1. Introduction
From a fundamental or basic science perspective, the molecular structure of a biomacromolecule is invaluable when it comes to deciphering and understanding mechanisms that are integral to life [
1]. Applications follow logically, which can include drug design and development. Ideally, atomic resolution structures are required. The focus of the current study is on membrane proteins, which represent approximately one-third of most genomes and are targets for close to half the drugs on the market [
2].
The experimental method most used to date for the high-resolution structure determination of membrane proteins is macromolecular X-ray crystallography (MX) (
https://blanco.biomol.uci.edu/mpstruc/, accessed on 7 August 2023). Single-particle cryogenic electron microscopy (cryo-EM) [
3] and computational methods such as AlphaFold [
4] increasingly play a role. MX is particularly attractive in that it has provided the highest-resolution structures to date, and it works with big and small proteins alike and with complexes. However, MX comes with its challenges, not least of which is the need for crystals. There is also the fact that to solve a structure by MX, both diffraction intensity and phase information are required [
5,
6]. The experimentally collected MX diffraction data provide intensity directly. Phases must be obtained indirectly, either using a model by molecular replacement (MR) or using heavy atoms or anomalous scatterers by isomorphous replacement and anomalous dispersion. To date, single-wavelength anomalous diffraction (SAD) phasing is the most frequently used method [
7].
Heavy atoms for use in phasing can be present intrinsically as part of the folded macromolecule as functional metal ions and metal-containing cofactors. As such, they can be regarded as enabling a form of native phasing. Heavy atoms can also be incorporated into the crystal as adventitious ions and heavy atom-containing compounds. Labelling protocols include co-crystallisation and crystal swishing and soaking [
8,
9]. It is also possible to replace the natural amino acids, methionine (Met) and cysteine (Cys), with seleno-labelled methionine (Se-Met) and cysteine (Se-Cys), which are the most used experimental phasing methods [
10]. Increasingly, the anomalous signal from the sulfur in methionine and cysteine is being exploited for label-free, native phasing [
11].
Crystals of membrane proteins are grown predominantly by either the in surfo or bilayer methods [
12]. The former approach was the first and the most successful crystallisation method [
13]. It uses surfactants to solubilise the membrane protein as a micellarised detergent-lipid-protein complex, which lends itself to crystal growth by methods commonly used with soluble proteins. Bilayer methods involve the use of bicelles or a lipid cubic mesophase as the medium in which crystals grow. The focus here is on the latter so-called in meso method, which has proven to be particularly useful with the medically important G protein-coupled receptors (GPCRs) [
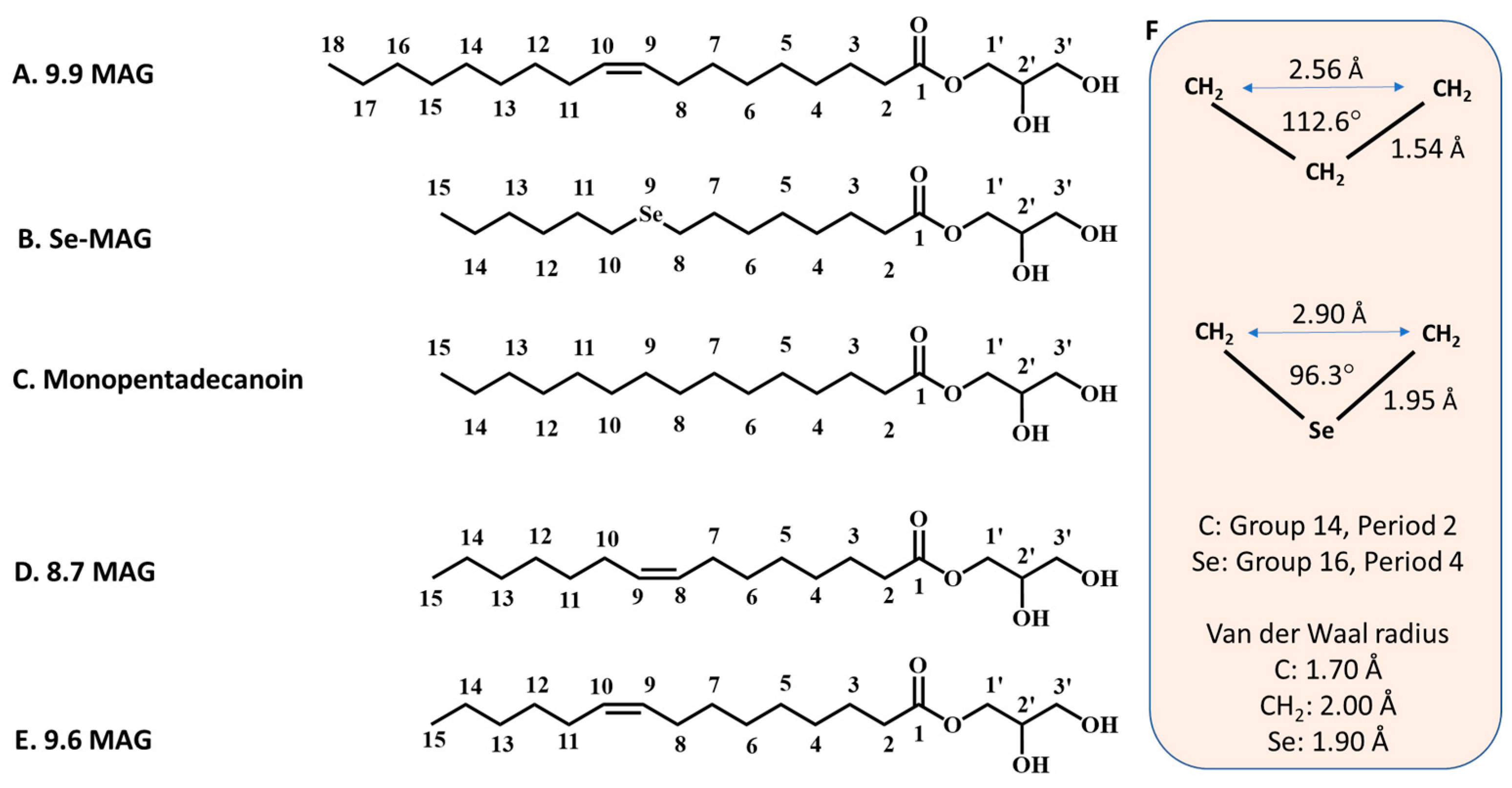
14]. The lipids most used to create the bilayered membrane of the cubic mesophase, in which the protein resides prior to crystallisation, are monoacylglycerols (MAGs) (
Figure 1). Monoolein (9.9 MAG) is by far the most popular MAG in this application. [
Cis monoenoic MAGs are described in shorthand using the N.T notation, where N and T refer, respectively, to the number of carbon atoms in the fatty acyl chain on either side of the
cis-olefin bond. Thus, for example, 8.7 MAG corresponds to monopentadecenoin, where the fatty acid, pentadecenoic acid, is (N + T = 8 + 7 =) 15 carbon atoms long with a
