

Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines
1
Department of Chemistry, Ball State University, Muncie, IN 47306, USA
2
Department of Chemistry, Purdue University, West Lafayette, IN 47907, USA
*
Author to whom correspondence should be addressed.
Crystals 2024, 14(2), 124; https://doi.org/10.3390/cryst14020124
Submission received: 27 December 2023
/
Revised: 17 January 2024
/
Accepted: 24 January 2024
/
Published: 26 January 2024
(This article belongs to the Special Issue Feature Papers in Crystals 2023)
Abstract
:Haloalkanes and amines are common halogen-bond (XB) donors and acceptors as well as typical reagents in nucleophilic substitution reactions. Thus, crystal engineering using these molecules requires an understanding of the interchange between these processes. Indeed, we previously reported that the interaction of quinuclidine (QN) with CHI3 in acetonitrile yielded co-crystals showing a XB network of these two constituents. In the current work, the interactions of QN with C2H5I or 1,4-diazabicyclo[2.2.2]octane (DABCO) with CH2I2 led to nucleophilic substitution producing I− anions and quaternary ammonium (QN-CH2CH3 or DABCO-CH2I+) cations. Moreover, the reaction of QN with CHI3 in dichloromethane afforded co-crystals containing XB networks of CHI3 with either Cl− or I− anions and QN-CH2Cl+ counter-ions. A similar reaction in acetone produced XB networks comprising CHI3, I− and QN-CH2COCH3+. These distinctions were rationalized through a computational analysis of XB complexes and the transition-state energies for the nucleophilic substitution. It indicated that the outcome of the reactions was determined mostly by the relative energies of the products. The co-crystals obtained in this work showed bonding between the cationic (DABCO-CH2I+, QN-CH2Cl+) or neutral (CHI3) XB donors and the anionic (I−, Cl−) or neutral (CHI3) acceptors. Their analysis showed comparable electron and energy densities at the XB bond critical points and similar XB energies regardless of the charges of the interacting species.

1. Introduction
Halogen bonding (XB), an attraction of electron-rich centers to electrophilic halogen substituents, has become one of the most valuable tools in crystal engineering over the last two decades [1]. The applications of this supramolecular interaction for the preparation of solid-state materials are related to its high directionality and the possibility of modulating its strength by changing the halogen atoms and the attached group [2,3]. Variations in the nature of halogen-containing molecules (XB donors) and nucleophiles (XB acceptors) allow for the variation of bond strength and length from the very weak interactions (<1 kcal/mol) with interatomic distances close to the van der Waals separations to those (40–50 kcal/mol) approaching the characteristics of covalent bonds [4,5]. The strength of the bonding is expected to increase with the rise of the positive potential on the XB donor and/or the negative potential on the XB acceptor (although the dispersion and polarization of both interacting species also should be taken into account) [6,7]. This suggests that the overall charges of the interacting species represent a vital factor in this supramolecular interaction and that cationic XB donors and anionic acceptors should form the strongest bonds. Yet, the majority of the XB complexes described up to now involve neutral XB donors and neutral or anionic XB acceptors, and even XB associations between ions of like charges were reported [2,3,8,9]. There is a rather limited number of experimental and/or theoretical investigations into XB ion pairs, e.g., those involving halogen-substituted pyridinium or imidazolium XB donors and halide anions [10,11,12,13,14,15,16]. There are only a few studies on the bonding involving XB donors and acceptors with various charges [17,18,19,20], especially with respect to the comparison of such bonding coexisting in the same crystal.
The structural characterization of ionic co-crystals derived from 3-halopyridinium halogenides and perfluoroiodobenzenes reported recently by Posavec et al. [20] provided an example of a system with co-existing halogen bonding involving cationic and neutral donors. All of them showed hydrogen bonds between halide anions and halopyridinium cations, as well as the halogen bonding of halides with iodoperfluorobenzenes, and, in some cases, halogen substituents in pyridinuium cations. The systems containing cationic and neutral species, which potentially could serve as XB donors, were also prepared through the controlled assembly of XB architectures incorporating di- and triiodobenzenes using the Menshutkin reaction [21]. However, these co-crystals showed halogen bonding only between neutral iodoperfluorobenzenes and Cl− anions (produced as a result of the reaction between 1,4-diazabicyclo[2.2.2]octane, DABCO, and dichloromethane solvent). The charges of the anions were balanced by another product of this nucleophilic substitution, i.e., a quaternary ammonium cation DABCO-CH2Cl+. Similar cations were formed in the dichloromethane solutions containing DABCO and CBr3NO2 [4]. The cationic nature suggests that a substantial positive potential is on the surface of its chlorine substituents. Yet, no halogen DABCO-CH3Cl+···Cl− bonding (or analogous XB of bromide with the bromosubstituted quaternary ammonium prepared in a similar way) were observed despite the fact that even dichloromethane is capable of forming halogen bonds [22]. DABCO-CH2Cl+ and DABCO-CH2I+ were also obtained in the copper(I)-mediated quaternization of DABCO in dichloromethane or in its mixture with diiodomethane [23]. In addition, halogen-substituted quaternary amines were reported in the publications describing the design of hybrid lead perovskite- or zinc chloride-based ferroelectrics [24,25]. The structures reported in these works comprised the halogen bonding between the iodine substituents of the ammonium cation and anionic species. However, these works were focused on the preparation and structural characterizations of the co-crystals (or their potential applications) without a comparison of the characteristics of halogen bonding involving differently charged species.
The interaction of DABCO with halomethanes resulting in the formation of quaternary ammonium cations also highlights the potential competition of halogen bonding and nucleophilic substitution reactions. Indeed, the halogen atoms in the halogenated alkanes are not only common XB donors but are also typical leaving groups in nucleophilic substitution reactions [26]. In turn, tertiary amines are both effective XB donors and nucleophiles. Thus, the development of a successful crystallization procedure involving these reagents requires an understanding of the interplay and competition between these processes. While several publications reported that the interaction of CH2X2 (X = Cl, Br or I) with DABCO (or other trialkylamines) resulted in facile nucleophilic substitutions (vide supra), the analogous interactions of trihalomethanes or tetrahalomethanes with the same nucleophiles led to the formation of XB associations between these strong XB donors and acceptors [4,27,28]. The absence of the products of nucleophilic substitutions in the systems with tri- and tetrahalomethanes is probably related to the higher barrier for such reactions between alkylamines and tri- or tetrahalomethanes compared with those with mono- and dihalomethanes. However, to the best of our knowledge, the energetics of these processes and the reason for such changeovers were not explored up to now.
In this work, we prepared five new ionic crystals (see Section 2 below) containing neutral and/or cationic XB donors obtained using DABCO or its 1-azabicyclo[2.2.2]octane analogue (quinuclidine, abbreviated hereinafter as QN) and anionic or neutral XB acceptors. It allowed us to examine the co-existing halogen bonding between XB donors and acceptors with different charges. Our goals were to compare the ability of two distinct halogen atoms, chlorine and iodine, to act as XB donors and, simultaneously, that of Cl− and I− anions to act as XB acceptors and to evaluate the effect of the charges of the interacting species on the characteristics of halogen bonding in these systems. Most importantly, we intended to rationalize the changeover between halogen bonding and nucleophilic substitution (Scheme 1) through computational analysis of the energetics of these processes (including the transition state energies for the corresponding SN2 reactions).
2. Materials and Methods
2.1. Materials
Commercially available triiodomethane, diidomethane, iodoethane, 1,4-diazabicyclo[2.2.2]octane and quinuclidine (from Sigma Aldrich, Burlington, MA, USA) were used without additional purification.
2.2. Crystallization and X-ray Structural Analysis
Single crystals of quaternary ammonium salts suitable for X-ray structural analysis were produced through the interaction of equimolar quantities of DABCO or QN and CH2I2, CHI3, or C2H5I in acetonitrile, dichloromethane, or acetone. In particular, an acetonitrile solution (5.0 mL) containing 268 mg (1.0 mmol) of CH2I2 was added to a solution containing 112 mg of DABCO (1.0 mmol) in 10 mL of acetonitrile. The mixture was slowly cooled down to −20 °C and kept at this temperature for several days. This resulted in the formation of the crystalline salt [DABCO-CH2I]I (1). X-ray structural analysis (vide infra) showed that it comprised DABCO-CH2I+ cations and I− anions. The analogous crystals of 1 (with the identical FT-IR spectrum shown in Figure S1 in the Supporting Information and the melting point of 156–159 °C (decomp.)) were obtained from an acetone solution of DABCO and CH2I2.
A dichloromethane solution (5.0 mL) containing 394 mg (1.0 mmol) of CHI3 was added to a solution containing 111 mg of QN (1.0 mmol) in 10 mL of CH2Cl2. The mixture was slowly cooled down to −20 °C and kept at this temperature for several days. This resulted in formation of crystalline materials containing a mixture of the crystals of [QN-CH2Cl]Cl·CHI3 (2) and [QN-CH2Cl]I·CHI3 (3).
An acetone solution containing 394 mg (1.0 mmol) of CHI3 was added to a solution containing 111 mg of QN (1.0 mmol) in 10 mL of acetone. The mixture was slowly cooled down to −20 °C and kept at this temperature for several days. This resulted in formation of a precipitate containing the crystals of [QN-CH2COCH3]I·CHI3 (4). They comprised CHI3 molecules, I− anions, and ammonium cations [QN-CH2COCH3] in which the DABCO moiety substituted one of the hydrogens in acetone.
Finally, a solution of 268 mg (1.0 mmol) of C2H5I and 111 mg of QN (1.0 mmol) in propionitrile was slowly cooled down to −20 °C and kept at this temperature for several days. This resulted in the formation of crystalline salt [QN-CH2CH3]I (5).
