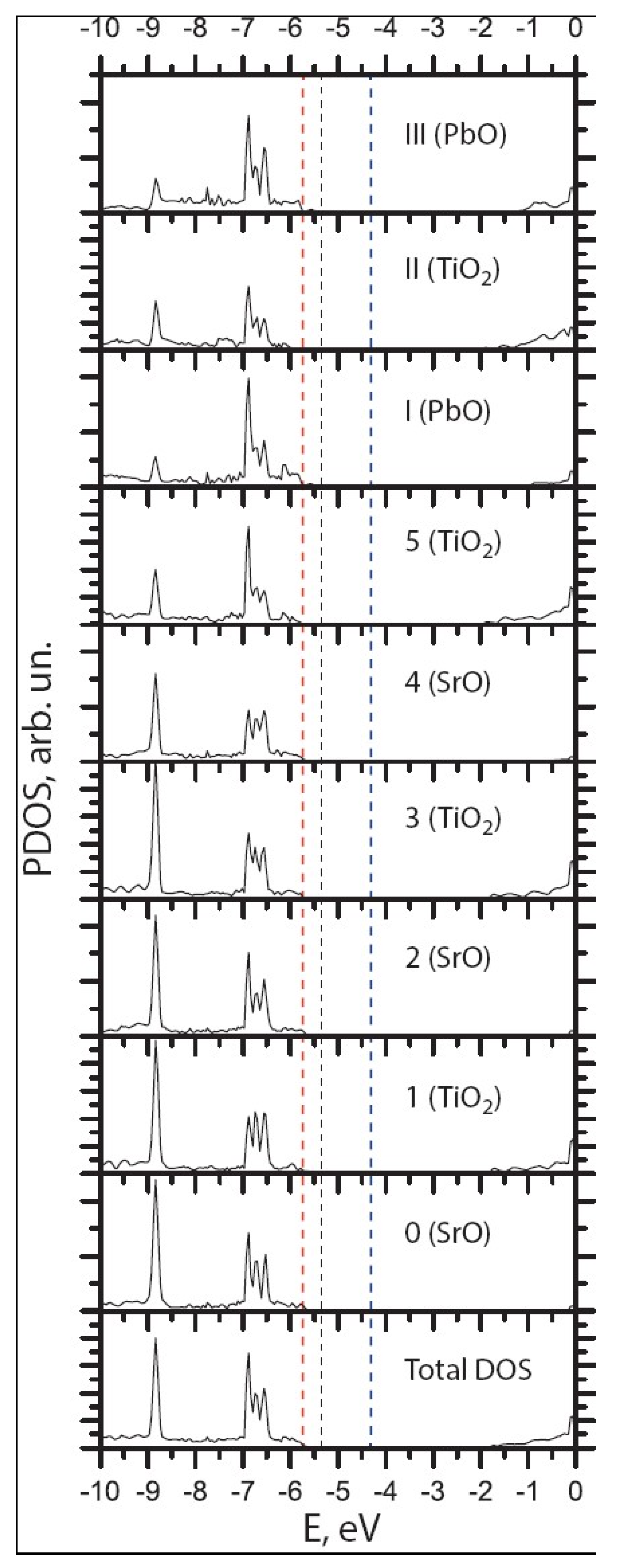
3.3. Ab Initio B3PW-Computed STO/PTO (001) Interfaces
3.4. Ab Initio B3PW-Computed SZO/PZO (001) Interfaces
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dawber, M.; Rabe, K.M.; Scott, J.F. Physics of thin-film ferroelectric oxides. Rev. Mod. Phys. 2005, 77, 1083–1130. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Vanderbilt, D. First-principles calculations of atomic and electronic structure of SrTiO3 (001) and (011) surfaces. Phys. Rev. B 2008, 77, 195408. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Vanderbilt, D. Ab initio calculations of BaTiO3 and PbTiO3 (001) and (011) surface structures. Phys. Rev. B 2007, 76, 155439. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Gabrusenoks, J.; Popov, A.I.; Jia, R. Comparative ab initio calculations of ReO3, SrZrO3, BaZrO3, PbZrO3 and CaZrO3 (001) surfaces. Crystals 2020, 10, 745. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Jia, R. Comparative Hybrid Hartree-Fock-DFT Calculations of WO2-Terminated Cubic WO3 as well as SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001) surfaces. Crystals 2021, 11, 455. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Popov, A.I.; Bocharov, D.; Chekhovska, A.; Jia, R. Ab initio computations of O and AO as well as ReO2, WO2 and BO2-terminated ReO3, WO3, BaTiO3, SrTiO3 and BaZrO3 (001) surfaces. Symmetry 2022, 14, 1050. [Google Scholar] [CrossRef]
- Zhang, R.; Hwang, G.S. First-principles mechanistic study of the initial growth of SrO by atomic layer deposition on TiO2-terminated SrTiO3 (001). J. Phys. Chem. C 2020, 124, 28116. [Google Scholar] [CrossRef]
- Sambrano, J.R.; Longo, V.M.; Longo, E.; Taft, C.A. Electronic and structural properties of the (001) SrZrO3 surface. J. Mol. Struct. THEOCHEM 2007, 813, 49–56. [Google Scholar] [CrossRef]
- Brik, M.G.; Ma, C.G.; Krasnenko, V. First-principles calculations of the structural and electronic properties of the cubic CaZrO3 (001) surfaces. Surf. Sci. 2013, 608, 146–153. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Kleperis, J.; Purans, J.; Popov, A.I.; Jia, R. Ab initio calculations of CaZrO3 (011) surfaces: Systematic trends in polar (011) surface calculations of ABO3 perovskites. J. Mater. Sci. 2020, 55, 203–217. [Google Scholar] [CrossRef]
- Li, W.; Landis, C.M.; Demkov, A. Domain morphology and electro-optic effect in Si-integrated epitaxial BaTiO3 films. Phys. Rev. Materials 2022, 6, 095203. [Google Scholar] [CrossRef]
- Erdman, N.; Poeppelmeier, K.R.; Asta, M.; Warschkov, O.; Ellis, D.E.; Marks, L.D. The structure and chemistry of the TiO2-rich surface of SrTiO3 (001). Nature 2002, 419, 55–58. [Google Scholar] [CrossRef]
- Celik, F.A. Electronic structure of two-dimensional-layered PbTiO3 perovskite crystal: An extended tight-binding study based on DFT. Bull. Mater. Sci. 2022, 45, 108. [Google Scholar] [CrossRef]
- Eglitis, R.I. Comparative first-principles calculations of SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001), (011) and (111) surfaces. Ferroelectrics 2015, 483, 53–67. [Google Scholar] [CrossRef]
- Zhong, M.; Zeng, W.; Liu, F.S.; Tang, B.; Liu, Q.J. First-principles study of the atomic structures, electronic properties, and surface stability of BaTiO3 (001) and (011) surfaces. Surf. Interf. Anal. 2019, 51, 1021–1032. [Google Scholar] [CrossRef]
- Costa-Amaral, R.; Gohda, Y. First-principles study of the adsorption of 3d transition metals on BaO- and TiO2-terminated cubic-phase BaTiO3 (001) surfaces. J. Chem. Phys. 2020, 152, 204701. [Google Scholar] [CrossRef] [PubMed]
- Eglitis, R.I.; Piskunov, S. First principles calculations of SrZrO3 bulk and ZrO2-terminated (001) surface F centers. Comput. Condens. Matter 2016, 7, 1–6. [Google Scholar] [CrossRef]
- Saghayezhian, M.; Sani, S.M.R.; Zhang, J.; Plummer, E.W. Rumpling and enhanced covalency at the SrTiO3 (001) surface. J. Phys. Chem. C 2019, 123, 8086–8091. [Google Scholar] [CrossRef]
- Kruchinin, S.P.; Eglitis, R.I.; Babak, V.P.; Vyshyvana, I.G.; Repetsky, S.P. Effects of Electron Correlation Inside Disordered Crystals. Crystals 2022, 12, 237. [Google Scholar] [CrossRef]
- Chun, H.J.; Lee, Y.; Kim, S.; Yoon, Y.; Kim, Y.; Park, S.C. Surface termination of BaTiO3 (111) single crystal: A combined DFT and XPS study. Appl. Surf. Sci. 2022, 578, 152018. [Google Scholar] [CrossRef]
