Terahertz Vibrations and Hydrogen-Bonded Networks in Crystals

Abstract
:
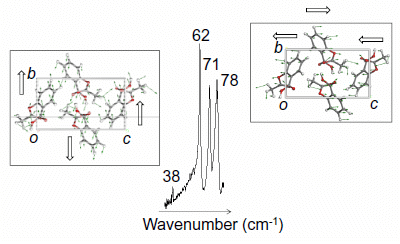{kind=link}
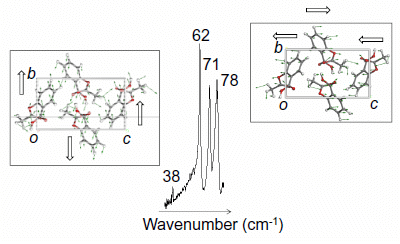{kind=link}
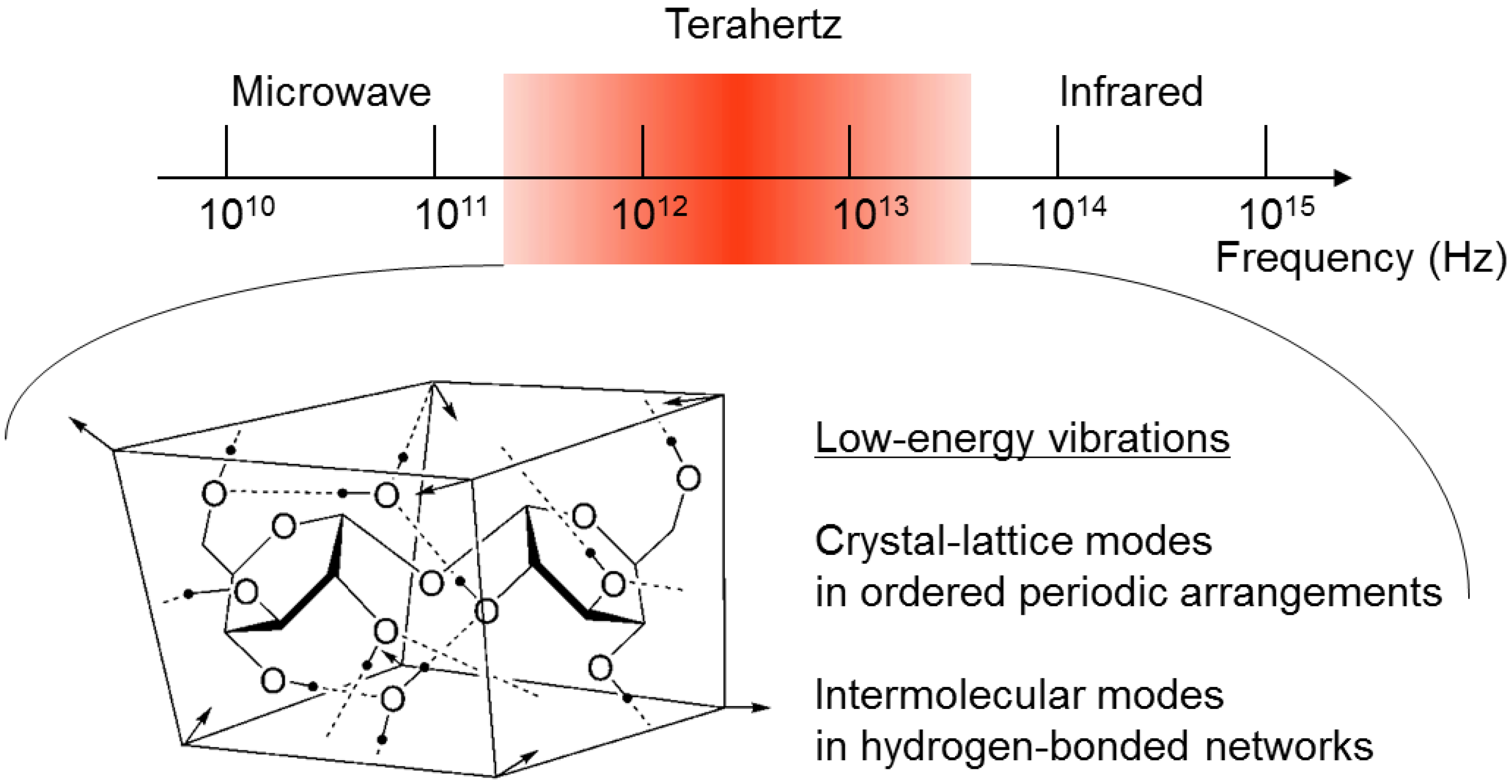{kind=link}
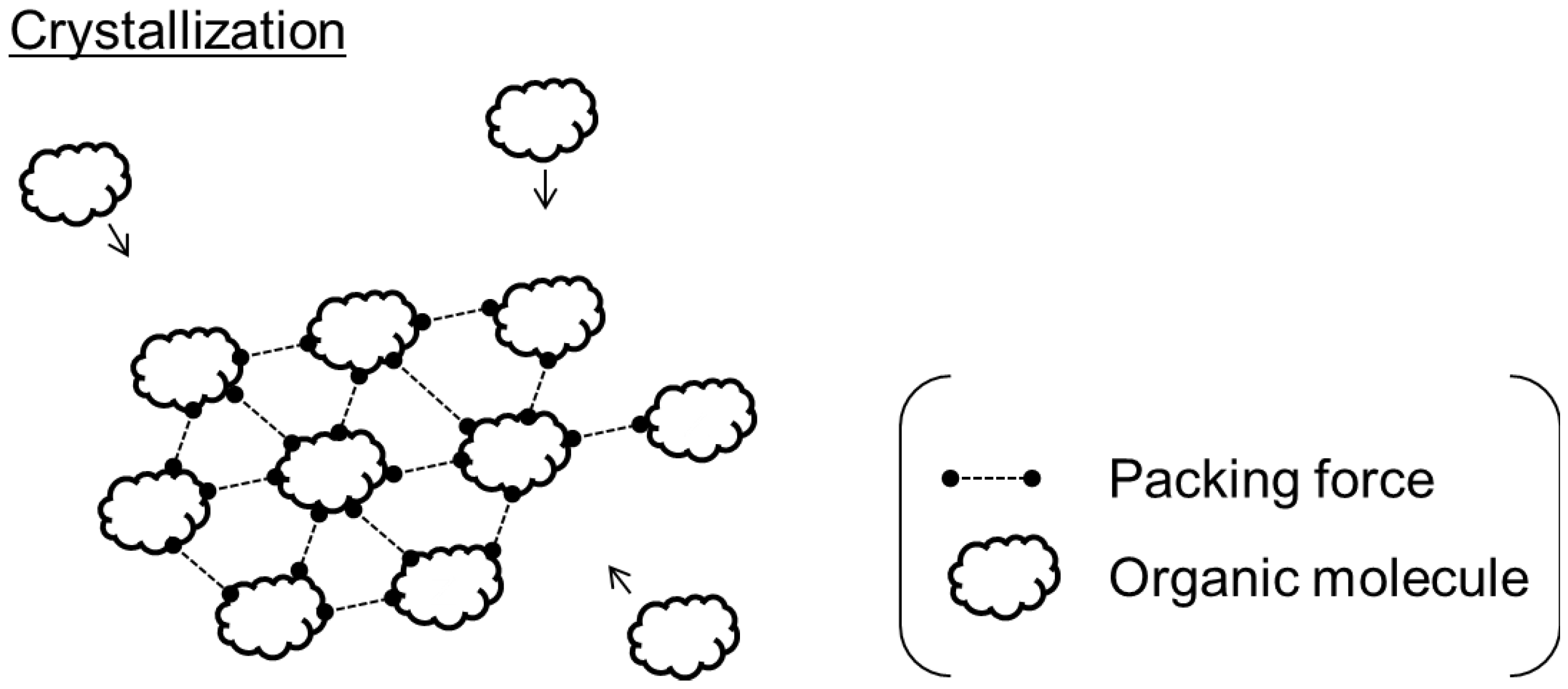{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

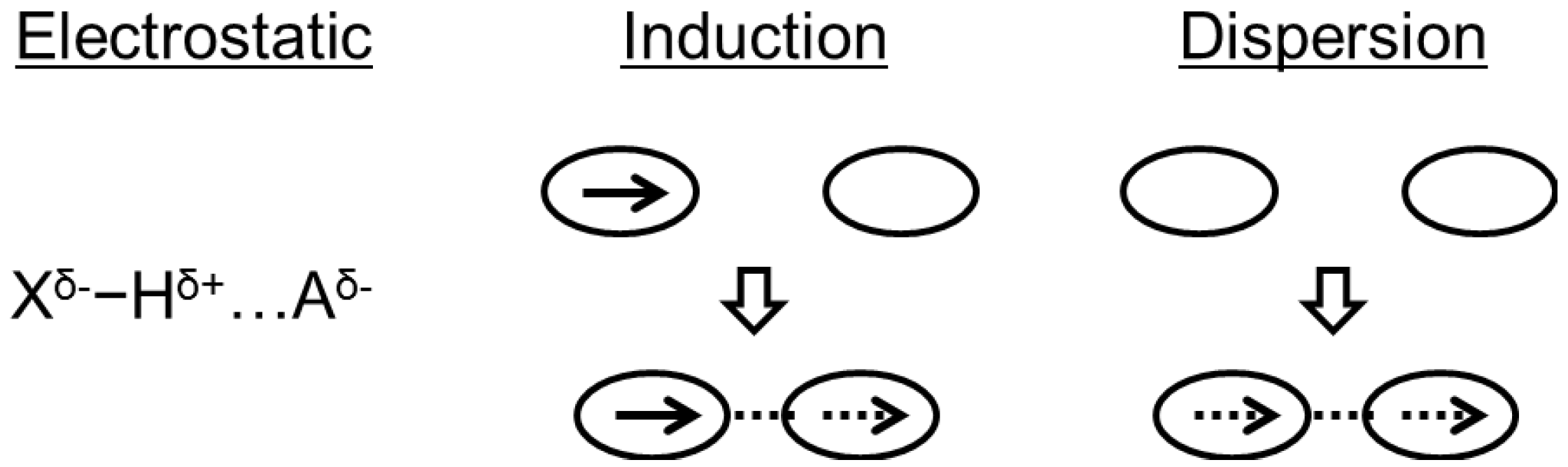
2. Crystal Packing Forces and Hydrogen-Bonded Networks



3. THz Vibrations of Hydrogen-Bonded Molecules


4. Computational Methods to Analyze the THz Spectra of Hydrogen-Bonded Molecules
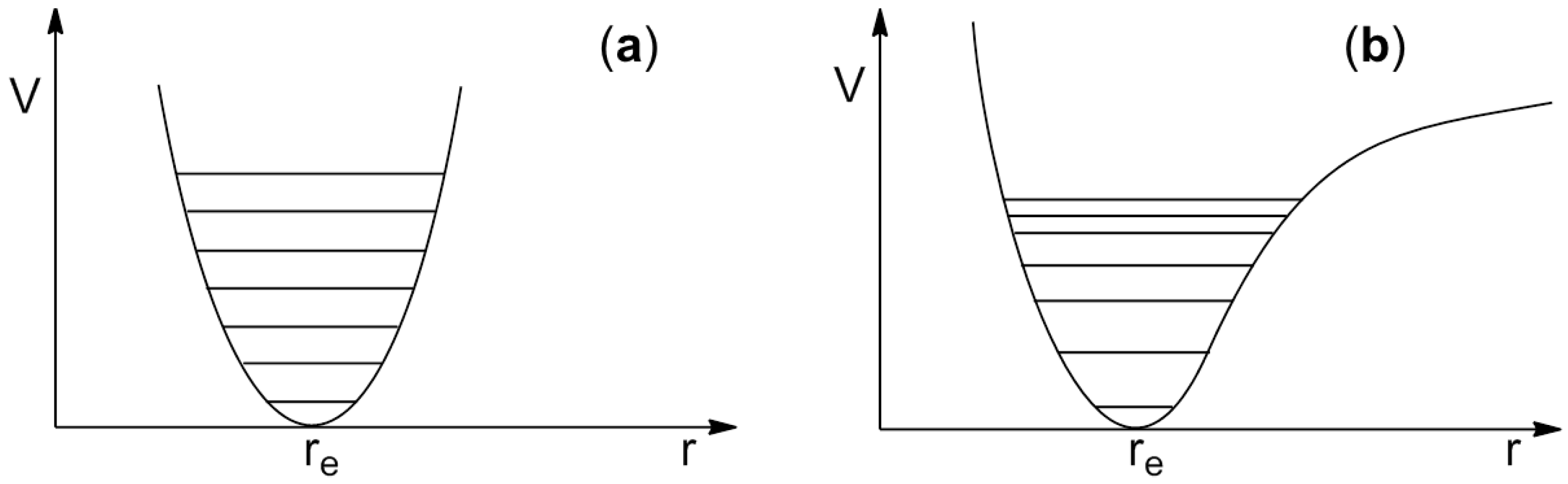
5. Dispersive Interactions and Anharmonicity



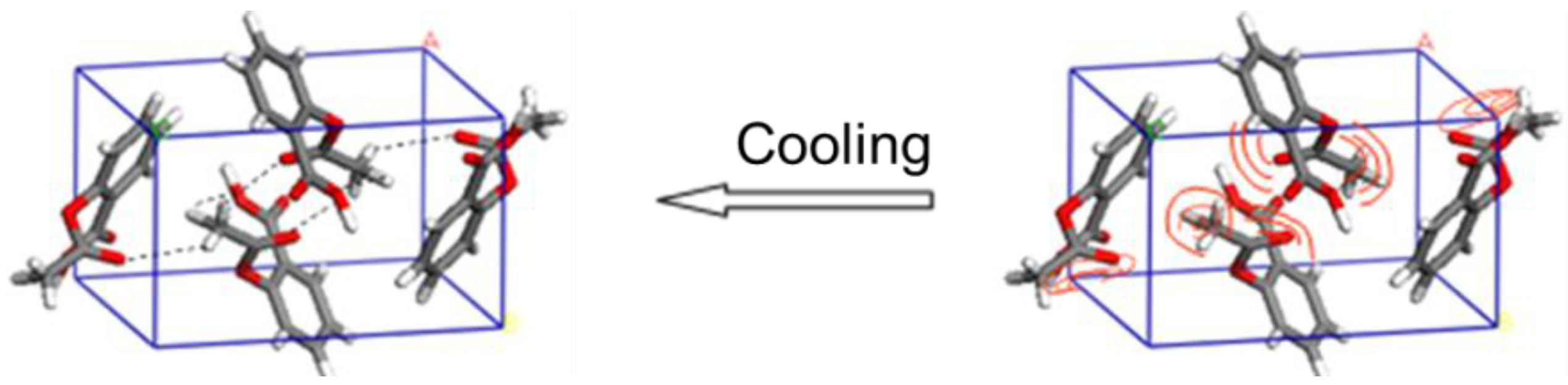
6. Temperature-Dependent THz Spectrum


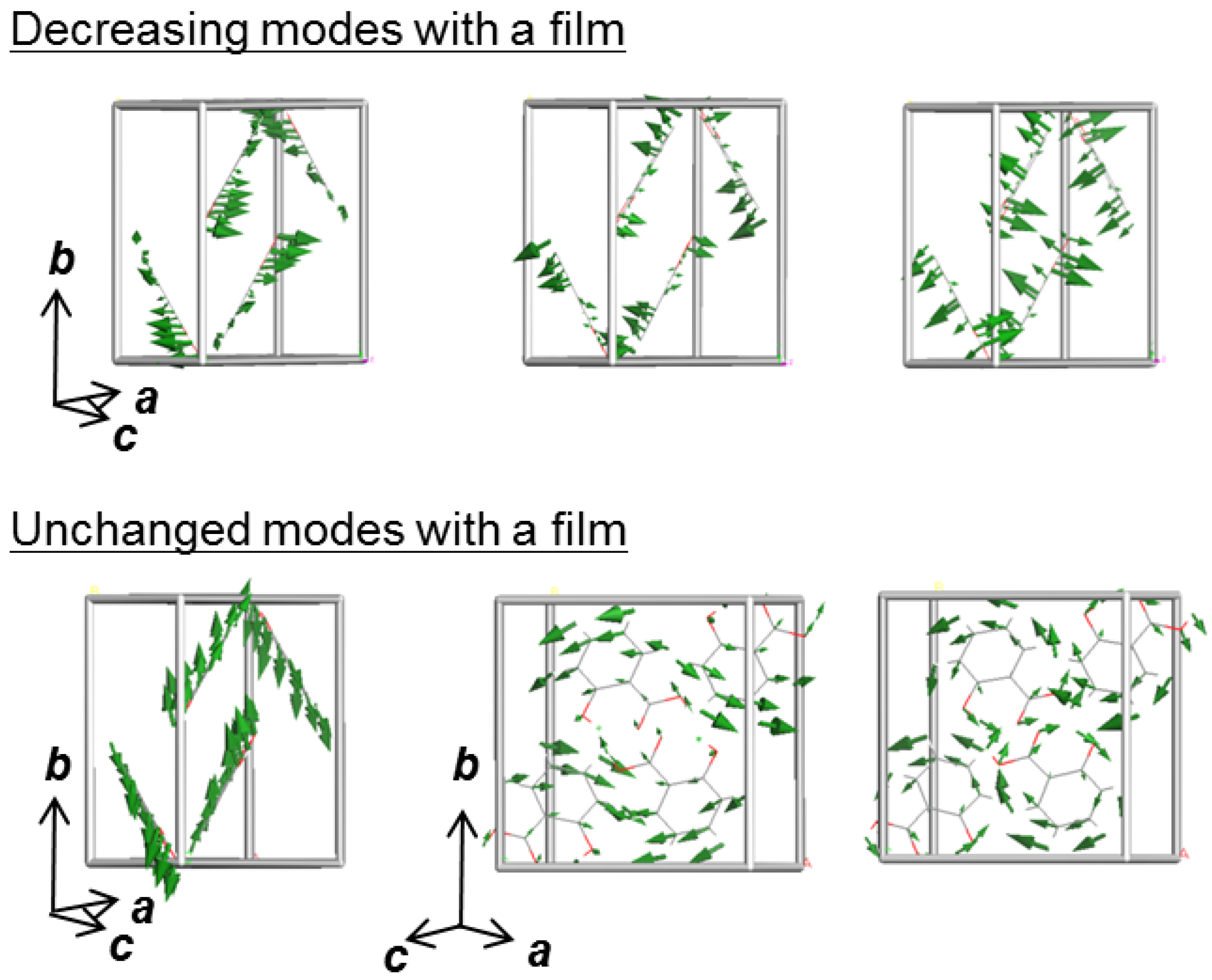
7. Polarization-Dependent THz Spectrum

8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mori, Y.; Takahashi, Y.; Iwai, T.; Yoshimura, M.; Yap, Y.K.; Sasaki, T. Slope nucleation method for the growth of high-quality 4-dimethylamino-methyl-4-stilbazolium-tosylate (DAST) crystals. Jpn. J. Appl. Phys. 2000, 39, L1006–L1008. [Google Scholar] [CrossRef]
- Zheng, X.; McLaughlin, C.V.; Cunningham, P.; Hayden, L.M. Organic broadband terahertz sources and sensors. J. Nanoelectron. Optoelectron. 2007, 2, 58–76. [Google Scholar] [CrossRef]
- Paul, O.; Beigang, R.; Rahm, M. Highly selective terahertz bandpass filters based on trapped mode excitation. Opt. Express 2009, 17, 18590–18595. [Google Scholar] [CrossRef]
- Theuer, M.; Beigang, R.; Grischkowsky, D.R. Adiabatic compression of terahertz waves using metal flares. Appl. Phys. Lett. 2010, 96, 191110:1–191110:3. [Google Scholar]
- Mendis, R.; Nag, A.; Chen, F.; Mittleman, D.M. A tunable universal terahertz filter using artificial dielectrics based on parallel-plate waveguides. Appl. Phys. Lett. 2010, 97, 131106:1–131106:3. [Google Scholar]
- Mendis, R.; Mittleman, D.M. A 2-D artificial dielectric with 0 < n < 1 for the terahertz region. IEEE Trans. Microw. Theory Tech. 2010, 58, 1993–1998. [Google Scholar] [CrossRef]
- Nagai, M.; Mukai, N.; Minowa, Y.; Ashida, M.; Takayanagi, J.; Ohtake, H. Achromatic THz wave plate composed of stacked parallel metal plates. Opt. Lett. 2014, 39, 146–149. [Google Scholar] [CrossRef]