cis double bond between carbon atom numbers 8 and 9.] Additive lipids can be included with monoolein in the mesophase to suit the needs of the target protein and for optimising crystallisation [
15]. In the case of GPCRs, cholesterol is a favourite additive lipid [
16,
17]. To date, phasing of structures generated with in meso-grown crystals has been obtained by MR and experimental methods that include single-wavelength anomalous diffraction (SAD), multi-wavelength anomalous diffraction (MAD), multi-wavelength isomorphous replacement with anomalous scattering (MIRAS), single isomorphous replacement (SIR), and single isomorphous replacement with anomalous scattering (SIRAS). Most of the in meso-based structure determination work has been performed at cryogenic temperatures using synchrotron radiation. However, some have been carried out at ambient temperatures with synchrotron [
18] and free-electron laser X-ray sources [
19,
20,
21,
22].
An interesting feature of the in meso method is that the lipids used to generate the hosting mesophase often end up as part of the crystal structure. Given the proposed mechanism of in meso crystallisation [
23], this makes sense. The bound lipid generally recapitulates the bilayer in which crystallisation occurs and the membrane from which the protein derived. The mesophase lipid can also be found in hydrophobic grooves in the protein that include the substrate, the product, and ligand-binding pockets [
24,
25,
26]. In such situations, the hosting lipids act as surrogates to demark functional parts of the protein. Lipids, such as cholesterol, doped into the mesophase to facilitate crystallisation and stabilise the protein can also appear as physiologically relevant bound lipids in the final structures. GPCRs are a notable case in point [
15,
16,
17].
Given the frequency with which mesophase MAG lipids find their way into the final crystal structure, we reasoned that a heavy atom-labelled MAG, if it co-crystallised with the protein, might be used for phasing purposes. An attractive feature of this method would be that all such proteins, which have a membrane domain, should associate with and possibly be labelled by the derivatised MAG. To test the proposal, a MAG analogue, in which one of the methylenes in its fatty acyl chain was replaced by selenium, was investigated. Henceforth, the seleno-labelled lipid is referred to as Se-MAG. The substitution was not expected to dramatically change the properties of the lipid given the similarities in geometries and electronic properties of the relevant moieties, -CH
2- and -Se- (
Figure 1).
In this study, a 50/50 mix of monoolein and Se-MAG proved optimal for cubic phase formation, and for in meso crystallisation, phasing and structure determination of two test membrane protein types. One was a bacterial outer membrane β-barrel protein, the alginate transporter, AlgE [
27]. The other was an inner membrane protein, the lipoprotein
N-acyltransferase, Lnt, which is approximately equal parts α-helix and β-strand [
25]. Both AlgE and Lnt structures were solved originally by the Se-SAD method [
25,
28]. In the current study, structures were determined to a resolution of 1.7 Å for AlgE and 1.9 Å for Lnt.
Given the ease with which the monoolein/Se-MAG lipid mixture can be prepared for use in crystallisation trials, the routine inclusion of this novel heavy atom-labelled MAG as a component of the hosting lipid may prove useful as a general strategy for the experimental phasing part of crystallographic structure determination of membrane proteins that crystallise via the in meso method.
