
Structural and Magnetic Properties of the {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 Heteronuclear Complex
by
 , ,
, ,
Fabio Santanni
1,2,*,
Laura Chelazzi
1,
Lorenzo Sorace
1,2,
Grigore A. Timco
3 and
Roberta Sessoli
1,2,* 1
Dipartimento di Chimica “Ugo Schiff”—DICUS, Università degli Studi di Firenze, Via della Lastruccia 3-13, 50019 Firenze, Italy
2
Consorzio Interuniversitario Nazionale di Scienza e Tecnologia dei Materiali—INSTM, Via G. Giusti 9, 50121 Firenze, Italy
3
Department of Chemistry, Photon Science Institute, The University of Manchester, Oxford Road, Manchester M13 9PL, UK
*
Authors to whom correspondence should be addressed.
Crystals 2023, 13(6), 901; https://doi.org/10.3390/cryst13060901
Submission received: 2 May 2023
/
Revised: 22 May 2023
/
Accepted: 27 May 2023
/
Published: 1 June 2023
(This article belongs to the Special Issue The Polyhedral Face of Coordination Chemistry)
Abstract
:Heterotopic ligands containing chemically different binding centers are appealing candidates for obtaining heteronuclear metal complexes. By exploiting this strategy, it is possible to introduce different paramagnetic centers characterized by specific anisotropic magnetic properties that make them distinguishable when weakly magnetically coupled. This molecular approach has great potential to yield multi-spin adducts capable of mimicking logical architectures necessary for quantum information processing (QIP), i.e., quantum logic gates. A possible route for including a single-ion magnetic center within a finite-sized heterometallic compound uses the asymmetric (1-pyridyl)-butane-1,3-dione (pybd) ligand reported in the literature for obtaining Cr3+−Cu2+ metallo-cages. To avoid the formation of cages, we adopted the cyclen (1,4,7,10-tetraazacyclododecane) ligand as a “capping” agent for the Cu2+ ions. We report here the structural and magnetic characterization of the unprecedented adduct {Cr(pybd)3[Cu(cyclen)]2}(BF4)4, whose structure is characterized by a central Cr3+ ion in a distorted octahedral coordination environment and two peripheral Cu2+ ions with square-pyramidal coordination geometries. As highlighted by Continuous Wave Electron Paramagnetic Resonance (EPR) spectroscopy and Direct Current (DC) magnetometry measurements, this adduct shows negligible intramolecular magnetic couplings, and it maintains the characteristic EPR signals of Cr3+ and Cu2+ moieties when diluted in frozen solutions.
1. Introduction
The qubit, the quantum analog of classical bits, bases its working principles on quantomechanical laws such as the superposition of states. In principle, it is possible to exploit two spin levels of an unpaired electron as the computational basis and encode the information in linear combinations of these states [1]. Magnetic molecules characterized by good spin coherence properties are appealing candidates for improving the quantum logical units of quantum computers [2,3]. They can work as qubits when two electronic spin levels are employed to encode the information (e.g., the mS = ±1/2 projections along the quantization axis of an S = 1/2 system) [4,5] or as qudits when more electronic and nuclear spin levels are involved [6,7].
When connected through specific channels (e.g., superconductive planar waveguides [8]), multi-qubit architectures capable of implementing more complex logic operators known as quantum logic gates can be achieved [9]. Likewise, it is possible to “connect” different molecular spins to mime such architectures present in modern quantum processors [10].
Beyond the tailoring of organic ligands and molecules to obtain multi-spin structures [11,12], many other conditions must be fulfilled. For instance, most logical operations and quantum algorithms require that qubits can act as entangled objects. In other words, an interaction between the two spins must be active, and the total state cannot be expressed as a simple product of the individual spin states. Besides this, entangled qubits must be individually addressable so that the selective manipulation of qubits results in the detectable variation of the counterparts quantum states. Magnetic interactions between the spin centers, dipolar or isotropic exchange in nature [11,12,13,14], can be exploited to achieve the required entanglement condition [15]. However, the magnetic coupling interaction should be small enough, typically of the order of 10−1–10−2 cm−1, to avoid the formation of a giant spin state. Two main strategies can be used to maintain the individual addressability of spins: (i) the introduction of specific ligands and organic scaffolds that guarantee a different relative orientation of the magnetic anisotropy tensors of spin centers [11,16] or (ii) the exploitation of different metal or spin centers within a single architecture, these being characterized by different magnetic anisotropy tensors [17,18,19].
Heterometallic coordination compounds are of high interest in various fields, spanning from catalysis [20,21,22,23] to molecular magnetism [24,25,26,27,28]. Obtaining such structures is not a trivial task, and a tailored design of employed ligands, eventually heterotopic ones [29,30,31], is often necessary. The use of heterometallic structures has been proposed as a potential strategy to implement quantum logic gates. As reported in [12], the entanglement condition among the different qubits within these architectures can be selectively enabled upon applying specific pulse sequences. In this sense, such a strategy is preferable to the first one since it allows us to mime what commonly happens in quantum circuits, where qubits act as single objects or entangled units upon necessity. However, two critical aspects can be identified for this strategy: (i) the obtainment of heterometallic multi-qubit systems is not trivial, nor are the design and synthetic efforts required [32]; (ii) it is difficult to predict the final quantum and magnetic properties of adducts, where parties can behave differently compared to their isolated forms.
Among the most investigated systems, molecular metallocages represent a significant part of reported systems [31,33,34]. Starting from the works of Brechin and coworkers on Cr-based cages built by the (1-pyridyl)-butane-1,3-dione (Hpybd) ligand [30,35,36,37], we focused on obtaining a heterometallic system by using the same ligand. Such a ligand allows for getting octahedral complexes of M3+ ions thanks to the coordinating nature of the diketonate core. Then, by further exploiting the peripheral pyridyl functions, it is also possible to coordinate up to three additional ions per M3+ unit. In our case, we exploited the ditopic nature of this ligand to obtain a heterometallic complex containing a central Cr3+ ion and Cu2+-based peripheral units. Differently from previous reports [30,35], we exploited the pyridyl units to coordinate a Cu2+ complex, i.e., the [Cu(cyclen)]2+ (cyclen = 1,4,7,10-tetraazacyclododecane). Using the cyclen moiety as a “capping agent” allowed us to get a finite-size complex that avoids forming polymers or cage-like structures.
In this work, we report the structural characterization of the {Cr(pybd)3[Cu(Cyclen)]2}(BF4)4 compound followed by powder X-ray diffraction studies and data analysis. Furthermore, we report the investigation of the static magnetic properties of the adduct performed by standard magnetometry and electron paramagnetic resonance (EPR) spectroscopy.