- Alam, N.N.; Malik, N.A.; Samat, M.H.; Hussin, N.H.; Jaafar, N.K.; Radzwan, A.; Mohyedin, M.Z.; Haq, B.U.; Ali, A.M.M.; Hassan, O.H.; et al. Underlaying mechanism of surface (001) cubic ATiO3 (A = Pb,Sn) in enhancing thermoelectric performance of thin-film applications using density functional theory. Surf. Interfaces 2021, 27, 101524. [Google Scholar] [CrossRef]
- Zhao, X.; Selloni, A. Structure and stability of NaTaO3 (001) and KTaO3 (001) surfaces. Phys. Rev. Mater. 2019, 3, 015801. [Google Scholar] [CrossRef]
- Kolpak, A.M.; Li, D.; Shao, R.; Rappe, A.M.; Bonnell, D.A. Evolution of the surface structure and thermodynamic stability of the BaTiO3 (001) surface. Phys. Rev. Lett. 2008, 101, 036102. [Google Scholar] [CrossRef] [PubMed]
- Eglitis, R.; Popov, A.I.; Purans, J.; Jia., R. First principles hybrid Hartree-Fock-DFT calculations of bulk and surface F centers in oxide perovskites and alkali-earth fluorides. Low Temp. Phys. 2020, 46, 1206–1212. [Google Scholar] [CrossRef]
- Gao, H.; Yue, Z.; Liu, Y.; Hu, J.; Li, X. A first-principles study on the multiferroic property of two-dimensional BaTiO3 (001) ultrathin film with surface Ba vacancy. Nanomaterials 2019, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Iles, N.; Finocchi, F.; Khodja, K.D. A systematic study of ideal and double layer reconstruction of ABO3 (001) surfaces (A=Sr, Ba: B=Ti, Zr) from first principles. J. Phys. Condens. Matter 2010, 22, 305001. [Google Scholar] [CrossRef]
- Borstel, G.; Eglitis, R.I.; Kotomin, E.A.; Heifets, E. Modelling of defects and surfaces in perovskite ferroelectrics. Phys. Status Solidi B 2003, 236, 253–264. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Zhang, L.; Chen, J.; Xing, X. PbTiO3-based perovskite ferroelectric and multiferroic thin films. Phys. Chem. Chem. Phys. 2017, 19, 17493–17515. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Popov, A.I. Systematic trends in (001) surface ab initio calculations of ABO3 perovskites. J. Saudi Chem. Soc. 2018, 22, 459–468. [Google Scholar] [CrossRef]
- Prasatkhetragarn, A.; Sareein, T.; Triamnak, N.; Yimnirun, R. Dielectric and ferroelectric properties of modified-BaTiO3 lead-free ceramics prepared by solid solution method. Ferroelectrics 2022, 586, 224–241. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio calculations of SrTiO3, BaTiO3, PbTiO3, CaTiO3, SrZrO3, PbZrO3 and BaZrO3 (001), (011) and (111) surfaces as well as F centers, polarons, KTN solid solutions and Nb impurities therein. Int. J. Mod. Phys. B 2014, 28, 1430009. [Google Scholar] [CrossRef]
- Zhang, M.; Lopato, E.M.; Ene, N.N.; Funni, S.D.; Du, T.; Jiang, K.; Bernard, S.; Salvador, P.A.; Rohrer, G.S. Synthesis and Structure of Ion-Exchange SrTiO3 Photocatalyst with Improved Reactivity for Hydrogen Evolution. Adv. Mater. Interfaces 2023, 10, 2202476. [Google Scholar] [CrossRef]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Tailor, N.K.; Abdi-Jalebi, M.; Gupta, V.; Hu, H.; Dar, M.I.; Li, G.; Satapathi, S. Recent progress in morphology optimization in perovskite solar cells. J. Mater. Chem. A 2020, 8, 21356–21386. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, G.; Li, Y.; Yang, T.; Hao, X.; Gong, W. High-performance PbZrO3-based antiferroelectric multilayer capacitors based on multiple enhancement strategy. Chem. Eng. J. 2022, 446, 136729. [Google Scholar] [CrossRef]
- Xu, R.; Zhu, Q.; Xu, Z.; Feng, Y.; Wei, X. PLZT antiferroelectric ceramics with promising energy storage and discharge performance for high power applications. J. Am. Ceram. Soc. 2020, 103, 1831–1838. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Kotomin, E.A.; Trepakov, V.A.; Kapphan, S.E.; Borstel, G. Quantum chemical modelling of electron polarons and “green” luminescence in PbTiO3 perovskite crystals. J. Phys. Condens. Matter. 2002, 14, L647. [Google Scholar] [CrossRef]
- Mete, E.; Shaltaf, R.; Ellialtioglu, S. Electronic and structural properties of a 4d perovskite: Cubic phase of SrZrO3. Phys. Rev. B 2003, 68, 035119. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, H.; Cui, S.; Bai, C. First-principles study of optical properties of SrZrO3 in cubic phase. Solid State Commun. 2008, 148, 472–475. [Google Scholar] [CrossRef]
- Krainyukova, N.V.; Hamalii, V.O.; Rusevich, L.L.; Kotomin, E.A.; Maier, J. Effect of ‘interplane’ contraction on the (001) surface of the model perovskite SrTiO3. Appl. Surf. Sci. 2023, 615, 156297. [Google Scholar] [CrossRef]
