
Dinuclear Molybdenum(VI) Complexes Based on Flexible Succinyl and Adipoyl Dihydrazones

1
Department of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, 10000 Zagreb, Croatia
2
Department of Chemistry and Biochemistry, School of Medicine, University of Zagreb, Šalata 3, 10000 Zagreb, Croatia
*
Author to whom correspondence should be addressed.
Crystals 2024, 14(2), 135; https://doi.org/10.3390/cryst14020135
Submission received: 5 January 2024
/
Revised: 23 January 2024
/
Accepted: 25 January 2024
/
Published: 29 January 2024
(This article belongs to the Special Issue The Polyhedral Face of Coordination Chemistry)
Abstract
:A series of molybdenum(VI) complexes with aryl-functionalized alkyl dihydrazones was prepared by the reaction of [MoO2(acac)2] and the appropriate dihydrazone in methanol. Their solid-state structures were elucidated via single-crystal X-ray diffraction (SC-XRD) and Fourier-transform infra-red (FTIR) spectroscopy, while the thermal stability of compounds was inspected by combined thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) experiments. The behaviour of complexes in DMSO-d6 solution was explored by nuclear magnetic resonance (NMR). The relevant data show that all complexes are dinuclear, with dihydrazones acting as ditopic hexadentate ligands. The in vitro cytotoxic activity of the prepared molybdenum(VI) complexes was evaluated on THP-1 and HepG2 cell lines, while their antibacterial activity was tested against Staphylococcus aureus, Enterococcus faecalis, Escherichia coli, and Moraxella catarrhalis bacteria. The majority of compounds proved to be non-cytotoxic, while some exhibited superior antibacterial activity in comparison to dihydrazone ligands.

1. Introduction
Over the last few decades, hydrazones have emerged as the privileged class of ligands owing to their stability, acid-base properties, and structural modularity [1]. Hydrazones are recognized for their remarkable coordination chemistry, which arises from their ability to act as flexible ligands, forming stable complexes with various metals [2]. This flexibility is attributed to the hydrazone’s nitrogen and oxygen atoms, which can engage in both chelating and bridging modes in metal–organic frameworks (MOFs) [3]. Such features make them suitable for the development of metal–organic assemblies for specialized applications related to, e.g., magnetism, catalysis [4], or biomedicine [5,6,7,8]. Furthermore, the prospect of E/Z isomerization, particularly as a response to different stimuli, such as light, pH, or heat, renders these systems suitable for the design of molecular switches or even more complex stimuli-responsive metal–organic architectures [9,10]. On the other hand, appropriate modification of the hydrazonic scaffolds can give rise to metal or covalent–organic frameworks for practical applications such as gas storage or separations [11].
By introducing more than one hydrazonic functionality within the same ligand molecule, and thus enhancing its coordination potential, one can target more complex metal-organic structures and functions as compared to the monohydrazone counterparts [12]. Multihydrazones, particularly those with alkyl chains, exhibit enhanced flexibility and can form more intricate and dynamic structures [13]. A notable example is the alkyl dihydrazone-based multinuclear Cu cages, which have been studied for their unique magnetic properties [14,15]. Structures of this type have also shown properties like magnetic refrigeration and slow magnetic relaxation, which could have a number of promising applications [16].
Complexation with appropriate metal cations is often beneficial when one aims to modulate the bioactivity of the related organic entities [17,18]. For monohydrazones, whose antibacterial, antifungal, and antitumor properties have been widely acclaimed [19,20,21,22], derivatization via metal cation coordination has proven to be a viable route towards complexes with altered or enhanced biological properties [23]. The flexible nature of succinyl and adipoyl dihydrazones and its influence on the properties of the corresponding complexes, coupled with the fact that dihydazones have been considerably less investigated as bioactive compounds [24], motivated us to explore the cytotoxic and antibacterial activity of dihydrazone-based Mo(VI) complexes and compare it with those of the free ligands [25].
Here, we present a solid-state and a solution study of a series of dinuclear molybdenum(VI) complexes with aryl-functionalized alkyl dihydrazones. Namely, we provide simple synthetic routes towards the title compounds, accompanied by their thorough solid-state analysis via single-crystal X-ray diffraction (SCXRD), Fourier-transform infra-red (FTIR) spectroscopy, and simultaneous thermogravimetry and differential scanning calorimetry (TGA-DSC). In DMSO-d6 solution, complexes were explored by nuclear magnetic resonance (NMR), unveiling symmetrical structures comparable with those established in the solid state. Finally, we focused on an evaluation of the cytotoxic and antibacterial activities of the obtained compounds against selected human cancer cell lines, and Gram-positive and Gram-negative bacterial strains, respectively.
2. Materials and Methods
2.1. Synthesis
Succinic dihydrazones of salicylaldehyde (H4L1), 2-hydroxy-1-naphtaldehyde(H4L2) 2,3-dihydroxybenzaldehyde (H4L3), and 2,4-dihydroxybenzaldehyde (H4L4), as well as adipic dihydrazones of salicylaldehyde (H4L5), 2-hydroxy-1-naphtaldehyde (H4L6) 2,3-dihydroxybenzaldehyde (H4L7), and 2,4-dihydroxybenzaldehyde (H4L8) were prepared by a previously published procedure [15]. [MoO2(acac)2] was synthesized by a well-established synthetic protocol [26]. Chemicals for the synthesis were purchased from TCI and used as received. Methanol, used in syntheses, was purchased from Kemika (Zagreb, Croatia).
In general, complexes were prepared by the reaction of two equivalents of [MoO2(acac)2] and one equivalent of selected dihydrazone suspended in methanol (Supplementary Materials, Scheme S1). The resulting suspensions were refluxed for two hours, during which the ligands slowly reacted. Upon cooling, the crystalline material that was deposited was filtered, washed with a small amount of cold methanol, and dried in air.
2.1.1. Synthesis of [Mo2O4(MeOH)2(L1)]∙2MeOH
Obtained by the reaction of 0.5 mmol (175 mg) of H4L1 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Orange powder. Yield: 0.32 g (88%). Anal. Calcd. For Mo2C22H30N4O8 (734.42): C, 35.98%, H, 4.12%, N, 7.63%, found: C, 35.98%, H, 4.12%, N, 7.86%. IR spectroscopy: 1615, 1599 υ(C=N); 1556, 1541 υ(C=C); 1338 υ(C–Oen); 1270 υ(C–Ophen); 1011 υ(C−OMeOH); 935 υsym(MoO2); 907 υasym(MoO2). TGA analysis: MeOH calcd. 17.45%, found 17.84%; MoO3 calcd. 39.20%, found 36.83%.
