

New Data on Crystal Phases in the System MgSO4–OC(NH2)2–H2O
by
 , ,
, ,
Rositsa Nikolova
*,
Vladislav Kostov-Kytin
 ,
,
Nadia Petrova
,
Krasimir Kossev
,
Rositsa Titorenkova
and
Gergana Velyanova
Institute of Mineralogy and Crystallography, Bulgarian Academy of Sciences, Akad. G. Bonchev bl. 107, 1113 Sofia, Bulgaria
*
Author to whom correspondence should be addressed.
Crystals 2024, 14(3), 227; https://doi.org/10.3390/cryst14030227
Submission received: 6 February 2024
/
Revised: 22 February 2024
/
Accepted: 25 February 2024
/
Published: 27 February 2024
(This article belongs to the Topic Advances in Inorganic Synthesis)
Abstract
:Urea complexes of magnesium sulfate have been intensively studied due to their application in many areas of life, including agricultural chemistry, pharmacy, medicine, etc. The aim of this study is to add new knowledge about the trends and consistencies in the preparation procedures of MgSO4·nOC(NH2)2·mH2O phases. A set of analytical methods was used to characterize their structure, thermal and spectroscopic properties. The conditions for obtaining the three complexes in pure form were specified and the crystal structures of MgSO4·OC(NH2)2·2H2O and MgSO4·OC(NH2)2·3H2O were determined. The spectroscopic data of the considered compounds were analysed with respect to their structural and chemical properties. Thermal analyses showed that both the melting point and the urea decomposition temperature depend on the OC(NH2)2: H2O ratio in the octahedral environment of the magnesium ion in the studied structures.
1. Introduction
Magnesium is one of the most abundant elements in nature. Magnesium makes up 2.4% of the Earth’s crust as a component of a number of rock-forming minerals [1]. The source of bioavailable magnesium is the hydrosphere, where the magnesium concentration is about 4.1 Clarke [2]. Magnesium salts dissolve well in water and, as a result, a variety of magnesium sulfate hydrates and complexes, easily accessible to living organisms, are formed. Magnesium is one of the major biogenic elements and, as part of over 300 enzyme systems, magnesium ions are involved in a number of biochemical reactions [3,4,5]. The essential role of magnesium determines the great research interest in its salts, hydrates and complexes. Magnesium compounds are widely used in almost all spheres of life, including agricultural chemistry, pharmacy, medicine, etc. [6,7,8,9]. Particularly important and promising are studies on the interaction of magnesium salts with amide-type ligands, since a large proportion of the low molecular weight substances present in plants and animals are of the amide type. One of the common amide ligands in nature is urea (OC(NH2)2), which forms compounds with many of the magnesium salts. In such compounds, urea moieties displace water molecules from the magnesium coordination surrounding, thereby reducing the hygroscopicity of such salts as magnesium chlorides, iodides, bromides, nitrates, tetrafluoroborates, chlorates, formats, dihydrogen phosphates and perrhenates [10,11,12,13,14,15,16,17,18,19,20]. Urea complexes of magnesium sulfate have been studied since the beginning of the last century. The phases reported so far are shown in Table 1. Some of the results in this field refers to studies of the MgSO4–OC(NH2)2–H2O system and/or relationship between synthesis conditions and stoichiometry of the obtained MgSO4·nOC(NH2)2·mH2O phases [21,22,23,24,25,26,27,28,29]. Others relate to optimisation of the processes for preparation of products for the agrochemical industry, which include urea and magnesium sulfate composites [26,27,28,29,30,31,32,33].
Among the results obtained, the following ones are relevant to the present study. Both starting reactants and final products of the studied system are very temperature sensitive and, even at the same ratio of starting reactants, a small difference in the temperature range leads to the formation of stoichiometrically different phases. For instance, when a solution of MgSO4 and OC(NH2)2 in molar ratio 1:1 is evaporated at 25 °C the product is MgSO4·OC(NH2)2·3H2O, whereas an evaporation at 30 °C or 35 °C leads to the formation of MgSO4·OC(NH2)2·2H2O [21,25]. Diversity of magnesium sulfate sources and their pre-activation treatment are allowable, as presented in the patents WO 2013/098367, US 2014/0360239 A1, and US 2016/0046534 A1. The authors describe conditions for obtaining mixtures of some of the above mentioned compounds using MgSO4 or MgSO4·7H2O as magnesium sources and reactivation temperatures varying in the range 40–95 °C. The main preparation methods are synthesis from solution and mechanoactivation, while US 2016/0046534 A1 reports procedures combining the features of both methods. All the compounds reported above have been obtained from solution, whereas only MgSO4·6OC(NH2)2·0.5H2O has been obtained by mechanoactivation as a pure end product. The solvent type plays a key role for the run products, thus MgSO4·OC(NH2)2·2H2O, MgSO4·OC(NH2)2·3H2O, MgSO4·4OC(NH2)2·H2O and MgSO4·6OC(NH2)2·0.5H2O have previously been obtained by evaporation from water solutions. However, in this case water is both a solvent and a reactant. In other cases, a methanol solution has been used as a medium e.g., MgSO4·5OC(NH2)2·2H2O and MgSO4·6OC(NH2)2·2H2O [23]. The latter compounds have been reported to be highly hygroscopic and unstable.
It should be noted that identification of the above-mentioned phases has been mainly based on chemical analysis and PXRD. Only MgSO4·4OC(NH2)2·H2O and MgSO4·6OC(NH2)2·0.5H2O have been identified by SCXRD [32,33]. The presence of any crystalline substance in pure or mixed products can only be unambiguously evidenced by the knowledge of its crystal structure. Typically, this is obtained by comparing calculated and experimental diffraction data. Moreover, crystal structures provide a basis for precise interpretation of the physico–chemical characteristics of the studied substances, which in turn allows optimization of methods for their syntheses.
The aims of this study str: (i) to add new knowledge about the trends and consistencies in the preparation procedures of the products obtained in the system MgSO4–OC(NH2)2–H2O; (ii) to determine the crystal structures of those MgSO4·nOC(NH2)2·mH2O phases that have not been sufficiently studied before and to compare them to the already known ones; (iii) to evaluate the influence of water and urea molecules on certain physico–chemical properties of the studied compounds, e.g., solubility and thermal stability, directly related to their application as fertilizers or soil additives.
2. Materials and Methods
2.1. Syntheses Conditions
The reagents used for the syntheses were: MgSO4 source—either MgSO4·7H2O (≥99%) from Sigma-Aldrich, St. Louis, MO, USA, or MgSO4·H2O prepared by appropriate thermal treatment of MgSO4·7H2O; CO(NH2)2 (Urea for analyses) from Merck and distilled water. The crystallization water of MgSO4·7H2O and MgSO4·H2O has also been taken into account in calculations of the recipes. Both magnesium sources are comparatively stable under normal conditions evidence also by their existence as mineral species—epsomite and kieserite respectively (https://www.mindat.org, 2 January 2024). In a typical synthesis the appropriate ratio of the three initial reagents were mixed with or without grinding according to the chosen strategy and then placed in thermostat and heated at the desired temperature until complete evaporation of the excess water.