- Theuer, M.; Torosyan, G.; Ellrich, F.; Jonuscheit, J.; Beigang, R. Terahertz imaging in industrial applications. TM Tech. Mess. 2009, 75, 64–70. [Google Scholar]
- Mittleman, D.M.; Jacobsen, R.H.; Neelamani, R.; Baraniuk, R.G.; Nuss, M.C. Gas sensing using terahertz time-domain spectroscopy. Appl. Phys. B 1998, 67, 379–390. [Google Scholar]
- Zandonella, C. Terahertz imaging: T-ray specs. Nature 2003, 424, 721–722. [Google Scholar] [CrossRef]
- Federici, J.F.; Schulkin, B.; Huang, F.; Gary, D.; Barat, R.; Oliveira, F.; Zimdars, D. THz imaging and sensing for security applications—Explosives, weapons and drugs. Semicond. Sci. Technol. 2005, 20, S266–S280. [Google Scholar] [CrossRef]
- Harsha, S.S.; Melinger, J.S.; Qadri, S.B.; Grischkowsky, D. Substrate independence of THz vibrational modes of polycrystalline thin films of molecular solids in waveguide THz-TDS. J. Appl. Phys. 2012, 111. [Google Scholar] [CrossRef]
- Yang, Y.; Harsha, S.S.; Shutler, A.J.; Grischkowsky, D.R. Identification of Genistein and Biochanin A by THz (far-infrared) vibrational spectra. J. Pharm. Biomed. Anal. 2012, 62, 177–181. [Google Scholar] [CrossRef]
- Koch, M. Terahertz communications: A 2020 vision. In Terahertz Frequency Detection and Identification of Materials and Objects (NATO Science for Peace and Security Series); Miles, R.E., Zhang, X.-C., Eisele, H., Krotkus, A., Eds.; Springer: Dordrecht, The Netherlands, 2007; Theme 4; pp. 325–338. [Google Scholar]
- Yang, Y.; Mandehgar, M.; Grischkowsky, D.R. Broadband THz pulse transmission through the atmosphere. IEEE Trans. THz Sci. Technol. 2011, 1, 264–273. [Google Scholar] [CrossRef]
- Koenig, S.; Lopez-Diaz, D.; Antes, J.; Boes, F.; Henneberger, R.; Leuther, A.; Tessmann, A.; Schmogrow, R.; Hillerkuss, D.; Palmer, R.; et al. Wireless sub-THz communication system with high data rate. Nat. Photon. 2013, 7, 977–981. [Google Scholar] [CrossRef]
- Mandehgar, M.; Yang, Y.; Grischkowsky, D. Atmosphere characterization for simulation of the two optimal wireless terahertz digital communication links. Opt. Lett. 2013, 38, 3437–3440. [Google Scholar] [CrossRef]