2.1.2. Synthesis of [Mo2O4(MeOH)2(L2)]
Obtained by the reaction of 0.5 mmol (225 mg) of H4L2 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Yellow-orange powder. Yield: 0.29 g (76%). Anal. Calcd. For Mo2C28H26N4O6 (770.45): C, 43.65%, H, 3.4%, N, 7.27%, found: C, 41.47%, H, 3.37%, N, 7.05%. IR spectroscopy: 1615, 1596 υ(C=N); 1550, 1534 υ(C=C); 1329 υ(C–Oen); 1278 υ(C–Ophen); 1012 υ(C−OMeOH); 934 υsym(MoO2); 907 υasym(MoO2). TGA analysis: MeOH calcd. 8.32%, found 9.80%; MoO3 calcd. 37.37%, found 34.26%.
2.1.3. Synthesis of [Mo2O4(MeOH)2(L3)]∙2MeOH
Obtained by the reaction of 0.5 mmol (191 mg) of H4L3 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Red powder. Yield: 0.33 g (93%). Anal. Calcd. For Mo2C20H22N4O8 (702.33): C, 34.2%, H, 3.16%, N, 7.98%, found: C, 33.52%, H, 3.29%, N, 7.98%. IR spectroscopy: 1603 υ(C=N); 1568, 1538 υ(C=C); 1340 υ(C–Oen); 1260 υ(C–Ophen); 1015 υ(C−OMeOH); 933 υsym(MoO2); 901 υasym(MoO2). TGA analysis: MeOH calcd. 16.72%, found 17.09%; MoO3 calcd. 37.56%, found 36.23%.
2.1.4. Synthesis of [Mo2O4(MeOH)2(L4)]
Obtained by the reaction of 0.5 mmol (191 mg) of H4L4 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Red powder. Yield: 0.29 g (76%). Anal. Calcd. For Mo2C22H30N4O10 (766.41): C, 34.48%, H, 3.95%, N, 7.31%, found: C, 35.17%, H, 4.11%, N, 7.31%. IR spectroscopy: 1609 υ(C=N); 1571, 1549 υ(C=C); 1334, 1315 υ(C–Oen, C–Ophen); 1014 υ(C−OMeOH); 944 υsym(MoO2); 873 υasym(MoO2). TGA analysis: MeOH calcd. 9.12%, found 8.54%; MoO3 calcd. 40.99%, found 39.99%.
2.1.5. Synthesis of [Mo2O4(MeOH)2(L5)]
Obtained by the reaction of 0.5 mmol (189 mg) of H4L5 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Orange powder. Yield: 0.28 g (80%). Anal. Calcd. For Mo2C22H26N4O6 (698.38): C, 37.84%, H, 3.75%, N, 8.02%, found: C, 38.22%, H, 3.6%, N, 7.94%. IR spectroscopy: 1611, 1596 υ(C=N); 1558, 1546 υ(C=C); 1321 υ(C–Oen); 1284, 1270 υ(C–Ophen); 1010 υ(C−OMeOH); 935 υsym(MoO2); 880 υasym(MoO2). TGA analysis: MeOH calcd. 9.18%, found 10.15%; MoO3 calcd. 41.22%, found 39.30%.
2.1.6. Synthesis of [Mo2O4(MeOH)2(L6)]
Obtained by the reaction of 0.5 mmol (239 mg) of H4L6 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Yellow-orange powder. Yield: 0.38 g (95%). Anal. Calcd. For Mo2C30H30N4O6 (798.5): C, 45.13%, H, 3.79%, N, 7.02%, found: C, 44.68%, H, 3.64%, N, 6.67%. IR spectroscopy: 1618, 1599 υ(C=N); 1551, 1533 υ(C=C); 1329 υ(C–Oen); 1277 υ(C–Ophen); 1000 υ(C−OMeOH); 937 υsym(MoO2); 904 υasym(MoO2). TGA analysis: MeOH calcd. 8.03%, found 9.78%; MoO3 calcd. 36.05%, found 35.89%.
2.1.7. Synthesis of [Mo2O4(MeOH)2(L7)]
Obtained by the reaction of 0.5 mmol (205 mg) of H4L7 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Red powder. Yield: 0.29 g (79%). Anal. Calcd. For Mo2C22H26N4O8 (730.38): C, 36.18%, H, 3.59%, N, 7.67%, found: C, 35.82%, H, 3.66%, N, 8.05%. IR spectroscopy: 1603 υ(C=N); 1569, 1551 υ(C=C); 1324 υ(C–Oen); 1263 υ(C–Ophen); 1035 υ(C−OMeOH); 939 υsym(MoO2); 907, 878 υasym(MoO2). TGA analysis: MeOH calcd. 8.77%, found 9.11%; MoO3 calcd. 39.42%, found 38.07%.
2.1.8. Synthesis of [Mo2O4(MeOH)2(L8)]∙2MeOH
Obtained by the reaction of 0.5 mmol (205 mg) of H4L8 and 1.0 mmol (326 mg) of [MoO2(acac)2] in 20 mL of methanol. Red powder. Yield: 0.33 g (84%). Anal. Calcd. For Mo2C24H34N4O10 (794.46): C, 36.28%, H, 4.31%, N, 7.05%, found: C, 37.01%, H, 4.44%, N, 7.33%. IR spectroscopy: 1596 υ(C=N); 1568, 1553 υ(C=C); 1333 υ(C–Oen); 1292 υ(C–Ophen); 1016 υ(C−OMeOH); 940 υsym(MoO2); 885 υasym(MoO2). TGA analysis: MeOH calcd. 16.13%, found 16.13%; MoO3 calcd. 36.24%, found 34.87%.
2.2. Methods
The chemical composition analysis for carbon, hydrogen, and nitrogen was conducted at the Analytical Services Laboratory, Ruđer Bošković Institute (Zagreb, Croatia). X-ray powder diffraction patterns of the samples were obtained using an Empyrean diffractometer (Malvern Panalytical, Almelo, The Netherlands) utilizing copper Kα radiation. Measurements were made using zero-background holders and the Bragg−Brentano setup, covering a 2θ range of 4° to 40°. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were acquired with a Nicolet iS50 spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The thermal behaviour of the samples was analysed using a Mettler Toledo TGA/DSC 3+ thermobalance (Mettler Toledo, Columbus, OH, USA), employing aluminium crucibles under a nitrogen flow of 50 mL per minute, across a temperature spanning from 25 °C to 600 °C at a heating rate of 10 °C per minute. Analysis of these experiments was performed using Mettler Toledo STARe Evaluation Software version 16.10.