2. Materials and Methods
2.1. Materials
Ammonium sulfate, chloramphenicol, ethylenediaminetetraacetic acid (EDTA), 4-(2-Hydroxyethyl)piperazineethane-1-sulfonic acid (HEPES), lithium sulfate, 2-(N-morpholino)ethanesulfonic acid (MES), 2-methyl-2,4-pentanediol (MPD), polyethylene glycol 400 (PEG400), phenylmethylsulfonyl fluoride (PMSF), tris(hydroxymethyl)aminomethane (tris), sodium hydroxide, Roche protease inhibitor tablets, sodium thiocyanate, potassium thiocyanate, sodium citrate, 50 kDa MWCO Amicon Ultra 15 concentrators, n-octyl tetraoxyethylene (C8E4), 1-bromohexane, 8-bromooctanoic acid, solketal, 4-dimethylaminopyridine, N,N′-dicyclohexylcarbodiimide, and urea (>99% purity) were purchased from Sigma-Aldrich. Glycerol, imidazole, isopropyl β-D-1-thiogalactopyranoside (IPTG), kanamycin, sodium chloride (NaCl), and Luria broth (LB) powder were obtained from ThermoFisher Scientific. Nickel nitrilotriacetic acid (Ni-NTA) Superflow resin was purchased from Qiagen. The ӒKTA fast protein liquid chromatograph (FPLC) and HiLoad 16/60 Superdex 200 column were obtained from GE Healthcare. The detergents 2,2-didecylpropane-1,3-bis-β-D-maltopyranoside (LMNG) and n-decyl-β-D-maltopyranoside (DM) were from Anatrace. Selenium powder, hydrazine monohydrate, sodium hydroxide, and hydrochloric acid were purchased from TCI Chemicals. All solvents used for Se-MAG synthesis and purification were purchased from local suppliers and were of the highest purity grade available. Water (resistivity 18.2 MΩ·cm) was purified by a Milli-Q Elix Water Purification System from Millipore. Gastight syringes (100 μL) were obtained from the Hamilton Company. The Xantus liquid and mesophase handling robot was from SIAS. Glass crystallisation plates were purchased from Marienfeld. Harvesting loops were from MiTeGen. Double-sided 140 μm thick spacers were obtained from Saunders Corp. The CX100 dry shipper and shipping case were from Molecular Dimensions.
2.2. Se-MAG Synthesis, Purification and Characterisation
To prepare Se-MAG
5, the requisite selena fatty acid, 8-(hexylselanyl)octanoic acid (9-selena pentadecanoic acid)
2, was synthesised, reacted with solketal
3, and the resulting acetonide
4 was deprotected. The stepwise synthesis is outlined in
Scheme 1.
Proton and carbon nuclear magnetic resonance (NMR) spectra were recorded on Varian 400/500 and 101 MHz instruments, respectively, with tetramethylsilane as the internal standard. Mass spectra (MS) were recorded by using electron spray ionisation on a Waters e2695 Separator’s module (Waters, Milford, MA, USA) mass spectrometer. Infrared (IR) spectra were recorded using a Perkin–Elmer–Fourier transform–infrared (FT)–IR Spectrum BX instrument (Model Spectrum BX; Shelton, CT, USA). Column chromatographic purification was carried out by silica gel (60–120 mesh) column chromatography. Reaction progress was monitored by thin-layer chromatography (TLC), which was performed on precoated silica gel 60 F254 from Merck (Darmstadt, Germany).
2.2.1. Synthesis of 8-(Hexylselanyl)octanoic Acid (9-Selena Pentadecanoic Acid) 2
The selenium-containing fatty acid
3 used for Se-MAG production was synthesised based on a reported method with slight modifications for the chosen chain length [
29]. A mixture of selenium (1 g, 12.7 mmol), sodium hydroxide (0.76 g, 19 mmol), and hydrazine hydrate (0.64 g, 13 mmol) in methanol (20 mL) was stirred for 6 h at 27 °C. To this mixture, 1-bromohexane
1 (2.09 g, 12.7 mmol) was added. After the reaction mixture turned yellow, sodium hydroxide pellets (1 g, 25.4 mmol) were added followed by hydrazine hydrate (0.76 g, 19.1 mmol), and the reaction mixture was stirred for another 3 h at 27 °C. 8-Bromoctanoic acid (1.43 g, 6.4 mmol) in methanol (10 mL) was added, and the total reaction mixture was stirred at 50 °C for 8 h. The temperature of the reaction was allowed to reach room temperature (27 °C) and the cooled mixture was poured into water (100 mL) and extracted with diethyl ether (2 × 30 mL). The aqueous layer was acidified with dilute HCl (2 M, 30 mL) and extracted with diethyl ether (2× 50 mL). The ether extract was washed with brine and water and dried over anhydrous sodium sulphate. Evaporation of the solvent gave crude selena alkanoic acid. Chromatography on silica gel with hexane-diethyl ether (96:4
v/
v) as the eluent gave pure 9-selena pentadecanoic acid
2 in 57% yield (1.12 g).
1H NMR (400 MHz (ppm), CDCl3): δ = 0.89 (t, 3H, J = 7.01 Hz); 1.32 (m, 16H); 1.6 (m, 2 H); 2.3 (t, 2H, J = 7.47 Hz); 2.56 (m, 4H).
MS: 307 [M-H].