- Ho, L.; Pepper, M.; Taday, P. Terahertz spectroscopy: Signature and fingerprints. Nat. Photon. 2008, 2, 541–543. [Google Scholar] [CrossRef]
- Ueno, Y.; Ajito, K. Analytical terahertz spectroscopy. Anal. Sci. 2008, 24, 185–192. [Google Scholar] [CrossRef]
- Davies, A.G.; Burnett, A.D.; Fan, W.; Linfield, E.H.; Cunningham, J.E. Terahertz spectroscopy of explosives and drugs. Mater. Today 2008, 11, 18–26. [Google Scholar]
- McGoverin, C.M.; Rades, T.; Gordon, K.C. Recent pharmaceutical applications of Raman and terahertz spectroscopies. J. Pharm. Sci. 2008, 97, 4598–4621. [Google Scholar] [CrossRef]
- Smith, R.M.; Arnold, M.A. Terahertz time-domain spectroscopy of solid samples: Principles, applications, and challenges. Appl. Spectrosc. Rev. 2011, 46, 636–679. [Google Scholar] [CrossRef]
- Theuer, M.; Harsha, S.S.; Molter, D.; Torosyan, G.; Beigang, R. Terahertz time-domain spectroscopy of gases, liquids, and solids. ChemPhysChem 2011, 12, 2695–2705. [Google Scholar] [CrossRef]
- McIntosh, A.I.; Yang, B.; Goldup, S.M.; Watkinson, M.; Donnan, R.S. Terahertz spectroscopy: A powerful new tool for the chmical sciences? Chem. Soc. Rev. 2012, 41, 2072–2082. [Google Scholar] [CrossRef]
- Sushko, O.; Dubrovka, R.; Donnan, R.S. Analysis of optical thickness determination of materials by THz-TDS. J. Phys. Conf. Ser. 2013, 472. [Google Scholar] [CrossRef]
- Ji, T.; Zhang, Z.; Zhao, H.; Chen, M.; Yu, X.; Xiao, T. A THz-TDS measurement method for multiple samples. Opt. Commun. 2014, 312, 292–295. [Google Scholar] [CrossRef]
- Hübers, H.-W. Towards THz integrated photonics. Nat. Photon. 2010, 4, 503–504. [Google Scholar] [CrossRef]
- Ferguson, B.; Zhang, X.-C. Materials for terahertz science and technology. Nat. Mater. 2002, 1, 26–33. [Google Scholar] [CrossRef]
- Miles, R.; Harrison, P.; Lippens, D. Terahertz Sources and Systems; NATO Science Series II; Kluwer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Siegel, P.H. Terahertz technology. IEEE Trans. Microw. Theory Tech. 2002, 50, 910–928. [Google Scholar] [CrossRef]
- Beard, M.C.; Turner, G.M.; Schmuttenmaer, C.A. Terahertz spectroscopy. J. Phys. Chem. B 2002, 106, 7146–7159. [Google Scholar] [CrossRef]