The single crystal structures were determined on a Bruker AXS D8 Quest CMOS diffractometer (Bruker AXS, LLC, Madison, WI, USA) with a fixed chi angle, a sealed fine-focus X-ray tube, a single crystal curved graphite incident beam monochromator, a Photon100 or PhotonII area detector and an Oxford Cryosystems low-temperature device. Data were collected at 150 K. Examination and data collection were performed with Mo Kα radiation (λ = 0.71073 Å). Reflections were indexed and processed, and the files were scaled and corrected for absorption using APEX3 or Apex4 [29]. The space groups were assigned using XPREP within the SHELXTL suite of programs [30]; the structures were solved through direct or dual methods and refined using full matrix least squares against F2 with all the reflections using Shelxl2018 or 2019 utilizing the graphical interface ShelXle [31,32,33]. If not specified otherwise, H atoms attached to carbon and nitrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C-H bond distances of 1.00, 0.99, and 0.98 Å for aliphatic C-H, CH2, and CH3 moieties, respectively. Methyl H atoms were allowed to rotate but not to tip to best fit the experimental electron density. Uiso(H) values were set to a multiple of Ueq(C), with 1.5 for CH3 and 1.2 for C–H and CH2 units, respectively. The cation in [DABCO-CH2CH3]I (5) is disordered across a mirror plane. Selected chemically equivalent bonds were restrained to be similar in length. One carbon atom, C5, was constrained to lie exactly on the mirror plane. Images and figures of X-ray structures were prepared using the program Mercury [34].
[DABCO-CH2I]I (1). Chemical formula C7H14IN2·I, M = 380.00 g/mol. Orthorhombic, Cmc21, a = 7.7371 (6) Å, b = 11.6169Å (8), c = 12.2399 (9) Å, V = 1100.14 (14) Å3, Z = 4, μ(MoKα) = 5.67 mm−1, 9943 reflections measured, 2050 unique (Rint = 0.027). The final R1 was 0.012 (I > 2σ(I)), and wR2 was 0.025 (all data).
[QN-CH2Cl]Cl·CHI3 (2). Chemical formula C8H15ClN·CHI3·Cl, M = 589.83 g/mol. Monoclinic, P21/n, a = 6.7531 (2) Å, b = 13.2065 (4), c = 17.5496 (5) Å, β = 99.9262 (12)°, V = 1541.73 (8) Å3, Z = 4, μ(MoKα) = 6.40 mm−1, 35,637 reflections measured, 5781 unique (Rint = 0.034). The final R1 was 0.021 (I > 2σ(I)), and wR2 was 0.040 (all data).
[QN-CH2COCH3]I·CHI3 (3). Chemical formula C8H15ClN·CHI3·I, M = 681.28g/mol. Orthorhombic, Pnma, a = 25.3615 (10) Å, b = 8.0459 (3) Å, c = 7.9252 (4) Å, V = 1617.19 (12)Å3, Z = 4, μ(MoKα) = 7.85 mm−1, 24,981 reflections measured, 3167 unique (Rint = 0.037). The final R1 was 0.022 (I > 2σ(I)), and wR2 was 0.045 (all data).
[QN-CH2COCH3]I·CHI3 (4). Chemical formula C10H18NO·CHI3·I, M = 688.87g/mol. Monoclinic, P21/m, a = 8.9024 (4) Å, b = 7.8802 (4) Å, c = 12.6430 (7) Å, β = 98.725 (2)° V = 876.68 (8) Å3, Z = 2, μ(MoKα) = 7.10 mm−1, 22,450 reflections measured, 3358 unique (Rint = 0.032). The final R1 was 0.019 (I > 2σ(I)) and wR2 was 0.042 (all data).
[QN-CH2CH3]I (5). Chemical formula C9H18N·I, M = 267.14 g/mol. Orthorhombic, Cmc21, a = 7.4861 (13) Å, b = 12.1185 (13) Å, c = 12.0879 (15) Å, V = 1096.6 (3) Å3, Z = 4, μ(MoKα) = 2.87 mm−1, 13,828 reflections measured, 2214 unique (Rint = 0.063). The final R1 was 0.028 (I > 2σ(I)), and R2 was 0.045 (all data).
2.3. Computations
Quantum mechanical calculations were carried out using the Gaussian 09 suite of programs [35]. Geometries of XB donors and complexes were optimized without constraints (starting from the geometries of the associations extracted from the X-ray structures) in the gas phase, dichloromethane, and acetonitrile through DFT calculations with the M06-2X functional and def2-tzvpp basis set [36,37]. An earlier analysis demonstrated that the M06-2X functional provided the most reliable results among the several density functionals in the analysis of XB complexes [38,39,40]. The def2tzvpp basis set does not include a diffuse function since previous analysis demonstrated that very similar results were obtained in the modeling of noncovalent interactions involving anions with the triple-ζ basis sets with and without diffuse functions [41]. The geometry optimizations in acetonitrile and in dichloromethane were performed using a polarizable continuum model [42]. The absence of imaginary vibrational frequencies confirmed that the optimized structures represent true minima. Values of binding energies were determined as follows: ΔE = Ecomp − (ER−X + EY) where Ecomp, ER−X, and EY are sums of the electronic energy and ZPE of the optimized complex, XB donor, and acceptor, respectively. Energies and atomic coordinates of the optimized complexes are listed in Table S1 in the Supporting Information.
The transition state for the model reactions between trimethylamine and halomethanes were identified using the opt = QST3 option in Gaussian 09. Their nature was confirmed through the presence of one imaginary frequency corresponding to the motion along the SN2 reaction coordinate (elongation of C-X and shortening of the N-C distances). The energies of TS, products, and XB complexes (Tables S2 and S3 in the Supporting Information) were determined relative to the sum of the energies of the reactants, i.e., (CH3)3N and halomethane.
Quantum theory of atoms in molecules (QTAIM) [43,44] and noncovalent interaction index (NCI) [45] analyses were performed using coordinates of the XB associations extracted from the X-ray structures using Multiwfn and visualized using VMD programs [46,47]. The NCI setting was as follows: isovalue = 0.5, color-coded with sgn(λ2)ρ, and in the range from −0.04 a.u. (blue, strong attractive interaction) to 0.02 a.u. (red, strong nonbonded overlap).
3. Results and Discussion
3.1. Preparation and X-ray Structural Characterization of the Quaternary Ammonium Salts
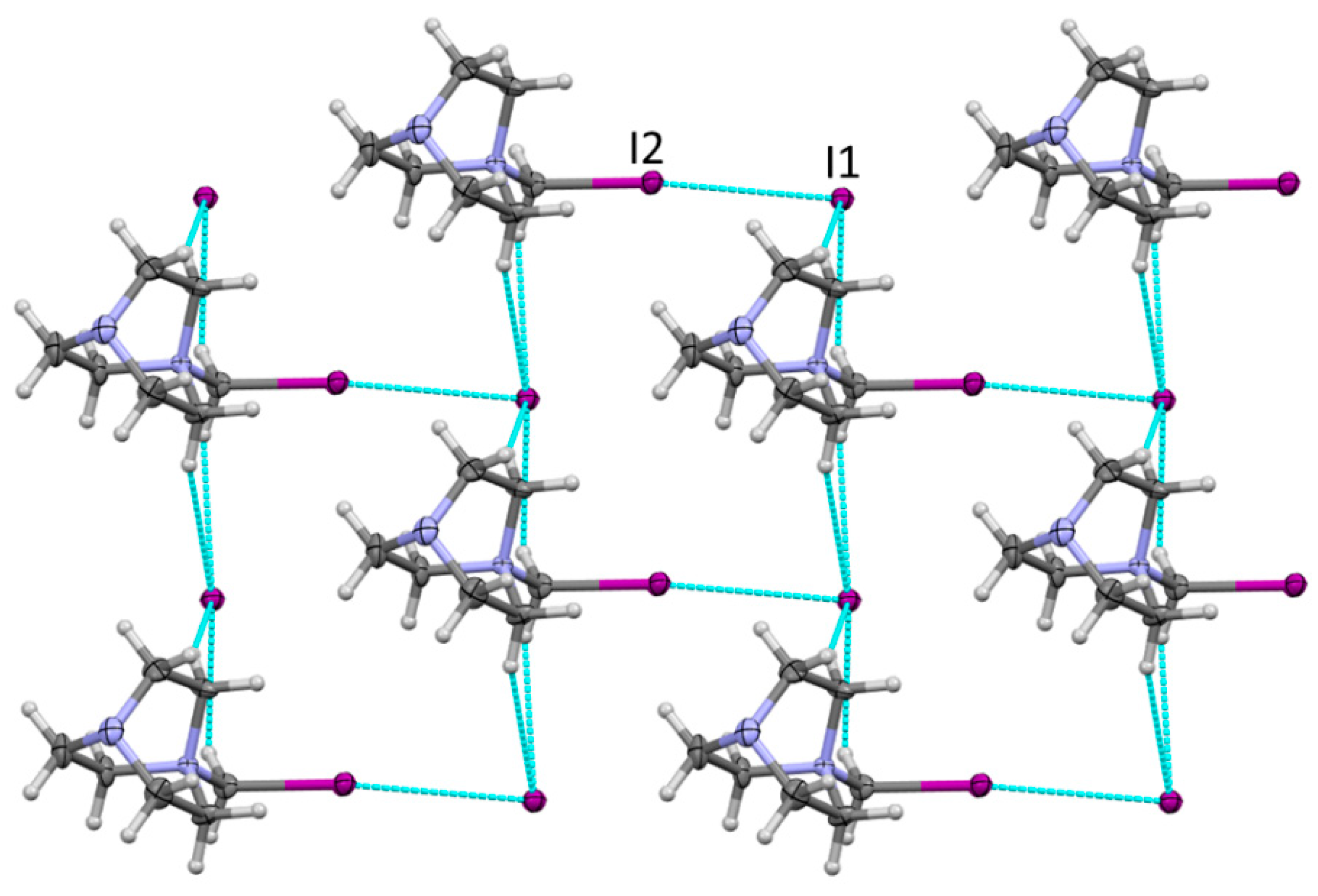
The mixing of equimolar quantities of DABCO and diiodomethane in acetonitrile resulted in the formation of a white crystalline material. The X-ray structural analysis of these crystals showed quaternary ammonium cations, DABCO-CH2I+, and iodide anions that had resulted from the Menshutkin reaction between DABCO and diiodomethane. Halogen and hydrogen bonding between these ions resulted in the formation of 2D layers, which are illustrated in Figure 1.
