Equivalence of Electron-Vibration Interaction and Charge-Induced Force Variations: A New O(1) Approach to an Old Problem

Abstract
: Calculating electron-vibration (vibronic) interaction constants is computationally expensive. For molecules containing N nuclei it involves solving the Schrödinger equation for nuclear configurations in addition to the cost of determining the vibrational modes. We show that quantum vibronic interactions are proportional to the classical atomic forces induced when the total charge of the system is varied. This enables the calculation of vibronic interaction constants from solutions of the Schrödinger equation. We demonstrate that the approach produces numerically accurate results by calculating the vibronic interaction constants for several molecules. We investigate the role of molecular vibrations in the Mott transition in κ-(BEDT-TTF)2Cu[N(CN)2]Br.1. Introduction
Electrons in molecules and solids feel two forces: a direct Coulomb interaction between themselves and a Coulombic interaction with the atomic nuclei. As the vibrations of the nuclei are quantized the interaction with the nuclei takes the form of the vibronic interaction. Electron-vibration interactions play important roles across science. In physics vibronic interactions can give rise to superconductivity [1], spin and charge density waves [2], polaron formation [3] and piezoelectricity [1]. In chemistry they impact electron-transfer processes [4], Jahn–Teller effects [5], spectroscopy [6,7], stereochemistry [5], activation of chemical reactions [5] and catalysis [5]. In biology the vibronic interactions play important roles in photoprotection [8,9], photosynthesis [10] and vision [11]. In engineering vibronic interactions play important roles in determining the efficiency of organic photovoltaic cells and organic light emitting diodes [12]. It is therefore clear that one of the central tasks for condensed matter theory and theoretical chemistry is to accurately and efficiently calculate vibronic interaction constants.
The Holstein Hamiltonian [3], which describes the vibronic coupling, is
An alternative is to consider Janak’s theorem [14] which states that
Conceptually it is useful to observe that in the frozen phonon and DFPT approaches one considers the problem from the point of view of the electrons. That is, one calculates the vibronic interaction constants by making a small perturbation to nuclei and considering the resultant change on the electron. However, in the approach one asks the question what happens to the nuclei when one puts a small additional charge in the system. In many cases one is only interested in the coupling of the vibrations to the electronic states closest to the Fermi level, however, if one were interested in interactions with other states, an extension of Janak’s theorem to other electronic states allows for the determination of these parameters straightforwardly within the method.
The primary observation reported here is that using Equation 4 the gi can be calculated by solving a single nuclear geometry. However in addition to pointing toward an efficient means for the calculation of the vibronic interaction, Equation 6 demonstrates a fundamental relationship between the electron-vibrational interaction and the charge induced geometrical relaxations. The former is generally viewed as a quantum-mechanical characteristic while the latter is viewed as a classical phenomena. Equation 6 simply states that the changes in the atomic Hellmann–Feynman forces due to charge addition or depletion depend on the same fundamental quantities as the electron-vibrational interaction. Below, we present results from this new O(1) method for calculation of the spin-vibron interaction (∂λμ/∂Qi).
2. Results
We now show that Equation 4 reproduces the values of the vibronic interaction constants calculated by the frozen phonon method for a number of small molecules. As discussed in the methods section, we have implemented our scheme for calculating electron-vibrational interaction constants within a density-functional-based computational code written by Pederson and collaborators [16–18] using the Perdew-Burke-Ernzerhof [19] exchange correlation functional.