2.2. Syntheses Approaches
The studied compounds are highly water-soluble complex salts. They are easily formed under various conditions and within a wide range of the synthesis medium parameters such as temperature and ratio of the initial reactants. In laboratory conditions, methods such as crystallization from solution [24,25] and more recently mechanochemistry [27,28,29,30,31] are used for their preparation. Techniques combining features of both methods have also been used. The reason for this is the high reactivity of the reagents, in which usually urea molecules displace water ones from the magnesium coordination surrounding and water is released. As a result, even short-term grinding at room temperature in an agate mortar of an initial batch consisting of the three starting components leads to the formation of a homogeneous slurry. By appropriate heat treatment it is possible to obtain from it a single well-crystallized phase of magnesium urea sulfate hydrate as the final product. This prior knowledge allowed outlining of two synthesis strategies.
On one hand, based on the efforts of previous researchers to obtain new knowledge for some of the studied compounds prepared from aqueous solution, triangular diagrams of crystallization (mole %) were constructed (see next section, Figure 1). Analysis of the data directed attention to synthesis conditions favourable for achieving the set goals—obtaining solely a pure phase in the final product. The conduct of the new syntheses showed that in some cases the conditions often exceed the limits of crystallization from solution and acquire characteristics of preparation by mechanoactivation, especially where the intermediate product resembles a slurry. Without claiming to be exhaustive in terms of precise delineation of the phase boundaries and the amount of yield, the obtained results build on the previous ones. The diagram in Figure 2 also indicates those conditions under which the highest percentage yields of the tested compounds were obtained. The diagrams have been constructed on results obtained from PXRD studied materials processed with appropriate software programs. These data reveal certain important trends in the relationship between the initial synthesis conditions and the obtained phases and can be used in future to optimize the procedures for preparation of desired final products. The following features are observed in this synthesis approach: short synthesis duration (24–96 h); imperfect tiny crystals; more often mixtures of crystalline and amorphous phases as run products.
On the other hand, syntheses aiming to obtain crystals suitable for single crystal X-ray analyses were planned. In this case, crystallization from a solution was preferred and used, because this technique ensures the production of sufficiently large and high-quality crystals, although it requires a longer duration (1–2 weeks). The results are important for conducting proper phase identification by simulating powder X-ray diffraction patterns based on the single crystal studies and semi-quantitative phase analysis in cases where the run-product contains more than one phase. The materials used for the subsequent single crystal, thermal and spectroscopic analyses were obtained mainly in this way.
2.3. Powder X-ray Diffraction (PXRD)
Powder XRD patterns of the studied samples were recorded on Empyrean Powder X-ray diffractometer (Malvern Panalytical, Netherlands) in the 2°–90° 2θ range, overall scanning time 15 min, using Cu radiation (λ = 1.5406 Å) and PIXcel3D detector. Phase identification was carried out using the HighScore Plus program [34] and/or through X-ray powder diffraction patterns simulated on the basis of the single-crystal studies for each of the studied complex salts using the PowderCell program [35]. The semi-quantitative phase analysis was also conducted in the same environments.
2.4. Single Crystal X-ray Diffraction
Colourless crystals of MgSO4·OC(NH2)2·2H2O and MgSO4·OC(NH2)2·3H2O were mounted on a glass capillary and used for single crystal diffraction analyses. The data collection experiments were performed on Bruker D8 Venture diffractometer. The determination of unit cell parameters, data integration, scaling and absorption corrections were carried out using APEX4 program package. The crystal structures were solved by direct methods (SHELXS-97/2013) and refined by full-matrix least-square procedures on F2 (SHELXL-97/2013). The hydrogens of the urea molecules were positioned geometrically, while the hydrogens of the water molecules were found by Fourier difference. All hydrogen atoms were refined by a riding model with U iso 1.2 times that of the attached nitrogen or oxygen atom. The experimental and structure refinement parameters are given in Table 1. Information about the structural data is available in the Cambridge Structural Database: CSD 2331202, CSD 2331203 for Mg(SO4)·OC(NH2)2.2H2O and Mg(SO4)·OC(NH2)2.3H2O, respectively.
2.5. Thermal Analyses
The differential scanning calorimetry (DSC) and thermogravimetry (TG) were carried out on the DSC-TG analyzer Setsys Evolution 2400, SETARAM at the following conditions: temperature range from 20 to 320 °C under Ar flow of 30 mL min−1, with a heating rate of 10 °C min−1 and 10–12 mg sample mass in a corundum crucible. Such temperature range has been chosen for practical reasons and namely before the occurrence of complete urea decomposition. A calibration run with empty crucible was applied for all experiments to base line slope correction of both DSC and TG signal. Simultaneous analysis of the evolved gases (EGA) was performed via mass spectrometry using a quadrupole mass spectrometer (QMS) OmniStar connected to the TG apparatus. The mass/charge (m/z) values programmed for detection in QMS were for the following gases: CH4 (16); NH3 (17); H2O (18), NO2 (28) and CO2 (44).
2.6. IR and Raman Spectroscopy
Infrared spectra were measured using Tensor 37 (Bruker) FT-IR spectrometer with 4 cm−1 spectral resolution after averaging 128 scans on standard KBr pallets in the spectral region 400–4000 cm−1 at room temperature. Raman scattering experiments were performed using LabRam HR (Horiba) spectrometer equipped with an Olympus BH41 microscope. The measurements were conducted using the 633-nm line of He-Ne laser in a backscattering geometry with an objective ×50. The spectra were collected in the range 100–4000 cm−1. Software package Origin 9 was used for spectral presentation.
3. Results and Discussion
3.1. Synthesis of Crystal Phases in the System MgSO4–OC(NH2)2–H2O
Figure 1 summarizes the results of previous synthesis [21,22,23,24] with respect to the experimental conditions and the type of phases obtained. In the diagram, we have not shown the products in the system at 25 °C because we could only find the abstract of the publication [25], which does not give any information about the ratio of the initial reagents. Figure 2 shows synthesis results of the present study. Symbols from the legend indicate the compounds obtained at 40 (blue color) and at 60 (red color) °C, respectively. Their positions in the diagrams correspond to the molar ratios of the reactants in the system. Figures showing the semi-quantitatively measured phase ratios (wt.%) are inserted immediately next to the symbols. In the studied system, maximum yields of urea complexes of magnesium sulfates are obtained under the following conditions:
- (i)
- nearly 100% MgSO4·6OC(NH2)2·0.5H2O—initial ratio MgSO4:OC(NH2)2:H2O~1:6:4 at 60 °C;
- (ii)
- nearly 100% MgSO4·OC(NH2)2·3H2O—initial ratio MgSO4:OC(NH2)2:H2O~1:1:2 at 60 °C;
- (iii)
- about 93% MgSO4·OC(NH2)2·2H2O—initial ratio MgSO4:OC(NH2)2:H2O~1:2:5 at 40 °C; and about 55% MgSO4·OC(NH2)2·H2O—initial ratio MgSO4:OC(NH2)2:H2O~1:2:1 at 60 °C.
In addition to the above-mentioned four magnesium urea sulfate hydrates, other compounds registered among the final products are three magnesium crystal hydrates, MgSO4·4H2O, MgSO4·5H2O, MgSO4·6H2O, and MgSO4·7H2O, with only the latter being used as a starting reagent, as well as (presumably unreacted) urea CO(NH2)2.
Such variety of products is due both to the high sensitivity of the studied system to the synthesis temperature and the ratio of the starting reagents, as well as to the high solubility of all considered phases. The latter allows easy structural arrangement and rearrangement of the primary and secondary building units with respect to the Mg polyhedral surrounding construction in the final products. Although the synthesis studies are not exhaustive, the achieved results clearly outline certain trends and consistencies. There is a direct dependence of the ratio of reactants and the chemical composition of the final products. Magnesium crystal hydrates crystallize as concomitant products in these parts of the system where there is an excess of SO42− anions. An increase in temperature leads to expansion of the fields of crystallization of the resulting phases that are of interest in this study, in direction and at the expense of the starting reactants.