- Hangyo, M.; Tani, M.; Nagashima, T. Terahertz time-domain spectroscopy of solids: A review. Int. J. Infrared Millim. Wave 2005, 26, 1661–1690. [Google Scholar] [CrossRef]
- Tonouchi, M. Cutting-edge terahertz technology. Nat. Photon. 2007, 1, 97–105. [Google Scholar] [CrossRef]
- Ellrich, F.; Weinland, T.; Theuer, M.; Jonuscheit, J.; Beigang, R. Fiber-coupled terahertz spectroscopy system. TM Tech. Mess. 2008, 75, 14–22. [Google Scholar]
- Ellrich, F.; Klier, J.; Jonuscheit, J.; Weinland, T.; Beigang, R. Terahertz waves—A new spectral band for industrial measurement techniques. TM Tech. Mess. 2010, 77, 452–461. [Google Scholar]
- Baxter, J.B.; Guglietta, G.W. Terahertz spectroscopy. Anal. Chem. 2011, 83, 4342–4368. [Google Scholar] [CrossRef]
- Wang, C.; Lu, B.; Lin, C.; Chen, Q.; Miao, L.; Deng, X.; Zhang, J. 0.34-THz wireless link based on high-order modulation for future wireless local area network applications. IEEE Trans. THz Sci. Technol. 2014, 4, 75–85. [Google Scholar] [CrossRef]
- Xu, H.; Wu, B.; Iverson, E.W.; Low, T.S.; Feng, M. 0.5 THz performance of a type-II DHBT with a doping-graded and constant-composition GaAsSb base. IEEE Electron Dev. Lett. 2014, 35, 24–26. [Google Scholar] [CrossRef]
- Smirnov, I.A.; Alekseev, E.A.; Ilyushin, V.V.; Margulés, L.; Motiyenko, R.A.; Drouin, B.J. Spectroscopy of the ground, first and second excited torsional states of acetaldehyde from 0.05 to 1.6 THz. J. Mol. Spectrosc. 2014, 295, 44–50. [Google Scholar] [CrossRef]
- Bosshard, C.; Sutter, K.; Prêtre, P.; Hulliger, J.; Flörsheimer, M.; Kaatz, P.; Günter, P. Organic Nonlinear Optical Materials (Advances in Nonlinear Optics); Gordon and Breach Science Publishers SA: Basel, Switzerland, 1995; Volume 1. [Google Scholar]
- Wong, M.S.; Pan, F.; Gramlich, V.; Bosshard, C.; Günter, P. Self-assembly of an acentric co-crystal of a highly hyperpolarizable merocyanine dye with optimized alignment for nonlinear optics. Adv. Mater. 1997, 9, 554–557. [Google Scholar]
- Bosshard, C.; Pan, F.; Wong, M.S.; Manetta, S.; Spreiter, R.; Cai, C.; Günter, P.; Gramlich, V. Nonlinear optical organic co-crystals of merocyanine dyes and phenolic derivatives with short hydrogen bonds. Chem. Phys. 1999, 245, 377–394. [Google Scholar] [CrossRef]