It is worthwhile to briefly comment on some of the approximations used during these numerics, which are not, formally, required by the method itself. All of the calculations below are performed within the Born–Oppenheimer approximation (BOA). It may not be immediately clear that giμ is finite in the BOA. The BOA is to assume that the many-body wavefunction Ψ({R}, {r}), which in general depends on the set of nuclear co-ordinates, {R}, and the set of electronic co-ordinates, {r}, is separable into a nuclear wavefunction ϕ({R}) and an electronic wavefunction ψ({R}, {r}). That is, Ψ({R}, {r}) = ϕ({R}) ⊗ ψ({R}, {r}). Physically this corresponds to two assumptions: (i) the electronic wavefunction depends on the nuclear positions, Rj, but not the momenta of the nuclei and (ii) the nuclear motion sees a smeared out potential from the speedy electrons. We then treat the nuclei as classical particles and so we only consider ψ({R}, {r}), and hence we calculate only the electronic density ρ({R}, {r}) within the DFT framework. It is the dependence of ρ({R}, {r}) on the nuclear coordinates (but not the nuclear momenta) that leads to finite giμ. Finally we note that the BOA does not give the exact solution to Equation 1 unless all of the giμ vanish. However for small giμ the BOA gives a good approximation to, for example, the exact solution of the two site Holstein model [3].
In Figure 1 we plot the calculated vibronic interactions for tetrathiafulvalene (TTF), tetracyanoquinodimethane (TCNQ), bis(ethylenedithio)-tetrathiafulvalene (BEDT-TTF), C60 and a number of diatomic and other small molecules calculated by the frozen phonon method against the vibronic interaction constants calculated by our method. It can be seen that the values calculated by either method are in good agrement with one another.
Note that, as the normal coordinate is the eigenvector of the dynamical matrix, the sign of Qi is not well defined. Therefore, although ∂E is negative definite, the sign of giμ is also not well defined. However, the product giμ∂Qi is well defined and describes the distortion caused by the addition of a charge. For example, for the stretching mode in a simple diatomic molecule, the sign of the product giμ∂Qi indicates whether the bond is lengthened or shortened when a charge is added.
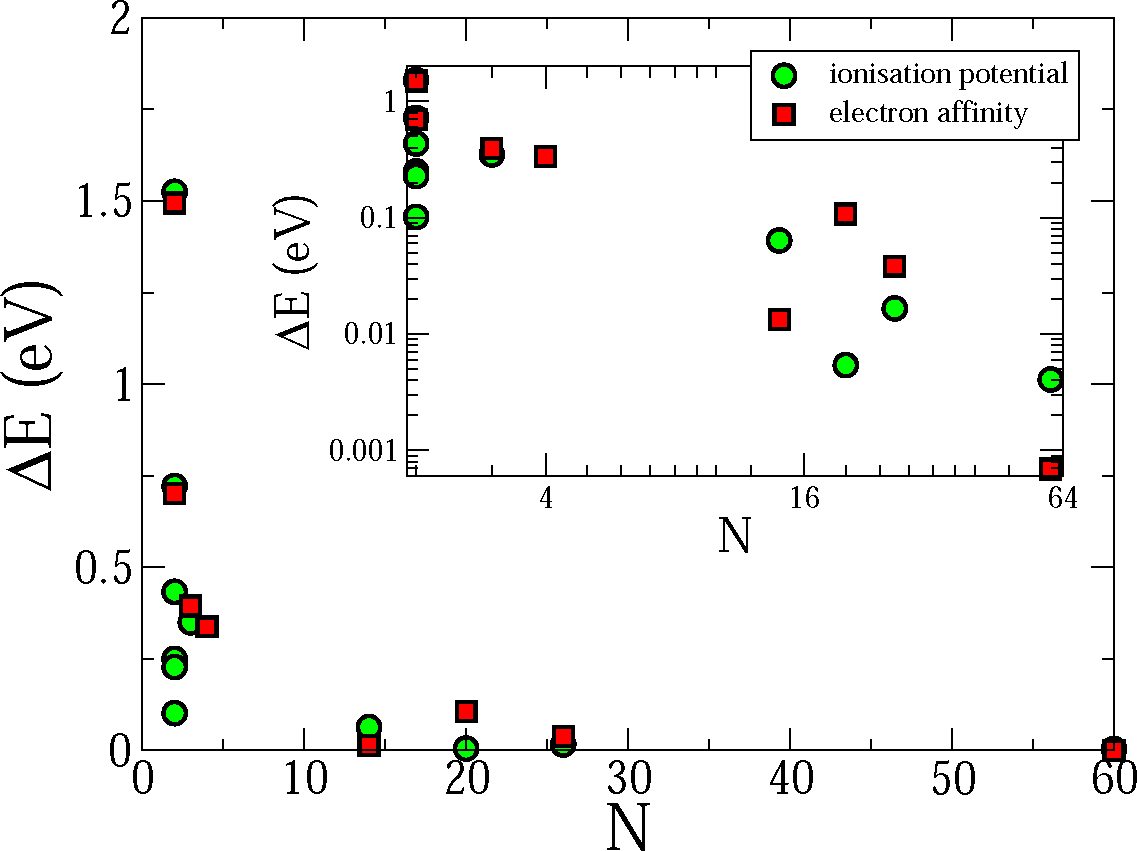
Because the calculation of vibronic interaction constants is now computationally inexpensive, it is possible to use the vibronic interaction to estimate other quantities. For example, the ionization energy (or electron affinity) of a molecule, as opposed to the vertical ionization energy (electron affinity), may be extracted from the information already calculated. To do this one determines the Hubbard U (a second derivative) from either the energy as a function of ∓δnμ and ∓2δnμ or from the appropriate eigenvalue as a function of ∓δnμ. As the vibronic interaction constants are known, the adiabatic ionization energy (electron affinity) can be calculated directly from Marcus–Hush theory [4]. In Figure 2 we plot the difference between the adiabatic ionization energies (electron affinities) calculated in this way and the correct result (within the PBE framework) for the molecules considered in Figure 1 as a function of the the number of the atoms in the molecules. While for small molecules this approximation is not accurate, the results for the larger molecules are excellent. Clearly, it is for larger molecules that this approximation is needed, as the computational power required to relax the geometry grows with the size of the molecule.