Single crystals of suitable quality of [Mo2O4(MeOH)2(L1)]∙2MeOH, [Mo2O4(H2O)2(L2)], [Mo2O4(MeOH)2(L3)]∙2MeOH, [Mo2O4(MeOH)2(L4)]∙2MeOH, [Mo2O4(MeOH)2(L5)], [Mo2O4(MeOH)2(L6)], [Mo2O4(MeOH)2(L7)], and [Mo2O4(MeOH)2(L8)]∙2MeOH were obtained from diluted methanol solutions. Crystallographic data for [Mo2O4(MeOH)2(L3)]∙2MeOH and [Mo2O4(MeOH)2(L4)]∙2MeOH were obtained using ω-scans on an Oxford Xcalibur diffractometer with a 4-circle kappa goniometer and a CCD Sapphire 3 detector, using Mo Kα radiation at room temperature. The remaining compounds’ data were gathered using a Rigaku XtaLAB Synergy-S diffractometer with a Dualflex source and a HyPix detector at 170(1) K. The data processing was carried out with the CrysAlis software suite [27]. Detailed crystal and intensity data collection and refinement parameters are presented in Supplementary Tables S1 and S2, and geometric data are reported in Tables S3–S7. Structure solution and refinement employed SHELXT [28] for dual space structure solution and SHELXL [29] for full-matrix least-squares refinement, treating non-hydrogen atoms anisotropically. Hydrogen atoms were modelled using geometrically idealized positions and the riding model, with their coordinates and distance constraints refined in later stages. The suite of SHELX programs was used within the Olex2 framework, [30] and Mercury 2021.3.0 software handled geometrical calculations and molecular graphics [31].
Nuclear Magnetic Resonance (NMR) spectroscopy was performed using a Bruker Avance III HD 400 MHz/54 mm Ascend spectrometer, equipped with a 5 mm PA BBI 1H/D BB Z-GRAD probehead. The range of experiments included 1D (1H, 13C-DEPTq) and 2D (COSY, 1H−13C HSQC, 1H−13C HMBC, 1H−15N HSQC, 1H−15N HMBC) techniques, conducted at room temperature. DMSO-d6 served as the solvent, with TMS as the internal standard for proton and carbon shifts, and nitrogen shifts using liquid ammonia as the standard.
In vitro biological evaluations were carried out to assess the cytotoxic effects of the complexes against cute monocytic leukaemia (THP-1) and hepatocellular carcinoma (HepG2) human cell lines, using the MTS assay as per established protocols [32]. The antibacterial efficacy of the complexes was tested against two Gram-positive bacteria (Staphylococcus aureus and Enterococcus faecalis) and two Gram-negative bacteria (Escherichia coli and Moraxella catarrhalis) using the broth microdilution method, adhering to CLSI guidelines [33,34].
3. Results and Discussion
3.1. Synthesis and Solid-State Characterization
Synthesis of the dioxomolybdenum(VI) complexes proceeded straightforwardly when reacting the methanolic solution of [MoO2(acac)2] with a suspension of ligands in methanol. The limiting factor of the reaction was the poor solubility of the ligands in methanol (and in any solvent other than DMSO, for that matter); thus, the reaction mixture had to be heated under reflux for two hours to guarantee reaction completion. Qualitatively, the solubility of the ligands in methanol can be described with the following scheme: H4L3, 7 ≈ H4L4, 8 > H4L1, 5 > H4L2, 6. This trend was also reflected in the reaction times, i.e., ligands derived from dihydroxyaldehydes reacted faster than those derived from salicylaldehyde and 2-hydroxynaphthaldehyde. Owing to the overall low solubility of the resulting complexes, the isolated products were, in all cases, obtained as fine powder, and the crystals were only obtained by crystallization from very diluted solutions. In the case of Mo(VI) complexes with H4L2 and H4L4 ligands, it should be noted that reactions yielded [Mo2O4(MeOH)2(L2)] and [Mo2O4(MeOH)2(L4)], while the (re)crystallization of that material from diluted methanolic solutions gave single crystals of [Mo2O4(H2O)2(L2)] and [Mo2O4(MeOH)2(L4)]∙2MeOH, respectively. This is confirmed by a comparison of the PXRD data (Supplementary Materials, Figures S19–S26) with the simulated powder patterns obtained from crystal structure data (vide infra).
The thermal behavior of the prepared bulk complexes (Supplementary Materials, Figures S11–S18) is somewhat independent of the ligand choice and, expectedly, dependent on the presence of uncoordinated methanol molecules in the crystal. Interestingly, all methanol molecules (both coordinated and uncoordinated) are driven out of the bulk phase concomitantly, i.e., no separate steps are observed. As expected for molybdenum(VI) hydrazone complexes, at high temperatures, the materials undergo a complex sequence of decompositions, finally yielding MoO3 above ~450 °C in an oxygen atmosphere. The mass fraction of the MoO3 obtained through TGA analysis is consistent with the expected Mo:ligand ratio of 2:1.
Single-crystal X-ray diffraction: A detailed survey of the relevant literature revealed only few examples of the molybdenum(VI) complexes with ligands of the type [35,36,37]. All complexes investigated in this study are dinuclear, with the succinyl- and adipoyl-type ligands acting as ditopic hexadentate ones. The two compartments of each ligand coordinate in the hydrazonato form [38], after the deprotonation of O1−H1 and O2−H2 functionalities. In this way, the ligands behave as tetraanions, with their two compartments binding the two MoO22+ units via O1, N1 and O2 donor atoms (Figure 1a,b and Figure 2a,b; Supplementary Materials, Figures S1 and S2). The remaining coordination sites are occupied by the two oxido atoms, and an oxygen OH atom of ancillary methanol or, in the case of the [Mo2O4(H2O)2(L2)] complex, water molecule (Figure 1b). Consequently, the coordination geometry of each Mo atom can be described as a distorted octahedral geometry, with the shortest distances being Mo=O ones, while those positioned trans to those are, expectedly, the longest within the coordination sphere. The Mo1 atom is, in all cases, shifted above the plane defined by the O1, N1, and O2 atoms towards the apical oxygen atom by 0.28–0.32 for the succinyl-type complexes, whereas for the adipoyl ones these distances are larger, between ca 0.31 and 0.36.
When considering the molecular structures of dinuclear complexes based on the succinyl-type of ligands, one observes that, except [Mo2O4(H2O)2(L2)], the remaining ones are conformationally fairly similar (Figure 1c). Namely, conformations of complexes are staggered such that the planes defined by the aldehyde residues are parallel and distanced by ca 0.66, 0.77, and 0.80 for [Mo2O4(MeOH)2(L4)], [Mo2O4(MeOH)2(L3)], and [Mo2O4(MeOH)2(L1)], respectively, whereas in the case of [Mo2O4(H2O)2(L2)], this distance is ca 0.34. Due to the longer bridge between the two compartments of the complex, conformational differences in the group of adipoyl-type complexes are larger (Figure 2c). Namely, the molecule of [Mo2O4(MeOH)2(L8)] is essentially planar, while [Mo2O4(MeOH)2(L5)], and [Mo2O4(MeOH)2(L7)] adopt staggered conformations. Moreover, in [Mo2O4(MeOH)2(L6)] and [Mo2O4(MeOH)2(L7)], planes defined by the aldehyde residues are not parallel but form angles of ca 32° and ca 26°, respectively.