Suitable samples for structural, IR and DSC-TG-MS analyses were obtained by crystallization from water solution under the following syntheses conditions:
- (i)
- MgSO4·OC(NH2)2·2H2O—MgSO4·7H2O:OC(NH2)2 = 1:1 at 80 °C;
- (ii)
- MgSO4·OC(NH2)2·3H2O—MgSO4·7H2O:OC(NH2)2 = 1:1 at 60 °C;
- (iii)
- MgSO4·4OC(NH2)2·H2O—MgSO4·7H2O:OC(NH2)2 = 1:4 at 60 °C;
- (iv)
- MgSO4·6OC(NH2)2·0.5H2O—MgSO4·7H2O:OC(NH2)2 = 1:6 at 60 °C.
3.2. Single Crystal Analyses
The experimental and structure refinement parameters for MgSO4·OC(NH2)2·2H2O and MgSO4·OC(NH2)2·3H2O are listed in Table 2. The molecular units of these compounds are shown in Figure 3 and their three-dimensional packing is shown in Figures S1 and S2. The crystal structures of MgSO4·4OC(NH2)2·H2O and MgSO4·6OC(NH2)2·0.5H2O have been published earlier [32,33]. MgSO4·OC(NH2)2·2H2O crystallizes in the triclinic crystal system, space group P-1. The magnesium ions in this compound form two types of octahedra. The octahedral units of the first type (MgO4(SO4)O2(H2O)) are connected by Mg–O-S-O–Mg bridges to neighboring octahedra of the same type to build chains parallel to [100]. The octahedral units of the second type (MgO2(SO4)O2(H2O)O2(urea)) connect the chains in [001] direction to form electroneutral layers parallel to (010). A schematic presentation of the described arrangement is shown in Figure 4. The water and urea molecules participate in a complex system of hydrogen bonding and stabilize the three-dimensional packing of the layers (Figure S1 and Table S1). The hydrogen atoms of the water molecules are only involved in intra-layer hydrogen bonds of the type O-H…O, whereas the hydrogen atoms of the urea molecules are involved in both intra-layer and inter-layer hydrogen bonds.
MgSO4·OC(NH2)2·3H2O crystalizes in a monoclinic crystal system, space group P21. In contrast to MgSO4·OC(NH2)2·2H2O, there is only one type of magnesium polyhedra (MgO2(SO4)O3(H2O)O1(Urea)) arranged in a folded electroneutral chain (Figure 4). The chains run parallel to [010] and their three-dimensional packing is stabilized by a network of hydrogen bonds in which all possible donors and acceptors are involved (Figure S2 and Table S2). Similar to the previously described compound, the water hydrogens stabilize the intra-chain arrangement, while the urea hydrogens participate in both intra-chain and inter-chain hydrogen bonding. MgSO4·4OC(NH2)2·H2O crystallizes in a monoclinic crystal system, space group P21/n, and exhibits structure where electroneutral magnesium octahedra (MgO4(urea)O1(H2O)O1(SO4)) occur as structure building units (Figure 4). MgSO4·6OC(NH2)2·0.5H2O crystallizes in an orthorhombic crystal system, space group Pccn. Complex (MgO6(urea))2+ cations and (SO4)2− anions are packed in such a way as to form the 3D crystal structure involving water molecules in the interstitials (Figure 4). The coordination polyhedra of Mg atoms for the known MgSO4·nOC(NH2)2·mH2O compounds are presented in Table S3. Similar to the magnesium sulfate hydrates’ structures found in the available structural databases (Cambridge Structural Database, Crystallography Open Database and American Mineralogist Crystal Structure Database), in the compounds studied here, the magnesium atom is in octahedral coordination. When the ligands number is less than six, the magnesium octahedra exhibit a tendency to polymerize, forming electroneutral units such as layers and chains or remaining as insular moieties in the structure (Figure 4). With six or more ligands in the compound, isolated complex magnesium cations are formed, the relatively large sizes of which predetermine the specific 3D arrangement of the structural units. Typical of the latter type compounds is the ionic nature of the bonds between the complex units. As for the structures made of electroneutral units, their tri-dimensional arrangement is stabilized by only hydrogen bonds.
3.3. Thermal Data
DSC-TG(DTG)-MS data of the compounds under study are presented in Figure 5. In the set temperature interval, the four studied compounds exhibit similar features of thermal behavior manifested in the following sequence: compound melting, crystallization water release, and urea decomposition. The first endothermic peak observed on the DSC curves for all of the studied compounds refers to the occurrence of melting processes at 62.0 °C, 87.5 °C, 91.7 °C and 128.2 °C. As can be seen, the melting temperature increases with decreasing of the crystallization water content for the corresponding compound. It should be mentioned that data on the melting temperature of MgSO4·6OC(NH2)2·0.5H2O obtained by mechano–chemical synthesis have been previously reported [27]. These authors stated that the melting effect occurs at 210 °C, although in the figure presented, a small endothermic peak could be detected at 127 °C (a temperature similar to that obtained in this study), which the authors did not attribute to any thermal process. The endo-effects positions and corresponding mass losses observed for MgSO4·OC(NH2)2·3H2O and MgSO4·OC(NH2)2·2H2O correlate well with those previously reported in US 2016/0046534 A1 patent data. The endothermal events occurring below 130 °C refer to the releasing of crystallization water molecules and their quantity per formula unit has been calculated on the basis of TG data. For MgSO4·OC(NH2)2·3H2O, one water molecule is released within the endothermal range, of which the maximum is at 116.6 °C. For MgSO4·4OC(NH2)2·H2O and MgSO4·6OC(NH2)2·0.5H2O, these effects occur at 124.4 °C and 128.2 °C and refer to the releasing of one or a half water molecules, respectively. All subsequent endothermal effects are related to the evolving of a variety of volatiles including CH4, NH3, H2O, NO2 and CO2. The results are indicative that, after 150 °C, the crystallization water release and urea decomposition occur simultaneously for all of the studied compounds. In general, the urea decomposition starts at various temperatures for each of the compounds: MgSO4·OC(NH2)2·3H2O—183.0 °C; MgSO4·OC(NH2)2·2H2O—184.8 °C, MgSO4·4OC(NH2)2·H2O—210.0 °C; MgSO4·6OC(NH2)2·0.5H2O—213.6 °C.
Based on these data, it could be concluded that the number of water molecules in the crystal structure is of particular importance for their thermal behavior and stability. It is evident that as the water content of the compound decreases, both melting point and urea decomposition temperature increase. The substitution of urea for water also plays a role in the structure stabilization, with the most stable structure being the one in which the magnesium surrounding involves solely urea moieties. Such a type of thermal stability trend has also been observed for other crystal hydrate adducts of urea [12,20].