It is important to stress that the calculation of the vibronic interaction constants is, in fact, intrinsically an problem as the dynamical matrix and the forces calculated on the geometries used to calculate the dynamical matrix together with the eigenvalue changes determined during the course of these calculations contain all the information required to calculate the giμ. However, significant computational complexities must be overcome to retrieve this information.
In the course of the above calculations, a particular practical benefit of the method became clear. It is well known that the calculated electron-vibrational interaction is rather sensitive to the size of the displacement used in a frozen phonon calculation. In general, a simple criterion for the size of the displacement (e.g., , where Δ is a small energy and k is the spring constant associated with the vibrational mode in question) does not produce uniformly reliable results for all possible vibrational frequencies in the frozen phonon calculations of the type discussed above. However, we have found that the results for the method are remarkably insensitive to the value of the “small” charge, δnμ, used in the calculation.
The calculation of isotope effects is an problem in our method. That is, only trivial matrix manipulation and no further solutions of the Schrödinger equation are required to calculate the vibronic interaction constants as the isotopic masses are varied. This contrasts to the frozen phonon method where, as the dynamical matrix and thus the normal modes change upon isotopic substitution, additional calculations are required.
A particularly interesting isotope effect is observed in the superconductor κ-(BEDT-TTF)2Cu[N(CN)2]Br. When the eight hydrogen atoms in the BEDT-TTF molecule are replaced by deuterium, the ground state is found to be a Mott insulator [20,21]. Recent efforts to calculate the Hubbard U [22–24] and intramolecular hopping integrals [23–25] for this material do not provide an explanation of this. This material is part of a larger family: κ-(BEDT-TTF)2X. Other well known members of the family included κ-(BEDT-TTF)2Cu[N(CN)2]Cl, which is a Mott insulator at ambient pressure and is driven metallic/superconducting by hydrostatic pressure, and κ-(BEDT-TTF)2Cu(SCN)2 which is metallic/superconducting at ambient pressure [21]. This has led to the idea of a unified phase diagram where “chemical pressure” (or hydrostatic pressure) can be used to tune the system across the Mott metal-insulator transition. Thus it is believed that κ-(BEDT-TTF)2Cu[N(CN)2]Br is very close to the Mott transition [20,21,26] and that deuteration produces a small negative chemical pressure [20,21,27,28]. However, the microscopic mechanism for this small negative chemical pressure (i.e., a small decrease in the ratio W/U, where W is the bandwidth and U is the effective repulsion between two electrons on the same dimer, (BEDT-TTF)2) has not previously been explained.