- Choubey, A.; Kwon, O.-P.; Jazbinsek, M.; Günter, P. High-quality organic single crystalline thin films for nonlinear optical applications by vapor growth. Cryst. Growth Des. 2007, 7, 402–405. [Google Scholar] [CrossRef]
- Günter, P. Nonlinear Optical Effects and Materials; Springer Series in Optical Sciences; Springer: Berlin, Germany, 2000; Volume 72. [Google Scholar]
- Kwon, O.-P.; Jazbinsek, M.; Yun, H.; Seo, J.-I.; Kim, E.-M.; Lee, Y.-S.; Günter, P. Pyrrole-based hydrazone organic nonlinear optical crystals and their polymorphs. Cryst. Growth Des. 2008, 8, 4021–4025. [Google Scholar] [CrossRef]
- Kwon, S.-J.; Jazbinsek, M.; Kwon, O-P.; Günter, P. Crystal growth and morphology control of OH1 organic electrooptic crystals. Cryst. Growth Des. 2010, 10, 1552–1558. [Google Scholar] [CrossRef]
- Jepsen, P.U.; Clark, S.J. Precise ab-initio prediction of terahertz vibrational modes in crystalline systems. Chem. Phys. Lett. 2007, 442, 275–280. [Google Scholar] [CrossRef]
- Oppenheim, K.C.; Korter, T.M.; Melinger, J.S.; Grischkowsky, D. Solid-state density functional theory investigation of the terahertz spectra of the structural isomers 1,2-dicyanobenzene and 1,3-dicyanobenzene. J. Phys. Chem. A 2010, 114, 12513–12521. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer-Verlag: New York, NY, USA, 1991. [Google Scholar]
- Hadži, D.; Bratos, S. Vibrational spectroscopy of the hydrogen bond. In The Hydrogen Bond: Recent Developments in Theory and Experiment; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland Publishing Company: Amsterdam, The Netherlands, 1976; Volume 2, pp. 565–612. [Google Scholar]
- Zeegers-Huyskens, T.; Huyskens, P.L. Intermolecular forces. In Intermolecular Forces; Huyskens, P.L., Luck, W.A.P., Zeegers-Huyskens, T., Eds.; Springer-Verlag: Berlin, Germany, 1991; pp. 1–30. [Google Scholar]
- Hamilton, W.C.; Ibers, J.A. Hydrogen Bonding in Solids; W.A. Benjamin: New York, NY, USA, 1968. [Google Scholar]