2. Materials and Methods
2.1. Synthesis
2.1.1. General Remarks
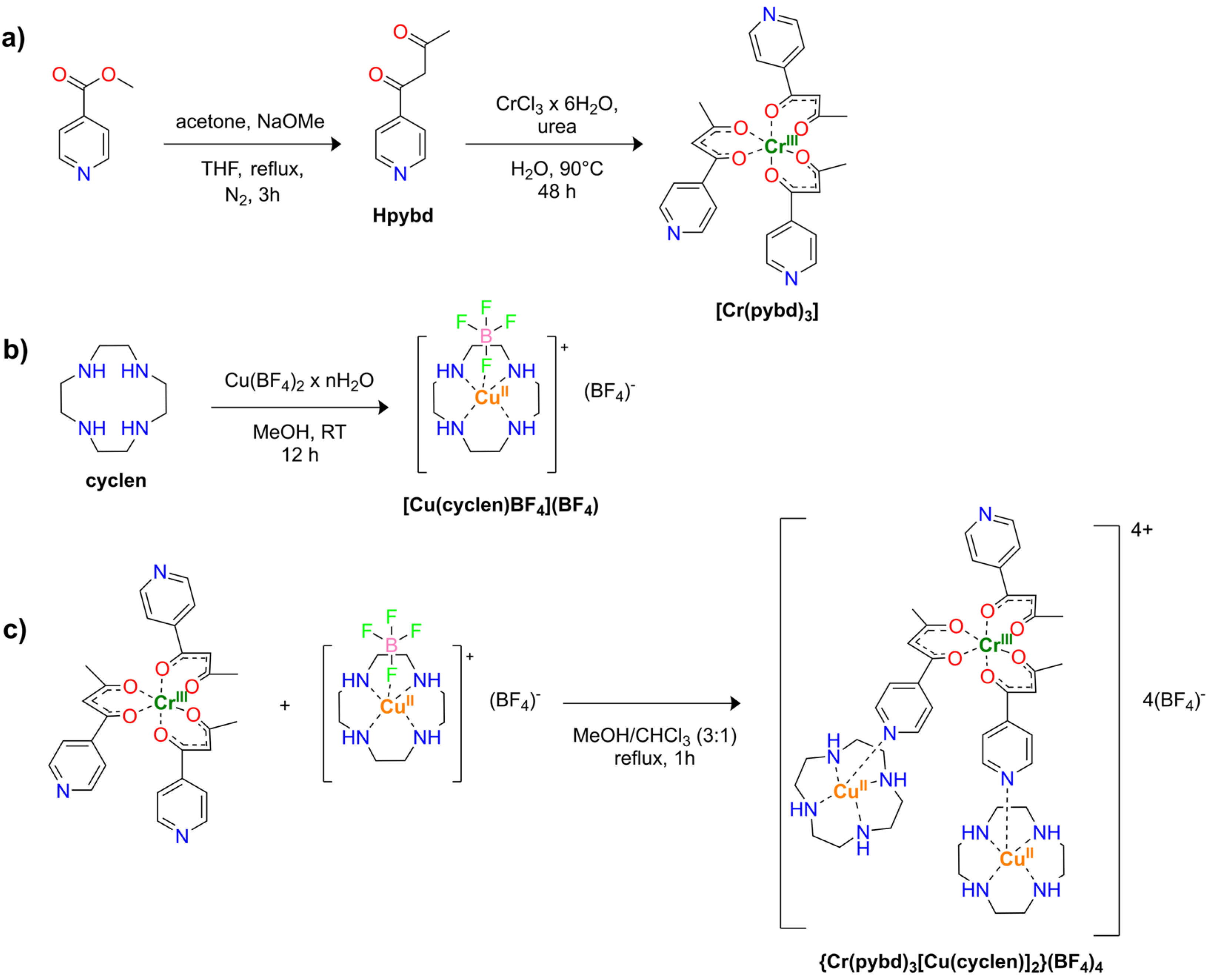
All the reagents and solvents were of reagent grade and employed without further purification. Compounds Hpybd and [Cr(pybd)3] were synthesized according to the literature [30]. More details about the synthesis of all the other compounds are reported in Section S1 of Supplementary Information (SI). A general reaction pathway is reported in Scheme 1 for clarity.
2.1.2. Synthesis of {Cr(pybd)3[Cu(cyclen)]2}(BF4)4
In a 50 mL two-neck flask equipped with a condenser, [Cu(cyclen)BF4]BF4 (235 mg, 0.6 mmol) was dissolved in 15 mL of MeOH and heated to reflux under magnetic stirring. A solution of [Cr(pybd)3] (100 mg, 0.2 mmol) in CHCl3 (5 mL) was added dropwise to the reaction mixture, and the solution refluxed for 1 h. Then, the solution was cooled to room temperature, filtered, and evaporated slowly. After three days, a crystalline product (218 mg, 85%) was collected by filtration and washed with CHCl3 (5 mL × 2) and Et2O (5 mL × 2). The grounded powder was employed for structural determination (see below). Elemental analysis (calc. for CrCu2C43H64N11O6B4F16): C, 38.45 (38.05); H, 4.37 (4.75); N; 11.29 (11.35). UV/Vis (λmax, nm): 279, 368, 587 (ε = 43 M−1 cm−1); see also Figure S1. FT-IR (ν, cm−1): 3325(w), 3306(s), 3129(vw), 2941(w), 2895(w), 1585(m), 1545(w), 1515(s), 1495(vw), 1429(m), 1394(s), 1322(vw), 1291(m), 1250(vw), 1217(w), 1081 (vs, b), 1057(vs, b) 974(m, b), 955(vw, b), 867(w), 849(w), 810(vw), 775(m), 701(s), 619(s) (Figure S2 of SI).
2.2. UV–Vis Spectroscopy
UV–Vis spectra were recorded on a Jasco V-670 double-beam spectrophotometer by using quartz cuvettes of 1 cm length. All experiments were performed on 0.25 M MeOH solutions of employed compounds. All experimental spectra are reported in Figures S1 and S3 of SI.
2.3. X-ray Structural Characterization
Single-crystal X-ray diffraction was employed to obtain the unit cell parameters. A single crystal was analyzed at 100 K using Cu-Kα radiation (λ = 1.54184 Å) on a Bruker Apex-II diffractometer equipped with pixel array detector PhotonII, controlled using APEX2 software [38].
Powder X-ray Diffraction (PXRD) was used for structure determination using unit cell parameters extracted from single crystal data. High-quality PXRD data were recorded in a 0.3 mm glass capillary at room temperature by using a Bruker New D8 Da Vinci diffractometer (Cu − Kα1 radiation = 1.54056 Å, 40 kV × 40 mA) equipped with a Bruker LYNXEYE-XE detector, scanning range 2θ = 3–70°, 0.02° increments of 2θ, and a counting time of 576 total time/step. Space group determination with EXPO2014 [39] resulted in space group P-1 with Z = 2. We then solved the structure by stimulated annealing using the EXPO2014 suite. Each annealing trial works on three runs, using a cooling rate (Tn/Tn−1) of 0.95. All the torsion angles were allowed to rotate freely during the refinement process while bond distances and angles were kept fixed. The best solution was chosen for Rietveld refinement, which was performed with the software TOPASv6 [40]. Background and peak shape were fitted using a shifted Chebyshev function with eight coefficients and a Pseudo-Voigt function, respectively. All atoms were isotropically refined with a typical thermal parameter depending on the element. All the hydrogen atoms were fixed in calculated positions. Crystal data and refinement parameters are reported in Table 1. The simulated pattern was obtained by using the software Mercury CSD 2022.2.0 (copyright CCDC, https://www.ccdc.cam.ac.uk/solutions/software/mercury, accessed: 26 May 2023) [41] by employing an FWHM value of 0.1 and an increment step size of 0.025°. Complete crystallographic data for the solved structures have been deposited in the Cambridge Crystallographic Data Centre with CCDC number 2260168.
2.4. DC Magnetometry
Direct current (DC) magnetic measurements were performed in the temperature range 2–300 K (B = 0.1 T) or in the field range 0–5 T (T = 2 K, 4 K, 8 K) using a Quantum Design Magnetic Properties Measuring System (QD-MPMS) equipped with a Superconductive Quantum Interference Device (SQUID). Magnetization data were corrected for the diamagnetic contributions of sample holders (previously measured using the same conditions) and of the sample itself, as deduced by using Pascal’s constant tables [42].
2.5. Electron Paramagnetic Resonance (EPR) Spectroscopy
CW X-band EPR spectra of all samples were recorded on a Bruker Elexsys E500 spectrometer equipped with an SHQ cavity (ν = 9.43 GHz). Low-temperature measurements were obtained using an Oxford Instruments ESR900 continuous-flow helium cryostat. All measured samples were 0.5 mM solutions of employed compounds in EtOH/MeOH 4:1. The solutions were introduced within a standard X-band quartz tube and rapidly frozen in a liquid nitrogen bath to obtain dispersions in glassy matrixes.