In the crystal the highest occupied molecular orbital (HOMO) of the BEDT-TTF molecule is doped with holes from the anion layer, therefore one expects [3] that, in the small polaron limit, on deuteration, the bandwidth will change by a factor
3. Methodological Details
To implement our method one simply recalculates the electronic structure with a small change in the charge in the orbital of interest. This allows the electronic orbitals to relax to account for the additional charge. However, in this process, the nuclear structure remains fixed as that of the equilibrium geometry of the charge neutral system. The forces on the molecule can be calculated using the Hellmann–Feynman theorem. As the dynamical matrix and vibrational eigenvectors are already known from the calculation of vibrational spectrum, the individual vibronic interaction constants can readily be calculated by projecting onto the basis of normal modes. To perform the electronic structure calculations we have followed the ansatz that the Kohn-Sham wavefunctions can be expanded in terms of a set of atom-centered gaussian-type orbitals. The methods for performing these calculations are discussed in Refs. [16–18,29–32].
The directional vibrational derivative (Equation 4) is determined in a standard way by taking the scalar product the energy gradient with the vibrational eigenvector. Provided a variationally optimized quantum-mechanical wavefunction is determined, Hellmann and Feynman have shown that this energy gradient associated with an atomic displacement is exactly equal to the electric field due to all other electronic and nuclear charges in the system. When finite-basis sets are used the energy gradient has an additional term, commonly referred to as the Pulay correction, which is required to determine the correct energy gradient. We emphasize that this additional term should be and is contained in our calculations. See Reference [16] for discussion and references regarding Hellmann–Feynman–Pulay forces.
4. Conclusion
Prior to concluding, a few additional remarks are appropriate with regard to assessing the simplifications garnered from the approach. First, it is worthwhile noting that if one determines the electron-vibrational interaction by performing 3N electronic structure calculations, it is in fact possible to determine the vibronic interactions associated with all the electronic states rather than only the LUMO and HOMO levels. If vibronic interactions from levels other than the LUMO or HOMO are desired, the approach discussed here can be modified by varying the charge of the the particular electronic state of interest. Since one is generally interested in vibronic interactions associated with frontier orbitals, the charge-variation approach discussed here should be more efficient for most applications. A more general statement is that the scaling associated with the charge variation method scales linearly with the number of electronic states needed for the model Hamiltonian. In the unlikely event that an electronic system has interesting electronic states, the traditional approach may then be as efficient as the approach.
A second issue is related to the total computational cost since the method requires that the vibrational modes be determined. In our work we have determined the dynamical matrix by numerically determining derivatives of the interatomic forces as a function of atomic displacement. This method, while reasonably standard, requires electronic structure calculations in the most general case. Therefore the most pessimistic assessment is that, given the equilibrium geometry, the determination of the vibronic interaction still requires calculations since one needs this many calculations to determine the vibrational eigenvectors. However, this should then be contrasted with the calculations required to obtain the same information by tradition approaches. One counterpoint to this assessment is that the dynamical matrix is required for other phenomena such as infrared and Raman spectra and one general goal should be to determine efficient post processing algorithms to determine the interaction between a bath of vibrons and the interesting aspects of their environment.
Another issue is the widely held hypothesis that vibrational energies for large systems can generally be determined faster than by using molecular dynamics followed by a Fourier transform of the velocity-velocity autocorrelation function. Once the vibrational frequencies are determined, a Fourier transform of the 3N-dimensional trajectory also allows for the determination of, at least, the non-degenerate vibrational eigenvectors. Coupled with such a strategy, it may indeed be possible find the vibronic interactions of an exceedingly large system with scaling below even if the cost of determining the vibrational degrees of freedom are included. However, the most rigourous and relevant statement is that the vibronic interaction may now be determined as quickly as the vibrational spectrum.
In summary we have demonstrated that by considering the forces on the nuclei due to the addition or subtraction of an arbitrarily small electronic charge one may calculate the vibronic interaction constants as an problem. This method was shown to be numerically accurate for a large number of small molecules.