3.4. IR and Raman Spectra
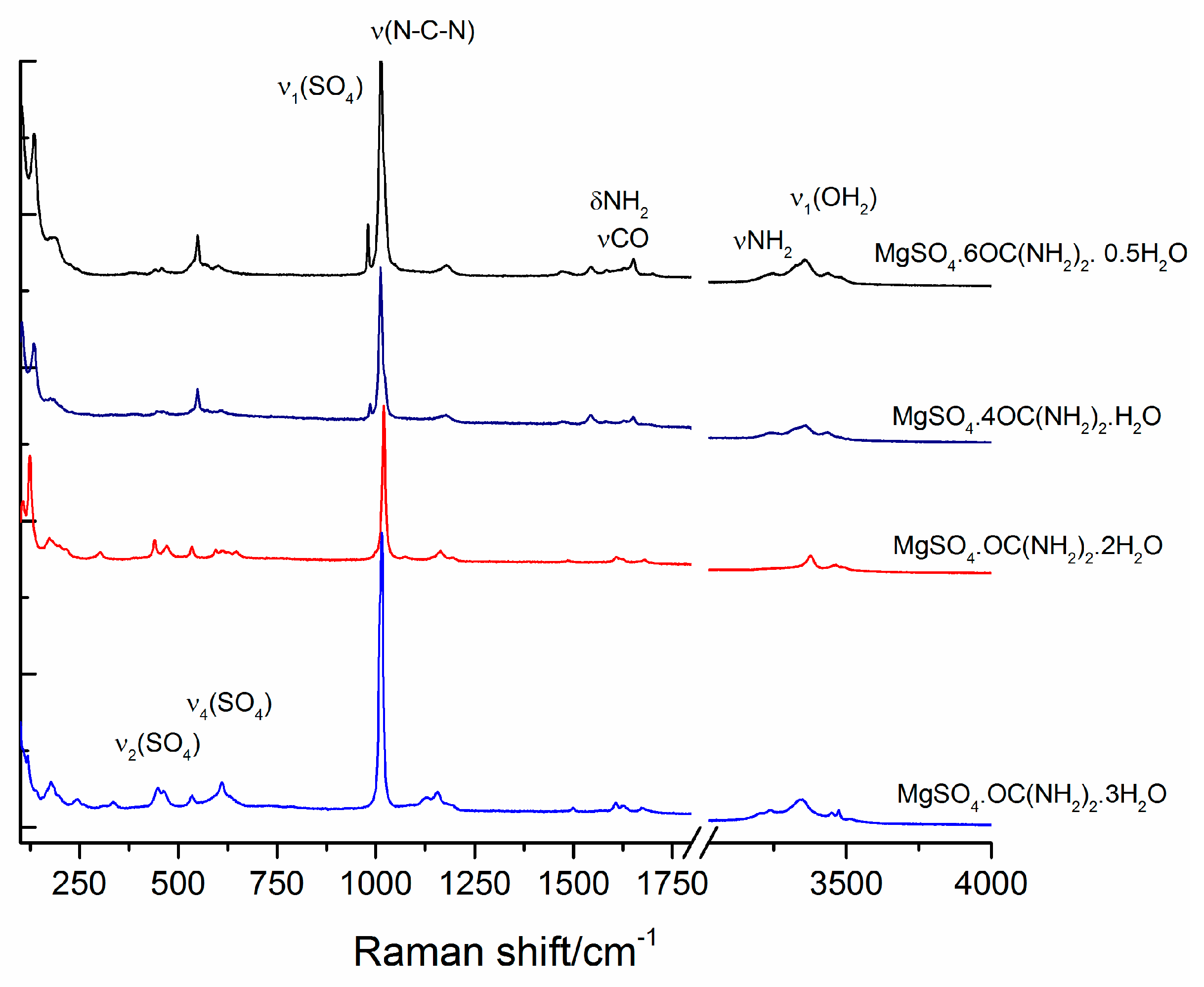
The infrared spectra of magnesium sulfates are dominated by the absorption peaks of sulfate group and water molecules and their peak positions and assignments have already been reported [1,36,37,38]. Experimental IR spectra of the studied compounds are presented in Figure 6. The peaks in the range of O-H stretching of water molecules are overlapped by the peaks of NH2 stretching of urea [20]. The samples of the compounds with six and four urea molecules exhibit three distinguishable peaks at 3480, 3350 and 3210 cm−1, whereas for the other two compounds with higher water contents these peaks merge into one broad band. In addition, the position of the peak arising from δ (CNH) is sensitive to the urea to water ratio. The position of this peak shifts from 1470 cm−1 in sample MgSO4·6OC(NH2)2·0.5H2O to 1480 cm−1 in a sample with higher water content (MgSO4·OC(NH2)2·3H2O). Another significant spectral difference can be detected in the region of ρ(OH2)rock + δ(OCN). Samples with high urea content reveal a peak at 783 cm−1, whereas another peak at around 750 cm−1 appears with increasing water content. Within the range 900–1150 cm−1, strong absorption of SO4 and NH2 groups dominates. It can be seen that the intensity ratio of the peaks at 1110 and 1150 cm−1 changes with the ratio of urea to water. The absorption near 1150 cm−1 is stronger for MgSO4·6OC(NH2)2·0.5H2O, whereas with the increase of water molecules in the rest of the compounds, the intensity of the peak at 1110 cm−1 increases. Raman spectra of the studied compounds are presented in Figure 7. The spectra are dominated by the strong peaks at 980, 1012, 1020 and 1025 cm−1 in the range of ν(SO4) and N-C-N stretching vibrations [39]. There are peaks at 980, 1013 and a shoulder at 1022 cm−1 in the spectrum of MgSO4·6OC(NH2)2·0.5H2O. With a decrease of urea for water substitution, the same peaks shift to 985, 1013 and 1025 cm−1 in sample MgSO4·4OC(NH2)2·H2O, to 997 and 1020 cm−1 in MgSO4·OC(NH2)2·2H2O, and to around 1015 cm−1 in MgSO4·OC(NH2)2·3H2O.
4. Conclusions
New data about the products prepared in the system MgSO4–OC(NH2)2–H2O are presented as follows:
- (i)
- Conditions for preparation of pure MgSO4·OC(NH2)2·2H2O, MgSO4·OC(NH2)2·3H2O, MgSO4·4OC(NH2)2·H2O and MgSO4·6OC(NH2)2·0.5H2O are specified (evaporation from dilute aqueous solutions) and of pure MgSO4·OC(NH2)2·3H2O and MgSO4·6OC(NH2)2·0.5H2O (upon mixing MgSO4·nH2O and urea in appropriate ratios and by applying mechanoactivation);
- (ii)
- The crystal structures of MgSO4·OC(NH2)2·2H2O and MgSO4·OC(NH2)2·3H2O are determined and their features are compared with previously published structures of MgSO4·4OC(NH2)2·H2O and MgSO4·6OC(NH2)2·0.5H2O;
- (iii)
- Thermal analysis establishes a direct relationship between the thermal stability of the studied compounds, as well as the decomposition temperature of urea and the OC(NH2)2:H2O ratio in the octahedral environment of the magnesium ion in the structures of the respective compounds;
- (iv)
- Certain spectroscopic characteristics (IR and Raman) of the studied compounds are reported for the first time and the results have been analysed with respect to the specific properties of the chemical composition.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst14030227/s1, Figures S1 and S2: 3D packing of MgSO4·OC(NH2)2·2H2O and MgSO4·OC(NH2)2·3H2O; Table S1: Distances (Å) and angles (°) describing hydrogen bonds in MgSO4·OC(NH2)2·2H2O; Table S2: Distances (Å) and angles (°) describing hydrogen bonds in MgSO4·OC(NH2)2·3H2O; Table S3: Coordination polyhedral of Mg atoms in the known MgSO4·nOC(NH2)2·mH2O compounds.
Author Contributions
Conceptualization, R.N. and V.K.-K.; methodology, R.N., V.K.-K., N.P., K.K. and R.T.; formal analysis, R.N., V.K.-K., N.P., R.T. and G.V.; writing—original draft preparation, R.N., V.K.-K., N.P., R.T. and K.K.; writing—review and editing, V.K.-K. and R.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Bulgarian National Science Fund, grant agreement KП-06-H64/4, 15 December 2022.