The geometric characteristics of the I···I contacts in these crystals are listed in Table 1. In particular, the distance between the iodine substituent in the ammonium cation and iodide of 3.670 Å was about 7% smaller than their van der Waals separation, which is consistent with the relatively weak halogen bonding (note that these RXY values were based on the Bondi’s radii [48], and larger contractions are implied if the recently proposed larger radii of halogens are used [49]). As such, these charge-assisted halogen bonds were slightly longer than the reported earlier XB lengths of about 3.50Å between neutral CHI3 and iodide anions [50,51,52].
Crystallization of DABCO analogue, QN, with CH3CH2I also resulted in the formation of the products of the Menshutkin reaction, which comprised the salt of iodide anions with QN-CH2CH3+ cations (crystal 5, see Figure S2 in the Supporting Information). In contrast, the previously reported crystallization of this amine with CHI3 in acetonitrile produced co-crystals comprising alternating chains in which CHI3 and DABCO were linked by hydrogen and halogen bonding [27]. The analogous slow crystallization of the mixture of CHI3 and QN in dichloromethane at −20 °C in the current work also resulted in the formation of the crystalline material. The careful examination of this material showed, however, that it contains two types of colorless crystals, i.e., monoclinic crystals (2) and orthorhombic crystals (3). X-ray structural analysis revealed that the monoclinic crystals (2) comprised triiodomethane together with the product of the Menshutkin reaction between DABCO and dicloromethane, i.e., QN-CH2Cl+ and Cl−. They showed halogen bonding between the two iodine substituents of the neutral XB donor, CHI3, and chlorides somewhat similar to the reported earlier interaction of chloride (obtained from the reaction of dichloromethane with DABCO) and iodoperfluorobenzenes (Figure 2).
The third iodine substituent forms a halogen bond with the equatorial area of the iodine substituent in another triiodomethane. In addition, the crystals of 2 showed short contacts between the chlorine substituents in QN-CH2Cl+ and Cl− anions. The latter also form short contacts with hydrogen substituents in QN-CH2Cl+. While these intermolecular interactions are also vital for the overall crystal architecture, the current work is focused on halogen bonding, and the hydrogen bonding was not examined in detail. The geometric characteristics of the Cl···Cl contacts are listed together with those of the I···Cl halogen bonds in Table 1. These data indicated that the interatomic Cl···Cl distance is about 4% shorter than the sum of the van der Waals radii of chlorine, and the C-Cl···Cl angle is close to 180°. As such, it is consistent with (weak) Cl···Cl halogen bonding. The RXY values in Table 1 suggest that this charge-assisted halogen bonding is weaker than that between neutral CHI3 and chloride in the crystals of 2.
X-ray analysis revealed that the orthorhombic crystals of 3 also comprised DABCO-CH2Cl+ cations and neutral CHI3 molecules (Figure 3). However, instead of Cl− anions in the crystals of 2, the charges of the ammonium cations in the crystals of 3 are balanced by the I− anions (the latter were probably produced by the electron transfer from DABCO to CHI3 in their XB complex accompanied by the dissociation of C-I bond in the resulting anion-radical.) The replacement of Cl− with the larger I− led to considerable changes in the crystal architecture. In the place of the XB networks formed by Cl− with both the cationic and neutral XB donors in 2, the crystals of 3 showed networks of halogen-bonded CHI3 and I− separated by layers of DABCO-CH2Cl+ cations. Specifically, halogen bonding between the three iodine substituents of CHI3 and iodide produced zigzag ladders (Figure 3) similar to those described previously between halide anions and triodo- or tribromomethane [52]. Geometric characteristics of these halogen bonds are listed in Table 1.
To further elucidate solvent effects in the formation of the XB network, we also carried out the co-crystallization of QN or DABCO with di- and triiodomethane in acetone. The mixing of the solutions of DABCO and CH2I2 produced the white precipitate of DABCO-CH2I+ with I−, which was analogous to that obtained in acetonitrile (vide supra). In comparison, the slow evaporation and cooling down of the solutions containing equimolar quantities of QN and CHI3 in acetone produced a non-homogenous mixture containing the yellow crystals of 4. The X-ray analysis showed that they comprised zigzag ladders formed by XB triiodomethane molecules and iodide anions, which were similar to those in the crystals of 3 (Figure 4).
The interatomic I···I separations in the XB networks in the crystals of 4 (Figure 4) were similar to those in the analogous ladder in the crystals of 3 as well as those in the charge-assisted I···I bonds in the crystals of 1. The XB networks in the crystals of 4 were separated by layers of the quaternary ammonium QN-CH2COCH3+ cations. The latter represented a product of the substitution of one of the hydrogen substituents in the acetonewith QN. This is probably related to the electron transfer from QN to CHI3 in their XB complex resulting in the formation of anion-radicals of CHI3 and cationic QN. Anion-radicals dissociate with the formation of halide anions and cationic QN attacks the enol form of acetone. A similar binding of acetone to amine was observed earlier in the acetone solutions containing iodoethane and 3-bromopyridine [16]. It was suggested that the resulting N-(2-oxopropyl)-3-bromopyridinium cations were presumably produced from the formation of an intermediate iodoacetone [16]. However, a discussion of the mechanism of its formation is beyond the scope of the current XB-focused work.
Overall, structural studies showed that the interaction of DABCO with CH2I2 (or CH3CH2I) resulted in fast nucleophilic substitution. The resulting iodine-substituted DABCO-CH2I+ formed charge-assisted halogen bonding with I− anions. The nature of the crystalline products formed by CHI3 and DABCO or QN varied with the solvent. In the unreactive acetonitrile, this interaction resulted in the formation of the co-crystals of DABCO with CHI3 comprising halogen- and hydrogen-bonded chains [27]. Mixing QN and CHI3 molecules in acetone produced crystals comprising the XB ladders of CHI3 and I−, with the quaternary ammonium QN-CH2COCH3+ counter-ions. In dichloromethane, two products were characterized. Both of them comprised quaternary ammonium cations QN-CH2Cl+ and CHI3 together with either chloride or iodide anions. Interestingly, while the crystals of 3 comprising iodide showed only halogen bonding between this anion and CHI3, the chloride anions in the crystals of 2 also showed short contact between the anions and chlorine substituents in QN-CH2Cl+ cations indicating charge-assisted Cl-Cl halogen bonding. To elucidate the distinctions in the reactivity and intermolecular bonding in these systems, we carried out computational analysis as follows.
3.2. Computational Analysis of XB in Cation/Anion, Neutral/Anion, and Neutral/Neutral Associations
X-ray structural analysis showed the presence of halogen bonding between cations and anions (i.e., QN-CH2Cl+···Cl− and DABCO-CH2I+···I−), between neutral molecules and anions (CHI3···I− and CHI3···Cl−), and between two neutral counterparts in the crystals under study. To compare the characteristics of these forms of bonding, we first used Bader’s quantum theory of atoms in molecules (QTAIM) [43,44]. This represents a powerful methodology for the classification and quantification of chemical bonding based on the topology of the electron densities and energies at bond critical points (BCPs) [43]. Similarly to the previous studies [22,53], this analysis was carried out using the coordinates of the XB complexes extracted from their X-ray structures. It revealed the presence of (3,−1) bond critical points (BCPs) along the C-X···Y bond path in all the XB associations under study (Figure 5).
The values of electron density ρ(r), the Laplacian ∇2ρ(r), and the energy density H(r) at these BCPs are listed in Table 2. They indicate that the electron densities at BCPs are all close to 0.01 a.u. and that the Laplacians are close to 0.03 a.u. regardless of the charges of the species involved in the bonding. These values are typical for noncovalent intermolecular bonding [44,54].
The small positive energy densities, H(r), are also consistent with the relatively weak bonding related primarily to electrostatic interaction. Importantly, previous QTAIM studies showed a strong correlation between the characteristics of BCPs and the strength of the interatomic bonding. For example, the linear relationships between the potential energy V(r) and Lagrangian kinetic energy G(r) densities at the bond critical points and the energies of the appropriate contacts, i.e., Eint = −0.67G(r) and Eint = 0.68V(r), were suggested by Tsirelson et al. [54]. As such, comparable values of the Eint as well as those of ρ(r) and H(r) at the BCPs for the cation–anion, neutral–anion and neutral–neutral associations in Table 2 suggest similar strengths of the corresponding forms of bonding. The conclusion about the comparable interaction strength observed across all these systems was confirmed through noncovalent interaction index (NCI) analysis (which determines whether an interaction is either one of bonding or one that is repulsive based on thedifferences in the reduced gradient of the density in the system compared with that observed for a homogenous electron gas) [45]. This showed essentially green areas with only a hint of a blue color at the locations of the BCPs. This corresponds to the very small negative values of sign(λ2) and thus the attractive noncovalent X···Y interactions in all these systems. (Note that the red areas in the cavities of DABCO and QN correspond to the positive values of sign(λ2), indicating a repulsive interaction.).
It is interesting to note that the suggestion about the comparable strengths of differently charged complexes seems inconsistent with the fact that the electrostatic attraction between cations and anions should be substantially larger than that taking place between neutral molecules and anions or that between two neutral molecules. Indeed, the surfaces of QN-CH2Cl+ and DABCO-CH2+ cations showed positive electrostatic potentials which varied between about 0.08 and 0.20 a.u. (Figure S3 in the Supporting Information). A closer look (by mapping these surfaces only in the positive range) revealed that the areas located around the hydrogen substituents are characterized by more positive potentials than those around the halogen atoms (Figure 6). Nevertheless, the potentials on the surfaces of the halogens along the extensions of the C-X bonds in these cations were slightly higher than on those around the equatorial areas. Most importantly, the values of +0.12 a.u. and 0.14 a.u. measured at these σ-holes in QN-CH2Cl+ and DABCO-CH2+, respectively, were much higher than the maximum potential of +0.043 a.u. on the surface of iodine in CHI3 (note that the polarization caused by the electron-rich counter-parts increases the positive potentials on the surfaces of the XB donors in their associations but is unlikely to substantially change the relative values of the maximum potentials). Furthermore, the potentials around the equatorial areas of the iodine in CHI3 are less negative than those around the iodide or chloride anions. This indicates that there is essentially no correlation between the characteristics of BCPs (and therefore the strength of XB) and the values of the electrostatic potentials on the surfaces of XB donors and acceptors. However, the characteristics of the short contacts in the solid-state associations could be distorted by crystal forces. To test this, we also compared the bonding in the corresponding optimized complexes.