3. Results and Discussion
3.1. Synthesis
The molecular complex [Cr(pybd)3] was synthesized following the strategy by Brechin et al. [30,35,36,37]. The reaction of CrCl3·6H2O with the Hpybd ligand was performed in water by using urea as the base for deprotonating the employed diketonate. Such a strategy is commonly used to avoid the formation of chromium oxides and hydroxides during the reaction since ammonia slowly develops by urea decomposition at 90 °C [43]. After purification, the [Cr(pybd)3] complex was reacted with three equivalents of the Cu2+ precursor: [Cu(cyclen)BF4]BF4, in a MeOH/CHCl3 6:1 solution. This procedure was used with the intent to introduce three Cu units per Cr center, as done in ref. [30], and to obtain the tetramer {CrCu3}. The reaction can be followed by observing the color changing from royal blue to greenish/blue. On cooling, dark crystals formed from the solution, and larger amounts of the product were obtained by slow evaporation of the reaction mixture. Notwithstanding our predictions and efforts to obtain a tetramer, it was only possible to get the trimeric specie {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 (see below). Interestingly, the obtained compound does not respect the expected Cr/Cu 1:3 stoichiometry even though a large excess of Cu2+ precursor is employed. This behavior could be imputed to the steric hindrance introduced by adding [Cu(cyclen)]2+ to the pyridyl units. Furthermore, it was only possible to obtain {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 by slow evaporation of the reaction mixture. Any other attempt to recrystallize it from solutions of different organic solvents and their mixtures (e.g., MeOH, CHCl3, DMSO, and DMF) led to the separation of individual Cr3+ and Cu2+ precursors.
3.2. Structural Characterization
As mentioned above, {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 (Figure 1) crystallizes from the reaction mixture upon cooling and subsequent evaporation of MeOH. Nevertheless, the plate-shaped crystals tend to pile along the c crystallographic axis. For this reason, cutting a single crystal suitable for X-ray structural determination was not possible. Hence, we employed the single-crystal diffraction tool to firstly estimate the cell parameters that were successively used for the structural refinement model employed on PXRD data (see Section 2). We report the comparison between the experimental and simulated PXRD patterns in Figure 2 as proof of the quality of the employed model. The plot evidences a remarkable agreement between experiment and simulation, both in terms of peak positions and relative intensities.
{Cr(pybd)3[Cu(cyclen)]2}(BF4)4 crystallizes in the P−1 space group (no. 2) with two {Cr(pybd)3[Cu(cyclen)]2}4+ cationic species and eight BF4− counterions per unit-cell. The asymmetric unit is represented by the {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 unit reported in Figure 1 (unit cell content shown in Figure S3 of SI). The crystal structure also presents solvent-accessible pores, occupying 5% of the unit cell volume with a volume of about 165 Å3 (Figure S4 of SI). The porous structure propagates along the a-axis direction.
In the asymmetric unit, the Cr3+ ion preserves the fac isomerism typical of [Cr(pydb)3] [30] and presents a distorted octahedral coordination geometry having mean Cr−O distances of 1.96 Å on the equatorial plane and Cr−O5 and Cr−O6 distances of 1.93 Å and 1.97 Å along the axial direction (see Figure S5a of SI). Furthermore, all the O2−Cr−O4, O1−Cr−O3, and O5−Cr−O6 angles are smaller than 180°, being of about 176.0°, 177.0°, and 179.0°, respectively (see Figure S5b of SI). Such a distorted octahedral geometry differs from the regular one reported for the [Cr(pydb)3] precursor, although it has been observed for [Cr(pydb)3]-based supramolecular cages [30,35,36]. In analogy to the Cr3+ ion, the two Cu2+ moieties present slightly distorted square-pyramidal geometry due to minor differences in the Cu−Ncyclen bond lengths (see Figure S6 of SI). In this case, the mean Cu−Ncyclen distance is 2.03 Å for both cyclen-based units, while the Cu−Npy distances are 2.15 Å and 2.17 Å for Cu1−N5 and Cu2−N10, respectively. These distances align with other square-pyramidal N-based copper complexes [44]. The mean Cr−Cu intramolecular distance is 8.9 Å. The smallest Cu···Cu intermolecular distance of 7.3 Å is measured between molecules in adjacent cells moving along the b-axis. The smallest intermolecular Cr···Cr distance is 7.36 Å, and it is measured between molecules in adjacent cells along the c axis. The structure presents four BF4− per molecular unit, three of them presenting short-contact interactions with the acidic N−H moieties of the two [Cu(cyclen)]2+ units per molecule. At the same time, the fourth is stabilized within the framework by similar interactions set with the cyclen unit of another molecule.
3.3. UV–Vis Spectroscopy Characterization
To check for the stability of the {[Cr(pybd)3][Cu(cyclen)]2}4+ adduct when in solution and confirm the coordination geometry of metal sites, we performed a UV–Vis spectroscopic investigation on 2.5 × 10−3 M solutions of [Cu(cyclen)]2+ and {[Cr(pybd)3][Cu(cyclen)]2}4+ in MeOH and compared their experimental spectra. To do that, we focused on the spectral region between 400 and 850 nm, where electronic transitions attributed to the copper(II) center are usually observed [44,45,46,47]. The graph in Figure 3 shows that the two complexes present an absorption band centered at about 600 nm. In particular, we observed absorption maxima at 587 nm (ε = 43 M−1 cm−1) and 601 nm (ε = 35 M−1 cm−1) for {[Cr(pybd)3][Cu(cyclen)]2}4+ and [Cu(cyclen)]2+, respectively. This spectral feature is imputed to a d-d transition of the copper center, and the observed energy is characteristic of pentacoordinate complexes having a square-pyramidal coordination geometry [44]. We further compared these UV–Vis spectra with an additional one obtained on a 2.5 × 10−3 M solution of [Cu(cyclen)]2+ in MeOH added in 100 equivalents of pyridine. This third spectrum shows a red shift of the absorption maximum to λ = 617 nm (ε = 35 M−1 cm−1), which is expected for stronger interactions with an axial ligand on the fifth position [48]. By comparing this spectrum with that obtained on {[Cr(pybd)3][Cu(cyclen)]2}4+, we can state that the coordination of the Cu2+ ion by the pyridyl unit of pybd ligand leads to a smaller crystal field interaction with respect to pure pyridine. This can be correlated to a weaker interaction with the unpaired doublet of the N donor atom, whose delocalization on the conjugated β-diketonate scaffold could be further affected by the electron-withdrawing effect of the coordinated Cr3+ ion.