- Regnault, N.; Xu, Y.; Li, M.R.; Ma, D.S.; Jovanovic, M.; Yazdani, A.; Parkin, S.S.P.; Felser, C.; Schoop, L.M.; Ong, N.P.; et al. Catalogue of flat-band stoichiometric materials. Nature 2022, 603, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Vivek, M.; Goniakowski, J.; Santander-Syro, A.; Gabay, M. Octahedral rotations and defect-driven metallicity at the (001) surface of CaTiO3. Phys. Rev. B 2023, 107, 045101. [Google Scholar] [CrossRef]
- Kotomin, E.A.; Piskunov, S.; Zhukovskii, Y.F.; Eglitis, R.I.; Gopejenko, A.; Ellis, D.E. The electronic properties of an oxygen vacancy at ZrO2-terminated (001) surfaces of a cubic PbZrO3: Computer simulations from the first principles. Phys. Chem. Chem. Phys. 2008, 10, 4258–4263. [Google Scholar] [CrossRef] [PubMed]
- Priyanga, S.; Mattur, M.N.; Nagappan, N.; Rath, S.; Thomas, T. Prediction of nature of band gap of perovskite oxides (ABO3) using a machine learning approach. J. Mater. 2022, 8, 937–948. [Google Scholar]
- Sophia, G.; Baranek, P.; Rérat, M.; Dovesi, R. The effect of composition on phonon softening in ABO3-type perovskites: DFT modelling. Phys. Chem. Chem. Phys. 2022, 24, 27064–27074. [Google Scholar] [CrossRef] [PubMed]
- Ananyev, M.V.; Farlenkov, A.S.; Zhigalina, O.M.; Khmelenin, D.N.; Atanova, A.V.; Basu, V.G. Antiphase Boundary Defects in Strontium-Doped Lanthanum Scandate. Phys. Status Solidi B 2022, 259, 2100376. [Google Scholar] [CrossRef]
- Paoletta, T.; Demkov, A.A. Pockels effect in low-temperature rhombohedral BaTiO3. Phys. Rev. B 2021, 103, 014303. [Google Scholar] [CrossRef]
- Meirzadeh, E.; Christensen, D.V.; Makagon, E.; Cohen, H.; Rosenhek-Goldian, I.; Morales, E.H.; Bhowmik, A.; Lastra, J.M.; Rappe, A.M.; Ehre, D.; et al. Surface Pyroelectricity in cubic SrTiO3. Adv. Mater. 2019, 31, 1904733. [Google Scholar] [CrossRef]
- Mathieu, C.; Lubin, C.; Doueff, G.L.; Cattelan, M.; Gemeiner, P.; Dkhil, B.; Salje, E.K.H.; Barret, N. Surface Proximity Effect, Imprint Memory of Ferroelectric Twins, and Tweed in the Paraelectric Phase of BaTiO3. Sci. Rep. 2018, 8, 13660. [Google Scholar] [CrossRef]
- Guedes, E.B.; Muff, S.; Brito, W.H.; Caputo, M.; Li, H.; Plumb, N.C.; Dil, J.H.; Radović, M. Universal Structural Influence on the 2D Electron Gas at SrTiO3 Surface. Adv. Sci. 2021, 8, 2100602. [Google Scholar] [CrossRef]
- Erdman, N.; Warschkow, O.; Asta, M.; Poeppelmeier, K.R.; Ellis, D.E.; Marks, L.D. Surface Structures of SrTiO3 (001): A TiO2-rich Reconstruction with a c (4 × 2) Unit Cell. J. Am. Chem. Soc. 2003, 125, 10050–10056. [Google Scholar] [CrossRef] [PubMed]
- Eglitis, R.I.; Kotomin, E.A.; Borstel, G. Quantum chemical modelling of perovskite solid solutions. J. Phys. Condens. Matter 2000, 12, L431–L434. [Google Scholar] [CrossRef]
- Solokha, V.; Garai, D.; Wilson, A.; Duncan, D.A.; Thakur, P.K.; Hingerl, K.; Zegenhagen, J. Water Splitting on Ti-Oxide-terminated SrTiO3 (001). J. Phys. Chem. C 2019, 123, 17232–17238. [Google Scholar] [CrossRef]
- Jia, W.; Vikhnin, V.S.; Liu, H.; Kapphan, S.; Eglitis, R.; Usvyat, D. Critical effects in optical response due to charge transfer vibronic excitions and their structure in perovskite-like systems. J. Lumin. 1999, 83–84, 109–113. [Google Scholar] [CrossRef]
- Slassi, A.; Hammi, M.; Rhazouani, O.E. Surface Relaxations, Surface Energies and Electronic Structures of BaSnO3 (001) Surfaces: Ab initio Calculations. J. Electron. Mater. 2017, 46, 4133–4139. [Google Scholar] [CrossRef]
- Wang, Y.X.; Arai, M. First-principles study of the (001) surface of cubic SrZrO3. Surf. Sci. 2007, 601, 4092–4096. [Google Scholar] [CrossRef]
- Bickel, N.; Schmidt, G.; Heinz, K.; Müller, K. Ferroelectric relaxation of the SrTiO3 (100) surface. Phys. Rev. Lett. 1993, 62, 2009–2012. [Google Scholar] [CrossRef]
- Hikita, T.; Hanada, T.; Kudo, M.; Kawai, M. Structure and electronic state of the TiO2 and SrO terminated SrTiO3 (100) surfaces. Surf. Sci. 1993, 287–288, 377–381. [Google Scholar] [CrossRef]
- Grigorjeva, L.; Millers, D.K.; Pankratov, V.; Williams, R.T.; Eglitis, R.I.; Kotomin, E.A.; Borstel, G. Experimental and theoretical studies of polaron optical properties in KNbO3 perovskite. Solid State Commun. 2004, 129, 691–696. [Google Scholar] [CrossRef]
- Heifets, E.; Dorfman, S.; Fuks, D.; Kotomin, E. Atomistic simulation of the [001] surface structure in BaTiO3. Thin Solid Films 1997, 296, 76–78. [Google Scholar] [CrossRef]
- Heifets, E.; Dorfman, S.; Fuks, D.; Kotomin, E.; Gordon, A. [001] Surface Structure in SrTiO3–Atomistic Study. Surf. Rev. Lett. 1998, 5, 341–345. [Google Scholar] [CrossRef]