Acknowledgments
This work was motivated by conversations with Greg Freebairn. We thank James Annett, Stephen Dougdale, Nikitas Gidopoulos, Barbara Montanari and Keith Refson for useful conversations. BJP is supported by the Australian Research Council under the Queen Elizabeth II scheme (project DP0878523). TB was supported by NSF NIRT-0304122. MRP carried out this work while at NRL and was supported in part by ONR and DoD-HPCMO CHSSI program. Some of the calculations were performed on the Australian National Computational Infrastructure and others were performed on the HPCMO computational platforms.
References and Notes
- Ziman, J.M. Electrons and Phonons; Oxford University Press: Oxford, UK, 1960. [Google Scholar]
- Grüner, G. Density Waves In Solids; Perseus Publishing: Cambridge, UK, 1994. [Google Scholar]
- Alexandrov, A.S.; Mott, N.F. Polarons and Biploarons; World Scientific: Singapore, 1995. [Google Scholar]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar]
- Bersuker, I.B. The Jahn-Teller Effect and Vibronic Interactions in Modern Chemistry; Plenum: New York, NY, USA, 1984. [Google Scholar]
- Fazzi, D.; Canesi, E.V.; Negri, F.; Bertarelli, C.; Castiglioni, C. Biradicaloid character of thiophene-based heterophenoquinones: The role of electron phonon coupling. ChemPhysChem 2010, 11, 3685–3695. [Google Scholar]
- Negri, F.; Orlandi, G. Vibronic Interactions in the Spectroscopy of Fullerenes: A Theoretical Investigation; Proceedings of the XIV International Symposium on Electron-Phonon Dynamics and Jahn-Teller Effect, Erice, Italy, 7–13 July 1998, Bevilacqua, G., Martinelli, L., Terzi, N., Eds.; World Scientific: Singapore, 1999; pp. 264–272. [Google Scholar]
- Olsen, S.; Riesz, J.; Mahadevan, I.; Coutts, A.; Bothma, J.P.; Powell, B.J.; McKenzie, R.H.; Smith, S.C.; Meredith, P. Convergent proton-transfer photocycles violate mirror-image symmetry in a key melanin monomer. J. Am. Chem. Soc. 2007, 129, 6672–6673. [Google Scholar]
- Powell, B.J. 5,6-Dihydroxyindole-2-carboxylic acid: A first principles density functional study. Chem. Phys. Lett. 2005, 402, 111–115. [Google Scholar]
- Reimers, J.R.; Hush, N.S. A unified description of the electrochemical, charge distribution, and spectroscopic properties of the special pair radical cation in bacterial photosynthesis. J. Am. Chem. Soc. 2004, 126, 4132–4144. [Google Scholar]
- Hahn, S.; Stock, G. Quantum-mechanical modeling of the femtosecond isomerization in Rhodopsin. J. Phys. Chem. B 2000, 104, 1146–1149. [Google Scholar]
- Jacko, A.C.; McKenzie, R.H.; Powell, B.J. Models of organometallic complexes for optoelectronic applications. J. Mater. Chem. 2010, 20, 10301–10317. [Google Scholar]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar]
- Janak, J.F. Proof that ∂E/∂ni = ∊ in density-functional theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar]
- The fact that (∂E/∂Qi)0 = 0 leads immediately to the sum rule ∑μ nμgiμ = 0.