Esterification of the selena fatty acid
2 with solketal
3 for the synthesis of acetonide
4 followed by deprotection to obtain the Se-MAG
5 was carried out as previously described [
30,
31].
2.2.2. Synthesis of (2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 8-(hexylselanyl)octanoate 4
In a 100 mL round-bottomed flask, solketal 3 (0.82 mg, 6.2 mmol) was dissolved in dichloromethane (15 mL) and DMAP (0.15 g, 1.24 mmol) was added at 0 °C. A mixture of DCC (0.71 g, 5.16 mmol) and 9-selena pentadecanoic acid 2 (1.06 g, 15.16 mmol) was added and dissolved in 20 mL dichloromethane. The ice bath was removed, and the reaction was stirred at room temperature (27 °C) for 24 h under nitrogen. The reaction mixture was filtered to remove the precipitate. The filtrate containing the desired product was washed with saturated aqueous sodium bicarbonate (50 mL) and water (3× 75 mL) and the organic layer was dried over anhydrous sodium sulphate and concentrated by rotary evaporation to obtain the crude product. The crude product was purified by silica gel (60–120 mesh) column chromatography using a hexane and ethyl acetate gradient to obtain pure acetonide 4 at a 70% yield (1.01 g).
1H NMR (400 MHz (ppm), CDCl3): δ = 0.88 (t, 3H, J = 7.01 Hz); 1.31 (m, 16H); 1.37 (s, 3H); 1.44 (s, 3H); 1.64 (m, 2H) 2.34 (t, 2H, J = 7.7 Hz); 2.54 (m, 4H); 3.74 (m, 1H); 4.09 (m, 2H); 4.16 (m, 1H); 4.31 (m, 1H).
13C NMR (100 MHz (ppm), CDCl3): δ = 13.68, 22.19, 23.51, 23.67, 24.44, 25.03, 26.32, 28.41, 28.62, 29.30, 29.39, 30.21, 30.28, 31.01, 33.68, 64.18, 65.97, 73.28, 109.45, 173.17.
MS: 445 (M+Na).
2.2.3. Synthesis of 2,3-Dihydroxypropyl 8-(Hexylselanyl)octanoate (Se-MAG) 5
The acetonide
4 (1 g, 2.4 mmol) was dissolved in methanol (60 mL) in a 250 mL round-bottomed flask, which was placed in an ice bath. To this cooled mixture, 2 M aqueous HCl (1.86 mL, 1.5 eq, 3.6 mmol) was added in one portion. After 15 min, the ice bath was removed, and the reaction mixture was stirred for 5 h at 20 °C while monitoring the progress of the reaction by TLC every hour. After 5 h, the conversion had maximised and the reaction mixture was diluted with 150 mL of diethyl ether, transferred to a separatory funnel, and washed with 100 mL of saturated aqueous sodium bicarbonate and extracted with diethyl ether (2 × 100 mL). The organic phase containing the product was dried over sodium sulphate and concentrated by rotary evaporation. The crude product was purified by silica gel column chromatography using a gradient of hexane and ethyl acetate to obtain the corresponding Se-MAG
5 in 64.6% yield (0.58 g). The product was concentrated and characterised using NMR (
Figures S1 and S2), IR, mass spectrometry (
Figure S3), TLC (
Figure S4), and ultraviolet-visible spectroscopic analysis (
Figure S5).
1H NMR (400 MHz (ppm), CDCl3): δ = 0.82 (t, J = 6.9 Hz, 3H), 1.37–1.17 (m, 12H), 1.63–1.52 (m, 6H), 2.15–2.03 (m, 2H), 2.33–2.25 (m, 2H), 2.48 (t, J = 7.2 Hz, 2H), 3.57–3.49 (m, 1H), 3.67–3.60 (m, 1H), 3.81–3.76 (m, 1H), 3.90–3.84 (m, 1H), 4.17–4.05 (m, 2H).
13C NMR (100 MHz (ppm), CDCl3): δ = 13.0, 22.21, 23.51, 23.67, 24.44, 28.38, 28.63, 29.29, 29.39, 30.19, 30.27, 31.0, 33.70, 64.16, 65.99, 73.32, 173.18.
IR (cm−1): 3420, 2880, 1720, 1528, 1315, 1214, 743.
MS: 405 [M+Na].
2.3. Lnt Expression and Purification
The wild-type (WT) gene for Lnt from Escherichia coli used for expression was in the pET28a vector (GenScript, Piscataway, NJ, USA). Expression was carried out in C43(DE3) cells. Cells were transformed with the Lnt plasmid and grown on kanamycin-supplemented lysogeny broth (LB) agar plates at 37 °C. After a 16 h incubation, the transformed colonies were suspended in 6 mL of LB. One milliliter of the cell suspension was used to inoculate 1 L of LB. Cultures were grown at 200 rpm and 37 °C to an OD600 of 1.6, then cooled on ice for ~20 min to 20 °C. To induce the expression of Lnt, isopropyl-β-D-thiogalactoside (IPTG) was added to a final concentration of 0.5 mM. Cells were grown for a further 20 h at 20 °C and 200 rpm post-induction. Cells were pelleted by centrifugation at 6000× g for 10 min at 4 °C, resuspended in Buffer A (20 mM Tris-HCl pH 8.0, 50 mM NaCl, 0.5 mM EDTA, 1 mM PMSF), and lysed on ice by passing the sample three times through an EmulsiFlex-C5 homogeniser (Avestin, Ottawa, ON, Canada) at 1200–1400 bar.