3.4. Magnetism and EPR Spectroscopy
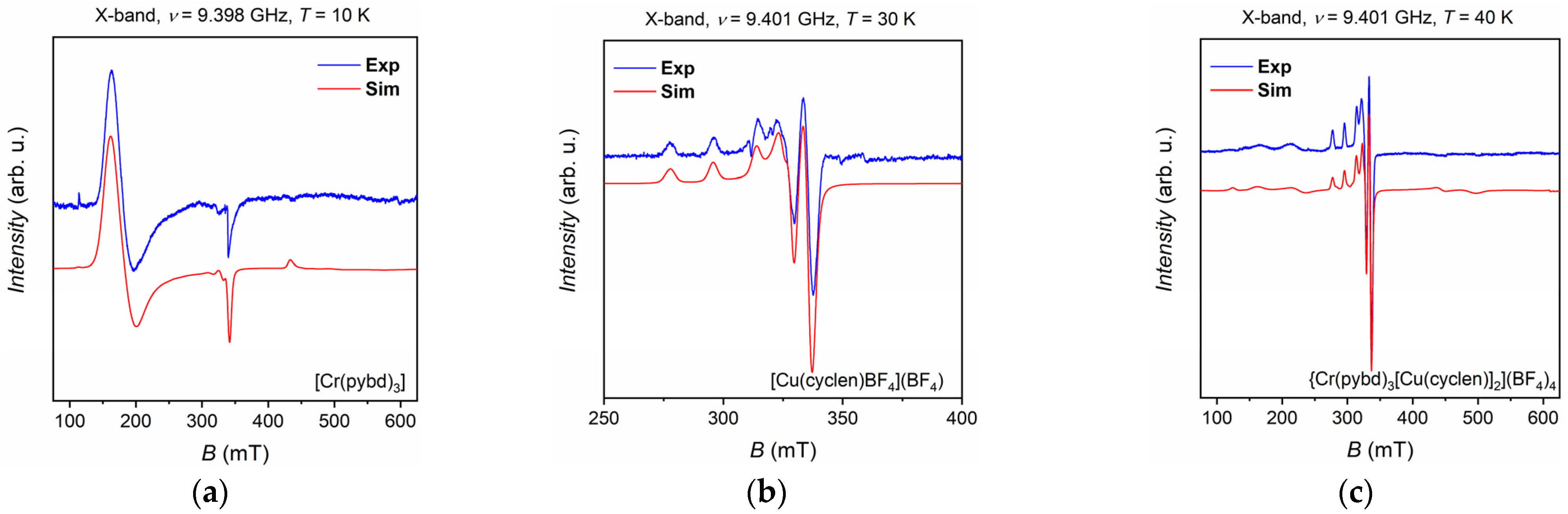
X-band (ν ≈ 9.4 GHz) continuous wave EPR measurements were performed to characterize and compare the magnetic anisotropy properties of the presented systems. To do that, we measured frozen solution of [Cr(pybd)3], [Cu(cyclen)]2+, and {Cr(pybd)3[Cu(cyclen)]2}4+ in EtOH/MeOH 4:1 (see Section 2). The experimental spectra are reported as blue lines in Figure 4a–c.
The [Cr(pybd)3] complex shows the characteristic EPR spectrum of an octahedrally coordinated Cr3+ ion (Figure 4a). The observed spectral features are attributed to spin transitions within the quartet basis state (S = 3/2) further split in two Kramers’ doublets (mS = ± 1/2; mS = ± 3/2) by the zero-field splitting (ZFS) interaction, which is characterized by axial (D) and rhombic (E) terms following the spin Hamiltonian (SH) in Equation (1).
The spectrum can be well simulated [49] with the SH parameters extracted from ref. [30] and reported in Table 1. The chromium complex is characterized by an easy-plane anisotropy (D = + 0.55 cm−1) leading to mS = ± 1/2 ground doublet. The E/D ratio of 0.045 confirms that the system has a weak rhombic distortion. Such values are in line with what has been reported in the literature for other Cr-acetylacetonate complexes, for which D values of about 0.6 cm−1 have been observed [50,51].
The [Cu(cyclen)]2+ compound showed the EPR spectrum in Figure 4b, which is closely in line with that expected for an S = 1/2 system characterized by an anisotropic axial g tensor with an axial hyperfine coupling interaction (A) between the spin and the copper nucleus (I = 3/2). A quantitative estimation of the magnetic anisotropy followed from the simulation of the experimental spectrum with the SH reported in Equation (2).
The values obtained for the components of g and A tensors (Table 2), for which g|| > g⏊ and A|| > A⏊ (with z = || and x = y = ⏊), are representative of a pentacoordinate Cu2+ complex [47,48] and confirm the experimental evidence given by the UV–Vis analysis (see above).
The X-band characterization of {Cr(pybd)3[Cu(cyclen)]2}4+ was performed at 40 K to avoid saturation of the Cu2+ signal and simultaneously detect that of the Cr3+ ion. The obtained spectrum (Figure 4c) slightly differs from the trivial sum of those of the two precursors. On the one hand, the low-field region between 100 mT and 25 mT, i.e., the most indicative one for the Cr3+ ion signal, largely differs from that of isolated [Cr(pybd)3]. On the other hand, the Cu2+ signal falling within the 250–400 mT region seems not to be affected by the presence of the Cr3+ ion. Based on this observation, we can say that there is no evidence of a clear isotropic exchange coupling interaction among the Cr and Cu sites. It follows that the unique modification of the chromium signal should be correlated to the variation of the SH parameters of the Cr3+ ion upon copper coordination of two pyridine rings of the pybd ligand, while those of Cu2+ remain unaltered. To validate such an assumption, we simulated the spectra by fixing the SH parameters of Cu2+ moieties and varying the D and E components of chromium using a SH given by the sum of those reported in Equations (1) and (2). The best simulation reported in Figure 4c was obtained by reducing both the D and E values to 0.21 cm−1 and 0.02 cm−1, respectively.
Consequently, the E/D ratio of the adduct increased to 0.095, highlighting a larger rhombicity of the system. This feature can be correlated to the distortion of the Cr3+ coordination sphere observed in the case of {Cr(pybd)3[Cu(cyclen)]2}4+ (see Structural Characterization section), which led to an increased rhombicity. It must be noted that, in our model, dipolar interactions among spins were not introduced directly, but they were included as contributions to the isotropic and anisotropic line-broadening. These interactions could be better highlighted and investigated by adopting advanced pulsed EPR experiments such as DEER (Double Electron-Electron Resonance) [52].