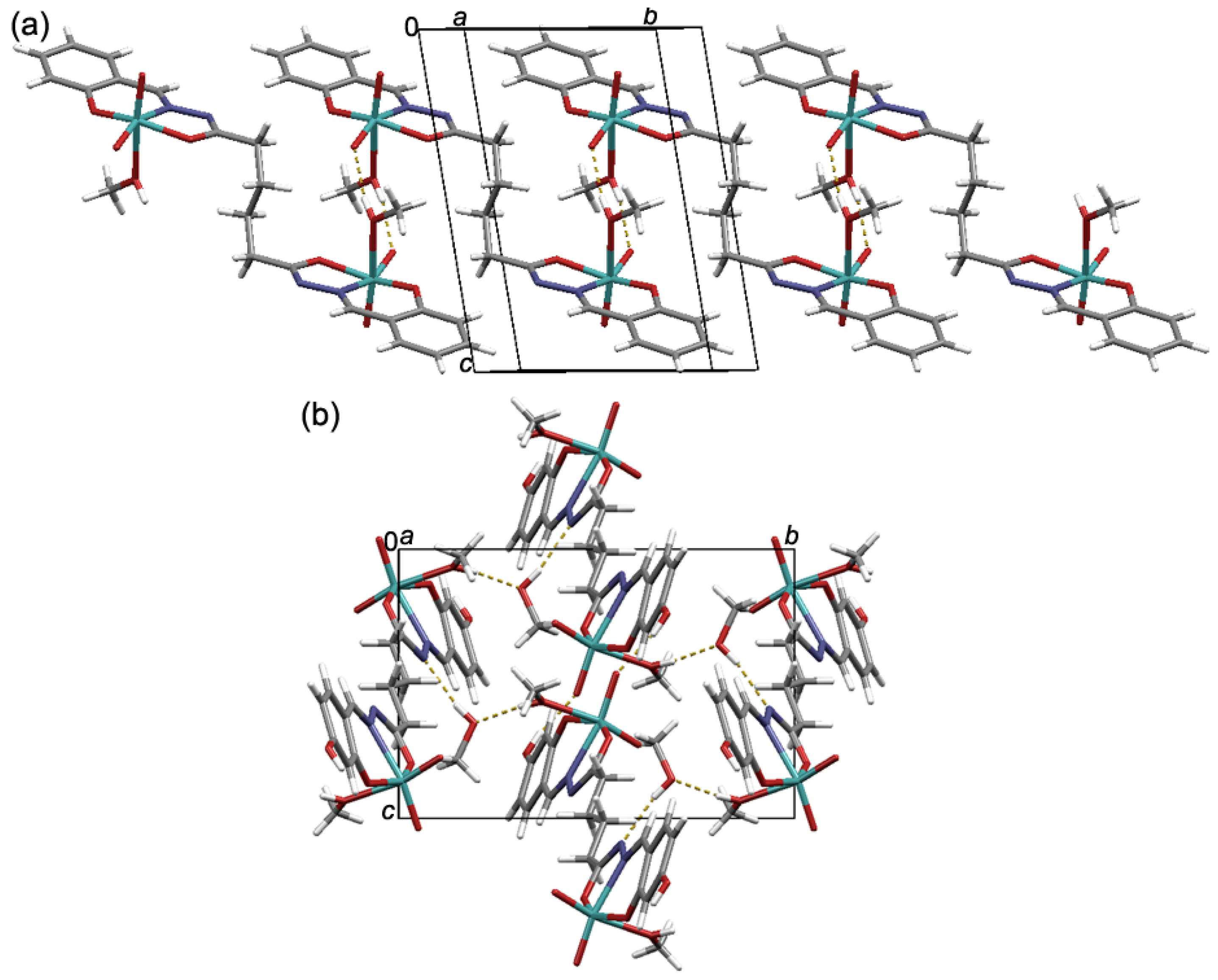
In solvates of the succinyl-type of complexes, [Mo2O4(MeOH)2(L1)]∙2MeOH, [Mo2O4(MeOH)2(L3)]∙2MeOH, and [Mo2O4(MeOH)2(L4)]∙2MeOH, non-coordinated methanol molecules bridge coordination entities by acting both as hydrogen bond donors and acceptors (Figure 3a and Figures S3–S5). The situation is different in [Mo2O4(H2O)2(L2)], where the coordinated water molecules act solely as hydrogen bond donors to connect neighbouring complexes via O–H···N and O–H···O hydrogen bonds (Figure 3b and Figure S6). The resulting supramolecular architectures are, in all cases, further supported and stabilized by a plethora of C–H···O hydrogen bonds. Similarly, in the crystal structures of adipoyl-type complexes, supramolecular interactions are mediated through the only available hydrogen bond donors, the coordinated methanol OH and aryl hydroxy OH group, while hydrogen bond acceptors are either imide nitrogen atom or the axial {MoO2}2+ oxygen atom (Figure 4 and Figures S7–S10). The presence of non-coordinated methanol molecules in the dual role of hydrogen bond donors and acceptors in [Mo2O4(MeOH)2(L8)]∙2MeOH makes the hydrogen-bonded network comparatively richer (Figure 4b and Figure S10). Finally, as in the case of succinyl-type complexes, crystal structures in adipoyl-type of complexes are stabilized by a collection of C–H···O hydrogen bonds.
3.2. NMR and FT-IR Spectroscopy
The chemical identities of the title complexes in DMSO-d6 solutions at room temperature were established via 1H, 13C, and 15N NMR spectroscopy (Supplementary Materials, Tables S8 and S9, Figures S27–S32). Relevant chemical shifts unveil that, in the DMSO solution, the explored dinuclear Mo(VI) complexes are symmetrical, since only one set of signals was observed in each case. Moreover, these complexes, unlike the related neutral ligands, in DMSO-d6 solutions, dominantly adopt one isomeric form. The absence of hydroxyaryl O1–H1 and hydrazonic N2–H2 protons in the spectra of complexes confirms that coordinated ligands are present in their tetraanionic form. Moreover, the significant deshielding of the N2 nitrogen chemical shift in the complexes, in comparison to neutral ligands, along with the slight deshielding of the C8 signal, confirms the enol-imino form of the coordinated ligands [39]. The 1H, 13C, and 15N chemical shifts in complexes are comparable to those of neutral ligands. Finally, owing to the strong donor nature of the DMSO, the title complexes undergo a solvent exchange reaction, which has been previously described in the literature for a similar type of Mo complex [40].