Data Availability Statement
The raw data are available upon request.
Acknowledgments
The authors acknowledge the CoE ‘National Centre of Mechatronics and Clean Technologies’, BG05M2OP001-1.001-0008-C01. for the single crystal experiments.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Patnaik, P. Handbook of Inorganic Chemicals, 1st ed.; The McGraw-Hill: New York, NY, USA, 2003; pp. 510–515. [Google Scholar]
- Bowen, H.J.M. Trace Elements in Biochemistry, 1st ed.; Academic Press: New York, NY, USA, 1996; pp. 241–250. [Google Scholar]
- Rogolino, D.; Carcelli, M.; Sechi, M.; Neamati, N. Viral enzymes containing magnesium: Metal binding as a successful strategy in drug design. Coord. Chem. Rev. 2012, 256, 3063–3086. [Google Scholar] [CrossRef]
- Seelig, M.S. Magnesium Deficiency in the Pathogenesis of Disease: Early Roots of Cardiovascular, Skeletal, and Renal Abnormalities, 1st ed.; Springer Science & Business Media: New York, NY, USA, 2012; pp. 3–20. [Google Scholar]
- Hsiao, C.; Tannenbaum, E.; VanDeusen, H.; Hershkovitz, E.; Perng, G.; Tannenbaum, A.; Williams, L.D. Chapter 1: Complexes of Nucleic Acids with Group I and Group II Cations. In Nucleic Acid-Metal Ion Interactions, 1st ed.; The RSC Publishihg: London, UK, 2008; pp. 1–38. [Google Scholar]
- Sanchez, P.A. Soil fertility and hunger in Africa. Science 2002, 295, 2019–2020. [Google Scholar] [CrossRef]
- Hawkesford, M.J.; Çakmak, İ.; Coskun, D.; De Kok, L.J.; Lambers, H.; Schjoerring, J.K.; White, P.J. Chapter 6: Functions of macronutrients. In Marschner’s Mineral Nutrition of Plants, 4th ed.; Academic Press: London, UK, 2023; pp. 201–281. [Google Scholar]
- Asa’ad, I.A. Magnesium and Drug Interactions. Iraqi Natl. J. Nurs. Spec. 2011, 24, 25–36. [Google Scholar]
- Remick, K.A.; Helmann, J.D. The elements of life: A biocentric tour of the periodic table. In Advances in Microbial Physiology, 1st ed.; Academic Press: London, UK, 2023; Volume 82, pp. 1–127. [Google Scholar]
- Hennings, E.; Schmidt, H.; Voigt, W. Crystal structures of hydrates of simple inorganic salts. I. Water-rich magnesium halide hydrates MgCl2·8H2O, MgCl2·12H2O, MgBr2·6H2O, MgBr2·9H2O, MgI2·8H2O and MgI2·9H2O. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2013, 69, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Rusev, R.; Tsvetanova, L.; Shivachev, B.; Kossev, K.; Nikolova, R. Ureates and hydrates of magnesium chloride, nitrate and tetrafluoroborate. Bulg. Chem. Commun. 2018, 50, 79–89. [Google Scholar]
- Kosev, K.; Petrova, N.; Georgieva, I.; Titorenkova, R.; Nikolova, R. Crystalline adducts of urea with magnesium iodide. J. Mol. Struct. 2021, 1224, 129009. [Google Scholar] [CrossRef]
- Lebioda, Ł.; Stadnicka, K.; Sliwinski, J. Hexakis (urea) magnesium bromide–urea (1/4). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1979, 35, 157–158. [Google Scholar] [CrossRef]
- Lebioda, Ł.; Lewiński, K. Structure of diaquatetrakis (urea) magnesium bromide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1980, 36, 693–695. [Google Scholar] [CrossRef]
- Kossev, K.; Tsvetanova, L.; Dimowa, L.; Nikolova, R.; Shivachev, B. Synthesis and crystal structure of magnesium chlorate dihydrate and magnesium chlorate hexahydrate. Bulg. Chem. Commun. 2013, 45, 543–548. [Google Scholar]
- Todorov, T.; Petrova, R.; Kossev, K.; Macícek, J.; Angelova, O. Hexakis (urea-O) magnesium Dichlorate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 927–929. [Google Scholar] [CrossRef]
- Yamagata, K.; Achiwa, N.; Hashimoto, M.; Koyano, N.; Iwata, Y.; Shibuya, I. Magnesium formate-urea (1/2). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1992, 48, 793–795. [Google Scholar] [CrossRef]
- Hayden, T.D.; Kim, E.E.; Eriks, K. Crystal structures of bis (urea) bis(dihydrogenphosphato) calcium-bis (urea) and its isomorphous magnesium analog, M[OC(NH2)2]2(H2PO4)2.2CO(NH2)2 (M = Ca, Mg). Inorg. Chem. 1982, 21, 4054–4058. [Google Scholar] [CrossRef]
- Angelova, O.; Macíček, J.; Petrova, R.; Todorov, T.; Mihailova, B. Structure of molecular adducts of inorganic salts. IV. Comparison of the Cd (ReO4)2·2Urea and Cd (ReO4)2·2H2O structures 1. Z. Für Krist. -Cryst. Mater. 1996, 211, 163–169. [Google Scholar] [CrossRef]
- Georgieva, I.; Kossev, K.; Titorenkova, R.; Petrova, N.; Zahariev, T.; Nikolova, R. Effect of urea on arrangement of novel Mg (II) perrhenate crystal structures and their optical properties: Experimental and theoretical insight. J. Solid State Chem. 2022, 312, 123263. [Google Scholar] [CrossRef]