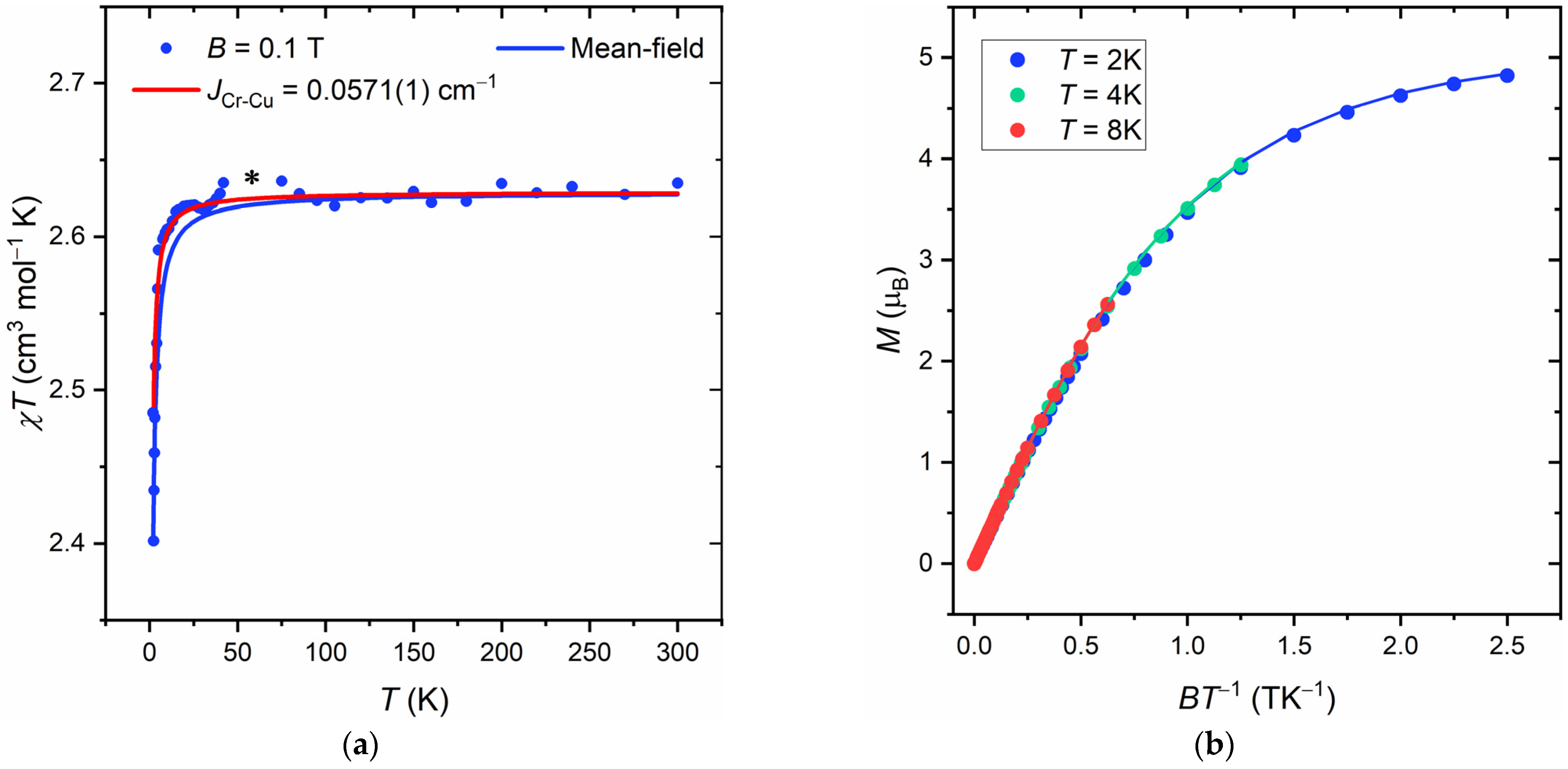
To further exclude that the lack of magnetic exchange interaction is not a consequence of structural changes occurring in the frozen solution, we measured crystalline powders of {Cr(pybd)3[Cu(cyclen)]2}(BF4)4. However, the EPR spectrum collected on the solid sample is significantly broadened because of intermolecular interactions (Figure S7 of SI), and it cannot pursue our scope. For this reason, we employed DC magnetometry (see Section 2) to get more insights into the magnetic interactions characteristic of the compound. The magnetic susceptibility (χ) data are reported in Figure 5a as χT product vs. T. Above 25 K, the χT value reaches a plateau around 2.62 cm3 mol−1 K, which is the sum of the Curie’s constants expected for the three paramagnetic centers characterized by the ZFS and g tensors anisotropies determined by our EPR experiments. Below this temperature, the χT value drops abruptly to 2.4 cm3 mol−1 K. This behavior can be correlated to two distinct factors, i.e., (i) a contribution linked to the ZFS of Cr3+ ion and (ii) a contribution due to intra- or intermolecular coupling interactions within the crystal lattice. In order to estimate the magnitude of such interactions, we performed a simultaneous fit of χT vs. T and M vs. B (Figure 5a,b) curves with the software PHI [53]. We used the set of SH parameters extracted from X-band measurements and kept them fixed, while the mean-field intermolecular interactions parameter (zJ) was used as the fitting parameter. The employed model provides a best-fit zJ value of 1.92(1)·10−2 cm−1. We repeated the same fitting procedure by replacing the mean-field interaction with an equivalent isotropic exchange coupling interaction between the two Cu2+ ions and the central Cr3+ and fitting the coupling constant JCr-Cu. The best JCr−Cu = −5.71(1)·10−2 cm−1 is of the same order of magnitude as the mean-field constant zJ¸and the fitted curve (red line in Figure 5) poorly reproduces the experimental χT points between 2 and 5 K.
This latter analysis, together with our EPR results reported for the diluted phase, suggests that, even if present, exchange coupling interactions are not detectable in the limit of the experimental conditions employed.
4. Conclusions
In this work, we reported the synthesis of the new molecular cluster {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 by exploiting the heterotopic properties of the pybd− ligand and using cyclen rings as capping agents to avoid the formation of molecular cages. The crystallographic structure has been resolved thanks to a combined approach involving single-crystal X-ray measurements and powder X-ray diffraction suites. Congruently to our elemental analysis results, the crystallographic characterization unveiled the structural peculiarity of the cluster, which is characterized by the presence of just two coordinated [Cu(cyclen)]2+ moieties over the three potential pyridyl units of the [Cr(pybd)3] core. Our X-band CW-EPR measurements on diluted frozen solutions highlighted the variation of the magnetic anisotropy properties of the Cr3+ center in {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 with respect to the monomeric [Cr(pybd)3] unit. The increased rhombic distortion in {Cr(pybd)3[Cu(cyclen)]2}(BF4)4, highlighted by an E/D ratio of 9.5·10−2, can be justified on the basis of crystallographic results, which pinpoints the more significant distortion of the Cr3+ coordination geometry in the trimer compared to the monomer. Furthermore, both EPR and magnetometry measurements highlighted a small coupling interaction between the central spin and the peripheral units, primarily relying on a dipolar coupling interaction between paramagnetic centers. This result is in contrast with what has been observed for Cu-Cr macromolecular cages obtained with the same [Cr(pybd)3] building block, for which a ferromagnetic coupling interaction of about 10−1 cm−1 has been reported [30,35]. However, while the Cu2+ ions included within cages are coordinated on equatorial positions by pyridyl units of the pybd− ligand, the same units point toward the axial position of the pentacoordinate [Cu(cyclen)]2+ units present in our cluster. In this respect, we could have expected a lower super-exchange interaction between the Cr3+ and Cu2+ ions in our cluster than in that of ref. [30] since here, the pyridyl unit points towards the doubly occupied of the copper ion, rather than the singly occupied . Unfortunately, the change in the orbital involved in the interaction reduces the interaction to a value smaller than the dipolar ones that is not detectable within the employed experimental conditions.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13060901/s1, more details about syntheses and employed chemicals; Figure S1: UV–Vis absorption spectra of equimolar solutions (0.25 mM) of all the investigated complexes; Figure S2: FT-IR spectra collected on [Cr(pybd)3]. [Cu(cyclen)BF4](BF4), and {Cr(dypb)3[Cu(cyclen)]}(BF4)2 powder samples; Figure S3: View of the crystal unit cell of {Cr(dypb)3[Cu(cyclen)]}(BF4)2 along with the a, b, and c crystallographic axes; Figure S4: View along the c axis of a 2 × 2 × 1.5 cell highlighting the porous nature of crystalline {Cr(dypb)3[Cu(cyclen)]}(BF4)2; Figure S5: (a) Generic view of the first coordination sphere of the Cr3+ ion reporting the six values of Cr−O distances. (b) Generic view of the first coordination sphere of the Cr3+ io reporting the three values of O−Cr−O angles; Figure S6: Generic view of the two Cu(cyclen) moieties highlighting the square-pyramidal coordination environment of the Cu2+ ion; Figure S7: X-band CW EPR spectrum of a {Cr(dypb)3[Cu(cyclen)]}(BF4)2 powder sample collected at 30 K.
Author Contributions
Conceptualization, F.S. and R.S.; formal analysis, F.S., L.C., G.A.T., L.S. and R.S.; investigation, F.S. and L.C.; data curation, F.S., L.C., G.A.T., L.S. and R.S.; writing—original draft preparation, F.S. and R.S.; writing—review and editing, F.S., L.C., G.A.T., L.S. and R.S.; supervision, L.S., G.A.T. and R.S.; project administration, R.S.; funding acquisition, R.S. All authors have read and agreed to the published version of the manuscript.
Funding
The support of EC through the EU Commission through the FETOPEN project FATMOLS (GA 862893) and Italian MIUR through Progetto Dipartimenti di Eccellenza 2023–2027 (CUP B97G22000740001 - DICUS 2.0) to the Department of Chemistry “Ugo Schiff” of the University of Florence and PNRR MUR project PE0000023-NQSTI is acknowledged.