- Pederson, M.R.; Jackson, K.A. Variational mesh for quantum-mechanical simulations. Phys. Rev. B 1990, 41, 7453–7461. [Google Scholar]
- Pederson, M.R.; Jackson, K.A. Pseudoenergies for simulations on metallic systems. Phys. Rev. B 1991, 43, 7312–7312. [Google Scholar]
- Pederson, M.R.; Jackson, K.A. Accurate forces in a local-orbital approach to the local-density approximation. Phys. Rev. B 1990, 42, 3276–3281. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar]
- Taniguchi, H.; Kanoda, K.; Kawamoto, A. Field switching of superconductor-insulator bistability in artificially tuned organics. Phys. Rev. B 2003, 67, 014510:1–014510:5. [Google Scholar]
- Powell, B.J.; McKenzie, R.H. Strong electronic correlations in superconducting organic charge transfer salts. J. Phys. Condens. Matter. 2006, 18, R827–R866. [Google Scholar]
- Scriven, E.; Powell, B.J. Toward the parametrization of the Hubbard model for salts of bis(ethylenedithio)tetrathiafulvalene: A density functional study of isolated molecules. Phys. Rev. B 2009, 80, 205107:1–205107:9. [Google Scholar]
- Scriven, E.; Powell, B.J. Effective Coulomb interactions within BEDT-TTF dimers. J. Chem. Phys. 2009, 130, 104508:1–104508:10. [Google Scholar]
- Nakmure, K.; Yoshimoto, Y.; Kosuga, T.; Arita, R.; Imada, M. Ab initio Derivation of Low-Energy Model for κ-ET Type Organic Conductors. J. Phys. Soc. Japan. 2009, 78, 083710:1–083710:4. [Google Scholar]
- Kandpal, H.C.; Opahle, I.; Zhang, Y.-Z.; Jeschke, H.O.; Valentí, R. Revision of Model Parameters for κ-Type Charge Transfer Salts: An Ab Initio Study. Phys. Rev. Lett. 2009, 103, 067004:1–067004:4. [Google Scholar]
- Faltermeier, D.; Barz, J.; Dumm, M.; Dressel, M.; Drichko, N.; Petrov, B.; Semkin, V.; Vlasova, R.; Meźière, C.; Batail, P. Bandwidth-controlled Mott transition in κ-(BEDT-TTF)2Cu[N(CN)2]Brx Cl1−x: Optical studies of localized charge excitations. Phys. Rev. B 2007, 76, 165113:1–165113:12. [Google Scholar]
- Kawasugi, Y.; Yamamoto, H.M.; Hosoda, M.; Tajima, N.; Fukunaga, T.; Tsukagoshi, K.; Kato, R. Strain-induced superconductor/insulator transition and field effect in a thin single crystal of molecular conductor. Appl. Phys. Lett. 2008, 92, 243508:1–243508:3. [Google Scholar]
- Bardin, A.A.; Burn, P.L.; Lo, S.C.; Powell, B.J. Superconductivity suppression and peak resistivity enhancement for thin crystals of κ-(BEDT-TTF)2Cu(SCN)2. Phys. Stat. Sol. B 2012. [Google Scholar] [CrossRef]
- Briley, A.; Pederson, M.R.; Jackson, K.A.; Patton, D.C.; Porezag, D.V. Vibrational frequencies and intensities of small molecules: All-electron, pseudopotential, and mixed-potential methodologies. Phys. Rev. B 1998, 58, 1786–1793. [Google Scholar]