- Whittaker, C.W.; Lundstrom, F.O.; Shim, J.H. The system magnesium sulfate-urea-water at 30°. J. Am. Chem. Soc. 1936, 58, 1975–1977. [Google Scholar] [CrossRef]
- Yee, J.Y.; Davis, R.O.E.; Hendricks, S.B. Double Compounds of Urea with Magnesium Nitrate and Magnesium Sulfate. J. Am. Chem. Soc. 1937, 59, 570–571. [Google Scholar] [CrossRef]
- Sulaimankulov, K.; Druzhinin, I.G.; Bergman, A.G. Interaction of urea with sulfates of divalent metals. i. isotherms 0, 30 and 45° systems: Water-urea-magnesium sulfate. J. Inorg. Chem. 1957, 2, 2668. (In Russian) [Google Scholar]
- Sulaimankulov, K. Compounds of Urea with Inorganic Salts, 1st ed.; Ilim: Frunse, Russia, 1971; p. 224. (In Russian) [Google Scholar]
- Zhang, F.; Wei, X.; Guo, Z.; Shi, Q. A study on the isothermal solubility of MgSO4-CO(NH2)2-H2O ternary system at 25 °C. Chin. J. Inorg. Chem. 1997, 13, 375–379. [Google Scholar]
- Mahadevan, C.K. Thermal parameters of MgSO4·7H2O and NiSO4·7H2O crystals added with urea and thiourea. Phys. B Condens. Matter 2008, 403, 3164–3167. [Google Scholar] [CrossRef]
- Honer, K.; Kalfaoglu, E.; Pico, C.; McCann, J.; Baltrusaitis, J. Mechanosynthesis of magnesium and calcium salt–urea ionic cocrystal fertilizer materials for improved nitrogen management. ACS Sustain. Chem. Eng. 2017, 5, 8546–8550. [Google Scholar] [CrossRef]
- Honer, K.; Pico, C.; Baltrusaitis, J. Reactive mechanosynthesis of urea ionic cocrystal fertilizer materials from abundant low solubility magnesium-and calcium-containing minerals. ACS Sustain. Chem. Eng. 2018, 6, 4680–4687. [Google Scholar] [CrossRef]
- Adlim, M.; Ramayani, R.F.I.; Khaldun, I.; Muzdalifah, F.; Sufardi, S.; Rahmaddiansyah, R. Fertilizing Properties of Urea-Magnesium Slowrelease Fertilizer Made of Rice-Husk-Ash Natural-Rubber Chitosan Composite. Rasayan J. Chem. 2021, 14, 1851–1859. [Google Scholar] [CrossRef]
- Maynard-Casely, H.E.; Brand, H.E.; Wilson, S.; Wallwork, K.S. Mineral diversity on Europa: Exploration of phases formed in the MgSO4–H2SO4–H2O ternary. ACS Earth Space Chem. 2021, 5, 1716–1725. [Google Scholar] [CrossRef]
- Adassooriya, N.M.; Mahanta, S.P.; Thakuria, R. Mechanochemistry as an emerging tool for the preparation of sustained release urea cocrystals as a nitrogen source. CrystEngComm 2022, 24, 1679–1689. [Google Scholar] [CrossRef]
- Todorov, T.; Petrova, R.; Kossev, K.; Macicek, J.; Angelova, O. Magnesium sulfate tetraurea monohydrate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 456–458. [Google Scholar] [CrossRef]
- Todorov, T.; Petrova, R.; Kossev, K.; Macicek, J.; Angelova, O. Magnesium sulfate hexaurea hemihydrate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1758–1760. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The HighScore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef]
- Kraus, W.; Nolze, G. POWDER CELL—A Program for the Representation and Manipulation of Crystal Structures and Calculation of the Resulting X-Ray Powder Patterns. J. Appl. Crystallogr. 1996, 29, 301–303. [Google Scholar] [CrossRef]
- Chukanov, N.V. Infrared spectra of mineral species, 1st ed.; Extended Library, Springer: Dordrecht, The Netherlands, 2014; pp. 21–1701. [Google Scholar]
- Buzgar, N.; Apopei, A.; Buzatu, A. Romanian Database of Raman Spectroscopy. Available online: http://rdrs.uaic.ro (accessed on 26 July 2023).
- Makreski, P.; Jovanovski, G.; Dimitrovska, S. Minerals from Macedonia: XIV. Identification of some sulfate minerals by vibrational (infrared and Raman) spectroscopy. Vib. Spectrosc. 2005, 39, 229–239. [Google Scholar] [CrossRef]
- Frost, R.; Kristof, J.; Rintoul, L.; Kloprogge, T. Raman spectroscopy of urea and urea-intercalated kaolinites at 77 K. Spectrochim. Acta Part A 2000, 56, 1681–1691. [Google Scholar] [CrossRef]
Figure 1.
Diagram of crystallization of magnesium compounds prepared in the system MgSO4–OC(NH2)2–H2O at 30 at 45 °C.
Figure 1.
Diagram of crystallization of magnesium compounds prepared in the system MgSO4–OC(NH2)2–H2O at 30 at 45 °C.

Figure 2.
Diagram of crystallization of magnesium compounds prepared in the system MgSO4–OC(NH2)2–H2O at 40 and 60 °C based on the results from this study.
Figure 2.
Diagram of crystallization of magnesium compounds prepared in the system MgSO4–OC(NH2)2–H2O at 40 and 60 °C based on the results from this study.
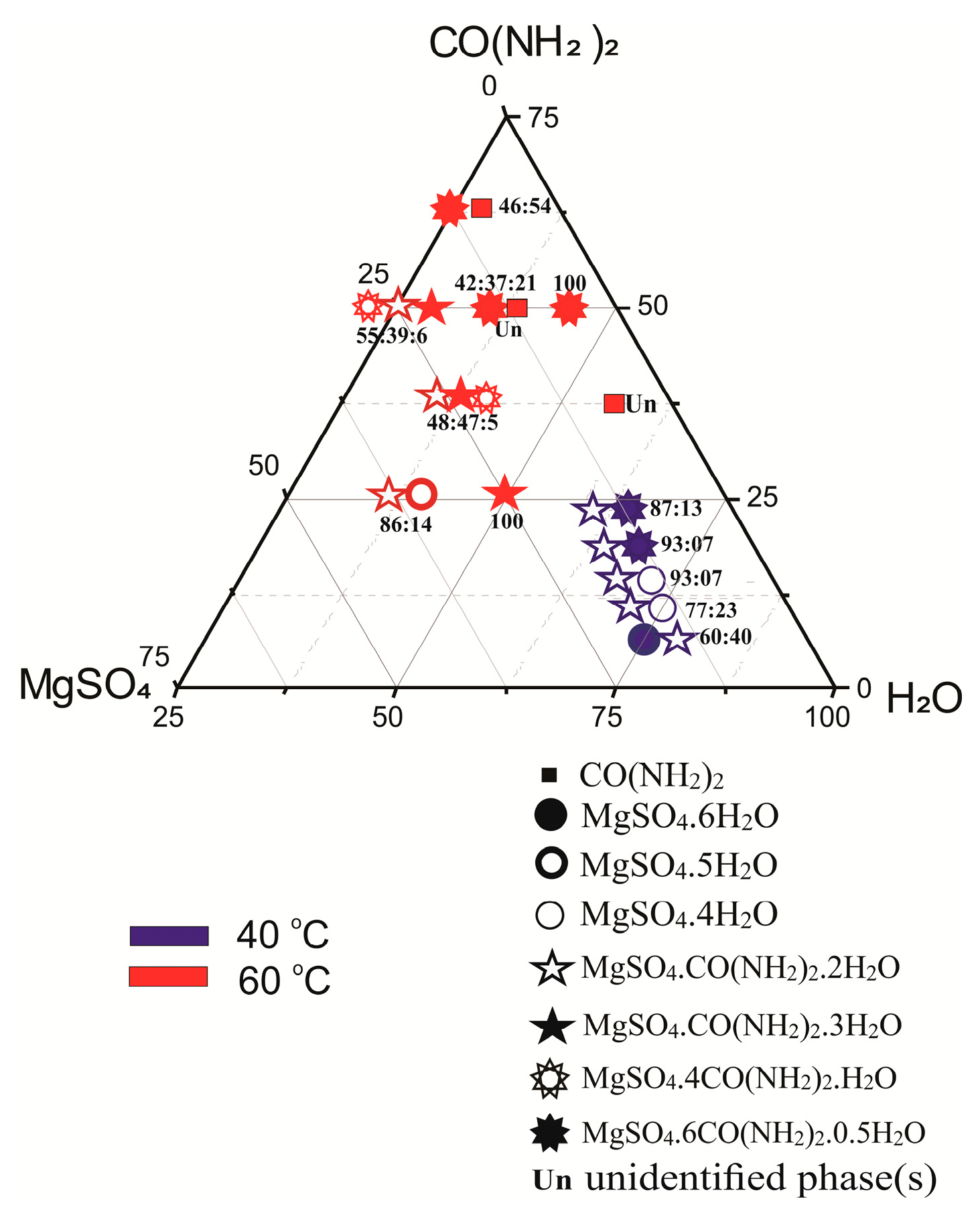
Figure 3.