Data Availability Statement
Data are available from authors upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Heinrich, A.J.; Oliver, W.D.; Vandersypen, L.M.K.; Ardavan, A.; Sessoli, R.; Loss, D.; Jayich, A.B.; Fernandez-Rossier, J.; Laucht, A.; Morello, A. Quantum-Coherent Nanoscience. Nat. Nanotechnol. 2021, 16, 1318–1329. [Google Scholar] [CrossRef]
- Atzori, M.; Sessoli, R. The Second Quantum Revolution: Role and Challenges of Molecular Chemistry. J. Am. Chem. Soc. 2019, 141, 11339–11352. [Google Scholar] [CrossRef] [PubMed]
- Gaita-Ariño, A.; Luis, F.; Hill, S.; Coronado, E. Molecular Spins for Quantum Computation. Nat. Chem. 2019, 11, 301–309. [Google Scholar] [CrossRef]
- Bonizzoni, C.; Ghirri, A.; Santanni, F.; Atzori, M.; Sorace, L.; Sessoli, R.; Affronte, M. Storage and Retrieval of Microwave Pulses with Molecular Spin Ensembles. npj Quantum Inf. 2020, 6, 68. [Google Scholar] [CrossRef]
- Bonizzoni, C.; Tincani, M.; Santanni, F.; Affronte, M. Machine-Learning-Assisted Manipulation and Readout of Molecular Spin Qubits. Phys. Rev. Appl. 2022, 18, 064074. [Google Scholar] [CrossRef]
- Chicco, S.; Chiesa, A.; Allodi, G.; Garlatti, E.; Atzori, M.; Sorace, L.; De Renzi, R.; Sessoli, R.; Carretta, S. Controlled Coherent Dynamics of [VO(TPP)], a Prototype Molecular Nuclear Qudit with an Electronic Ancilla. Chem. Sci. 2021, 12, 12046–12055. [Google Scholar] [CrossRef]
- Chizzini, M.; Crippa, L.; Zaccardi, L.; Macaluso, E.; Carretta, S.; Chiesa, A.; Santini, P. Quantum Error Correction with Molecular Spin Qudits. Phys. Chem. Chem. Phys. 2022, 24, 20030–20039. [Google Scholar] [CrossRef]
- Hazra, S.; Bhattacharjee, A.; Chand, M.; Salunkhe, K.V.; Gopalakrishnan, S.; Patankar, M.P.; Vijay, R. Ring-Resonator-Based Coupling Architecture for Enhanced Connectivity in a Superconducting Multiqubit Network. Phys. Rev. Appl. 2021, 16, 024018. [Google Scholar] [CrossRef]
- Nielsen, M.A.; Chuang, I.L. Quantum Computation and Quantum Information; Cambridge University Press: Cambridge, UK, 2010; ISBN 9780511976667. [Google Scholar]
- Lockyer, S.J.; Chiesa, A.; Timco, G.A.; McInnes, E.J.L.; Bennett, T.S.; Vitorica-Yrezebal, I.J.; Carretta, S.; Winpenny, R.E.P. Targeting Molecular Quantum Memory with Embedded Error Correction. Chem. Sci. 2021, 12, 9104–9113. [Google Scholar] [CrossRef]
- Ranieri, D.; Santanni, F.; Privitera, A.; Albino, A.; Salvadori, E.; Chiesa, M.; Totti, F.; Sorace, L.; Sessoli, R. An Exchange Coupled Meso—Meso Linked Vanadyl Porphyrin Dimer for Quantum Information Processing. Chem. Sci. 2023, 14, 61–69. [Google Scholar] [CrossRef]
- Ferrando-Soria, J.; Moreno Pineda, E.; Chiesa, A.; Fernandez, A.; Magee, S.A.; Carretta, S.; Santini, P.; Vitorica-Yrezabal, I.J.; Tuna, F.; Timco, G.A.; et al. A Modular Design of Molecular Qubits to Implement Universal Quantum Gates. Nat. Commun. 2016, 7, 11377. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Chiesa, A.; Morra, E.; Chiesa, M.; Sorace, L.; Carretta, S.; Sessoli, R. A Two-Qubit Molecular Architecture for Electron-Mediated Nuclear Quantum Simulation. Chem. Sci. 2018, 9, 6183–6192. [Google Scholar] [CrossRef] [PubMed]
- Borilovic, I.; Alonso, P.J.; Roubeau, O.; Aromí, G. A Bis-Vanadyl Coordination Complex as a 2-Qubit Quantum Gate. Chem. Commun. 2020, 56, 3139–3142. [Google Scholar] [CrossRef] [PubMed]
- Troiani, F.; Affronte, M. Molecular Spins for Quantum Information Technologies. Chem. Soc. Rev. 2011, 40, 3119. [Google Scholar] [CrossRef]
- Nakazawa, S.; Nishida, S.; Ise, T.; Yoshino, T.; Mori, N.; Rahimi, R.D.; Sato, K.; Morita, Y.; Toyota, K.; Shiomi, D.; et al. A Synthetic Two-Spin Quantum Bit: G -Engineered Exchange-Coupled Biradical Designed for Controlled-NOT Gate Operations. Angew. Chem. Int. Ed. 2012, 51, 9860–9864. [Google Scholar] [CrossRef]
- Maniaki, D.; Garay-Ruiz, D.; Barrios, L.A.; Martins, D.O.T.A.; Aguilà, D.; Tuna, F.; Reta, D.; Roubeau, O.; Bo, C.; Aromí, G. Unparalleled Selectivity and Electronic Structure of Heterometallic [LnLn′Ln] Molecules as 3-Qubit Quantum Gates. Chem. Sci. 2022, 13, 5574–5581. [Google Scholar] [CrossRef]
- Macaluso, E.; Rubín, M.; Aguilà, D.; Chiesa, A.; Barrios, L.A.; Martínez, J.I.; Alonso, P.J.; Roubeau, O.; Luis, F.; Aromí, G.; et al. A Heterometallic [LnLn′Ln] Lanthanide Complex as a Qubit with Embedded Quantum Error Correction. Chem. Sci. 2020, 11, 10337–10343. [Google Scholar] [CrossRef]
- Aguilà, D.; Barrios, L.A.; Velasco, V.; Roubeau, O.; Repollés, A.; Alonso, P.J.; Sesé, J.; Teat, S.J.; Luis, F.; Aromí, G. Heterodimetallic [LnLn′] Lanthanide Complexes: Toward a Chemical Design of Two-Qubit Molecular Spin Quantum Gates. J. Am. Chem. Soc. 2014, 136, 14215–14222. [Google Scholar] [CrossRef]
- Mata, J.A.; Hahn, F.E.; Peris, E. Heterometallic Complexes, Tandem Catalysis and Catalytic Cooperativity. Chem. Sci. 2014, 5, 1723–1732. [Google Scholar] [CrossRef]
- Maldonado, C.S.; de la Rosa, J.R.; Lucio-Ortiz, C.J.; Sandoval-Rangel, L.; Martínez-Vargas, D.X.; Sánchez, R.A.L. Applications of Heterometallic Complexes in Catalysis. In Direct Synthesis of Metal Complexes; Elsevier: Amsterdam, The Netherlands, 2018; pp. 369–377. [Google Scholar]
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic Catalysis Based on Heterometallic Complexes and Clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Gao, W.-X.; Lin, L.; Jin, G.-X. Recent Advances in the Construction and Applications of Heterometallic Macrocycles and Cages. Coord. Chem. Rev. 2017, 344, 323–344. [Google Scholar] [CrossRef]
- Uber, J.S.; Estrader, M.; Garcia, J.; Lloyd-Williams, P.; Sadurní, A.; Dengler, D.; van Slageren, J.; Chilton, N.F.; Roubeau, O.; Teat, S.J.; et al. Molecules Designed to Contain Two Weakly Coupled Spins with a Photoswitchable Spacer. Chem. Eur. J. 2017, 23, 13648–13659. [Google Scholar] [CrossRef]
- Wernsdorfer, W.; Mailly, D.; Timco, G.A.; Winpenny, R.E.P. Resonant Photon Absorption and Hole Burning in {Cr7Ni} Antiferromagnetic Rings. Phys. Rev. B 2005, 72, 060409. [Google Scholar] [CrossRef]