- Hadži, D. Theoretical Treatments of Hydrogen Bonding; Wiley: Chichester, UK, 1997. [Google Scholar]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Covalent nature of the strong homonuclear hydrogen bond. Study of the O–H···O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Sutor, D.J. The O–H···O hydrogen bond in crystals. Nature 1962, 195, 68–69. [Google Scholar] [CrossRef]
- Taylor, R.; Kennard, O. Crystallographic evidence for the existence of C–H···O, C–H···N, and C–H···Cl hydrogen bonds. J. Am. Chem. Soc. 1982, 104, 5063–5070. [Google Scholar] [CrossRef]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; SSCI, Inc.: West Lafayette, IN, USA, 1999. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Threlfall, T.L. Analysis of organic polymorphs. A review. Analyst 1995, 120, 2435–2460. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal gazing: Structure prediction and polymorphism. Science 1997, 278, 404–405. [Google Scholar] [CrossRef]
- Upadhya, P.C.; Nguyen, K.L.; Shen, Y.C.; Obradovic, J.; Fukushige, K.; Griffiths, R.; Gladden, L.F.; Davies, A.G.; Linfield, E.H. Characterization of crystalline phase-transformations in theophylline by time-domain terahertz spectroscopy. Spectrosc. Lett. 2006, 39, 215–224. [Google Scholar] [CrossRef]
- Day, G.M.; Zeitler, J.A.; Jones, W.; Rades, T.; Taday, P.F. Understanding the influence of polymorphism on phonon spectra: Lattice dynamics calculations and terahertz spectroscopy of carbamazepine. J. Phys. Chem. B 2006, 110, 447–456. [Google Scholar] [CrossRef]
- Nguyen, K.L.; Friščić, T.; Day, G.M.; Gladden, L.F.; Jones, W. Terahertz time-domain spectroscopy and the quantitative monitoring of mechanochemical cocrystal formation. Nat. Mater. 2007, 6, 206–209. [Google Scholar] [CrossRef]
- Heinz, A.; Gordon, K.C.; McGoverin, C.M.; Rades, T.; Strachan, C.J. Understanding the solid-state forms of fenofibrate—A spectroscopic and computational study. Eur. J. Pharm. Biopharm. 2009, 71, 100–108. [Google Scholar] [CrossRef]
- King, M.D.; Davis, E.A.; Smith, T.M.; Korter, T.M. Importance of accurate spectral simulations for the analysis of terahertz spectra: Citric acid anhydrate and monohydrate. J. Phys. Chem. A 2011, 115, 11039–11044. [Google Scholar] [CrossRef]