- Eglitis, R.I. Comparative ab initio calculations of SrTiO3 and CaTiO3 polar (111) surfaces. Phys. Stat. Sol. B 2015, 252, 635–642. [Google Scholar] [CrossRef]
- Al-Aqtash, N.; Alsaad, A.; Sabirianov, R. Ferroelectric properties of BaZrO3/PbZrO3 and SrZrO3/PbZrO3 superlattices: An ab-initio study. J. Appl. Phys. 2014, 116, 074112. [Google Scholar] [CrossRef]
- Sorokine, A.; Bocharov, D.; Piskunov, S.; Kashcheyevs, V. Electronic charge redistribution in LaAlO3 (001) thin films deposited at SrTiO3 substrate: First-principles analysis and the role of stoichiometry. Phys. Rev. B 2012, 86, 155410. [Google Scholar] [CrossRef]
- Qi, H.; Chen, X.; Benckiser, E.; Wu, M.; Cristiani, G.; Logvenov, G.; Keimer, B.; Kaiser, V. Formation mechanism of Ruddlesden-Popper faults in compressive-strained ABO3 perovskite superlattices. Nanoscale 2021, 13, 20663–20669. [Google Scholar] [CrossRef] [PubMed]
- Aso, R.; Kan, D.; Shimakawa, Y.; Kurata, H. Atomic level observation of octahedral distortions at the perovskite oxide heterointerface. Sci. Rep. 2013, 3, 2214. [Google Scholar] [CrossRef]
- Raza, S.; Zhang, R.; Zhang, N.; Li, Z.; Liu, L.; Zhang, F.; Wang, D.; Jia, C.L. ATiO3/TiO (A=Pb, Sn) superlattice: Bridging ferroelectricity and conductivity. Comput. Condens. Matter 2020, 25, e00491. [Google Scholar] [CrossRef]
- Eglitis, R.; Kruchinin, S.P. Ab initio calculations of ABO3 perovskite (001), (011) and (111) nano-surfaces, interfaces and defects. Mod. Phys. Lett. B 2020, 34, 2040057. [Google Scholar] [CrossRef]
- Fredrickson, K.D.; Demkov, A.A. Switchable conductivity at the ferroelectric interface: Nonpolar oxides. Phys. Rev. B 2015, 91, 115126. [Google Scholar] [CrossRef]
- Wang, J.B.N.J.; Neaton, J.B.; Zheng, H.; Nagarajan, V.; Ogale, S.B.; Liu, B.; Viehland, D.; Vaithyanathan, V.; Schlom, D.G.; Waghmare, U.W.; et al. Epitaxial BiFeO3 multiferroic thin film heterostructures. Science 2003, 299, 1719–1722. [Google Scholar] [CrossRef]
- Bi, Z.; Uberuaga, B.P.; Vernon, L.J.; Fu, E.; Wang, Y.; Li, N.; Wang, H.; Misra, A.; Jia, Q.X. Radiation damage in heteroepitaxial BaTiO3 thin films on SrTiO3 under Ne ion irradiation. J. Appl. Phys. 2013, 113, 023513. [Google Scholar] [CrossRef]
- Eglitis, R.I. Ab initio calculations of CaZrO3, BaZrO3, PbTiO3 and SrTiO3 (001), (011) and (111) surfaces as well as their (001) interfaces. Integr. Ferroelectr. 2019, 196, 7–15. [Google Scholar] [CrossRef]
- Stepkova, V.; Marton, P.; Setter, N.; Hlinka, J. Closed-circuit domain quadruplets in BaTiO3 nanorods embedded in a SrTiO3 film. Phys. Rev. B 2014, 89, 060101. [Google Scholar] [CrossRef]
- Piyanzina, I.I.; Eyert, V.; Lysogorskiy, Y.V.; Tayurskii, D.A.; Kopp, T. Oxygen vacancies and hydrogen doping in LaAlO3/SrTiO3 heterostructures: Electronic properties and impact on surface and interface reconstruction. J. Phys. Condens. Matter 2019, 31, 295601. [Google Scholar] [CrossRef]
- Okamoto, S.; Millis, A.J.; Spaldin, N.A. Lattice relaxation in oxide heterostructures: LaTiO3/SrTiO3 superlattices. Phys. Rev. Lett. 2006, 97, 056802. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, L.; Kim, J.; Stern, A.; Wang, H.; Zhang, K.; Yan, X.; Li, L.; Liu, H.; Zhao, G.; et al. Discovery of a magnetic conductive interface in PbZr0.2Ti0.8O3/SrTiO3 heterostructures. Nat. Commun. 2018, 9, 685. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Piskunov, S.; Popov, A.I.; Purans, J.; Bocharov, D.; Jia, R. Systematic Trends in Hybrid-DFT Computations of BaTiO3/SrTiO3, PbTiO3/SrTiO3 and PbZrO3/SrZrO3 (001) Hetero Structures. Condensed Matter 2022, 7, 70. [Google Scholar] [CrossRef]
- Wang, X.R.; Li, C.J.; Lu, W.M.; Paudel, T.R.; Leusink, D.P.; Hoek, M.; Poccia, N.; Vailionis, A.; Venkatesan, T.; Loey, J.M.D.; et al. Imaging and control of ferromagnetism in LaMnO3/SrTiO3 heterostructures. Science 2015, 349, 716–719. [Google Scholar] [CrossRef]
- Liu, M.; Ma, C.; Collins, G.; Liu, J.; Chen, C.; Dai, C.; Lin, Y.; Shui, L.; Xiang, F.; Wang, H.; et al. Interface Engineered BaTiO3/SrTiO3 Heterostructures with Optimized High-Frequency Dielectric Properties. ACS Appl. Mater. Interfaces 2012, 4, 5761–5765. [Google Scholar] [CrossRef]
- Tchiomo, A.P.N.; Braun, W.; Doyle, B.P.; Sigle, W.; Aken, P.V.; Mannart, J.; Ngabonziza, P. High-temperature-grown buffer layer boosts electron mobility in epitaxial La-doped BaSnO3/SrZrO3 heterostructures. APL Mater. 2019, 7, 041119. [Google Scholar] [CrossRef]