The optimization of the XB association involving the charged species in the gas phase produced complexes in which the XB lengths were considerably shorter than the corresponding values determined in the X-ray structures (Table 3). Furthermore, the optimization starting from the geometry of the experimental XB association QN-CH2Cl+···Cl− produced a complex in which the Cl− anion was located at the side of the QN core (Figure S4 in the Supporting Information).
While the interatomic distances and the QTAIM characteristics in the solid-state structures suggest comparable magnitudes of all the halogen bonds, the binding energies between the cations and anions in the complexes optimized in the gas phase were much higher than those between the neutral XB donor and anions, which in turn are higher than those between neutral species. Apparently, the gas phase is not an appropriate medium for modeling the interactions of the charged species.
Optimizations of the XB complexes (starting from the experimental geometries) using the PCM model and CH2Cl2 or CH3CN as solvents produced local minima showing halogen bonding similar to that in the solid state (note, however, that the calculated structures in which anions are located near the hydrogen substituents on the side of cationic DABCO or QN had lower energies than the corresponding XB complexes, see Figure S4 in the Supporting Information). The interatomic separations in the optimized XB complexes involving charged species were substantially longer than those obtained in the gas phase. As such, they were much closer to that observed in the solid state. In fact, most of the values obtained in CH3CN were within about 0.1 Å of the X-ray structural values. These data confirmed that computations with the moderately polar solvents as media provide substantially better models of the experimental solid-state XB complexes involving charged species (the distances in the neutral–neutral complexes were essentially the same in all three media). Furthermore, binding energies in the cation–anion complexes obtained in these solvents were comparable to those found for the associations between neutral XB donors and anions. Moreover, the binding energies between the neutral CHI3 molecules in these media were only slightly lower than those in the charged associations. These data support the results of QTAIM analysis indicating the comparable strength of the interactions between differently charged species in the solid-state associations.
Finally, to clarify the reasons for the switch from the formation of XB complexes to SN2 reactions, we evaluated the energetics of the intermolecular (halogen and hydrogen) bonding together with the products and transition states for the nucleophilic substitutions involving trimethylamine (as a model of the ternary alkylamine) and various halomethanes (Equations (1)–(3)).
(CH3)3N + CHnXm ⇄ [(CH3)3N···X-CHnXm−1]
(CH3)3N + CHnXm ⇄ [(CH3)3N···H-CHnXm−1]
(CH3)3N + CHnXm → (CH3)3N-CHnXm−1+ + X−
The results of these computations (which were carried out using dichloromethane as the medium) are listed in Table S3 in the Supporting Information and are illustrated in Figure 7.
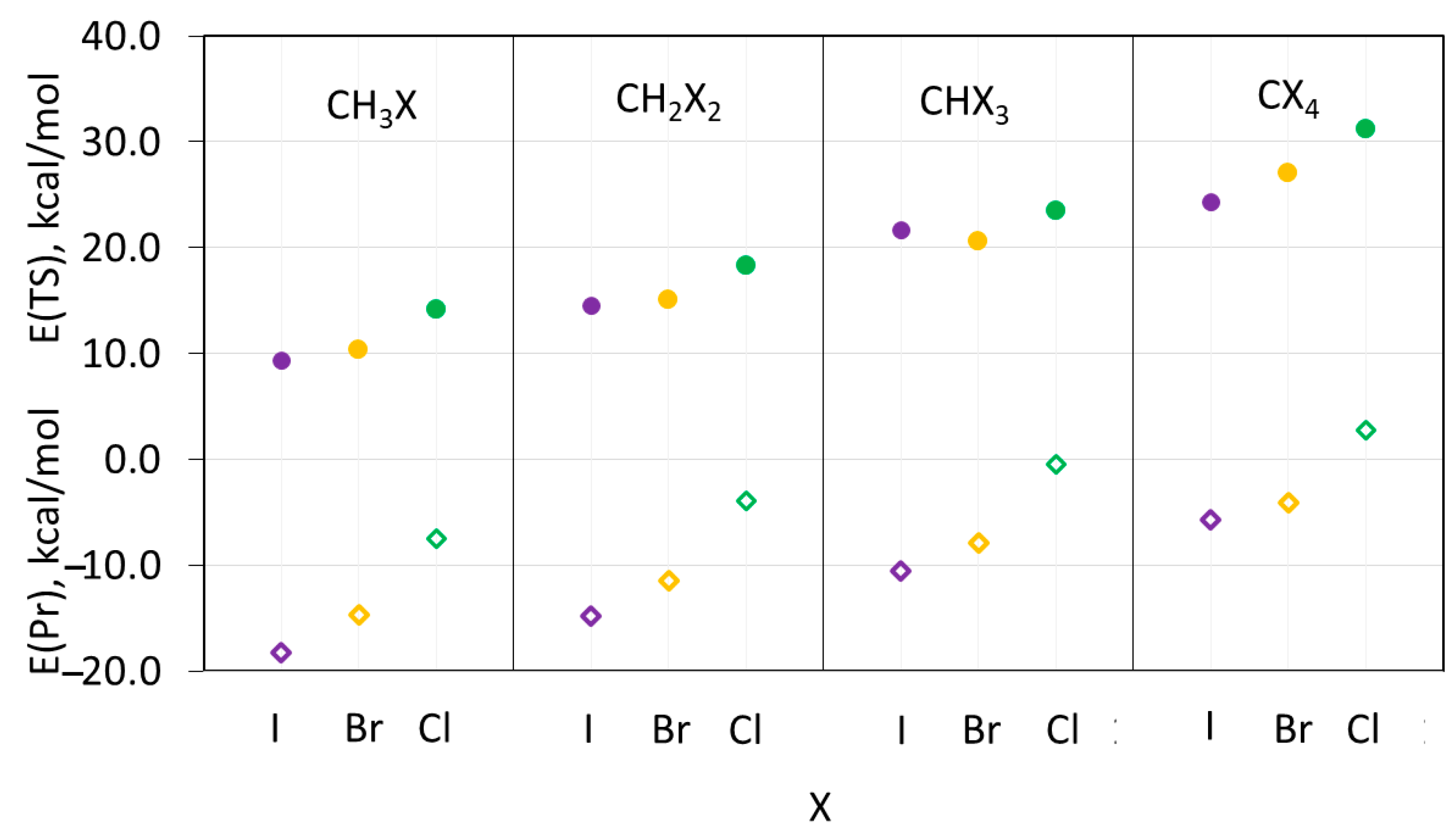
Overall, the energies of the XB and HB complexes in Table S3 and Figure 7 are consistent with the earlier reported data [22,52]. In particular, the stability of the XB complexes of the halomethanes with the same number of halogen substituents increased from chloro- to bromo- to iodo-containing species (Figure 7). On the other hand, the strength of hydrogen bonding with such molecules was not very sensitive to the nature of the halogen atom. In addition, the strength of both types of intermolecular bonding increased with the increase in the number of halogen substituents. These calculations indicated that not only CH2Cl2 but also CH3Cl form stable XB and HB complexes with the ternary amines. Most relevant for the current study, however, is the fact that the TS energies increase with the increase in the number of halogen substituents (Figure 8).
Furthermore, for the reactions of halomethanes with the same numbers of halogen substituents, the TS energies increase, in most cases, in the order of I < Br < Cl. The fact that the reactions involving halomethanes with the smallest substituents, which comprise chlorine, are characterized by the highest TS energies suggests that the steric hindrance for the nucleophilic attack is not the determining factor in these processes. Moreover, the C–X bond dissociation energies (BDEs) in halomethanes increase in the order of I < Br < Cl, which is consistent with the increase in TS energies. However, the BDE values in these species decrease with the increase in the number of the halogen substituents (e.g., either from 81.7 kcal/mol to 77.6 kcal/mol and 72.7 kcal/mol in CH2Cl2, CHCl3, and CCL4, respectively, or from 67.0 kcal/mol to 59.1 kcal/mol in CHBr3 and CBr4, respectively) [56]. This trend contrasts the variations in the TS energies, which increase with the rise in the number of halogen substituents. This distinction suggests that the BDEs of the C–X bonds are also not the determining factor of the TS energies. This also excludes the possibility of the SN1 nucleophilic substitution mechanism for these systems. On the other hand, Figure 8 demonstrates that the increase in the TS energies follows the increase in the energies of the products of the nucleophilic substitution. While this correlation is characterized by the R2 of only 0.75 (Figure S5 in the Supporting information), it suggests that the product energies play a critical role in the interchange between the formation of XB and the HB complexes and the nucleophilic reactions in these systems. In addition, the formation of the stronger XB complexes between amines and tri- and tetrahalomethane (Figure 7) can also hinder nucleophilic substitution [57].
4. Conclusions
The interaction of DABCO or QN with di- and triiodomethane resulted in the formation of ionic salts, thereby showing halogen bonding between cationic and anionic, neutral and anionic, or two neutral counterparts. The QTAIM analysis of the solid-state complexes (Table 2) showed comparable characteristics of halogen bonding regardless of the charges of the XB donors and acceptors. The energetics of the XB complexes, which were optimized in more or less polar solvents, were consistent with this conclusion. Computational analysis also revealed that the changeover from the nucleophilic substitution to the formation of XB complexes with the increase in the number of the halogen substituents from two to three is not determined by the increase in the steric hindrance for an SN2 reaction or by the changes in BDE of the halomethanes. Instead, it is apparently related to the changes in the stability of the products of the nucleophilic reactions.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst14020124/s1, Figure S1: IR spectra of [DABCO-CH2I]I. Figure S2: X-ray structure of 5. Figure S3: ESP of [DABCO-CH2I]+ and [QN-CH2Cl]+ shown in −02 to 0.2 a.u. range. Figure S4: Optimized hydrogen-bonded [QN-CH2Cl]+·Cl− and [DABCO-I]+·I− complexes. Figure S5: Correlation between energies of TSs and products. Table S1: Energies of the XB complexes and their components. Table S2: Energies of XB complexes and their components for CHmXn + N(CH3)3 systems. Table S3: Relative energies of the XB and HB complexes, TS and products of the SN2 reactions between CHmXn, and N(CH3)3. Atomic coordinates of the optimized XB complexes.