- Porezag, D.V.; Pederson, M.R. Infrared intensities and Raman-scattering activities within density-functional theory. Phys. Rev. B 1996, 54, 7830–7836. [Google Scholar]
- Quong, A.A.; Pederson, M.R.; Feldman, J.L. First principles determination of the interatomic force-constant tensor of the fullerene molecule. Solid Stat. Commun. 1993, 87, 535–539. [Google Scholar]
- Porezag, D.V. Development of ab initio and Approximate Density Functional Methods and their Application to Complex Fullerene Systems. Ph.D. Thesis, Technische Universitaet, Chemnitz, Germany, 1997. [Google Scholar]



{kind=link}
{kind=link}
| Protonated
| Deuterated
| ||||
|---|---|---|---|---|---|
| ωi [1/cm] | Mi [amu] | gih [Ha/Bohr] | ωi [1/cm] | Mi [amu] | gih [Ha/Bohr] |
| 31.1 | 7.93 | 4.84 | 30.4 | 9.75 | 5.08 |
| 43.6 | 10.92 | 0.52 | 42.4 | 11.88 | 0.49 |
| 48.3 | 13.65 | 0.28 | 47.4 | 14.3 | 0.3 |
| 100.8 | 16.38 | 0.1 | 99.4 | 16.24 | 0.1 |
| 110.8 | 20.37 | 0.11 | 109.8 | 20.14 | 0.11 |
| 148.3 | 10.93 | 0.38 | 144.8 | 10.47 | 0.36 |
| 170.5 | 13.44 | 0.1 | 168 | 13.81 | 0.11 |
| 191.5 | 12.42 | 0.57 | 190.7 | 12.07 | 0.53 |
| 209.6 | 8.49 | 0.1 | 200.8 | 5.34 | 0.25 |
| 227.8 | 10.75 | 0.36 | 216.8 | 3.54 | 0.15 |
| 246.6 | 9.13 | 2 × 10−3 | 223.9 | 7.87 | 0.37 |
| 253.5 | 2.97 | 0.1 | 235.3 | 6.45 | 0.09 |
| 266.3 | 2.63 | 0.08 | 246 | 12.37 | 0.03 |
| 290.4 | 5.53 | 5 × 10−4 | 279.5 | 11.4 | 0.01 |
| 302.5 | 9.93 | 0.21 | 296.7 | 14.58 | 0.19 |
| 340.4 | 13.67 | 0.01 | 328.3 | 6.79 | 0.01 |
| 347.4 | 12.17 | 0.01 | 337.2 | 6.64 | 0.01 |
| 352.8 | 9.33 | 0.01 | 343.3 | 20.13 | 0.01 |
| 385.4 | 21.83 | 4 × 10−3 | 383.8 | 21.16 | 0.01 |
| 433.9 | 16.26 | 0.21 | 403.6 | 5.68 | 0.02 |
| 439.6 | 14.81 | 0.19 | 405.8 | 5.85 | 0.03 |
| 447.5 | 4.33 | 0.05 | 433.4 | 19.66 | 0.2 |
| 448.9 | 3.36 | 0.01 | 439.2 | 15.24 | 0.2 |
| 457.3 | 15.11 | 0.16 | 455.3 | 24.86 | 0.17 |
| 466.1 | 11.46 | 0.01 | 463.9 | 11.5 | 0.03 |
| 468.2 | 7.44 | 0.02 | 464.9 | 11.86 | 0.02 |
| 482.8 | 25.38 | 0.73 | 482.6 | 25.38 | 0.73 |
| 635.2 | 4.84 | 0.01 | 592.5 | 5.71 | 2 × 10−3 |
| 638.1 | 4.67 | 0.01 | 593.3 | 5.57 | 3 × 10−3 |
| 670.6 | 4.36 | 0.01 | 618.1 | 5.35 | 0.01 |
| 673.9 | 4.43 | 0.01 | 622 | 5.37 | 0.01 |
| 751.4 | 4.54 | 4 × 10−3 | 677.7 | 3.95 | 4 × 10−3 |
| 755.9 | 4.47 | 2 × 10−3 | 678.9 | 3.88 | 2 × 10−3 |
| 756.6 | 12.59 | 0.01 | 723.6 | 2.91 | 0.01 |
| 838.2 | 9.06 | 5 × 10−3 | 725.9 | 2.9 | 0.02 |
| 857.2 | 7.48 | 0.07 | 756.7 | 13.15 | 0.01 |
| 863.6 | 7.35 | 0.04 | 777 | 2.21 | 0.02 |
| 869.4 | 2.34 | 0.01 | 777.5 | 2.24 | 0.03 |
| 876.1 | 2.55 | 5 × 10−3 | 788.9 | 2.51 | 4 × 10−4 |