- Wysocki, L.; Yang, L.; Gunkel, F.; Dittmann, R.; Loosdrecht, P.H.M.; Lindfors-Vrejoiu, I. Validity of magnetotransport detection of skyrmions in epitaxial SrRuO3 heterostructures. Phys. Rev. Mater. 2020, 4, 054402. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, F.; Chen, M.; Lan, Z.; Ren, Z.; Tian, H.; Yang, K. Tuning Interfacial Magnetic Ordering via Polarization Control in Ferroelectric SrTiO3/PbTiO3 Heterostructure. ACS Appl. Mater. Interfaces 2018, 10, 10536–10542. [Google Scholar] [CrossRef] [PubMed]
- Piskunov, S.; Eglitis, R.I. Comparative ab initio calculations of SrTiO3/BaTiO3 and SrZrO3/PbZrO3 (001) heterostructures. Nucl. Instr. Methods B 2016, 374, 20–23. [Google Scholar] [CrossRef]
- Mahjoub, R.; Nagarajan, V.; Junquera, J. Structural and electronic properties of monodomain ultrathin PbTiO3/SrTiO3/PbTiO3/SrRuO3 heterostructures: A first-principles approach. J. Appl. Phys. 2020, 128, 244102. [Google Scholar] [CrossRef]
- Ko, L.D.; Hsin, T.; Lai, Y.H.; Ho, S.Z.; Zheng, Y.; Huang, R.; Pan, H.; Chen, Y.C.; Chu, Y.H. High-stability transparent flexible energy storage based on PbZrO3/muscovite heterostructure. Nano Energy 2021, 87, 106149. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Q.; Qi, R.; Shen, H.; Sui, F.; Yang, J.; Bai, W.; Tang, X.; Chen, X.; Fu, Z.; et al. High Energy Storage Performance of PZO/PTO Multilayer via Interface Engineering. ACS Appl. Mater. Interfaces 2023, 15, 7157–7164. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Piskunov, S.; Zhukovskii, Y.F. Ab initio calculations of PbTiO3/SrTiO3 (001) heterostructures. Phys. Stat. Sol. C 2016, 13, 913–920. [Google Scholar]
- Wei, H.; Yang, C.; Wu, Y.; Cao, B.; Lorenz, M.; Grundmann, M. From energy harvesting to topological insulating behavior: ABO3-type epitaxial thin films and superlattices. J. Mater. Chem. C 2020, 8, 15575–15596. [Google Scholar] [CrossRef]
- Nazir, S.; Schwingenschlőg, U. Strain effects on the spin polarized electron gas in ABO3/SrTiO3 (A = Pr, Nd and B = Al,Ga) heterostructures. Appl. Phys. Lett. 2013, 102, 141604. [Google Scholar] [CrossRef]
- Khazraie, A.; Elfimov, I.; Foyevtsova, K.; Sawatzky, G.A. Potential superconducting interfaces in polar ABO3/Ba(Sr)BiO3 heterostructures. Phys. Rev. B 2020, 101, 035135. [Google Scholar] [CrossRef]
- Piskunov, S.; Eglitis, R.I. First principles hybrid DFT calculations of BaTiO3/SrTiO3 (001) interface. Solid State Ion. 2015, 274, 29–33. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.; Kim, Y.S.; Lee, J.; Kim, L.; Jung, D. Large nonlinear dielectric properties of artificial BaTiO3/SrTiO3 superlattices. Appl. Phys. Lett. 2002, 80, 3581–3583. [Google Scholar] [CrossRef]
- Johnston, K.; Huang, X.; Neaton, J.B.; Rabe, K.M. First-principles study of symmetry lowering and polarization in BaTiO3/SrTiO3 superlattices with in-plane expansion. Phys. Rev. B 2005, 71, 100103(R). [Google Scholar] [CrossRef]
- Neaton, J.B.; Rabe, K.M. Theory of polarization enhancement in epitaxial BaTiO3/SrTiO3 superlattices. Appl. Phys. Lett. 2003, 82, 1586–1588. [Google Scholar] [CrossRef]
- Dawber, M.; Lichtensteiger, C.; Cantoni, M.; Veithen, M.; Ghosez, P.; Johnston, K.; Rabe, K.M.; Triscone, J.M. Unusual behavior of the ferroelectric polarization in PbTiO3/SrTiO3 superlattices. Phys. Rev. Lett. 2005, 95, 177601. [Google Scholar] [CrossRef] [PubMed]
- Ohtomo, A.; Hwang, H.Y. A high-mobility electron gas at the LaAlO3/SrTiO3 heterointerface. Nature 2004, 427, 423–426. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Wang, Y.; Yuan, L.; Tang, W. Anomalous change of electronic properties for uniaxial-strained LaAlO3/SrTiO3 (001) heterostructures. J. Appl. Phys. 2023, 133, 024303. [Google Scholar] [CrossRef]
- Cancellieri, C.; Mishchenko, A.S.; Aschauer, U.; Filippeti, A.; Faber, C.; Barišić, O.S.; Rogalev, V.A.; Schmitt, T.; Nagaosa, N.; Strocov, V.N. Polaronic metal state at the LaAlO3/SrTiO3 interface. Nat. Commun. 2016, 7, 10386. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, Z.; Li, M.; Wang, S.; Ren, L.; Jin, K. Photoresponsive properties at (001), (111) and (110) LaAlO3/SrTiO3 interfaces. J. Phys. Condens. Matter 2020, 32, 135002. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, Z.; Wang, S.; Wei, X.; Chen, C.; Jin, K. Magnetism control by doping in LaAlO3/SrTiO3 heterointerfaces. ACS Appl. Mater. Interfaces 2018, 10, 14209–14213. [Google Scholar] [CrossRef]