Author Contributions
Conceptualization and methodology, S.V.R.; crystal preparation, O.G. and S.V.R.; X-ray crystallographic analysis, M.Z.; quantum mechanical computations and data analysis, S.V.R.; writing—original draft preparation, S.V.R.; writing—review and editing, O.G., M.Z., and S.V.R.; visualization, S.V.R.; supervision, S.V.R.; funding acquisition, S.V.R. and M.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Division of Chemistry of the National Science Foundation, Grant No. CHE-2003603. X-ray measurements were supported by the National Science Foundation through the Major Research Instrumentation Program, Grant No. CHE 1625543 (funding for the single crystal X-ray diffractometer).
Data Availability Statement
Complete crystallographic data, in the CIF format, have been deposited with the Cambridge Crystallographic Data Centre. CCDC 2314969–2314972 and 2321200 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge through www.ccdc.cam.ac.uk/data_request/cif.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Metrangolo, P.; Resnati, G.; Poilati, T.; Biella, S. Halogen bonding in crystal engineering. Struct. Bond. 2008, 126, 105–136. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, C.; Hines, R.; Zeller, M.; Rosokha, S.V. Continuum of covalent to intermolecular bonding in the halogen-bonded complexes of 1,4-diazabicyclo[2.2.2]octane with bromine-containing electrophiles. Chem. Commun. 2018, 54, 8060–8063. [Google Scholar] [CrossRef] [PubMed]
- Loy, C.; Zeller, M.; Rosokha, S.V. Halogen bonding in the complexes of brominated electrophiles with chloride anions: From a weak supramolecular interaction to a covalent Br–Cl bond. Crystals 2020, 10, 1075. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electrostatics and polarization in σ- and π-hole noncovalent interactions: An overview. ChemPhysChem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Holthoff, J.M.; Weiss, R.; Rosokha, S.V.; Huber, S.M. Anti-electrostatic halogen bonding between Ions of Like Charges. Chem. Eur. J. 2021, 27, 16530–16542. [Google Scholar] [CrossRef]
- Loy, C.; Holthoff, J.M.; Weiss, R.; Huber, S.M.; Rosokha, S.V. Anti-electrostatic” halogen bonding in solution. Chem. Sci. 2021, 12, 8246–8251. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willett, R.D.; Twamley, B. The role of charge assisted arylhalogen-halide ion interactions in the structures of the dibromopyridinium halide salts. J. Mol. Struct. 2009, 918, 116–122. [Google Scholar] [CrossRef]
- Cametti, M.; Raatikainen, K.; Metrangolo, P.; Pilati, T.; Terraneo, G.; Resnati, G. 2-Iodo-imidazolium receptor binds oxoanions via charge-assisted halogen bonding. Org. Biomol. Chem. 2012, 10, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Kassl, C.J.; Swenson, D.C.; Pigge, F.C. Charge-assisted halogen bonding in bromo- and iodophenylpyridinium chlorides. Cryst. Growth Des. 2015, 15, 4571–4580. [Google Scholar] [CrossRef]
- Zapata, F.; Caballero, A.; White, N.G.; Claridge, T.D.W.; Costa, P.J.; Felix, V.; Beer, P.D. Fluorescent charge-assisted halogen-bonding macrocyclic halo-imidazolium receptors for anion recognition and sensing in aqueous media. J. Am. Chem. Soc. 2012, 134, 11533–11541. [Google Scholar] [CrossRef]
- Fotovic, L.; Stilinovic, V. Halogenide anions as halogen and hydrogen bond acceptors in iodopyridinium halogenides. CrystEngComm 2020, 22, 4039–4046. [Google Scholar] [CrossRef]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate chiral bis(imidazolium)-based halogen-bond donors: Synthesis and applications in enantioselective recognition and catalysis. Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [Google Scholar] [CrossRef]
- Fotovic, L.; Stilinovic, V. Halogen Bonding in N-Alkyl-3-halogenopyridinium Salts. Crystals 2021, 11, 1240. [Google Scholar] [CrossRef]
- Fotovic, L.; Bedekovic, N.; Stilinovic, V. Evaluation of Halogenopyridinium Cations as Halogen Bond Donors. Cryst. Growth Des. 2021, 21, 6889–6901. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, G.; Xu, Z.; Chen, Z.; Wang, J.; Shi, J.; Zhu, W. Halogen bonding in differently charged complexes: Basic profile, essential interaction terms and intrinsic σ-Hole. Phys. Chem. Chem. Phys. 2019, 21, 15106–15119. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Z.; Lu, Y.; Xu, Z.; Wu, W.; Zhu, W.; Peng, C.; Liu, H. Halogen bonding interactions in ion pairs versus conventional charge-assisted and neutral halogen bonds: A theoretical study based on imidazolium species. RSC Adv. 2015, 5, 74284–74294. [Google Scholar] [CrossRef]
- Posavec, L.; Nemec, V.; Stilinović, V.; Cinčić, D. Halogen and hydrogen bond motifs in ionic cocrystals derived from 3-halopyridinium halogenides and perfluorinated iodobenzenes. Cryst. Growth Des. 2021, 21, 6044–6050. [Google Scholar] [CrossRef]
- Pfrunder, M.C.; Micallef, A.S.; Rintoul, L.; Arnold, D.P.; Davy, K.J.P.; McMurtrie, J. Exploitation of the Menshutkin reaction for the controlled assembly of halogen bonded architectures incorporating 1,2-diiodotetrafluorobenzene and 1,3,5-triiodotrifluorobenzene. Cryst. Growth Des. 2012, 12, 714–724. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Kinzhalov, M.A.; Novikov, A.S.; Ananyev, I.V.; Romanova, A.A.; Boyarskiy, V.P.; Haukka, M.; Kukushkin, V.Y. H2C(X)–X···X– (X = Cl, Br) Halogen bonding of dihalomethanes. Cryst. Growth Des. 2017, 17, 1353–1362. [Google Scholar] [CrossRef]
- Gustafsson, B.; Håkansson, M.; Jagner, S. Copper(I)-mediated quaternisation of 1,4-diazabicyclo[2.2.2]octane (DABCO). Crystal structure of bis{(1-chloromethyl-4-aza-1-azoniabi cyclo [2.2.2]octane)-μ-chloro-chlorocopper(I)}. Inorg. Chim. Acta 2005, 358, 1309–1312. [Google Scholar] [CrossRef]
- Hua, X.-N.; Liao, W.-Q.; Tang, Y.-Y.; Li, P.-F.; Shi, P.-P.; Zhao, D.; Xiong, R.-G. A Room-temperature hybrid lead iodide perovskite ferroelectric. J. Am. Chem. Soc. 2018, 140, 12296–12302. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liao, W.-Q.; Ai, Y.; Li, J.; Deng, S.; Hou, Y.; Tang, Y.-Y. Precise molecular design toward organic-inorganic zinc chloride abx3 ferroelectrics. J. Am. Chem. Soc. 2020, 142, 6236–6243. [Google Scholar] [CrossRef]
- March, J. Advanced Organic Chemistry. In Reaction Mechanisms and Structure, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1992. [Google Scholar]
- Adeniyi, E.; Grounds, O.; Stephens, Z.; Zeller, M.; Rosokha, S.V. Thermodynamics and spectroscopy of halogen- and hydrogen-bonded complexes of haloforms with aromatic and aliphatic amines. Molecules 2022, 27, 6124. [Google Scholar] [CrossRef]
- Blackstock, S.C.; Lorand, J.P.; Kochi, J.K. Charge-transfer interactions of amines with tetrahalomethanes. x-ray crystal structures of the donor-acceptor complexes of quinuclidine and diazabicyclo [2.2.2]Octane with carbon tetrabromide. J. Org. Chem. 1987, 52, 1451–1460. [Google Scholar] [CrossRef]
- Bruker. Apex3 v2019.1-0, SAINT V8.40A.; Bruker AXS Inc.: Madison, WI, USA, 2013/2014. [Google Scholar]
- SHELXTL Suite of Programs. Version 6.14, 2000-2003, Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G. Crystal Structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Hübschle, C.; Sheldrick, G.; Dittrich, B. ShelXle: A Qt Graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design an assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xu, Z.; Zhu, W. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Inf. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef]
- Bauza, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the reliability of pure and hybrid DFT methods for the evaluation of halogen, chalcogen, and pnicogen bonds involving anionic and neutral electron donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Is the use of diffuse functions essential for the properly description of noncovalent interactions involving anions? J. Phys. Chem. A 2013, 117, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Popelier, P.L.A. The QTAIM perspective of chemical bonding. In The Chemical Bond; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; p. 271. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Chernyshov, I.Y.; Ananyev, I.V.; Pidko, E.A. Revisiting van der Waals radii: From comprehensive structural analysis to knowledge-based classification of interatomic contacts. Chemphyschem 2020, 21, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Holl, S. Interaction in molecular crystals. Part 168. σ-donor/acceptor complexes {HCI3···XJ} (X− = Cl−, Br−, I−) of triiodomethane in tetraphenylphosphonium halides. Z. Naturforsch. B 2014, 56, 152–163. [Google Scholar] [CrossRef]
- du Mont, W.-W.; Stenzel, V.; Jeske, J.; Jones, P.G.; Sebald, A.; Pohl, S.; Saak, W.; Baetcher, M. Destructive or cooperative attack of iodide anions on alkyltriiodophosphonium cations: Elimination of iodine in solution and layer structures in the solid state. Inorg. Chem. 1994, 33, 1502–1505. [Google Scholar] [CrossRef]
- Watson, B.; Grounds, O.; Borley, W.; Rosokha, S.V. Resolving halogen vs hydrogen bonding dichotomy in solutions: Intermolecular complexes of trihalomethanes with halide and pseudohalide anions. Phys. Chem. Chem. Phys. 2018, 2, 21999–22007. [Google Scholar] [CrossRef]
- Mertsalov, D.F.; Gomila, R.M.; Zaytsev, V.P.; Grigoriev, M.S.; Nikitina, E.V.; Zubkov, F.I.; Frontera, A. On the importance of halogen bonding interactions in two x-ray structures containing all four (F, Cl, Br, I) halogen atoms. Crystals 2021, 11, 1406. [Google Scholar] [CrossRef]
- Miller, D.K.; Loy, C.; Rosokha, S.V. Examining a transition from supramolecular halogen bonding to covalent bonds: Topological analysis of electron densities and energies in the complexes of bromosubstituted electrophiles. ACS Omega 2021, 6, 23588–23597. [Google Scholar] [CrossRef]
- Bertolotti, F.; Shishkina, A.V.; Forni, A.; Gervasio, G.; Stash, A.I.; Tsirelson, V.G. Intermolecular bonding features in solid iodine. Cryst. Growth Des. 2014, 14, 3587–3595. [Google Scholar] [CrossRef]
- Isse, A.A.; Lin, C.Y.; Coote, M.L.; Gennaro, A. Estimation of standard reduction potentials of halogen atoms and alkyl halides. J. Phys. Chem. B 2011, 115, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Kennepohl, P. Catalytic activation via pi-backbonding in halogen bonds. Faraday Discuss. 2023, 244, 241–251. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Structures of the XB donors and acceptors and the potential modes of their interactions.