Molecular units of: (a) MgSO4·OC(NH2)2·2H2O [(i) 1-x, 1-y, 1-z; (ii) –x, 1-x, 2-z; (iii) x-1, y, z (vi) 1-x, 1-y, 2-z] and (b) MgSO4·OC(NH2)2·3H2O [(i) 1-x, ½+y, 2-z]. The hydrogen atoms are marked with blue circles.
Figure 3.
Molecular units of: (a) MgSO4·OC(NH2)2·2H2O [(i) 1-x, 1-y, 1-z; (ii) –x, 1-x, 2-z; (iii) x-1, y, z (vi) 1-x, 1-y, 2-z] and (b) MgSO4·OC(NH2)2·3H2O [(i) 1-x, ½+y, 2-z]. The hydrogen atoms are marked with blue circles.
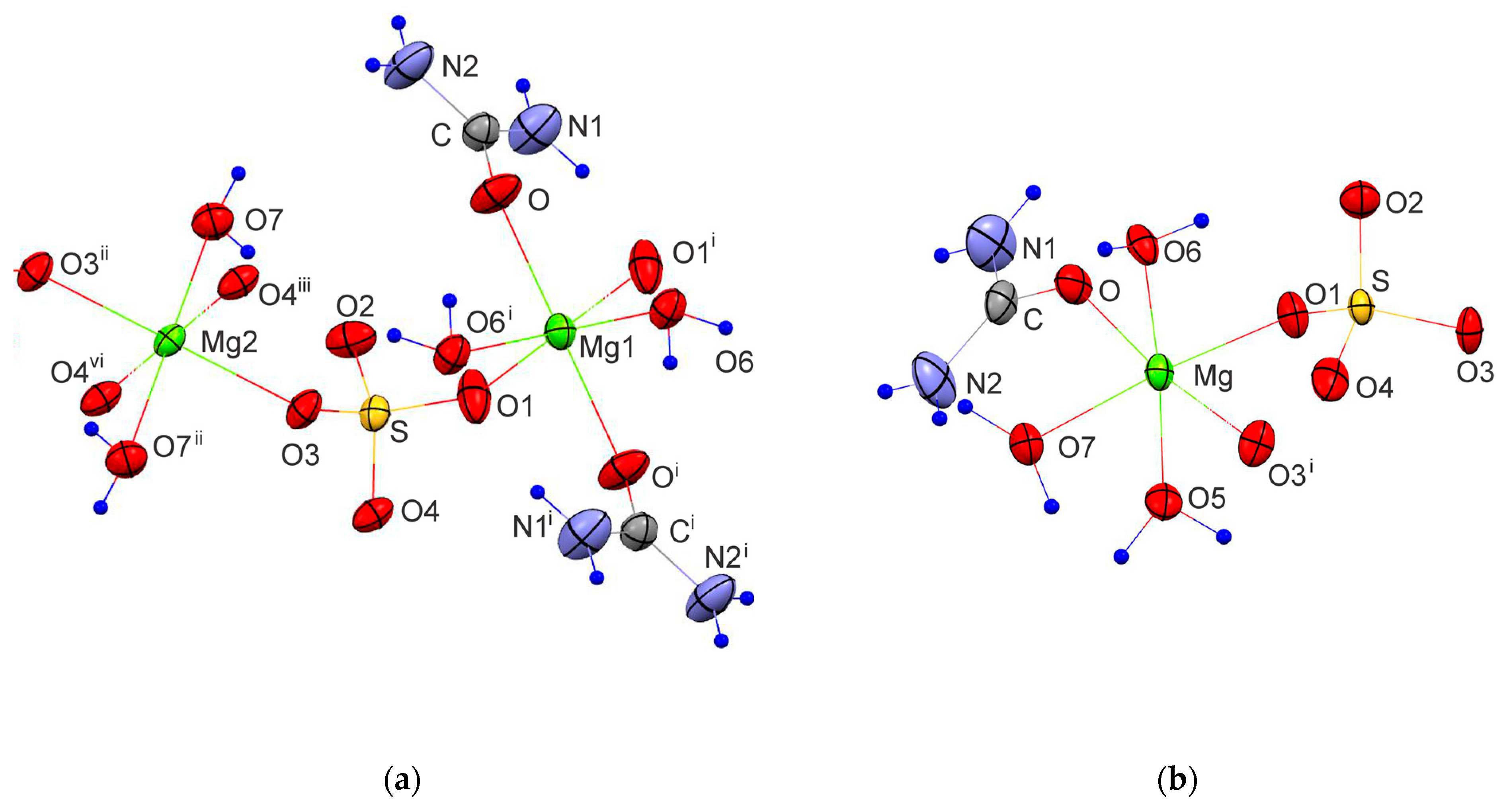
Figure 4.
Schematic presentation of atomic arrangement for the Urea complexes of MgSO4: (1) MgSO4·OC(NH2)2·2H2O, (2) MgSO4·OC(NH2)2·3H2O, (3) MgSO4·4OC(NH2)2·H2O and (4) MgSO4·6OC(NH2)2·0.5H2O. Urea and water oxygens are labelled as Ou and Ow, respectively.
Figure 4.
Schematic presentation of atomic arrangement for the Urea complexes of MgSO4: (1) MgSO4·OC(NH2)2·2H2O, (2) MgSO4·OC(NH2)2·3H2O, (3) MgSO4·4OC(NH2)2·H2O and (4) MgSO4·6OC(NH2)2·0.5H2O. Urea and water oxygens are labelled as Ou and Ow, respectively.

Figure 5.
DSC-TG(DTG)-MS data of the studied compounds. The numbers indicate molecular weight of the corresponding gas as follows: 16 for CH4; 17 for NH3; 18 for H2O; 28 for NO2, and 44 for CO2.
Figure 5.
DSC-TG(DTG)-MS data of the studied compounds. The numbers indicate molecular weight of the corresponding gas as follows: 16 for CH4; 17 for NH3; 18 for H2O; 28 for NO2, and 44 for CO2.

Figure 6.
Infrared spectra of the studied compounds.