- Zhong, W.; Vanderbilt, D.; Rabe, K.M. First-principles theory of ferroelectric phase transitions for perovskites: The case of BaTiO3. Phys. Rev. B 1995, 52, 6301. [Google Scholar] [CrossRef] [PubMed]
- King-Smith, R.D.; Vanderbilt, D. First-principles investigation of ferroelectricity in perovskite compounds. Phys. Rev. B 1994, 49, 5828. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Vanderbilt, D.; Rabe, K.M. Phase Transitions in BaTiO3 from First Principles. Phys. Rev. Lett. 1994, 73, 1861. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Vanderbilt, D. Competing structural instabilities in cubic perovskites. Phys. Rev. Lett. 1995, 74, 2587. [Google Scholar] [CrossRef]
- Cohen, R.E. Origin of ferroelectricity in perovskite oxides. Nature 1992, 358, 136–138. [Google Scholar] [CrossRef]
- Cohen, R.E.; Krakauer, H. Electronic structure studies of the differences in ferroelectric behavior of BaTiO3 and PbTiO3. Ferroelectrics 1992, 136, 65–83. [Google Scholar] [CrossRef]
- Zelezny, V.; Chvostova, D.; Simek, D.; Maca, F.; Masek, J.; Setter, N.; Huang, Y.H. The variation of PbTiO3 bandgap at ferroelectric phase transition. J. Phys. Condens. Matter 2016, 28, 025501. [Google Scholar] [CrossRef]
- Oliveira, M.C.; Ribeiro, R.A.P.; Longo, E.; Bomio, M.R.D.; Motta, F.V.; Lazaro, S.R.D. Temperature dependence on phase evolution in BaTiO3 polytypes studied using ab initio calculations. Int. J. Quantum Chem. 2020, 120, e26054. [Google Scholar] [CrossRef]
- Nova, T.F.; Disa, A.S.; Fechner, M.; Cavalleri, A. Metastable ferroelectricity in optically strained SrTiO3. Science 2019, 364, 1075–1079. [Google Scholar] [CrossRef]
- Vanderbilt, D. First-principles theory of structural phase transitions in cubic perovskites. J. Korean Phys. Soc. 1998, 32, S103–S106. [Google Scholar]
- Pilania, G.; Tan, D.Q.; Cao, Y.; Venkataramani, V.S.; Chen, Q.; Ramprasad, R. Ab initio study of antiferroelectric PbZrO3 (001) surfaces. J. Mater. Sci. 2009, 44, 5249–5255. [Google Scholar] [CrossRef]
- Bellaiche, L.; Garcia, A.; Vanderbilt, D. Finite temperature properties of Pb(Zr1-xTix)O3 alloys from first principles. Phys. Rev. Lett. 2000, 84, 5427. [Google Scholar] [CrossRef] [PubMed]
- Ligny, D.D.; Richet, P. High-temperature heat capacity and thermal expansion of perovskites. Phys. Rev. B 1996, 53, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Evarestov, R.A.; Bandura, A.V.; Alexandrov, V.E. Hybrid HF-DFT comparative study of SrZrO3 and SrTiO3 (001) surface properties. Phys. Status Solidi B 2006, 243, 2756–2763. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ziesche, P.; Eschrig, H. Electronic Structure of Solids 1991; Akademie Verlag: Berlin, Germany, 1991. [Google Scholar]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL-2017 User Manual; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Vassilyeva, A.F.; Eglitis, R.I.; Kotomin, E.A.; Dauletbekova, A.K. Ab initio calculations of MgF2 (001) and (011) surface structure. Phys. B Condens. Matter 2010, 405, 2125–2127. [Google Scholar] [CrossRef]
- Shi, H.; Chang, L.; Jia, R.; Eglitis, R.I. Ab initio calculations of the transfer and aggregation of F centers in CaF2. J. Phys. Chem. C 2012, 116, 4832–4839. [Google Scholar] [CrossRef]
- Robertson, J. Band offsets of wide-band-gap oxides and implications for future electronic devices. J. Vac. Sci. Technol. B 2000, 18, 1785–1791. [Google Scholar] [CrossRef]
- Lisitsyn, V.M.; Lisitsyna, L.A.; Popov, A.I.; Kotomin, E.A.; Abuova, F.U.; Akilbekov, A.; Maier, J. Stabilization of primary mobile radiation defects in MgF2 crystals. Nucl. Instr. Methods B 2016, 374, 24–28. [Google Scholar] [CrossRef]
- Rubloff, G.W. Far-Ultraviolet Reflectence Spectra and the Electronic Structure of Ionic Crystals. Phys. Rev. B 1972, 5, 662–684. [Google Scholar] [CrossRef]
- Slater, J.C. A Simplification of the Hartree-Fock Method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Roetti, C.; Pisani, C.; Saunders, V.R. The Periodic Hartree-Fock Method and Its Implementation in the Crystal Code. Phys. Stat. Sol. B 2000, 217, 63–88. [Google Scholar] [CrossRef]
- Dirac, P.A.M. Note on Exchange Phenomena in the Thomas Atom. Proc. Camb. Phil. Soc. 1930, 26, 376–385. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Boettger, J.C. Nonconvergence of surface energies obtained from thin-film calculations. Phys. Rev. B 1994, 49, 16798. [Google Scholar] [CrossRef]
- Fiorentini, V.; Methfessel, M. Extracting convergent surface energies from slab calculations. J. Phys. Condens. Matter 1996, 8, 6525. [Google Scholar] [CrossRef]