Figure 1.
Fragment of the X-ray structure of 1 showing the 2D network formed by halogen and the bonding between DABCO-CH2I+ and iodide. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine. Blue lines show contacts shorter than van der Waals separations.
Figure 1.
Fragment of the X-ray structure of 1 showing the 2D network formed by halogen and the bonding between DABCO-CH2I+ and iodide. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine. Blue lines show contacts shorter than van der Waals separations.
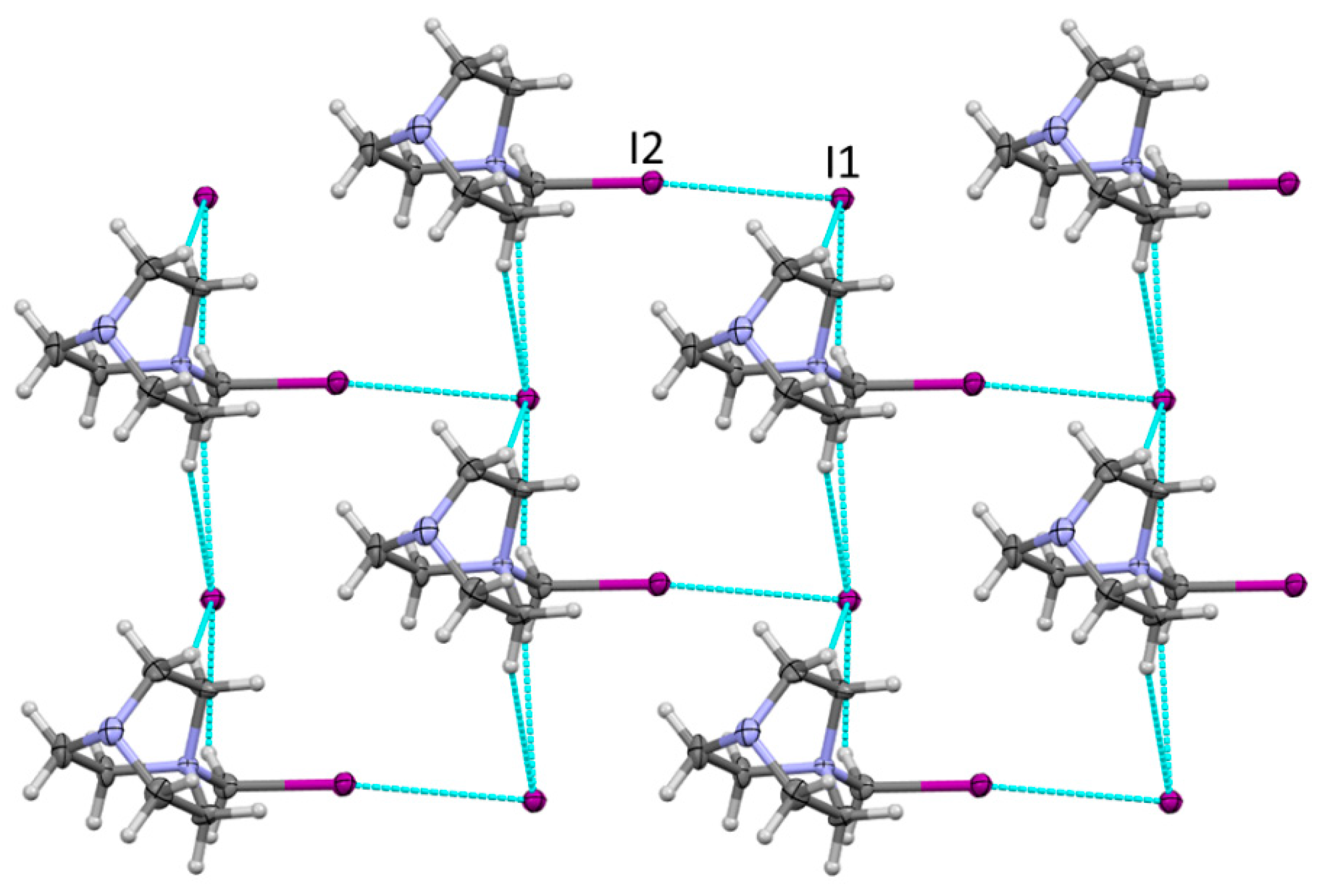
Figure 2.
Fragment of the X-ray structure of 2 showing XB and HB between Cl−, QN-CH2Cl+, and CHI3. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine, green—chlorine.
Figure 2.
Fragment of the X-ray structure of 2 showing XB and HB between Cl−, QN-CH2Cl+, and CHI3. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine, green—chlorine.
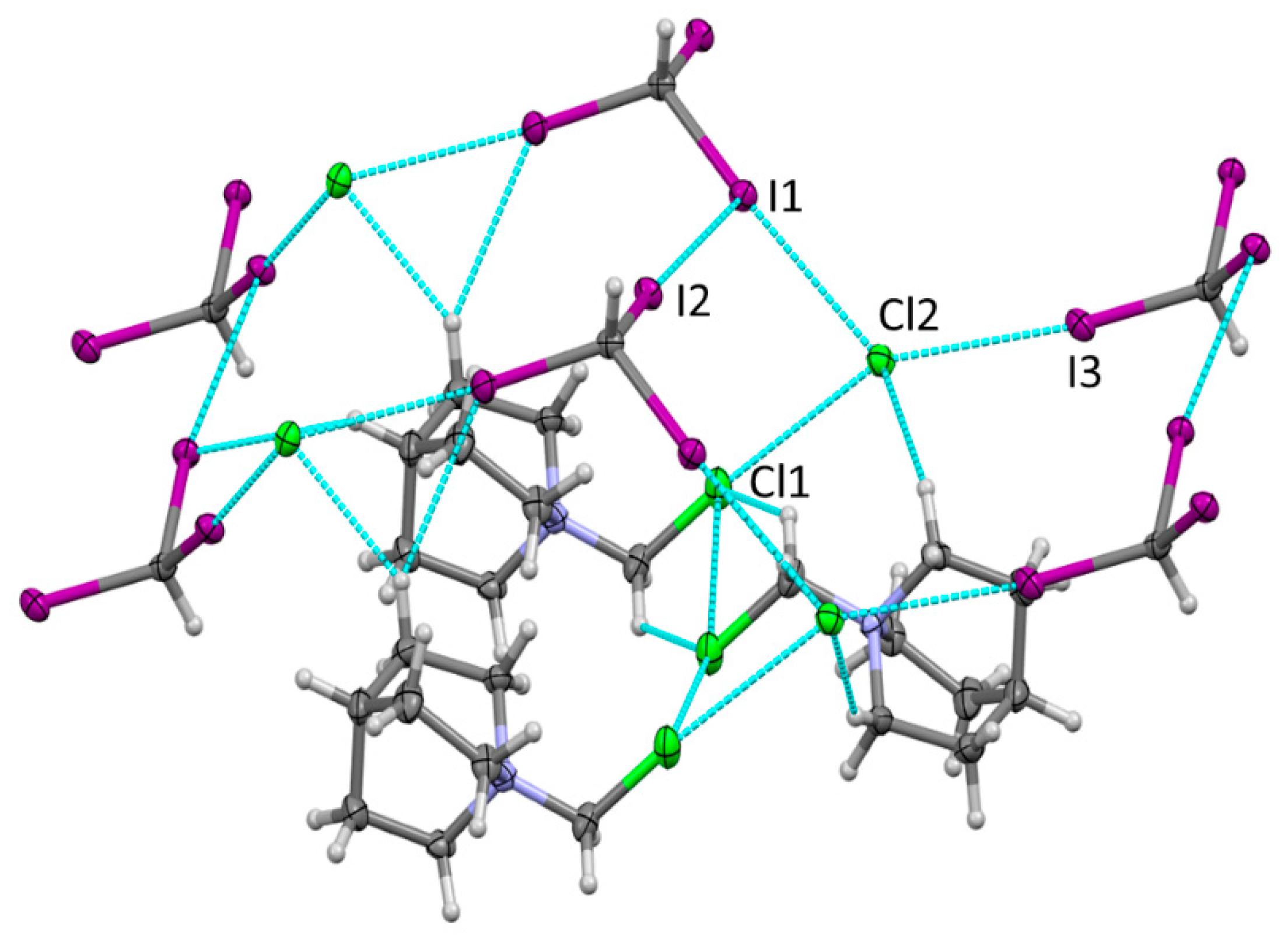
Figure 3.
Fragment of the X-ray structure of 3 showing the zigzag ladder formed by XB between CHI3 and I−, as well as networks of QN-CH2Cl+ cations. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine, green—chlorine.
Figure 3.
Fragment of the X-ray structure of 3 showing the zigzag ladder formed by XB between CHI3 and I−, as well as networks of QN-CH2Cl+ cations. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine, green—chlorine.