| 897.2 | 1.81 | 0.03 | 792 | 2.48 | 0.02 |
| 901.7 | 1.76 | 0.03 | 811.8 | 5.62 | 0.01 |
| 960.7 | 11.33 | 8 × 10−4 | 813 | 5.78 | 3 × 10−3 |
| 967.4 | 9.47 | 4 × 10−3 | 845.7 | 12.39 | 1 × 10−3 |
| 980.9 | 2.77 | 3 × 10−3 | 862.5 | 13.1 | 0.06 |
| 981.4 | 7.49 | 0.01 | 868 | 13.06 | 0.03 |
| 986.1 | 2.85 | 5 × 10−4 | 922.9 | 2.67 | 0.04 |
| 1, 108.5 | 1.04 | 1 × 10−3 | 925.1 | 2.66 | 0.04 |
| 1, 109.9 | 1.04 | 0.01 | 956.1 | 9.26 | 3 × 10−3 |
| 1, 156.8 | 1.23 | 0.03 | 961.8 | 8.62 | 1 × 10−3 |
| 1, 158.9 | 1.24 | 0.03 | 980.6 | 11.98 | 3 × 10−3 |
| 1, 243.8 | 1.26 | 1 × 10−3 | 992.7 | 3.61 | 3 × 10−3 |
| 1, 245 | 1.25 | 0.01 | 993.6 | 3.65 | 4 × 10−3 |
| 1, 270.5 | 1.21 | 0.07 | 1, 014.7 | 2.8 | 0.04 |
| 1, 273.8 | 1.21 | 0.07 | 1, 015 | 2.83 | 0.07 |
| 1, 405.1 | 1.07 | 0.01 | 1, 034.4 | 2.33 | 0.01 |
| 1, 408.1 | 1.07 | 0.01 | 1, 034.4 | 2.33 | 0.01 |
| 1, 410.4 | 1.07 | 0.01 | 1, 091.3 | 4.57 | 0.06 |
| 1, 415.5 | 1.08 | 0.01 | 1, 097.1 | 4.65 | 0.06 |
| 1, 505.4 | 11.88 | 0.47 | 1, 505.2 | 12.03 | 0.47 |
| 1, 524.2 | 11.72 | 0.01 | 1, 523.7 | 12.04 | 0.01 |
| 1, 551.2 | 11.87 | 0.09 | 1, 550.9 | 12.03 | 0.09 |
| 2, 993.4 | 1.05 | 4 × 10−3 | 2, 172.9 | 2.19 | 3 × 10−3 |
| 2, 999.9 | 1.05 | 0.01 | 2, 176.9 | 2.19 | 3 × 10−3 |
| 3, 000 | 1.05 | 3 × 10−3 | 2, 179.4 | 2.21 | 0.01 |
| 3, 004.3 | 1.05 | 0.01 | 2, 182.7 | 2.21 | 0.01 |
| 3, 048.7 | 1.1 | 7 × 10−4 | 2, 261.8 | 2.38 | 2 × 10−3 |
| 3, 053.2 | 1.1 | 6 × 10−4 | 2, 265.4 | 2.38 | 3 × 10−3 |
| 3, 060.8 | 1.1 | 5 × 10−4 | 2, 269.1 | 2.37 | 3 × 10−4 |
| 3, 064.5 | 1.1 | 5 × 10−4 | 2, 272.1 | 2.37 | 7 × 10−4 |
© 2012 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Powell, B.J.; Baruah, T.; Pederson, M.R. Equivalence of Electron-Vibration Interaction and Charge-Induced Force Variations: A New O(1) Approach to an Old Problem. Crystals 2012, 2, 236-247. https://doi.org/10.3390/cryst2020236
Powell BJ, Baruah T, Pederson MR. Equivalence of Electron-Vibration Interaction and Charge-Induced Force Variations: A New O(1) Approach to an Old Problem. Crystals. 2012; 2(2):236-247. https://doi.org/10.3390/cryst2020236
Chicago/Turabian StylePowell, Benjamin J., Tunna Baruah, and Mark R. Pederson. 2012. "Equivalence of Electron-Vibration Interaction and Charge-Induced Force Variations: A New O(1) Approach to an Old Problem" Crystals 2, no. 2: 236-247. https://doi.org/10.3390/cryst2020236
APA StylePowell, B. J., Baruah, T., & Pederson, M. R. (2012). Equivalence of Electron-Vibration Interaction and Charge-Induced Force Variations: A New O(1) Approach to an Old Problem. Crystals, 2(2), 236-247. https://doi.org/10.3390/cryst2020236