- Dey, A.; Acharya, J.; Chandrasekhar, V. Heterometallic 3d–4f Complexes as Single-Molecule Magnets. Chem. Asian J. 2019, 14, 4433–4453. [Google Scholar] [CrossRef]
- Larsen, E.M.H.; Bonde, N.A.; Weihe, H.; Ollivier, J.; Vosch, T.; Lohmiller, T.; Holldack, K.; Schnegg, A.; Perfetti, M.; Bendix, J. Experimental Assignment of Long-Range Magnetic Communication through Pd & Pt Metallophilic Contacts. Chem. Sci. 2023, 14, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Funes, A.V.; Perfetti, M.; Kern, M.; Rußegger, N.; Carrella, L.; Rentschler, E.; Slageren, J.; Alborés, P. Single Molecule Magnet Features in the Butterfly [CoIII2LnIII 2] Pivalate Family with Alcohol-Amine Ligands. Eur. J. Inorg. Chem. 2021, 2021, 3191–3210. [Google Scholar] [CrossRef]
- Belli Dell’Amico, D.; Ciattini, S.; Fioravanti, L.; Labella, L.; Marchetti, F.; Mattei, C.A.; Samaritani, S. The Heterotopic Divergent Ligand N-Oxide-4,4′-Bipyridine (BipyMO) as Directing-Agent in the Synthesis of Oligo- or Polynuclear Heterometallic Complexes. Polyhedron 2018, 139, 107–115. [Google Scholar] [CrossRef]
- Sanz, S.; O’Connor, H.M.; Pineda, E.M.; Pedersen, K.S.; Nichol, G.S.; Mønsted, O.; Weihe, H.; Piligkos, S.; McInnes, E.J.L.; Lusby, P.J.; et al. [CrIII8MII6]12+ Coordination Cubes (MII = Cu, Co). Angew. Chem. Int. Ed. 2015, 54, 6761–6764. [Google Scholar] [CrossRef]
- Wise, M.D.; Holstein, J.J.; Pattison, P.; Besnard, C.; Solari, E.; Scopelliti, R.; Bricogne, G.; Severin, K. Large, Heterometallic Coordination Cages Based on Ditopic Metallo-Ligands with 3-Pyridyl Donor Groups. Chem. Sci. 2015, 6, 1004–1010. [Google Scholar] [CrossRef]
- Timco, G.A.; Carretta, S.; Troiani, F.; Tuna, F.; Pritchard, R.J.; Muryn, C.A.; McInnes, E.J.L.; Ghirri, A.; Candini, A.; Santini, P.; et al. Engineering the Coupling between Molecular Spin Qubits by Coordination Chemistry. Nat. Nanotechnol. 2009, 4, 173–178. [Google Scholar] [CrossRef]
- Durot, S.; Taesch, J.; Heitz, V. Multiporphyrinic Cages: Architectures and Functions. Chem. Rev. 2014, 114, 8542–8578. [Google Scholar] [CrossRef]
- Pilgrim, B.S.; Champness, N.R. Metal-Organic Frameworks and Metal-Organic Cages—A Perspective. Chempluschem 2020, 85, 1842–1856. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, H.M.; Sanz, S.; Pitak, M.B.; Coles, S.J.; Nichol, G.S.; Piligkos, S.; Lusby, P.J.; Brechin, E.K. [CrIII8MII6]N+ (MII = Cu, Co) Face-Centred, Metallosupramolecular Cubes. CrystEngComm 2016, 18, 4914–4920. [Google Scholar] [CrossRef]
- O’Connor, H.M.; Sanz, S.; Scott, A.J.; Pitak, M.B.; Klooster, W.T.; Coles, S.J.; Chilton, N.F.; McInnes, E.J.L.; Lusby, P.J.; Weihe, H.; et al. [CrIII8MII6]N+ Heterometallic Coordination Cubes. Molecules 2021, 26, 757. [Google Scholar] [CrossRef] [PubMed]
- Sanz, S.; O’Connor, H.M.; Martí-Centelles, V.; Comar, P.; Pitak, M.B.; Coles, S.J.; Lorusso, G.; Palacios, E.; Evangelisti, M.; Baldansuren, A.; et al. [MIII2MII3]N+ Trigonal Bipyramidal Cages Based on Diamagnetic and Paramagnetic Metalloligands. Chem. Sci. 2017, 8, 5526–5535. [Google Scholar] [CrossRef] [PubMed]
- Bruker. Bruker APEX 2; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A Kit of Tools for Phasing Crystal Structures from Powder Data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An Optimization Program Integrating Computer Algebra and Crystallographic Objects Written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Bain, G.A.; Berry, J.F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532. [Google Scholar] [CrossRef]
- Glidewell, C. Metal Acetylacetonate Complexes: Preparation and Characterization. In Inorganic Experiments; Woollins, J.E., Ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Faggi, E.; Gavara, R.; Bolte, M.; Fajarí, L.; Juliá, L.; Rodríguez, L.; Alfonso, I. Copper(Ii) Complexes of Macrocyclic and Open-Chain Pseudopeptidic Ligands: Synthesis, Characterization and Interaction with Dicarboxylates. Dalton Trans. 2015, 44, 12700–12710. [Google Scholar] [CrossRef]
- Flores-Rojas, G.G.; Ruiu, A.; Vonlanthen, M.; Rojas-Montoya, S.M.; Martínez-Serrano, R.D.; Morales-Morales, D.; Rivera, E. Synthesis and Characterization of Cyclen Cored Photoactive Star Compounds and Their Cu(I) and Cu(II) Complexes. Effect of the Valence and Ligand Size on Their Molar Extinction Coefficient. Inorg. Chim. Acta 2020, 513, 119927. [Google Scholar] [CrossRef]
- Tosato, M.; Dalla Tiezza, M.; May, N.V.; Isse, A.A.; Nardella, S.; Orian, L.; Verona, M.; Vaccarin, C.; Alker, A.; Mäcke, H.; et al. Copper Coordination Chemistry of Sulfur Pendant Cyclen Derivatives: An Attempt to Hinder the Reductive-Induced Demetalation in 64/67 Cu Radiopharmaceuticals. Inorg. Chem. 2021, 60, 11530–11547. [Google Scholar] [CrossRef] [PubMed]
- El Ghachtouli, S.; Cadiou, C.; Déchamps-Olivier, I.; Chuburu, F.; Aplincourt, M.; Roisnel, T. (Cyclen– and Cyclam–Pyridine)Copper Complexes: The Role of the Pyridine Moiety in CuII and CuI Stabilisation. Eur. J. Inorg. Chem. 2006, 2006, 3472–3481. [Google Scholar] [CrossRef]
- Lacerda, S.; Campello, M.P.; Santos, I.C.; Santos, I.; Delgado, R. Study of the Cyclen Derivative 2-[1,4,7,10-Tetraazacyclododecan-1-Yl]-Ethanethiol and Its Complexation Behaviour towards d-Transition Metal Ions. Polyhedron 2007, 26, 3763–3773. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, R.P.; Di Bilio, A.J.; Riggi, F. EPR Investigation of Chromium(III) Complexes: Analysis of Their Frozen Solution and Magnetically Dilute Powder Spectra. Chem. Phys. 1991, 151, 323–333. [Google Scholar] [CrossRef]
- Elbers, G.; Remme, S.; Lehmann, G. EPR of Chromium(3+) in Tris(Acetylacetonato)Gallium(III) Single Crystals. Inorg. Chem. 1986, 25, 896–897. [Google Scholar] [CrossRef]
- Jeschke, G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [Google Scholar] [CrossRef]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. PHI: A Powerful New Program for the Analysis of Anisotropic Monomeric and Exchange-Coupled Polynuclear d- and f -Block Complexes. J. Comput. Chem. 2013, 34, 1164–1175. [Google Scholar] [CrossRef]
Scheme 1.