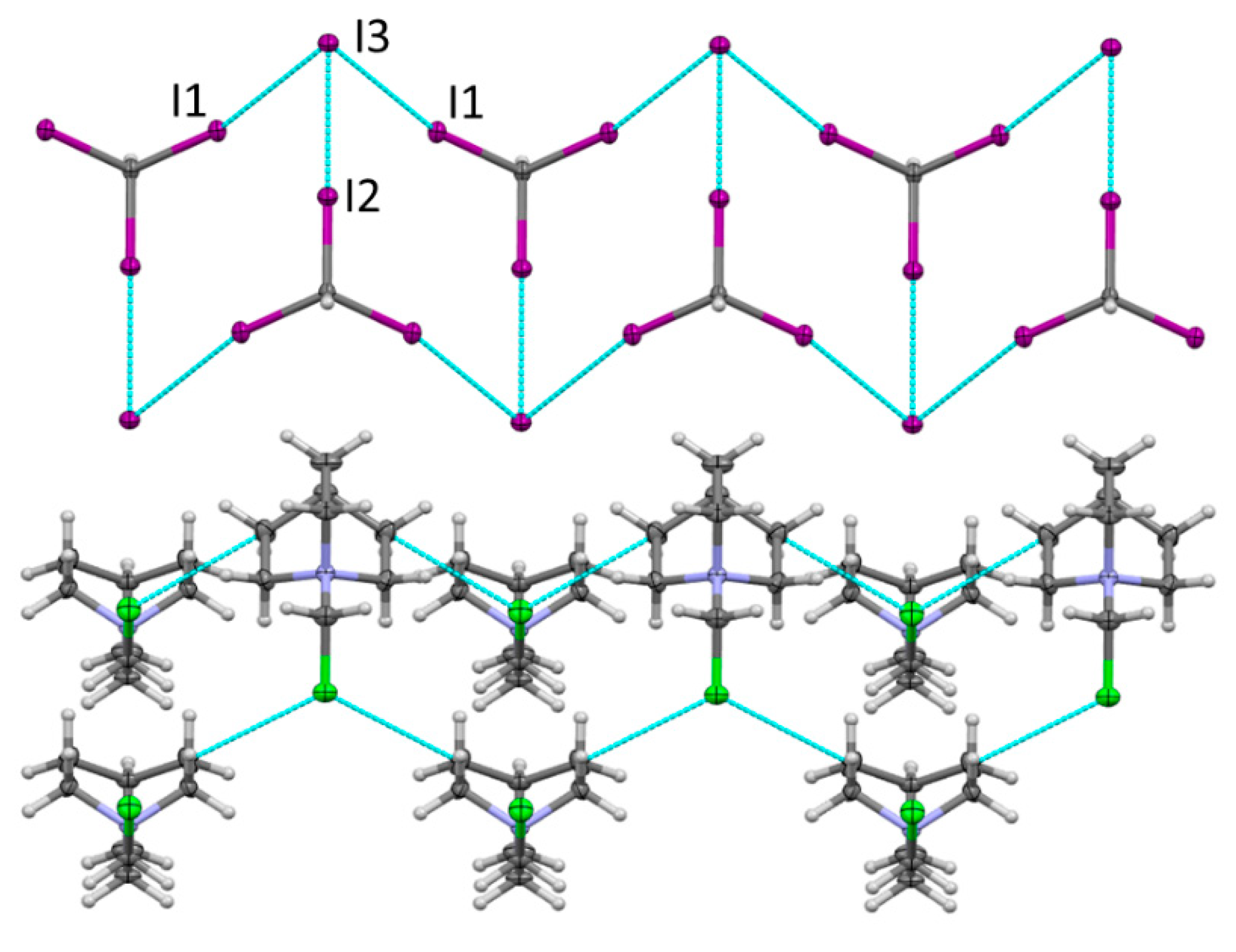
Figure 4.
Fragment of the crystal structure of 4 showing zigzag ladders formed by halogen bonding of CHI3 and iodide separated by layers of QN-CH2COCH3+ cations. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine, green—chlorine.
Figure 4.
Fragment of the crystal structure of 4 showing zigzag ladders formed by halogen bonding of CHI3 and iodide separated by layers of QN-CH2COCH3+ cations. Color code: dark gray—carbon, light gray—hydrogen, blue—nitrogen, purple—iodine, green—chlorine.
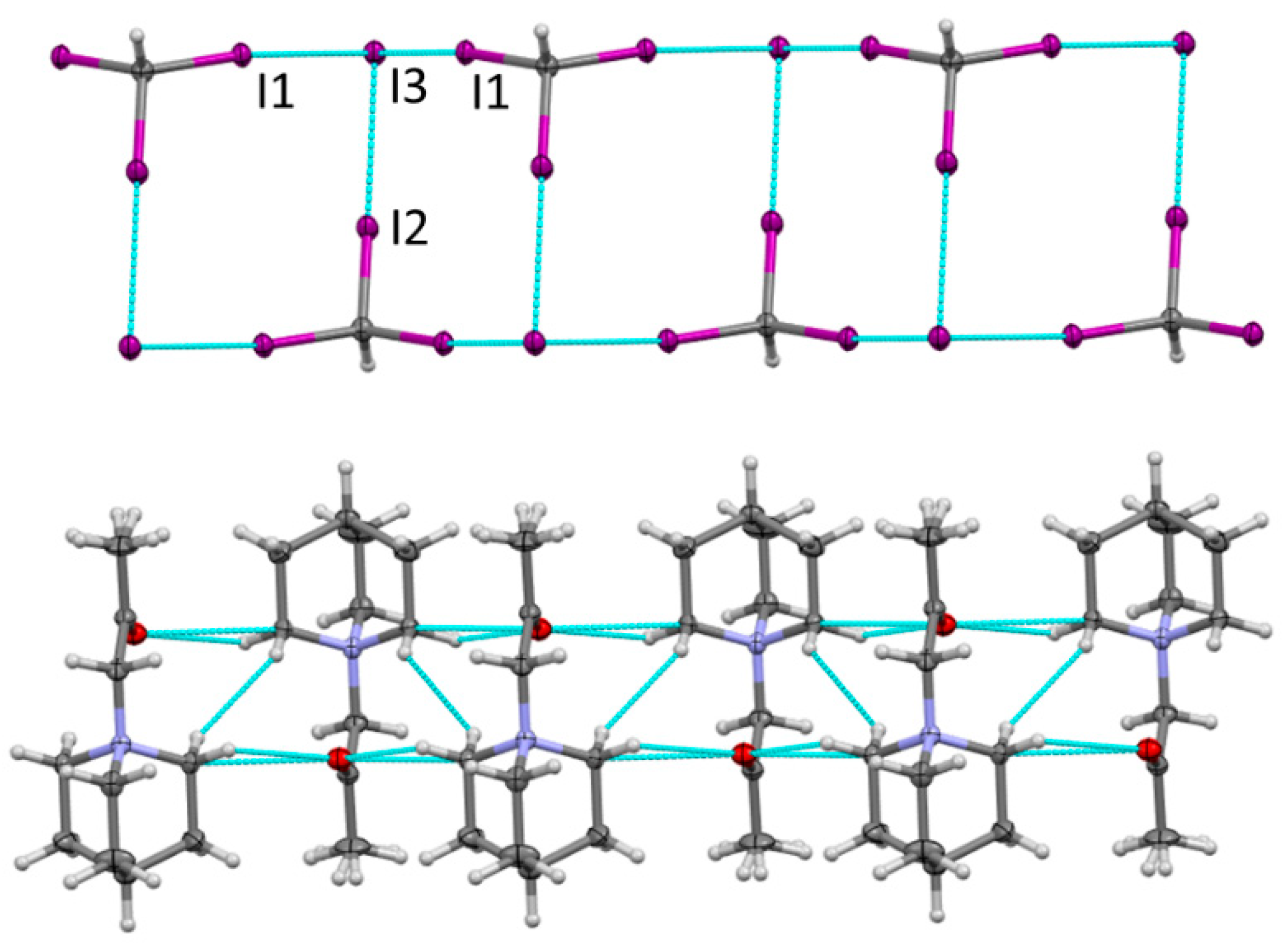
Figure 5.
Superposition of the results of the QTAIM and NCI analyses of DABCO-CH2I+···I− (A), QN-CH2Cl+···Cl− (B), CHI3···I− (C), CHI3···Cl− (D), and CHI3···CHI3 (E). The bond paths and BCPs (QTAIM) are shown as orange lines and spheres, respectively, and blue-green areas indicate bonding interactions (NCI).
Figure 5.
Superposition of the results of the QTAIM and NCI analyses of DABCO-CH2I+···I− (A), QN-CH2Cl+···Cl− (B), CHI3···I− (C), CHI3···Cl− (D), and CHI3···CHI3 (E). The bond paths and BCPs (QTAIM) are shown as orange lines and spheres, respectively, and blue-green areas indicate bonding interactions (NCI).

Figure 6.
Surface electrostatic potentials (at an isovalue of 0.001 in atomic units) of XB donors CHI3 (A), QN-CH2Cl+ (B), and DABCO-CH2I+ (C).
Figure 6.
Surface electrostatic potentials (at an isovalue of 0.001 in atomic units) of XB donors CHI3 (A), QN-CH2Cl+ (B), and DABCO-CH2I+ (C).