To produce pure Lnt, membranes were obtained by centrifuging the lysed cell homogenate at 120,000× g for 45 min at 4 °C. The membrane pellet was resuspended in Buffer B (20 mM HEPES-NaOH pH 7.5, 200 mM NaCl, 10% (v/v) glycerol, 1 mM PMSF) using a Dounce homogeniser at a ratio of 5 mL buffer per gram of membrane pellet. To solubilise the membranes, LMNG was added to a final concentration of 2% (w/v) and the sample was incubated for 45 min at 20–22 °C on a rotating wheel mixer at 12 rpm. The suspension was centrifuged at 60,000× g for 45 min at 4 °C to remove any unsolubilised material. The supernatant was incubated with 4 mL Ni resin (Qiagen, Hilden, Germany) pre-equilibrated in Buffer B with 20 mM imidazole and 0.05% (w/v) LMNG. The sample was incubated with the Ni resin on a rotating wheel mixer at 12 rpm for 1 h at 4 °C. The suspension was loaded into an empty chromatography column and the liquid was allowed to drain from the resin under gravity. The resin was washed with 80 mL of Buffer C (20 mM HEPES-NaOH pH 7.5, 800 mM NaCl, 40 mM imidazole, 10% (v/v) glycerol, 0.5 mM PMSF, 0.05% (w/v) LMNG) and the bound protein eluted with 15 mL of Buffer D (40 mM sodium citrate pH 6.0, 200 mM NaCl, 400 mM imidazole, 10% (v/v) glycerol, 0.5 mM PMSF, 0.1% (w/v) LMNG). The Lnt protein was further purified using a HiLoad 16/60 Superdex 200 column (GE Healthcare, Hatfield, UK) equilibrated in Buffer E (20 mM sodium citrate pH 6.0, 200 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) LMNG) at 4 °C. The purified protein was concentrated using a 50 kDa molecular weight cut-off (MWCO) Amicon Ultra 15 concentrator (Millipore, Burlington, MA, USA USA) to ≥13 mg/mL. Purified protein was aliquoted, snap-frozen in liquid nitrogen, and stored at −72 °C until required.
2.4. AlgE Expression and Purification
To express AlgE, the gene for the WT protein from Pseudomonas aeruginosa in the pET28a plasmid was transformed into BL21 RIL(DE3) cells and grown on kanamycin- and chloramphenicol-supplemented LB agar plates at 37 °C for 16 h. A single colony was used to inoculate a 10 mL LB starter culture and grown at 37 °C and 180 rpm for 16 h. The next day, 1 L of LB was inoculated with 10 mL of starter culture. The cells were grown at 37 °C and 180 rpm to an OD600 of 0.6. The flask was transferred to an incubator at 30 °C, and the expression of AlgE was induced with 1 mM IPTG. The cells were grown for a further 4 h at 30 °C and 180 rpm after adding IPTG. The cells were harvested at 6000× g for 10 min at 4 °C.
The cell mass from 1 L of culture was resuspended in 35 mL of Buffer F (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 Roche protease inhibitor tablet). The cells were lysed on ice with 3 passes through an Emulsiflex-C5 cell disruptor (Avestin, Ottawa, ON, CanadaCanada) at 1200–1400 bar. The lysed cells were centrifuged at 100,000× g for 20 min at 4 °C. The pellet containing the AlgE protein in inclusion bodies was washed with 15 mL of Buffer F and centrifuged again at 100,000× g for 20 min at 4 °C. The pellet was resuspended in 25 mL of Buffer G (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 8 M urea) and mixed on a wheel mixer at 12 rpm for 3 h at 20–22 °C to solubilise inclusion bodies. The sample was centrifuged at 30,000× g for 45 min at 4 °C to remove insoluble material. The supernatant was slowly added, with mixing, into 500 mL of Buffer H (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.5% (w/v) DM) pre-warmed to 37 °C. This mixture was incubated at 37 °C and 130 rpm for 16 h to refold the AlgE protein. To the 500 mL of Buffer H, 5 mL of Ni resin (Qiagen, Hilden, Germany) was added and incubated at 4 °C on a rotating wheel mixer at 12 rpm for 1 h. The suspension was loaded into an empty chromatography column and the liquid was allowed to drain from the Ni resin under gravity. The Ni resin was washed with 30 mL of Buffer H and the bound AlgE was eluted with Buffer H containing 60 mM imidazole. The protein was further purified using a HiLoad 16/60 Superdex 200 column (GE Healthcare, Hatfield, UK) equilibrated in Buffer I (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.45% (v/v) n-octyl tetraoxyethylene (C8E4)). The purified protein was concentrated using a 50 kDa molecular weight cut-off (MWCO) Amicon Ultra 15 concentrator (Millipore, Burlington, MA, USA) to ≥35 mg/mL. The purified protein was aliquoted, snap-frozen in liquid nitrogen, and stored at −72 °C until required.