Synthetic schemes showing the general strategies employed for the obtaining of reported compounds. (a) Synthesis of Hpybd and [Cr(pybd)3]. (b) Synthesis of [Cu(cyclen)BF4](BF4). (c) Synthesis of the macromolecular adduct {Cr(pybd)3[Cu(cyclen)]2}(BF4)4.
Scheme 1.
Synthetic schemes showing the general strategies employed for the obtaining of reported compounds. (a) Synthesis of Hpybd and [Cr(pybd)3]. (b) Synthesis of [Cu(cyclen)BF4](BF4). (c) Synthesis of the macromolecular adduct {Cr(pybd)3[Cu(cyclen)]2}(BF4)4.

Figure 1.
View of the molecular structure of {Cr(pybd)3[Cu(cyclen)]2}(BF4)4. Hydrogen atoms are omitted for clarity. Color code: Cr = green; Cu = copper; F = yellow/green; O = red; N = blue; C = grey; B = pink.
Figure 1.
View of the molecular structure of {Cr(pybd)3[Cu(cyclen)]2}(BF4)4. Hydrogen atoms are omitted for clarity. Color code: Cr = green; Cu = copper; F = yellow/green; O = red; N = blue; C = grey; B = pink.

Figure 2.
Comparison between experimental and simulated PXRD patterns obtained for microcrystalline {[Cr(pybd)3][Cu(cyclen)]2}(BF4)4.
Figure 2.
Comparison between experimental and simulated PXRD patterns obtained for microcrystalline {[Cr(pybd)3][Cu(cyclen)]2}(BF4)4.

Figure 3.
UV–Vis absorption spectra collected on 2.5 × 10−3 M MeOH solutions of [Cu(cyclen)]2+, [Cu(cyclen)]2+ + 100 eq of pyridine (py), and {Cr(pybd)3[Cu(cyclen)]2}4+.
Figure 3.
UV–Vis absorption spectra collected on 2.5 × 10−3 M MeOH solutions of [Cu(cyclen)]2+, [Cu(cyclen)]2+ + 100 eq of pyridine (py), and {Cr(pybd)3[Cu(cyclen)]2}4+.

Figure 4.
(a) Experimental and simulated X-band EPR spectra of [Cr(pybd)3] 0.5 mM in EtOH/MeOH 4:1; (b) Experimental and simulated X-band EPR spectra of [Cu(cyclen)]2+ 0.5 mM in EtOH/MeOH 4:1; (c) Experimental and simulated X-band EPR spectra of {Cr(pybd)3[Cu(cyclen)]2}4+ 0.5 mM in EtOH/MeOH 4:1.
Figure 4.
(a) Experimental and simulated X-band EPR spectra of [Cr(pybd)3] 0.5 mM in EtOH/MeOH 4:1; (b) Experimental and simulated X-band EPR spectra of [Cu(cyclen)]2+ 0.5 mM in EtOH/MeOH 4:1; (c) Experimental and simulated X-band EPR spectra of {Cr(pybd)3[Cu(cyclen)]2}4+ 0.5 mM in EtOH/MeOH 4:1.

Figure 5.
(a) χT vs. T plot obtained for {[Cr(pybd)3][Cu(cyclen)]2}(BF4)4 at B = 0.1 T. An asterisk is placed in correspondence of a signal anomaly due to oxygen impurity within the measurement chamber; (b) M vs. BT−1 plot obtained for {[Cr(pybd)3][Cu(cyclen)]2}(BF4)4 at T = 2, 4, and 8 K. Solid lines within the plot correspond to the fitting outcomes.
Figure 5.
(a) χT vs. T plot obtained for {[Cr(pybd)3][Cu(cyclen)]2}(BF4)4 at B = 0.1 T. An asterisk is placed in correspondence of a signal anomaly due to oxygen impurity within the measurement chamber; (b) M vs. BT−1 plot obtained for {[Cr(pybd)3][Cu(cyclen)]2}(BF4)4 at T = 2, 4, and 8 K. Solid lines within the plot correspond to the fitting outcomes.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystallographic data and refinement parameters.
| Molecular Formula | C42CrCu2N12O6,4(BF4) |
|---|---|
| Mr | 1294.8 |
| T(K) | RT |
| λ (Å) | 1.54056 |
| crystal system, space group | triclinic, P-1 |
| unit cell dimensions (Å, °) | a = 12.5014(5), b = 15.2007(8), c = 18.4058(8) α = 105.05(4), β = 90.228(5), γ = 95.988(3) |
| volume (Å3) | 3357.5(3) |
| Z, Dx (g/cm−3) | 2, 1.325 |
| μ (mm−1) | 0.906 |
| Rwp (%) | 4.66 |
| GOOFs | 1.52 |
Table 2.
List of best-simulation parameters extracted for X-band spectra of 0.5 mM frozen solutions in EtOH/MeOH 4:1. In the case of the Cr-Cu adduct, we did not consider the relative orientation of magnetic reference frameworks of each site.
Table 2.
List of best-simulation parameters extracted for X-band spectra of 0.5 mM frozen solutions in EtOH/MeOH 4:1. In the case of the Cr-Cu adduct, we did not consider the relative orientation of magnetic reference frameworks of each site.
| Parameters | [Cr(pybd)3] | [Cu(cyclen)BF4](BF4) | {Cr(pybd)3[Cu(cyclen)]}(BF4)4 | |
|---|---|---|---|---|
| gx | 1.97(1) | 2.047(2) | 1.97(1) a | 2.050(2) b |
| gy | 1.97(1) | 2.047(2) | 1.97(1) a | 2.050(2) b |
| gz | 1.97(1) | 2.204(2) | 1.97(1) a | 2.206(2) b |
| D (cm−1) | 0.55(5) | - | 0.21(1) a | - |
| E (cm−1) | 0.025(1) | - | 0.02(1) a | - |
| Ax (10−4 cm−1) | - | 17(2) | - | 18(2) b |
| Ay (10−4 cm−1) | - | 17(2) | - | 18(2) b |
| Az (10−4 cm−1) | - | 182(2) | - | 184(2) b |
a = set of parameters extracted for the Cr3+ ion; b = set of parameters extracted for the two equivalent Cu2+ sites.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Santanni, F.; Chelazzi, L.; Sorace, L.; Timco, G.A.; Sessoli, R. Structural and Magnetic Properties of the {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 Heteronuclear Complex. Crystals 2023, 13, 901. https://doi.org/10.3390/cryst13060901
AMA Style
Santanni F, Chelazzi L, Sorace L, Timco GA, Sessoli R. Structural and Magnetic Properties of the {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 Heteronuclear Complex. Crystals. 2023; 13(6):901. https://doi.org/10.3390/cryst13060901
Chicago/Turabian StyleSantanni, Fabio, Laura Chelazzi, Lorenzo Sorace, Grigore A. Timco, and Roberta Sessoli. 2023. "Structural and Magnetic Properties of the {Cr(pybd)3[Cu(cyclen)]2}(BF4)4 Heteronuclear Complex" Crystals 13, no. 6: 901. https://doi.org/10.3390/cryst13060901
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.