- Delaney, S.P.; Witko, E.M.; Smith, T.M.; Korter, T.M. Investigating tautomeric polymorphism in crystalline anthranilic acid using terahertz spectroscopy and solid-state density functional theory. J. Phys. Chem. A 2012, 116, 8051–8057. [Google Scholar] [CrossRef]
- Charron, D.M.; Ajito, K.; Kim, J.-Y.; Ueno, Y. Chemical mapping of pharmaceutical cocrystals using terahertz spectroscopic imaging. Anal. Chem. 2013, 85, 1980–1984. [Google Scholar] [CrossRef]
- Delaney, S.P.; Pan, D.; Yin, S.X.; Smith, T.M.; Korter, T.M. Evaluating the roles of conformational strain and cohesive binding in crystalline polymorphs of aripiprazole. Cryst. Growth Des. 2013, 13, 2943–2952. [Google Scholar] [CrossRef]
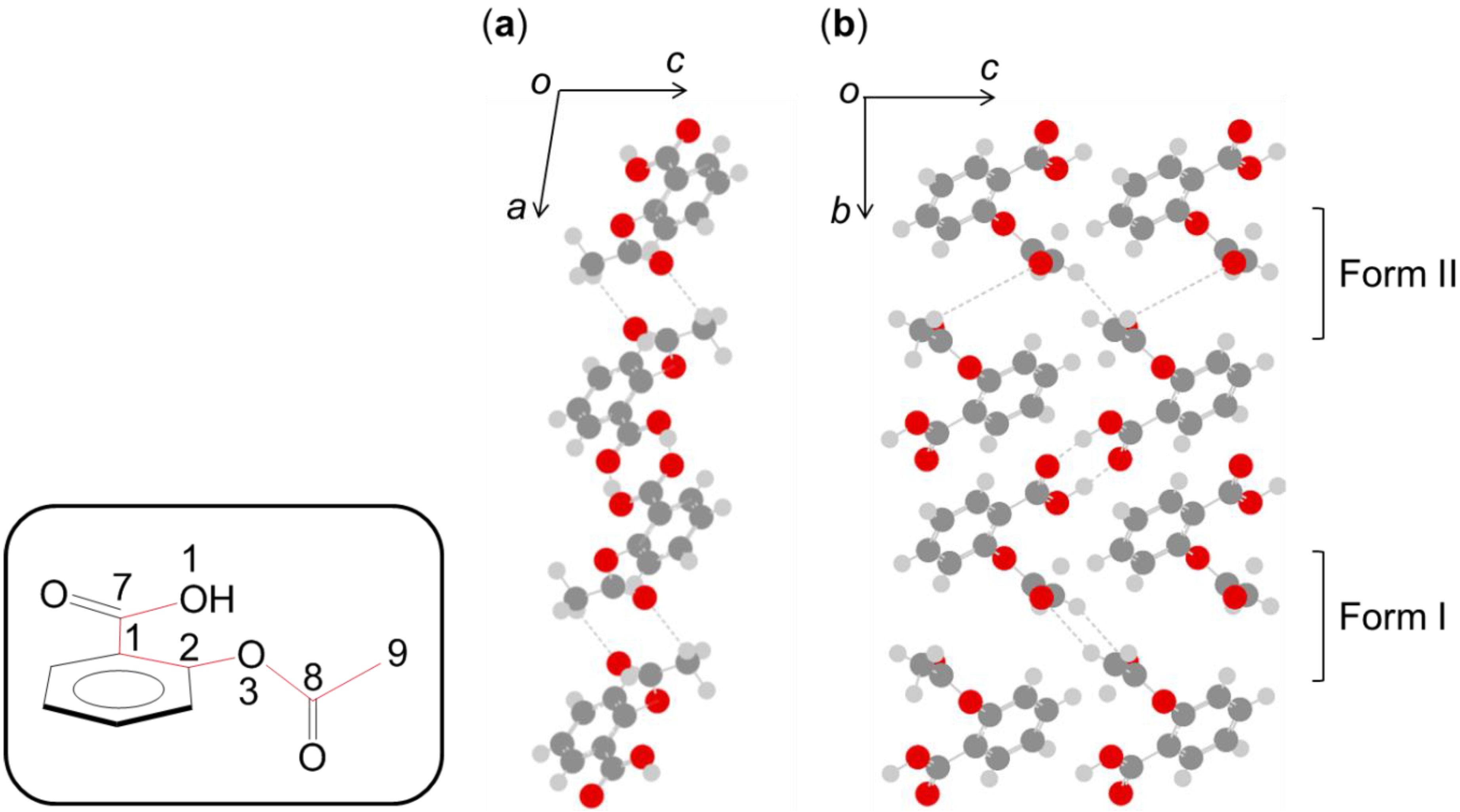
- Takahashi, M.; Ishikawa, Y. Translational vibrations between chains of hydrogen-bonded molecules in solid-state aspirin form I. Chem. Phys. Lett. 2013, 576, 21–25. [Google Scholar] [CrossRef]
- Nangia, A. Conformational polymorphism in organic crystals. Acc. Chem. Res. 2008, 41, 595–604. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef]
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Laus, G.; Wieser, J.; Griesser, U.J. Packing polymorphism of a conformationally flexible molecule (aprepitant). New J. Chem. 2008, 32, 1677–1685. [Google Scholar] [CrossRef]
- Brittain, H.G. Polymorphism and solvatomorphism 2007. J. Pharm. Sci. 2009, 98, 1617–1642. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Islam, K.; Kirchner, M.T.; Mehta, G.; Row, T.N.G. In situ cryocrystallization of diphenyl ether: C–H···π mediated polymorphic forms. J. Am. Chem. Soc. 2004, 126, 12274–12275. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Schaffner, S.; Shardlow, E.J. Polymorphs of 4ʹ-(hex-5-ynyloxy)-2,2ʹ:6ʹ,2″-terpyridine: Structural diversity arising from weak intermolecular interactions in the solid state. CrystEngComm 2005, 7, 599–602. [Google Scholar] [CrossRef]
- Omondi, B.; Fernandes, M.A.; Layh, M.; Levendis, D.C.; Look, J.L.; Mkwizu, T.S.P. Polymorphism and phase transformations in 2,6-disubstituted N-phenylformamides: The influence of hydrogen bonding, chloro-methyl exchange, intermolecular interactions and disorder. CrystEngComm 2005, 7, 690–700. [Google Scholar] [CrossRef]