- Monkhorst, H.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in Fortran 77, 2nd ed; Cambridge University Press: Cambridge, MA, USA, 1997. [Google Scholar]
- Mayer, I. Bond Order and Valence: Relations to Mulliken’s Population Analysis. Int. J. Quantum Chem. 1984, 26, 151–154. [Google Scholar] [CrossRef]
- Bochicchio, R.C.; Reale, H.F. On the nature of crystalline bonding: Extension of statistical population analysis to two- and three- dimensional crystalline systems. J. Phys. B 1993, 26, 4871–4883. [Google Scholar] [CrossRef]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1984, 82, 299–310. [Google Scholar] [CrossRef]
- Edwards, J.W.; Speiser, R.; Johnston, H.L. Structure of Barium Titanate at Elevated Temperatures. J. Amer. Chem. Soc. 1951, 73, 2934–2935. [Google Scholar] [CrossRef]
- Mabud, S.A.; Glazer, A.M. Lattice parameters and birefringence in PbTiO3 single crystals. J. Appl. Cryst. 1979, 12, 49–53. [Google Scholar] [CrossRef]
- Okazaki, A.; Scheel, H.J.; Müller, K.A. The lattice constant vs. temperature relation around the 105 K transition of a flux-grown SrTiO3 crystal. Phase Trans. 1985, 5, 207–218. [Google Scholar]
- Kennedy, B.J.; Howard, C.J. High-temperature phase transition in SrZrO3. Phys. Rev. B 1999, 59, 4023–4027. [Google Scholar] [CrossRef]
- Aoyagi, S.; Kuroiwa, Y.; Sawada, A.; Tanaka, H.; Nishibori, E.; Takata, M.; Sakata, M. Direct observation of covalency between O and disordered Pb in cubic PbZrO3. J. Phys. Soc. Japan 2002, 71, 2353–2356. [Google Scholar] [CrossRef]
- Piskunov, S.; Kotomin, E.A.; Heifets, E.; Maier, J.; Eglitis, R.I.; Borstel, G. Hybrid DFT calculations of the atomic and electronic structure for ABO3 perovskite (001) surfaces. Surf. Sci. 2005, 575, 75–88. [Google Scholar] [CrossRef]
- Wemple, S.H. Polarization Fluctuations and the Optical-Absorption Edge in BaTiO3. Phys. Rev. B 1970, 2, 2679–2689. [Google Scholar] [CrossRef]
- Benthem, K.; Elsässer, C.; French, R.H. Bulk electronic structure of SrTiO3: Experiment and theory. J. Appl. Phys. 2001, 90, 6156–6164. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ab initio Method | BZO [29] | MgF2 [119] | CaF2 [120] |
| Experiment | 5.3 [121] | 13.0 [122] | 12.1 [123] |
| B3LYP | 4.79 | 9.42 | 10.85 |
| B3PW | 4.93 | 9.48 | 10.96 |
| HF | 12.96 | 19.65 | 20.77 |
| PWGGA | 3.24 | 6.94 | 8.51 |
| Material | BTO | PTO | STO | SZO | PZO |
|---|---|---|---|---|---|
| Computed | 4.008 | 3.936 | 3.904 | 4.195 | 4.220 |
| Experiment | 4.004 [137] | 3.97 [138] | 3.898 [139] | 4.154 [140] | 4.1614 [141] |
| Perovskite | Structure at Room Temperature | B3PW Γ-Γ Gap | Exp. Γ-Γ Gap |
|---|---|---|---|
| PTO | Tetragonal phase | 4.32 eV | 3.4 eV [121] |
| BTO | Tetragonal ↔ orthorhombic phase at a temperature of 278 K | 3.55 eV | 3.38 eV (// c); 3.27 (⊥ c) [143] |
| STO | Cubic phase | 3.96 eV | 3.75 eV [144] |
| Ion | Property | BTO | PTO | STO | SZO | PZO |
|---|---|---|---|---|---|---|
| A | Q | 1.79 | 1.35 | 1.87 | 1.88 | 1.30 |
| P | −0.034 | 0.016 | −0.010 | −0.008 | −0.020 | |
| O | Q | −1.39 | −1.23 | −1.41 | −1.33 | −1.12 |
| P | 0.100 | 0.098 | 0.088 | 0.084 | 0.100 | |
| B | Q | 2.36 | 2.34 | 2.35 | 2.13 | 2.07 |
| Layer | Prop. | STO | BTO1 | BTO2 | BTO3 | BTO4 | BTO5 | BTO6 | BTO7 | BTO8 | BTO9 | BTO10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X | QTi | 2.30 | ||||||||||
| QO | −1.27 | |||||||||||
| Qplane | −0.23 | |||||||||||
| Δz | −3.92 | |||||||||||
| IX | QBa | 1.75 | 1.76 | |||||||||
| QO | −1.48 | −1.37 | ||||||||||
| Qplane | 0.27 | 0.39 | ||||||||||
| Δz | −2.32 | 4.60 | ||||||||||
| VIII | QTi | 2.30 | 2.37 | 2.36 | ||||||||
| QO | −1.26 | −1.41 | −1.36 | |||||||||
| Qplane | −0.23 | −0.44 | −0.36 | |||||||||
| Δz | −3.84 | 3.65 | 0.65 | |||||||||
| VII | QBa | 1.75 | 1.76 | 1.80 | 1.79 | |||||||
| QO | −1.48 | −1.37 | −1.42 | −1.40 | ||||||||
| Qplane | 0.26 | 0.39 | 0.38 | 0.39 | ||||||||
| Δz | −2.21 | 4.62 | 0.68 | 2.12 | ||||||||
| VI | QTi | 2.30 | 2.37 | 2.36 | 2.36 | 2.36 | ||||||
| QO | −1.26 | −1.40 | −1.36 | −1.38 | −1.37 | |||||||
| Qplane | −0.23 | −0.43 | −0.36 | −0.40 | −0.38 | |||||||
| Δz | −3.70 | 3.69 | 0.74 | 1.88 | 1.50 | |||||||
| V | QBa | 1.75 | 1.76 | 1.80 | 1.79 | 1.79 | 1.79 | |||||
| QO | −1.49 | −1.38 | −1.42 | −1.40 | −1.41 | −1.41 | ||||||
| Qplane | 0.26 | 0.38 | 0.37 | 0.39 | 0.39 | 0.39 | ||||||
| Δz | −2.07 | 4.68 | 0.76 | 2.15 | 1.40 | 1.71 | ||||||