FTIR-ATR spectra (Supplementary Materials, Figures S33–S40) show features characteristic of dioxomolybdenum(VI) hydrazone complexes. In contrast to the respective ligands [15], absorption peaks corresponding to C=O stretching vibrations (usually found at ~1650 cm−1) and those related to N-H stretching vibrations (at around ~3200 cm−1) are absent in the spectra of prepared complexes. Thus, it can be concluded that the ligand is (tetra)deprotonated and in enol-imino form. However, the high-wave-number region of the spectra is rich, owing to the O–H stretching vibrations of crystal and coordinated methanol molecules, and in the case of complexes derived from H4L3, H4L4, H4L7, and H4L8, additional phenolic O–H bands. A significant shift to lower wavenumbers of the C=N stretching bands (~1590–1600 cm−1) compared to those of the free ligands (~1610–1630 cm−1), together with the absence of the C=O band, corroborates the coordination of the {MoO2}2+ core by the ONO pincer-like compartment of the ligand in its hydrazonato form. The C-O stretching vibrations of the enolato and phenolato fragments can be found at ~1330 and ~1270 cm−1, respectively. All complexes exhibit distinct bands at ~930–940 cm −1 and ~880–910 cm−1, typical for dioxomolybdenum(VI) complexes, corresponding to symmetric and asymmetric {MoO2}2+ core stretching vibrations, respectively [41]. Moreover, the spectra display bands indicative of coordinated methanol molecules, i.e., C–O stretching vibrations at ca. 1010–1030 cm−1. Taken together, the spectroscopic data are consistent with the identities and structures of the prepared complexes obtained from SC-XRD experiments.
3.3. In Vitro Cytotoxic and Antibacterial Activity
The here-reported molybdenum(VI) complexes were tested for their cytotoxic activity against HepG2 and THP-1 cells, while their antibacterial activity was evaluated on S. aureus, E. faecalis, E. coli, and M. catarrhalis bacterial strains. The bioassay results are summarized in Table 1.
According to the results (Table 1), all of the investigated complexes were found to be non-cytotoxic towards HepG2 cells, and, except [Mo2O4(MeOH)2(L1)], [Mo2O4(MeOH)2(L2)] and [Mo2O4(MeOH)2(L6)], which displayed only weak to moderate cytotoxicity, did not show any cytotoxic effects against THP-1. The results, given in Table 1, also reveal that complexes in general exhibited poor or no antibacterial activity, while some of them with MICs equal to 32 μg mL−1 demonstrated mild activity. Only [Mo2O4(MeOH)2(L2)] and [Mo2O4(MeOH)2(L6)], with minimum inhibitory concentrations equal to 4 and 2 μg mL−1, respectively, were proved to possess appreciable antibacterial potential towards M. catarrhalis.
Apart from [Mo2O4(MeOH)2(L1)], [Mo2O4(MeOH)2(L2)], and [Mo2O4(MeOH)2(L6)], chelation in general did not have a large influence on the cytotoxic properties of the examined compounds [15]. However, some differences in the antibacterial properties of Mo(VI) complexes in comparison with the corresponding free ligands were established. On the one hand, a noticeable reduction in antibacterial properties after the complexation to molybdenum was only observed for the anti-E. faecalis activity of H4L5, as well as for the anti-E. faecalis and anti-E. coli activities of H4L6. On the other hand, the bactericidal potency of [Mo2O4(MeOH)2(L2)] and [Mo2O4(MeOH)2(L7)] complexes was demonstrated, along with the increased anti-M. catarrhalis activity of [Mo2O4(MeOH)2(L6)], when compared to the neutral ligands.
4. Conclusions
A series of novel molybdenum(VI) complexes with succinyl and adipoyl dihydrazones was synthesized, characterized, and evaluated with respect to their biological activity. Conventional solution synthesis proved to be a straightforward route towards the title complexes, although it was to some extent limited by the solubility of the ligands. The structures of the complexes were explored via single-crystal X-ray diffraction and FTIR spectroscopy in the solid state, while their behavior in the DMSO-d6 solution was studied by the NMR technique. Structural studies unveiled that all complexes are dinuclear, with tetraanions of dihydrazones acting as flexible ditopic hexadentate ligands, both in the solid state and in DMSO solution. The complexes generally showed non-cytotoxic behavior towards HepG2 cells and varying degrees of cytotoxicity against THP-1 cells. Some exhibited mild antibacterial activity, with notable effectiveness against Moraxella catarrhalis. The results highlight the moderate improvement in antimicrobial properties compared to the respective ligands. These findings, combined with the facile modulation of the complexes of this type, could serve as a fruitful platform for the future development of this type of compound as potentially bioactive compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst14020135/s1, Scheme S1: Reaction scheme; Tables S1 and S2: General and crystallographic data; Tables S3–S7: Selected geometric data; Figures S1–S10: Crystal structure representations; Figures S11–S18: TGA/DSC data; Figures S19–S26: Comparison of PXRD patterns; Figures S27–S32: NMR spectra Tables S8 and S9: The NMR numbering schemes; 1H and 13C and 15N assignments; Figures S33–S40: ATR FT-IR spectra.
Author Contributions
Investigation, formal analysis, writing—original draft preparation, E.T., V.D., K.P. and M.R.; visualization, E.T. and M.R.; writing—review and editing, E.T., V.D., K.P. and M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been fully supported by Croatian Science Foundation under the project (IP-2016-06-4221). We acknowledge the support of project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund–Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016).
Data Availability Statement
Crystallographic data sets for the structures [Mo2O4(MeOH)2(L1)]∙2MeOH, [Mo2O4(H2O)2(L2)], [Mo2O4(MeOH)2(L3)]∙2MeOH, [Mo2O4(MeOH)2(L4)]∙2MeOH, [Mo2O4(MeOH)2(L5)], [Mo2O4(MeOH)2(L6)], [Mo2O4(MeOH)2(L7)] and [Mo2O4(MeOH)2(L8)]∙2MeOH are available through the Cambridge Structural Database with deposition numbers CCDC 2314177-2314184. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/ (accessed on 26 January 2024).