2.5. Crystallisation
For in meso crystallisation trials, 8 µL of molten 9.9 MAG was homogenised with 8 µL of molten Se-MAG in a lipid mixing device consisting of two 100 μL Hamilton syringes connected by a narrow-bore coupler [
32]. The lipid mixture was transferred to one of the syringes. Next, 16 µL of either the Lnt or AlgE protein solution was added to the empty syringe and the protein was reconstituted into the bilayer of the cubic mesophase by repeated passaging of the contents of the two syringes back and forth through the coupler at 20–22 °C [
33,
34]. Crystallisation trials were set up by dispensing 50 nL of the protein-laden mesophase onto a silanised 96-well glass sandwich plate and covered by 800 nL of the precipitant solution using an in meso robot [
33]. The glass plates were sealed and stored in an incubator/imager (RockImager RI1500, Formulatrix, Bedford, MA, USA) at 20 °C for crystal growth. For Lnt, the screen consisted of 0.1 M MES-NaOH pH 6.0, 8% (
v/
v) MPD, and 0.05–0.4 M sodium or potassium thiocyanate. For AlgE, screening was carried out with precipitant screens containing 34–41% (
v/
v) PEG400, 100 mM LiSO
4, and 0.1 M sodium citrate at pH 5.6 and 6.0 and 34–41% (
v/
v) PEG400, 25–300 mM (NH
4)
2SO
4, and 0.1 M sodium citrate at pH 5.6 and 6.0. For harvesting crystals of Lnt and AlgE between 21 and 30 days post-setup, MiTeGen loops were used and the crystals were snap-cooled in liquid nitrogen without added cryoprotectant. The crystals were shipped to the Swiss Light Source in UniPucks contained in a CX100 dry shipper and shipping case.
2.6. Macromolecular X-ray Crystallography
2.6.1. Data Collection
X-ray diffraction data were acquired at the protein crystallography beamline X06SA-PXI at the Swiss Light Source (SLS), Villigen, Switzerland. Data were collected with a 40 × 40 µm
2 micro-focused X-ray beam at wavelengths of 0.97794 Å and 0.97939 Å for AlgE and Lnt, respectively, in a cryo-stream at 100 K using DA+ SLS data acquisition software suite [
35]. Measurements were carried out in steps of 0.1 and 0.2° at a speed of 0.1 s/step using an EIGER 16 M detector in shutterless mode and a SmarGon multi-axis goniometer (SMARGON–Microrobot, SmarAct, Oldenburg, Germany). For AlgE, seventy-two 360° ω scans were collected from 24 crystals. Each scan was carried out with multiple SmarGon χ angles in the 0° to 30° range. The crystal was translated for each χ angle to minimise radiation damage. A similar data collection strategy was used for Lnt with reduced ω scan ranges due to the limited crystal size. A total of 144 ω scans were recorded using 76 crystals (
Table 1). The radiation dose was kept below 5 MGy at each crystal position.
2.6.2. Data Processing, Selection, and Merging
X-ray diffraction data were processed using XDS with scaling and merging carried out with XSCALE [
36,
37]. Isomorphous datasets were selected using XDSCC12 [
38]. The selection was first performed using a multi-dimensional scaling procedure, which identifies datasets with large non-isomorphism relative to clusters of other datasets. Further selection was based on ΔCC
1/2, which measures the influence a set of reflections has on the overall CC
1/2 of the merged data. In this way, 17 datasets of 72 obtained from crystals of AlgE were then merged to a final complete data set of 1.7 Å in space group
P2
1212. For Lnt, 41 datasets of 144 were merged to a final complete dataset of 1.9 Å in space group in the Results section. Data collection, processing, and validation statistics are provided in the Results section.
2.6.3. Structure Determination and Refinement
Experimental phasing was carried out by the Se-SAD method using anomalous diffraction datasets from crystals of AlgE and Lnt. Heavy atom location, structure phasing, and density modification were performed using the
Crank2 [
39] interface of SHELXC/D [
40] and produced interpretable electron density maps for both proteins. From the experimentally phased maps,
Buccaneer [
41] was used for initial model building. Models were completed manually using
Coot [
42].
Phenix.refine [
43] was used during the refinement of all structures. Phasing and refinement statistics are reported in the Results section. Figures of molecular structures were generated using
PyMOL (version 1.8.4.0; Schrödinger, LLC) [
44].
2.7. Small-Angle X-ray Scattering (SAXS)
SAXS data were collected with samples in 1-mm-diameter glass X-ray capillaries on beamline X10SA-PXII (SLS) at room temperature (22 °C), as previously described [
45,
46]. Two-dimensional X-ray powder diffraction patterns were generated with a 75 μm × 15 μm X-ray beam at a wavelength of 1 Å (12.398 keV) and were recorded on an EIGER2 16 M detector at frame rates of 10 and 100 Hz and at sample-to-detector distances ranging from 20 to 100 cm. Patterns were indexed using radial averaged intensity versus 2 theta (I/2θ) plots.
4. Discussion
The purpose of this study was to determine if a MAG, modified to include a selenium atom in place of methylene in its acyl chain, could be used for experimental phasing of X-ray structures obtained from crystals of membrane proteins grown by the in meso method. The results show convincingly that phasing is indeed possible. It was demonstrated with two contrasting membrane proteins, AlgE and Lnt. For this method to work, the labelled lipid used to create the mesophase from which crystals grow must appear as a structured lipid in the final crystal structure. In other words, the lipid must co-crystallise with the protein. This is what was observed. Indeed, most in meso crystal structures, of which there are close to a thousand on record in the Protein Data Bank [
45,
54], with enough resolution show multiple structured lipids in the final solved structure.