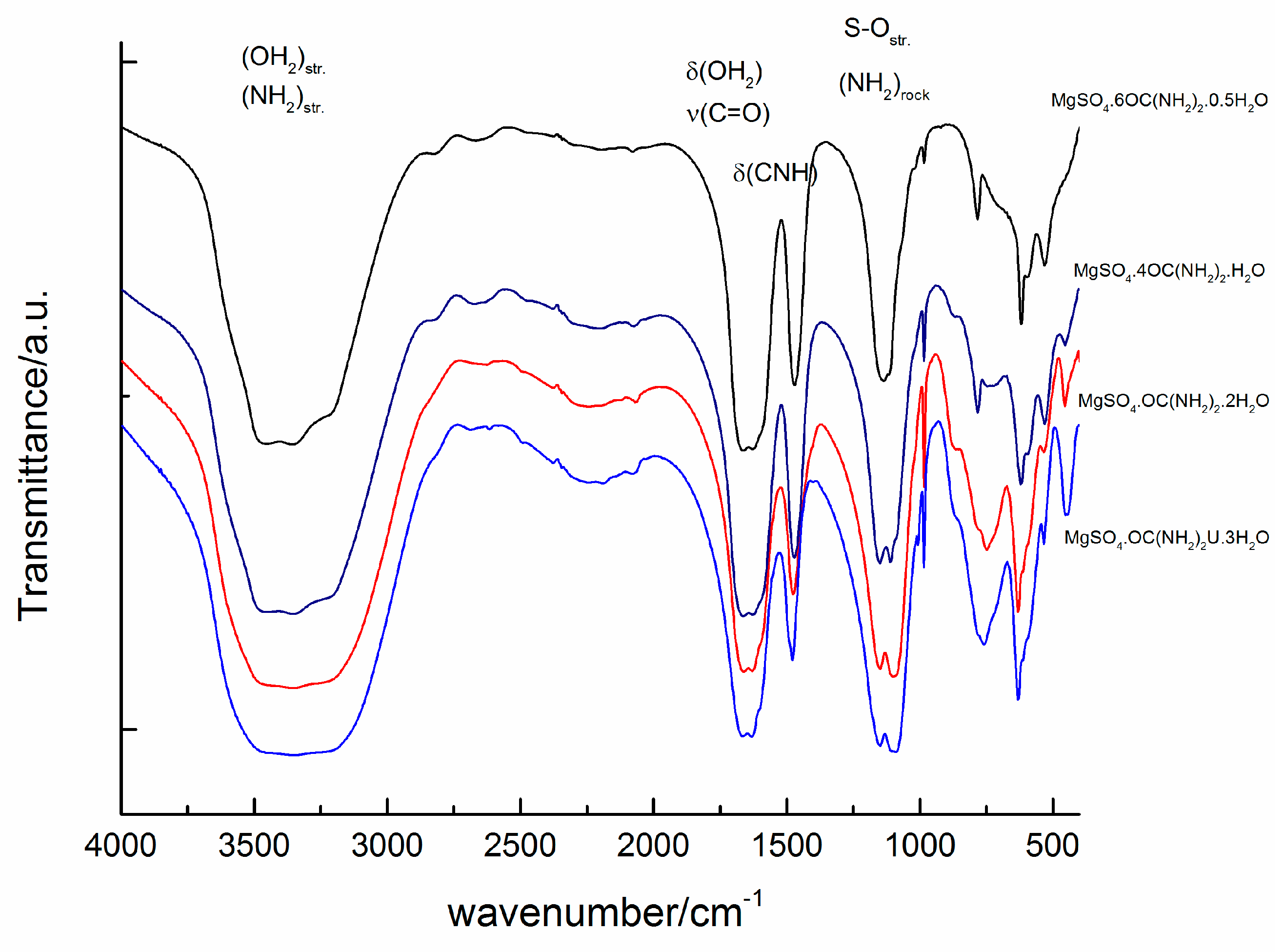
Figure 7.
Raman spectra of studied compounds.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Reported MgSO4·nOC(NH2)2·mH2O phases.
| Compound | Synthesis | Identification | Structural Data | References |
|---|---|---|---|---|
| MgSO4·OC(NH2)2·2H2O | Solution, MgSO4·7H2O + nUrea + mH2O/25 °C | Chem. analyses of obtained solid, IR | Powder diffraction pattern | [25,27] |
| MgSO4·OC(NH2)2·3H2O | Solution, MgSO4·7H2O + nUrea + mH2O/30 °C | Chem. analyses of obtained solid | Powder diffraction pattern | [21,23,24] |
| MgSO4·4OC(NH2)2·H2O | Solution, MgSO4·7H2O + 4Urea + mH2O/22 °C (RT) | Single crystal and powder XRD Crystal structure determined | P21/n a = 9.345(2), b = 14.683(4), c = 11.244(3) β = 93.07(2) | [24,32] |
| MgSO4·6OC(NH2)2·0.5H2O | Mechanoactivation, MgSO4·7H2O + 6Urea/22 °C (RT) Solution, MgSO4·7H2O + 6Urea + mH2O/22 °C (RT) | Single crystal and powder XRD Crystal structure determined | Pnma a = 15.316(5), b = 19.798(5), c = 14.484(2) | [28,33] |
| MgSO4·5OC(NH2)2·2H2O | Solution, MgSO4·7H2O + nUrea + 95% methyl alcohol/30 °C | Single crystal and powder XRD Unit cell parameters only | XRD Pnma a = 17.32, b = 11.40, c = 9.61 | [22] |
| MgSO4·6OC(NH2)2·2H2O | Solution, MgSO4·7H2O + nUrea + 95% methyl alcohol/25 °C | Single crystal and powder XRD Unit cell parameters only | XRD Pccn a = 16.20, b = 19.97, c = 14.38 | [22] |
Table 2.
Most important data collection and refinement parameters for the studied compounds.
| Chemical Formula | Mg(SO4)·OC(NH2)2·2H2O | Mg(SO4)·OC(NH2)2·3H2O |
|---|---|---|
| Formula weight | 216.46 | 234.48 |
| Temperature (K) | 290(2) | 290(2) |
| Crystal system | Triclinic | Monoclinic |
| Space group | P-1 | P 21 |
| a (Å) | 5.2140(8) | 5.2452(11) |
| b (Å) | 7.7014(9) | 7.7080(14) |
| c (Å) | 10.4275(16) | 10.230(2) |
| α (°) | 68.758(13) | 90 |
| β (°) | 77.177(13) | 94.291(8) |
| γ (°) | 71.498(12) | 90 |
| Volume (Å3) | 367.38(10) | 412.43(14) |
| Z | 2 | 2 |
| ρcalc (g/cm3) | 1.957 | 1.888 |
| μ (mm−1) | 0.534 | 0.492 |
| Crystal size (mm3) | 0.03 × 0.02 × 0.02 | 0.03 × 0.03 × 0.01 |
| Radiation, λ (Å) | MoKα λ = 0.71073 | MoKα λ = 0.71073 |
| Θ range for data collection (°) | 2.111 to 25.345 | 3.313 to 26.004 |
| Limiting indices | −6 ≤ h ≤ 6, −9 ≤ k ≤ 9, −12 ≤ l ≤ 12 | −6 ≤ h ≤ 6, −9 ≤ k ≤ 9, −12 ≤ l ≤ 12 |
| Reflections collected /unique | 10667/1347 (R(int) = 0.0564) | 6912/1618 (R(int) = 0.0608) |
| Data/restraints/parameters | 1347/0/112 | 1476/1/119 |
| Goodness-of-fit on F2 | 1.239 | 0.914 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0673, wR2 = 0.1474 | R1 = 0.0371 wR2 = 0.0994 |
| Final R indexes [all data] | R1 = 0.0762 wR2 = 0.1511 | R1 = 0.0379 wR2 = 0.1003 |
| Largest diff. peak/hole (e Å−3) | 0.870/−0.448 | 0.372/−0.590 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nikolova, R.; Kostov-Kytin, V.; Petrova, N.; Kossev, K.; Titorenkova, R.; Velyanova, G. New Data on Crystal Phases in the System MgSO4–OC(NH2)2–H2O. Crystals 2024, 14, 227. https://doi.org/10.3390/cryst14030227
AMA Style
Nikolova R, Kostov-Kytin V, Petrova N, Kossev K, Titorenkova R, Velyanova G. New Data on Crystal Phases in the System MgSO4–OC(NH2)2–H2O. Crystals. 2024; 14(3):227. https://doi.org/10.3390/cryst14030227
Chicago/Turabian StyleNikolova, Rositsa, Vladislav Kostov-Kytin, Nadia Petrova, Krasimir Kossev, Rositsa Titorenkova, and Gergana Velyanova. 2024. "New Data on Crystal Phases in the System MgSO4–OC(NH2)2–H2O" Crystals 14, no. 3: 227. https://doi.org/10.3390/cryst14030227
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.