Acknowledgments
We are grateful to Ljubica Ljubić, Kristina Prezelj, for the assistance in the synthetic procedures and Nikola Cindro for the help with the NMR measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mali, S.N.; Thorat, B.R.; Gupta, D.R.; Pandey, A. Mini-Review of the Importance of Hydrazides and Their Derivatives—Synthesis and Biological Activity. Eng. Proc. 2021, 11, 21. [Google Scholar]
- Tatum, L.A.; Su, X.; Aprahamian, I. Simple Hydrazone Building Blocks for Complicated Functional Materials. Acc. Chem. Res. 2014, 47, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Romo, F.J.; Doonan, C.J.; Furukawa, H.; Oisaki, K.; Yaghi, O.M. Crystalline covalent organic frameworks with hydrazone linkages. J. Am. Chem. Soc. 2011, 133, 11478–11481. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.K.; Plutenko, M.O.; Schachner, J.A.; Haukka, M.; Mösch-Zanetti, N.C.; Fritsky, I.O. Dioxomolybdenum(VI) complexes of hydrazone phenolate ligands—Syntheses and activities in catalytic oxidation reactions. J. Indian Chem. Soc. 2021, 98, 100006. [Google Scholar] [CrossRef]
- Guskos, N.; Likodimos, V.; Glenis, S.; Typek, J.; Wabia, M.; Paschalidis, D.G.; Tossidis, I.; Lin, C.L. Magnetic properties of rare-earth hydrazone compounds. J. Magn. Magn. Mater. 2004, 272, 1067–1069. [Google Scholar] [CrossRef]
- Liu, R.; Cui, J.; Ding, T.; Liu, Y.; Liang, H. Research Progress on the Biological Activities of Metal Complexes Bearing Polycyclic Aromatic Hydrazones. Molecules 2022, 27, 8393. [Google Scholar] [CrossRef] [PubMed]
- Tupolova, Y.P.; Popov, L.D.; Vlasenko, V.G.; Gishko, K.B.; Kapustina, A.A.; Berejnaya, A.G.; Golubeva, Y.A.; Klyushova, L.S.; Lider, E.V.; Lazarenko, V.A.; et al. Crystal structure and cytotoxic activity of Cu(ii) complexes with bis-benzoxazolylhydrazone of 2,6-diacetylpyridine. New J. Chem. 2023, 47, 14972–14985. [Google Scholar] [CrossRef]
- Tupolova, Y.P.; Shcherbakov, I.N.; Popov, L.D.; Vlasenko, V.G.; Gishko, K.B.; Kapustina, A.A.; Berezhnaya, A.G.; Golubeva, Y.A.; Klyushova, L.S.; Lider, E.V.; et al. Copper coordination compounds based on bis-quinolylhydrazone of 2,6-diacetylpyridine: Synthesis, structure and cytotoxic activity. Polyhedron 2023, 233, 116292. [Google Scholar] [CrossRef]
- Su, X.; Aprahamian, I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963–1981. [Google Scholar] [CrossRef]
- Zavalishin, M.N.; Gamov, G.A.; Pimenov, O.A.; Pogonin, A.E.; Aleksandriiskii, V.V.; Usoltsev, S.D.; Marfin, Y.S. Pyridoxal 5′-phosphate 2-methyl-3-furoylhydrazone as a selective sensor for Zn2+ ions in water and drug samples. J. Photochem. Photobiol. A 2022, 432, 114112. [Google Scholar] [CrossRef]
- Bagherian, N.; Karimi, A.R.; Amini, A. Chemically stable porous crystalline macromolecule hydrazone-linked covalent organic framework for CO2 capture. Colloids Surf. A 2021, 613, 126078. [Google Scholar] [CrossRef]
- Golla, U.; Adhikary, A.; Mondal, A.K.; Tomar, R.S.; Konar, S. Synthesis, structure, magnetic and biological activity studies of bis-hydrazone derived Cu(ii) and Co(ii) coordination compounds. Dalton Trans. 2016, 45, 11849–11863. [Google Scholar] [CrossRef]
- Wang, M.; Cheng, C.; Chunbo, L.; Wu, D.; Song, J.; Wang, J.; Zhou, X.; Xiang, H.; Liu, J. Smart, chiral, and non-conjugated cyclohexane-based bissalicylaldehyde hydrazides: Multi-stimuli-responsive, turn-on, ratiometric, and thermochromic fluorescence, single crystal structures, and DFT calculations. J. Mater. Chem. C 2019, 7, 6767–6778. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, S.; Shen, Y.; Zou, H.; Liu, D.; Liang, F. Copper(II) Clusters of Two Pairs of 2,3-Dihydroxybutanedioyl Dihydrazones: Synthesis, Structure, and Magnetic Properties. Eur. J. Inorg. Chem. 2014, 2014, 5783–5792. [Google Scholar] [CrossRef]
- Chen, Z.; Shen, Y.; Li, L.; Zou, H.; Fu, X.; Liu, Z.; Wang, K.; Liang, F. High-nuclearity heterometallic clusters with both an anion and a cation sandwiched by planar cluster units: Synthesis, structure and properties. Dalton Trans. 2017, 46, 15032–15039. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.K.; Jena, H.S.; Malviya, A.; Konar, S. Lanthanide-Directed Fabrication of Four Tetranuclear Quadruple Stranded Helicates Showing Magnetic Refrigeration and Slow Magnetic Relaxation. Inorg. Chem. 2016, 55, 5237–5244. [Google Scholar] [CrossRef] [PubMed]
- Schattschneider, C.; Doniz Kettenmann, S.; Hinojosa, S.; Heinrich, J.; Kulak, N. Biological activity of amphiphilic metal complexes. Coord. Chem. Rev. 2019, 385, 191–207. [Google Scholar] [CrossRef]
- Liang, J.; Sun, D.; Yang, Y.; Li, M.; Li, H.; Chen, L. Discovery of metal-based complexes as promising antimicrobial agents. Eur. J. Med. Chem. 2021, 224, 113696. [Google Scholar] [CrossRef]
- Verma, G.; Marella, A.; Shaquiquzzaman, M.; Akhtar, M.; Ali, M.R.; Alam, M.M. A review exploring biological activities of hydrazones. J. Pharm. Bioallied Sci. 2014, 6, 69–80. [Google Scholar]
- Le Goff, G.; Ouazzani, J. Natural hydrazine-containing compounds: Biosynthesis, isolation, biological activities and synthesis. Bioorg. Med. Chem. 2014, 22, 6529–6544. [Google Scholar] [CrossRef]
- Kumar, P.; Narasimhan, B. Hydrazides/hydrazones as antimicrobial and anticancer agents in the new millennium. Mini Rev. Med. Chem. 2013, 13, 971–987. [Google Scholar] [CrossRef] [PubMed]
- Popiołek, Ł. Hydrazide-hydrazones as potential antimicrobial agents: Overview of the literature since 2010. Med. Chem. Res. 2017, 26, 287–301. [Google Scholar] [CrossRef]
- Arora, T.; Devi, J.; Boora, A.; Taxak, B.; Rani, S. Synthesis and characterization of hydrazones and their transition metal complexes: Antimicrobial, antituberculosis and antioxidant activity. Res. Chem. Intermed. 2023, 49, 4819–4843. [Google Scholar] [CrossRef]
- Ullah, H.; Previtali, V.; Mihigo, H.B.; Twamley, B.; Rauf, M.K.; Javed, F.; Waseem, A.; Baker, R.J.; Rozas, I. Structure-activity relationships of new Organotin(IV) anticancer agents and their cytotoxicity profile on HL-60, MCF-7 and HeLa human cancer cell lines. Eur. J. Med. Chem. 2019, 181, 111544. [Google Scholar] [CrossRef] [PubMed]
- Topić, E.; Damjanović, V.; Pičuljan, K.; Vrdoljak, V.; Rubčić, M. Succinyl and Adipoyl Dihydrazones: A Solid-State, Solution and Antibacterial Study. Crystals 2022, 12, 1175. [Google Scholar] [CrossRef]
- Gherke, H., Jr.; Veal, J. Acetylacetonate complexes of molybdenum (V) and molybdenum (VI). I. Inorg. Chim. Acta 1969, 3, 623–627. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, version 1.171.41.92a; Rigaku Oxford Diffraction: Oxford, UK, 2020.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallograph. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallograph. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallograph. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallograph. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Rubčić, M.; Pisk, J.; Pičuljan, K.; Damjanović, V.; Lovrić, J.; Vrdoljak, V. Symmetrical disubstituted carbohydrazides: From solid-state structures to cytotoxic and antibacterial activity. J. Mol. Struct. 2019, 1178, 222–228. [Google Scholar] [CrossRef]
- CLSI Document M07-A8; Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009.