The Se-MAG used in this study can be viewed as an isostere of monopentadecanoin, a MAG with a fatty acid 15 carbon atoms long in ester linkage to a glyceryl head group (
Figure 1). In the case of Se-MAG, the methylene at position 9 in the pentadecenoyl chain of monopentadenanoin is replaced by a selenium atom. It was not clear at the outset if this Se-MAG, in its pure and fully hydrated form, would stabilise in the cubic phase at room temperature (~20 °C) nor indeed if it would be suitable for direct use in in meso crystallisation. It transpired that neat Se-MAG, when fully hydrated, formed the L
α phase and not the cubic mesophase at RT. In this sense, therefore, Se-MAG resembles its
cis monoenoic equivalents, 8.7 MAG and 9.6 MAG [the
trans monoenoic forms of 8.7 MAG and 9.6 MAG will have a mesophase behaviour analogous to that of monopentadecanoin], which have a fatty acyl chain 15 carbon atoms long with a double bond between methylenes at positions 8 and 9, and 9 and 10, respectively [
45]. Both monoenoic MAGs are predicted to form liquid crystalline phases when fully hydrated at RT. We know from separate work that certain MAGs are miscible with one another and that mixtures can be prepared with physical properties that reflect the percentage of each in the mix [
15,
50,
55]. This approach was taken with Se-MAG. Thus, by combining it with monoolein in a 50/50 mix, the mixture (Se-MAG*) displayed the desired property of forming the cubic phase when fully hydrated at RT. Importantly, Se-MAG* also proved capable of supporting the crystallisation of membrane proteins by the in meso method.
Since we had no prior experience with Se-MAG, it was not known if this labelled lipid would preferentially co-crystallise with the membrane protein or if the unlabelled monoolein would competitively displace Se-MAG during crystallisation. The results show anomalous signals from selenium associated with most structured lipids in the solved structures. Interestingly, at some of the MAG binding sites, two anomalous signals were recorded. We interpret this as MAG molecules binding to the protein at the same site in two different orientations, one with the head group toward the periplasmic side of the protein and the other with it facing the cytoplasmic side.
The success of SAD phasing depends on the anomalous signal strength at each site and the total anomalous signal [
56,
57]. In meso crystallisation generally ensures that structured MAGs decorate the membrane protein. If these lipids are 100% labelled MAG, then collectively they should provide enough phasing signal. However, the 50:50 mixture of Se-MAG and monoolein, combined with partial disorder and partial occupancies of most bound Se-MAGs, reduce the observable anomalous signal and render the sub-structure determination challenging. Therefore, similar to challenging native SAD cases, higher-multiplicity data are needed for Se-MAG compared to Se-Met phasing where most Se-Mets are ordered with 100% occupancies. A total of 6120° of data with a multiplicity of 46 and 8040° with a multiplicity of 80 were used for AlgE and Lnt phasing, respectively. In the case of both AlgE and Lnt phasing processes, a few well-ordered Se-MAGs and preferentially occupied Se-MAG sites bootstrapped the initial sub-structure determination and subsequent sub-structure completion. The relatively high solvent content of in meso-grown crystals also helped in the density modification and experimental map improvement.
The selenium atom in Se-MAG is located toward the middle of the fatty acyl chain. It is known that the degree of disorder, as in the frequency of
cis/
trans isomerizations, increases toward the methyl end of the chain [
58]. This would suggest that placing the selenium further up the chain closer to the glyceryl head group would put it in a more ordered part of the chain and thus be more likely to give a useful anomalous signal. Therefore, instead of having the selenium atom at position 9 in the chain, as in Se-MAG, placing it at position 7 or 5 might produce a more effective labelling agent. Another possibility for increasing the effectiveness of the labelled MAG would be to increase the number of selenium atoms in the chain. These could be contiguous, or methylene, ethylene, or propylene interrupted. Challenges with any of these proposed modifications may arise during synthesis and subsequently with chemical stability. Mesophase behaviour must also be considered. Changing the selenium atom by just one position in the chain, from 9 to 8 for example, may alter phase behaviour in the same way as changing the position of a double bond in the chain does [
45,
46,
47,
59,
60,
61]. It may be that by suitably repositioning the selenium atom in Se-MAG its phase behaviour is altered such that when fully hydrated at RT the pure lipid forms the cubic mesophase. In this case, combining it with monoolein, as performed in this study, would no longer be necessary. It would also mean that the anomalous signal would be as strong as it possibly could be since every lipid molecule in the mesophase used for crystallisation will carry a selenium atom.