| IV | QTi | 2.30 | 2.37 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | ||||
| QO | −1.26 | −1.40 | −1.36 | −1.38 | −1.37 | −1.38 | −1.37 | |||||
| Qplane | −0.22 | −0.43 | −0.36 | −0.40 | −0.38 | −0.39 | −0.39 | |||||
| Δz | −3.55 | 3.73 | 0.83 | 1.92 | 1.57 | 1.57 | 1.63 | |||||
| III | QBa | 1.75 | 1.76 | 1.80 | 1.79 | 1.79 | 1.79 | 1.79 | 1.79 | |||
| QO | −1.49 | −1.39 | −1.43 | −1.41 | −1.41 | −1.41 | −1.40 | −1.41 | ||||
| Qplane | 0.25 | 0.38 | 0.37 | 0.38 | 0.39 | 0.38 | 0.39 | 0.38 | ||||
| Δz | −1.84 | 4.77 | 0.89 | 2.27 | 1.48 | 1.79 | 1.53 | 1.68 | ||||
| II | QTi | 2.29 | 2.37 | 2.35 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | ||
| QO | −1.25 | −1.40 | −1.35 | −1.38 | −1.37 | −1.38 | −1.37 | −1.38 | −1.37 | |||
| Qplane | −0.21 | −0.42 | −0.35 | −0.39 | −0.37 | −0.39 | −0.38 | −0.39 | −0.39 | |||
| Δz | −3.20 | 3.83 | 1.03 | 2.04 | 1.72 | 1.69 | 1.75 | 1.60 | 1.71 | |||
| I | QBa | 1.75 | 1.76 | 1.80 | 1.79 | 1.79 | 1.79 | 1.79 | 1.79 | 1.79 | 1.79 | |
| QO | −1.51 | −1.40 | −1.44 | −1.43 | −1.42 | −1.42 | −1.42 | −1.42 | −1.41 | −1.42 | ||
| Qplane | 0.23 | 0.36 | 0.35 | 0.36 | 0.37 | 0.37 | 0.38 | 0.37 | 0.38 | 0.37 | ||
| Δz | −1.54 | 4.87 | 1.02 | 2.35 | 1.53 | 1.84 | 1.55 | 1.70 | 1.51 | 1.64 | ||
| 5 | QTi | 2.29 | 2.37 | 2.35 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 |
| QO | −1.29 | −1.41 | −1.38 | −1.40 | −1.39 | −1.40 | −1.40 | −1.40 | −1.40 | −1.40 | −1.40 | |
| Qplane | −0.30 | −0.45 | −0.40 | −0.44 | −0.42 | −0.44 | −0.43 | −0.44 | −0.44 | −0.45 | −0.44 | |
| Δz | −5.95 | 1.96 | −1.13 | −0.11 | −0.46 | −0.47 | −0.43 | −0.56 | −0.48 | −0.61 | −0.53 | |
| 4 | QSr | 1.85 | 1.88 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 |
| QO | −1.37 | −1.41 | −1.39 | −1.38 | −1.38 | −1.37 | −1.38 | −1.37 | −1.37 | −1.36 | −1.37 | |
| Qplane | 0.48 | 0.47 | 0.48 | 0.50 | 0.49 | 0.50 | 0.50 | 0.51 | 0.50 | 0.51 | 0.50 | |
| Δz | 4.13 | −1.32 | 0.60 | −0.43 | −0.10 | −0.39 | −0.26 | −0.43 | −0.33 | −0.47 | −0.37 | |
| 3 | QTi | 2.35 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 |
| QO | −1.38 | −1.42 | −1.41 | −1.42 | −1.41 | −1.42 | −1.42 | −1.43 | −1.42 | −1.43 | −1.42 | |
| Qplane | −0.42 | −0.48 | −0.45 | −0.48 | −0.47 | −0.49 | −0.48 | −0.49 | −0.48 | −0.49 | −0.49 | |
| Δz | −0.96 | 0.27 | −0.20 | −0.25 | −0.21 | −0.38 | −0.29 | −0.44 | −0.35 | −0.48 | −0.40 | |
| 2 | QSr | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 |
| QO | −1.42 | −1.40 | −1.41 | −1.39 | −1.40 | −1.39 | −1.39 | −1.38 | −1.39 | −1.38 | −1.39 | |
| Qplane | 0.45 | 0.47 | 0.46 | 0.48 | 0.47 | 0.49 | 0.48 | 0.49 | 0.48 | 0.49 | 0.49 | |
| Δz | 0.88 | −0.35 | 0.12 | −0.29 | −0.15 | −0.37 | −0.26 | −0.42 | −0.32 | −0.45 | −0.37 | |
| 1 | QTi | 2.35 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 | 2.36 |
| QO | −1.40 | −1.42 | −1.41 | −1.42 | −1.42 | −1.42 | −1.42 | −1.43 | −1.42 | −1.43 | −1.42 | |
| Qplane | −0.44 | −0.47 | −0.46 | −0.48 | −0.47 | −0.49 | −0.48 | −0.49 | −0.48 | −0.49 | −0.49 | |
| Δz | 0.14 | −0.09 | −0.01 | −0.27 | −0.17 | −0.36 | −0.26 | −0.42 | −0.33 | −0.46 | −0.37 | |
| 0 | QSr | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 | 1.87 |
| QO | −1.42 | −1.40 | −1.41 | −1.39 | −1.40 | −1.39 | −1.39 | −1.38 | −1.39 | −1.38 | −1.39 | |
| Qplane | 0.45 | 0.47 | 0.46 | 0.48 | 0.47 | 0.49 | 0.48 | 0.49 | 0.48 | 0.49 | 0.49 | |
| Δz | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| δ | 2.58 | 3.47 | 2.33 | 3.29 | 2.16 | 3.25 | 2.10 | 3.24 | 2.06 | 3.22 | 2.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eglitis, R.I.; Bocharov, D.; Piskunov, S.; Jia, R. Review of First Principles Simulations of STO/BTO, STO/PTO, and SZO/PZO (001) Heterostructures. Crystals 2023, 13, 799. https://doi.org/10.3390/cryst13050799
Eglitis RI, Bocharov D, Piskunov S, Jia R. Review of First Principles Simulations of STO/BTO, STO/PTO, and SZO/PZO (001) Heterostructures. Crystals. 2023; 13(5):799. https://doi.org/10.3390/cryst13050799
Chicago/Turabian StyleEglitis, Roberts I., Dmitry Bocharov, Sergey Piskunov, and Ran Jia. 2023. "Review of First Principles Simulations of STO/BTO, STO/PTO, and SZO/PZO (001) Heterostructures" Crystals 13, no. 5: 799. https://doi.org/10.3390/cryst13050799
APA StyleEglitis, R. I., Bocharov, D., Piskunov, S., & Jia, R. (2023). Review of First Principles Simulations of STO/BTO, STO/PTO, and SZO/PZO (001) Heterostructures. Crystals, 13(5), 799. https://doi.org/10.3390/cryst13050799