- Ngan, N.K.; Lo, K.M.; Wong, C.S.R. Dinuclear and polynuclear dioxomolybdenum(VI) Schiff base complexes: Synthesis, structural elucidation, spectroscopic characterization, electrochemistry and catalytic property. Polyhedron 2012, 33, 235. [Google Scholar] [CrossRef]
- Kurbah, S.D.; Kumar, A.; Shangpung, S.; Syiemlieh, I.; Khongjoh, I.; Lal, R.A. Synthesis, Characterization, and Fluorescence Chemosensor Properties of a cis-Dioxomolybdenum(VI) Complex Containing Multidentate Hydrazone Ligands. Z. Anorg. Allg. Chem. 2017, 643, 794–801. [Google Scholar] [CrossRef]
- Kurbah, S.D.; Kumar, A.; Syiemlieh, I.; Asthana, M.; Lal, R.A. Bimetallic cis-dioxomolybdenum(VI) complex containing hydrazone ligand: Syntheses, crystal structure and catalytic studies. Inorg. Chem. Commun. 2017, 86, 39–43. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Hrenar, T.; Rubčić, M.; Pavlović, G.; Friganović, T.; Cindrić, M. Ligand-Modulated Nuclearity and Geometry in Nickel(II) Hydrazone Complexes: From Mononuclear Complexes to Acetato- and/or Phenoxido-Bridged Clusters. Int. J. Mol. Sci. 2023, 24, 1909. [Google Scholar] [CrossRef]
- Maurya, M.R.; Saini, N.; Avecilla, F. Effect of N-based additive on the optimization of liquid phase oxidation of bicyclic, cyclic and aromatic alcohols catalyzed by dioxidomolybdenum(vi) and oxidoperoxidomolybdenum(vi) complexes. RSC Adv. 2015, 5, 101076. [Google Scholar] [CrossRef]
- Pisk, J.; Rubčić, M.; Kuzman, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Molybdenum(vi) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: An experimental and theoretical approach. New J. Chem. 2019, 43, 5531–5542. [Google Scholar] [CrossRef]
- Berg, J.M.; Holm, R.H. Structure proofs of ligated and polymeric dioxomolybdenum(VI)-tridentate complexes: MoO2(C5H3N-2,6-(CH2S)2)(C4H8SO) and [MoO2(C5H3N-2,6-(CH2O)2)]n. Inorg. Chem. 1983, 22, 1768–1771. [Google Scholar] [CrossRef]
Figure 1.
Molecular structures of: (a) [Mo2O4(MeOH)2(L1)]∙2MeOH and (b) [Mo2O4(H2O)2(L2)] with the atom numbering schemes. The central aliphatic C2 fragments of molecules lie on the inversion center. (c) Overlay of the molecular structures of [Mo2O4(MeOH)2(L1)] (pink), [Mo2O4(H2O)2(L2)] (yellow), [Mo2O4(MeOH)2(L3)] (blue), and [Mo2O4(MeOH)2(L4)] (gray). In (a,b) displacement ellipsoids are drawn at the 50% probability level, while the hydrogen atoms are presented as spheres of arbitrary small radii. In (a), hydrogen bonds are highlighted by yellow dashed lines.
Figure 1.
Molecular structures of: (a) [Mo2O4(MeOH)2(L1)]∙2MeOH and (b) [Mo2O4(H2O)2(L2)] with the atom numbering schemes. The central aliphatic C2 fragments of molecules lie on the inversion center. (c) Overlay of the molecular structures of [Mo2O4(MeOH)2(L1)] (pink), [Mo2O4(H2O)2(L2)] (yellow), [Mo2O4(MeOH)2(L3)] (blue), and [Mo2O4(MeOH)2(L4)] (gray). In (a,b) displacement ellipsoids are drawn at the 50% probability level, while the hydrogen atoms are presented as spheres of arbitrary small radii. In (a), hydrogen bonds are highlighted by yellow dashed lines.
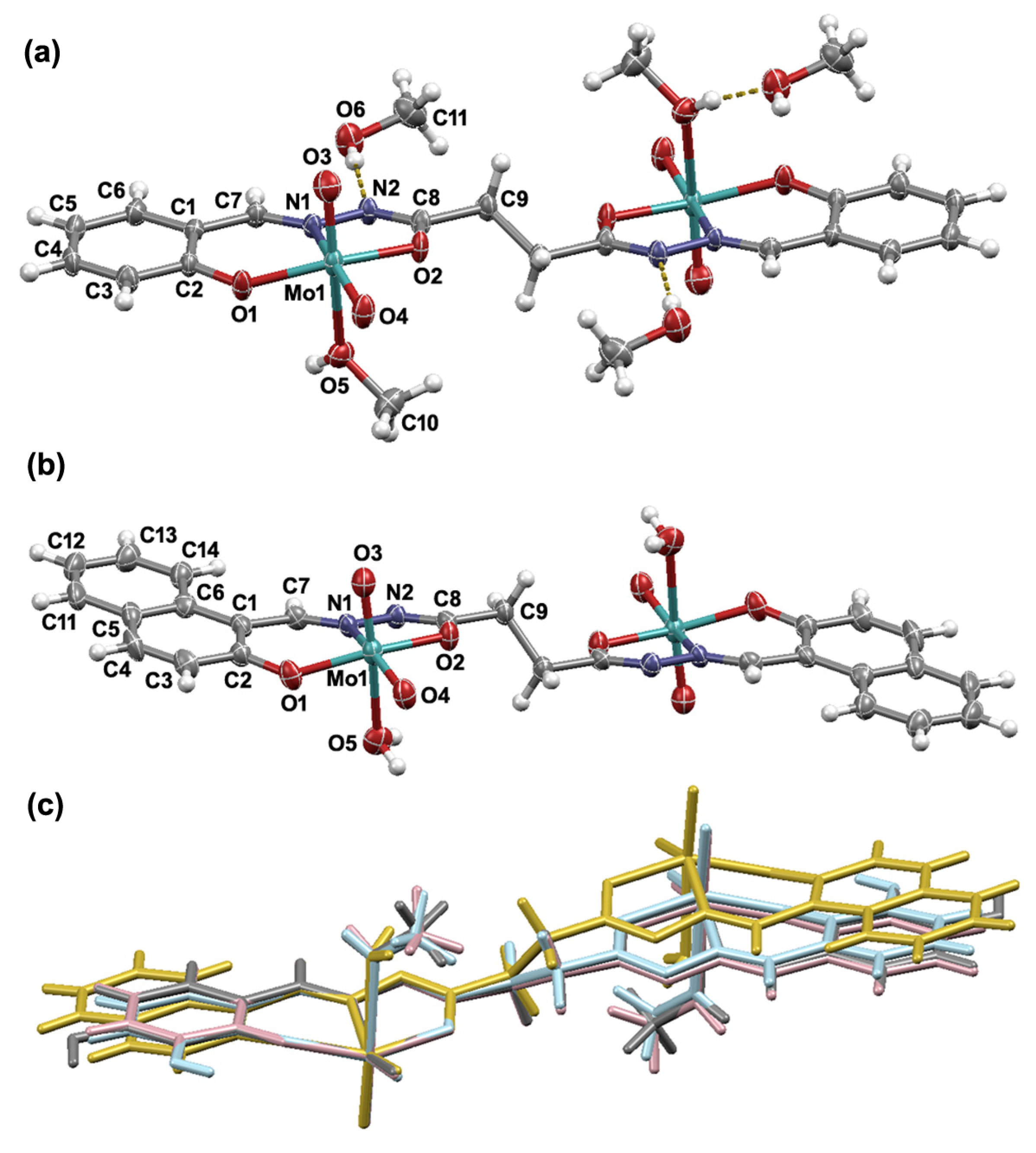
Figure 2.