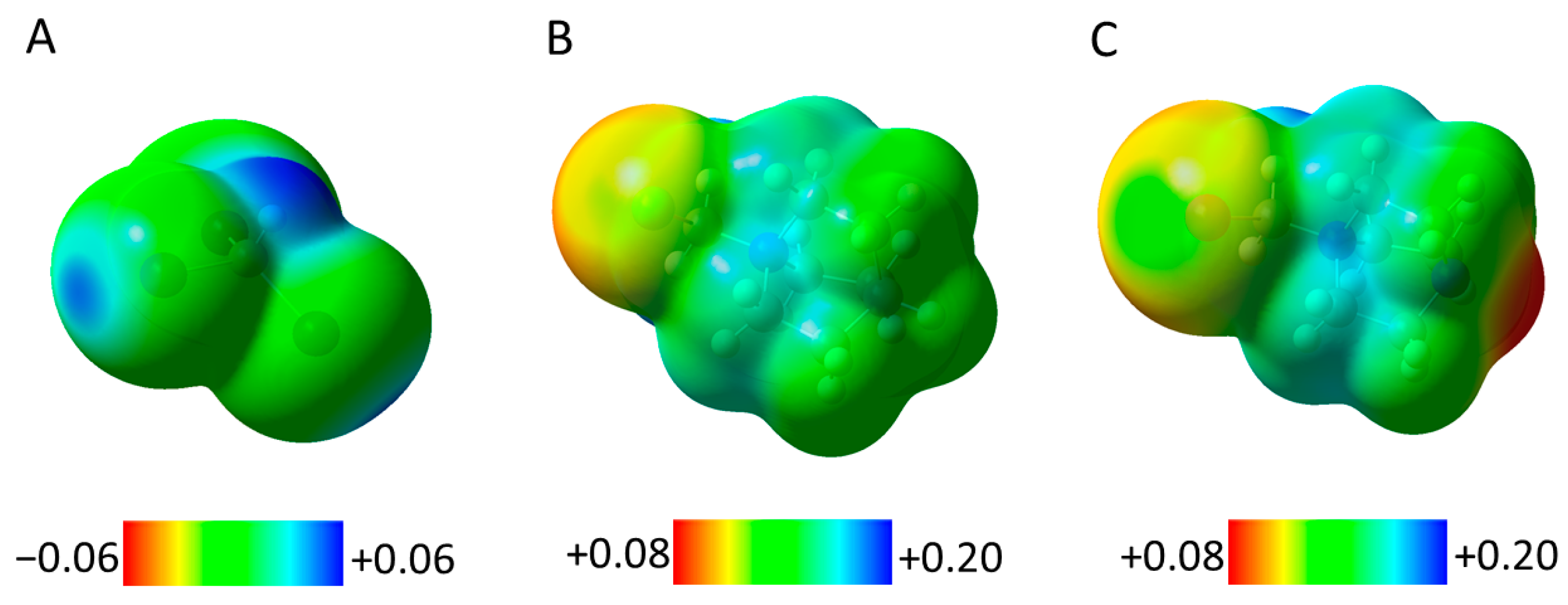
Figure 7.
Binding energies in the XB (filled circles) and HB (open rhombics) complexes between (CH3)3N and halomethanes CHnXm. Color code: purple—iodine, yellow—bromine, green—chlorine.
Figure 7.
Binding energies in the XB (filled circles) and HB (open rhombics) complexes between (CH3)3N and halomethanes CHnXm. Color code: purple—iodine, yellow—bromine, green—chlorine.
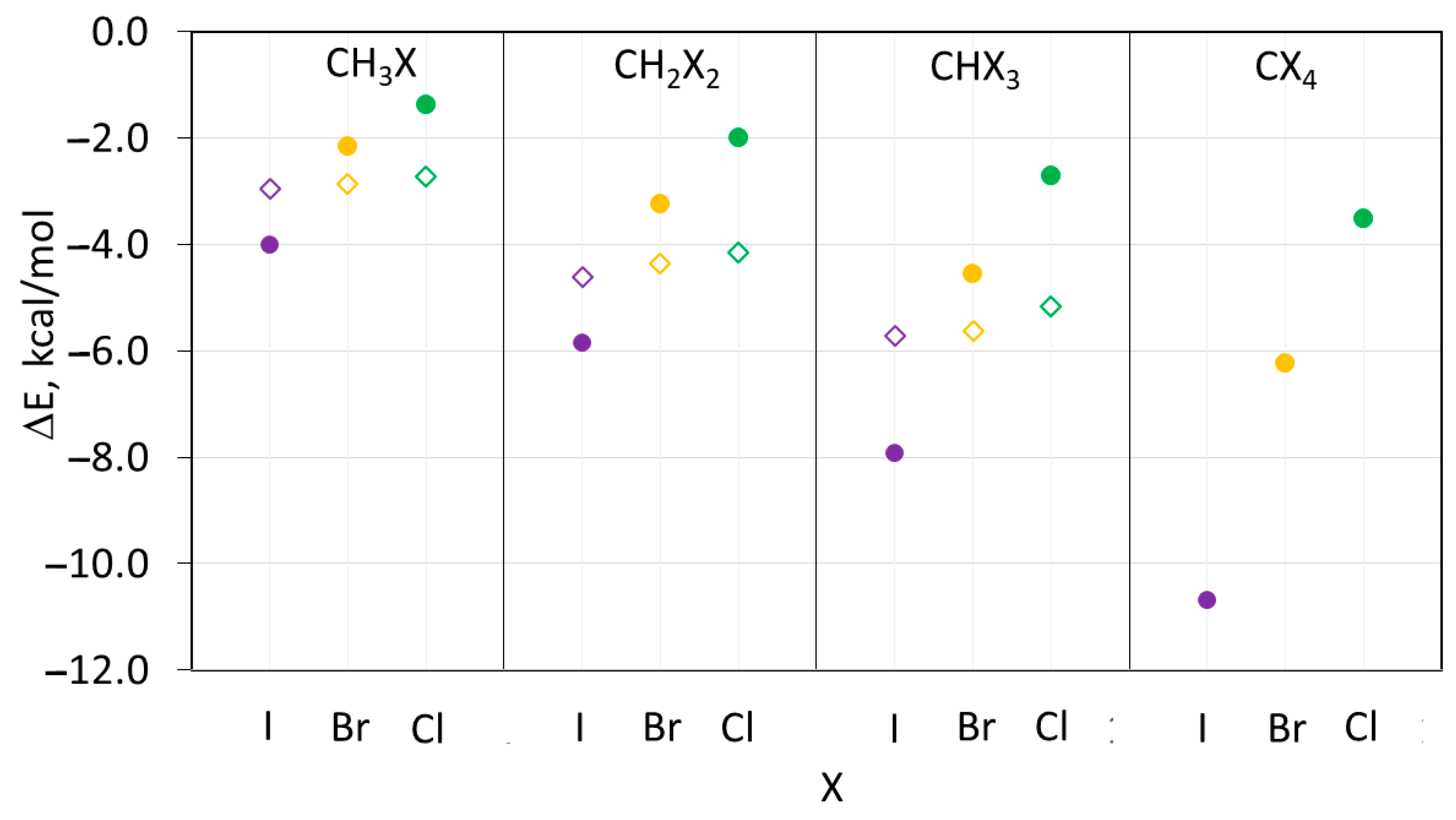
Figure 8.
Energies (relative to reactants) of the transition states (filled circles) and products (open rhombics) of the SN2 reaction between (CH3)3N and halomethanes CHnXm. Color code: purple—iodine, yellow—bromine, green—chlorine.
Figure 8.
Energies (relative to reactants) of the transition states (filled circles) and products (open rhombics) of the SN2 reaction between (CH3)3N and halomethanes CHnXm. Color code: purple—iodine, yellow—bromine, green—chlorine.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Characteristics of halogen bonding in solid-state associations.
| Crystal | Contact | d(X···Y), Å | ∠(C-X-Y), deg | RXY a |
|---|---|---|---|---|
| 1 | C-I2+···I1− | 3.670 | 160.6 | 0.93 |
| 2 | C-I3···Cl2− | 3.317 | 171.2 | 0.89 |
| C-I1···Cl2− | 3.276 | 171.4 | 0.88 | |
| C-Cl1+···Cl2− | 3.361 | 175.7 | 0.96 | |
| C-I1···I2-C | 3.637 | 170.6 | 0.91 | |
| 3 | C-I2···I3− | 3.581 | 167.7 | 0.90 |
| C-I1···I3− | 3.652 | 163.4 | 0.92 | |
| 4 | C-I1···I3− | 3.644 | 176.5 | 0.92 |
| C-I2···I3− | 3.648 | 160.2 | 0.92 |
a Normalized interatomic separations, RXY = dX···Y/(rX + rY), where rX and ry are van der Waals radii.
Table 2.
Electron and energy densities, ρ(r) and H(r), and Laplacians of density, ∇2ρ(r), at BCPs (all in a.u.), and the corresponding interaction energies in the solid-state complexes.
Table 2.
Electron and energy densities, ρ(r) and H(r), and Laplacians of density, ∇2ρ(r), at BCPs (all in a.u.), and the corresponding interaction energies in the solid-state complexes.
| Complex a | ρ(r) | ∇2ρ(r) | H(r) | G(r) | V(r) | Eint b | Eint c |
|---|---|---|---|---|---|---|---|
| DABCO-CH2I+···I− | 0.0123 | 0.0285 | 0.0006 | 0.0065 | −0.0059 | −2.7 | −2.5 |
| QN-CH2Cl+···Cl− | 0.0094 | 0.0316 | 0.0014 | 0.0065 | −0.0051 | −2.7 | −2.2 |
| CHI3···I− | 0.0129 | 0.0297 | 0.0006 | 0.0069 | −0.0063 | −2.9 | −2.7 |
| CHI3···Cl− | 0.0157 | 0.0447 | 0.0011 | 0.0101 | −0.0090 | −4.3 | −3.9 |
| CHI3···CHI3 | 0.0122 | 0.0311 | 0.0008 | 0.0070 | −0.0063 | −3.0 | −2.7 |
Table 3.
Halogen bond lengths and energies in the optimized complexes.
| Gas Phase | Dichloromethane | Acetonitrile | ||||
|---|---|---|---|---|---|---|
| Complex | dX···Y, Å | ΔE, kcal/mol | dX···Y, Å | ΔE, kcal/mol | dX···Y, Å | ΔE, kcal/mol |
| DABCO-CH2I+···I− | 3.006 | −78.6 | 3.506 | −10.4 | 3.596 | −5.2 |
| QN-CH2Cl+···Cl− | N/A a | −95.3 | 3.216 | −6.7 | 3.324 | −2.6 |
| CHI3···I− | 3.166 | −18.3 | 3.513 | −4.6 | 3.557 | −3.9 |
| CHI3···Cl− | 2.733 | −28.0 | 3.057 | −6.8 | 3.108 | −5.7 |
| CHI3···CHI3 | 3.757 | −2.0 | 3.790 | −1.9 | 3.754 | −1.8 |
a The optimization produced a complex without the Cl···Cl halogen bonding (see text).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Grounds, O.; Zeller, M.; Rosokha, S.V. Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines. Crystals 2024, 14, 124. https://doi.org/10.3390/cryst14020124
AMA Style
Grounds O, Zeller M, Rosokha SV. Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines. Crystals. 2024; 14(2):124. https://doi.org/10.3390/cryst14020124
Chicago/Turabian StyleGrounds, Olivia, Matthias Zeller, and Sergiy V. Rosokha. 2024. "Halogen Bonding versus Nucleophilic Substitution in the Co-Crystallization of Halomethanes and Amines" Crystals 14, no. 2: 124. https://doi.org/10.3390/cryst14020124
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.