While Se-Met is the most popular labelling agent employed for de novo phasing, other non-amino acid Se-containing compounds have been used successfully with membrane proteins. A recent review by Hanashima et al. [
62] describes the application of heavy atom-labelled detergents and lipids for locating them in and for the experimental phasing of membrane protein crystal structures. Thus, for example, the common detergent dodecyl-β-D-maltoside (DDM) was modified to include a selenium atom in the sugar head group and used to phase the structures of a leukotriene C4 synthase and a pentameric ligand-gated ion channel. The review also refers to immunostimulatory α-galactosylceramide (α-GalCer) ligands for the non-classical major histocompatibility protein, CD1d. Selenium was incorporated singly at two different positions in the fatty acyl chain of the ligand with the aim of helping better locate the chain in the binding pocket of CD1d. Importantly, the heavy atom labels did not significantly alter the signalling properties of the ligand. The Se-labelled α-GalCers were referred to in the original study as being potentially useful for defining ligand–receptor interactions in greater detail [
63]. Their use for phasing is expected to be limited, however, given that a single lipid, and thus two selenium atoms, are bound per CD1d molecule.
Recently, seleno-urea has been employed to Se-SAD phase a single crystal of the membrane enzyme, glycerol 3-phosphate acyltransferase (PlsY), grown by the in meso method [
64]. Crystals of the enzyme in the mesophase were labelled by the soaking method. As expected, the seleno-urea was associated with the exposed, more polar extramembrane parts of the protein. Importantly, neither the mesophase nor the protein was perturbed by the labelling procedure, which makes seleno-urea a highly attractive de novo phasing agent.
It would appear therefore that de novo phasing of membrane protein crystal structures with selenium as the signal-producing heavy atom has a sizable array of diverse and benign compounds and labelling strategies from which to choose. Furthermore, while molecular replacement, complemented by computational approaches such as AlphaFold [
4] and RoseTTAFold [
65], is now the go-to phasing method, there will always be instances where experimental phasing is required. In such cases, phasing with selenium-labelled analogues of small organics, ligands, detergents, and/or lipids, represents an attractive and proven option.
5. Conclusions
Se-MAG works as an additive lipid in combination with monoolein to crystallise membrane proteins by the in meso method and for the experimental phasing of crystallographic structures.
The test proteins used in this study are known to be stable and give crystals by the in meso method that diffract to reasonably high resolutions. The acid test has yet to be performed when the phasing strategy introduced here is evaluated with less well-behaved membrane proteins whose structures cannot be phased by molecular replacement and where experimental phasing methods must be resorted to. In such cases, the Se-MAG/monoolein mix (Se-MAG*) described here is certainly worth trying.
Anecdotally, we found that compared to monoolein alone, Se-MAG* gave bigger crystals of AlgE and better diffraction in the case of AlgE and Lnt. The exact molecular mechanism by which this comes about is not known. It must reflect to some degree on the identity and microstructure of the mesophase that the lipid mixture generates and in which crystallisation occurs. How the mesophase responds to the different components of the precipitant and the protein solutions, which include detergents, buffers, salts, and the protein itself, will depend on the chemical identity of the lipids creating the mesophase [
15]. As a result of these personal observations with Lnt and AlgE, whenever we run into issues crystallising a membrane protein by the in meso method using monoolein, we turn to Se-MAG* as an alternative in anticipation that bigger and better crystals will emerge sooner. With a stock of the Se-MAG/monoolein mixture in store, running such crystallisation trials is no more difficult to conduct than it is with monoolein alone. In addition, the heavy atom is already in place which may prove useful should phasing be required.
There are two challenging aspects to using Se-MAG for experimental phasing. The first is the need for high multiplicity, which requires large amounts of quality diffraction data and the crystals for generating that data. The second challenge concerns accessing the Se-MAG. To our knowledge, Se-MAGs are not available commercially. However, custom synthesis is always an option. It is also possible to perform the synthesis in-house in the researcher’s own laboratory if they have suitable facilities and skilled personnel. Collaborating with a synthetic organic chemist is another option. Full details of Se-MAG synthesis, purification, and characterisation are included in the Materials and Methods and
Supplementary Information.
Work is needed to identify new Se-MAG lipids that, upon full hydration at ambient temperatures and without added monoolein, form the cubic mesophase. The advantage of such a lipid includes the fact that it can be used directly without the need to mix it with another MAG such as monoolein. Further, all co-crystallised MAGs, as opposed to just the Se-MAG component in the mix, would contribute anomalous signals to facilitate phasing. To find such a lipid will involve a focused Se-MAG synthesis and mesophase characterisation study. This is reminiscent of an investigation undertaken to identify
cis monounsaturated MAGs with which to screen host lipids for optimal in meso crystallisation [
66]. The approach was successful, giving rise to a number of high-profile structures, one of which was the β
2 adrenergic receptor-Gs protein complex structure, which took centre stage in the 2012 Nobel Prize in Chemistry [
17].
AlphaFold [
4] and RoseTTAFold [
65] models are increasingly being used for phasing structures by molecular replacement [
67,
68]. However, computational methods are not always reliable, and structures solved by experimental phasing methods will be required for some time to come. Se-MAG* has proven to be a useful phasing agent as demonstrated in this study. Selenium has good anomalous strength, and the bioisosteric Se-for-CH
2 substitution in the lipid acyl chain is considered relatively benign in terms of mesophase behaviour. Accordingly, Se-MAGs are a very reasonable option for use in in meso crystallisation trials when predicted and homologous structures fail to provide the requisite crystallographic phasing information.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}