Molecular structures of: (a) [Mo2O4(MeOH)2(L5)] and (b) [Mo2O4(MeOH)2(L8)]∙2MeOH with the atom numbering schemes. The central aliphatic C4 fragments of molecules sit on the inversion center. (c) Overlay of the molecular structures of [Mo2O4(MeOH)2(L5)] (yellow), [Mo2O4(H2O)2(L6)] (grey), [Mo2O4(MeOH)2(L7)] (red), and [Mo2O4(MeOH)2(L8)] (blue). In (a,b) displacement ellipsoids are drawn at the 50% probability level, while the hydrogen atoms are presented as spheres of arbitrary small radii. In (a), hydrogen bonds are highlighted by yellow dashed lines.
Figure 2.
Molecular structures of: (a) [Mo2O4(MeOH)2(L5)] and (b) [Mo2O4(MeOH)2(L8)]∙2MeOH with the atom numbering schemes. The central aliphatic C4 fragments of molecules sit on the inversion center. (c) Overlay of the molecular structures of [Mo2O4(MeOH)2(L5)] (yellow), [Mo2O4(H2O)2(L6)] (grey), [Mo2O4(MeOH)2(L7)] (red), and [Mo2O4(MeOH)2(L8)] (blue). In (a,b) displacement ellipsoids are drawn at the 50% probability level, while the hydrogen atoms are presented as spheres of arbitrary small radii. In (a), hydrogen bonds are highlighted by yellow dashed lines.

Figure 3.
Supramolecular architectures found in: (a) [Mo2O4(MeOH)2(L1)]∙2MeOH and (b) [Mo2O4(H2O)2(L2)]. Hydrogen bonds are highlighted as yellow dashed lines.
Figure 3.
Supramolecular architectures found in: (a) [Mo2O4(MeOH)2(L1)]∙2MeOH and (b) [Mo2O4(H2O)2(L2)]. Hydrogen bonds are highlighted as yellow dashed lines.

Figure 4.
Crystal packing in: (a) [Mo2O4(MeOH)2(L5)] and (b) [Mo2O4(MeOH)2(L8)]∙2MeOH. Hydrogen bonds are highlighted as yellow dashed lines.
Figure 4.
Crystal packing in: (a) [Mo2O4(MeOH)2(L5)] and (b) [Mo2O4(MeOH)2(L8)]∙2MeOH. Hydrogen bonds are highlighted as yellow dashed lines.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The IC50 values and minimum inhibitory concentrations (MIC) of the Mo(VI) complexes.
| Compound | IC50 (μmol L−1) | MIC (μg mL−1) | ||||
|---|---|---|---|---|---|---|
| THP-1 | HepG2 | S. aureus | E. faecalis | E. coli | M. catarrhalis | |
| 1-Mo | 19.58 | >100 | >256 | 64 | >256 | 32 |
| 2-Mo | 7.04 | >100 | 32 | 32 | 32 | 4 |
| 3-Mo | >100 | >100 | 256 | 256 | 128 | 128 |
| 4-Mo | >100 | >100 | >256 | 128 | >256 | >256 |
| 5-Mo | >100 | >100 | >256 | >256 | >256 | >256 |
| 6-Mo | 9.78 | 57.98 | >256 | 128 | 128 | 2 |
| 7-Mo | >100 | >100 | >256 | 32 | 64 | 64 |
| 8-Mo | >100 | >100 | >256 | >256 | >256 | >256 |
| staurosporine | 0.10 | 7.98 | – | – | – | – |
| azithromycin | – | – | 1 | 8 | 0.50 | 0.125 |
1-Mo = [Mo2O4(MeOH)2(L1)]; 2-Mo = [Mo2O4(MeOH)2(L2)]; 3-Mo = [Mo2O4(MeOH)2(L3)]; 4-Mo = [Mo2O4(MeOH)2(L4)]; 5-Mo = [Mo2O4(MeOH)2(L5)]; 6-Mo = [Mo2O4(MeOH)2(L6)]; 7-Mo = [Mo2O4(MeOH)2(L7)]; and 8-Mo = [Mo2O4(MeOH)2(L8)].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Topić, E.; Damjanović, V.; Pičuljan, K.; Rubčić, M. Dinuclear Molybdenum(VI) Complexes Based on Flexible Succinyl and Adipoyl Dihydrazones. Crystals 2024, 14, 135. https://doi.org/10.3390/cryst14020135
AMA Style
Topić E, Damjanović V, Pičuljan K, Rubčić M. Dinuclear Molybdenum(VI) Complexes Based on Flexible Succinyl and Adipoyl Dihydrazones. Crystals. 2024; 14(2):135. https://doi.org/10.3390/cryst14020135
Chicago/Turabian StyleTopić, Edi, Vladimir Damjanović, Katarina Pičuljan, and Mirta Rubčić. 2024. "Dinuclear Molybdenum(VI) Complexes Based on Flexible Succinyl and Adipoyl Dihydrazones" Crystals 14, no. 2: 135. https://doi.org/10.3390/cryst14020135
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.