- Chopra, D.; Nagarajan, K.; Row, T.N.G. Polymorphism in 1-(4-fluorophenyl)-3,6,6-trimethyl-2-phenyl-1,5,6,7-tetrahydro-4H-indol-4-one: A subtle interplay of weak intermolecular interactions. Cryst. Growth Des. 2005, 5, 1035–1039. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Nagarajan, K.; Row, T.N.G. Solvent mediated centric/non-centric polymorph pairs of an indole derivative: Subtle variation of C–H···O hydrogen bonds and C–H···π interactions. CrystEngComm 2006, 8, 482–488. [Google Scholar] [CrossRef]
- Hulme, A.T.; Johnston, A.; Florence, A.J.; Fernandes, P.; Shankland, K.; Bedford, C.T.; Welch, G.W.A.; Sadiq, G.; Haynes, D.A.; Motherwell, W.D.S.; et al. Search for a predicted hydrogen bonding motif—A multidisciplinary investigation into the polymorphism of 3-azabicyclo[3.3.1]nonane-2,4-dione. J. Am. Chem. Soc. 2007, 129, 3649–3657. [Google Scholar] [CrossRef] [Green Version]
- Sardo, M.; Amado, A.M.; Ribeiro-Claro, P.J.A. Hydrogen bonding in nitrofurantoin polymorphs: A computation-assisted spectroscopic study. J. Raman Spectrosc. 2009, 40, 1956–1965. [Google Scholar] [CrossRef]
- Yu, F.; Yang, G.; Su, Z. The effect of multiple weak interactions on the charge transport ability in polymorphs. Synth. Met. 2011, 161, 1073–1078. [Google Scholar] [CrossRef]
- Tewari, A.K.; Singh, V.P.; Dubey, R.; Puerta, C.; Valerga, P.; Verma, R. Importance of weak interactions in developing 1,3-bis(4,6-dimethyl-1H-nicotinonitrile-1-yl)1,3-dioxy propane polymorphs. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 1267–1275. [Google Scholar] [CrossRef]
- Flakus, H.T.; Hachła, B. Polarized IR spectra of the hydrogen bond in two different oxindole polymorphs with cyclic dimers in their lattices. J. Phys. Chem. A 2011, 115, 12150–12160. [Google Scholar] [CrossRef]
- Kumar, S.S.; Nangia, A. A new conformational polymorph of N-acetyl-L-cysteine. The role of S–H···O and C–H···O interactions. CrystEngComm 2013, 15, 6498–6505. [Google Scholar] [CrossRef]
- Vishweshwar, P.; McMahon, J.A.; Oliveira, M.; Peterson, M.L.; Zaworotko, M.J. The predictably elusive form II of aspirin. J. Am. Chem. Soc. 2005, 127, 16802–16803. [Google Scholar] [CrossRef]
- Wheatley, P.J. The crystal and molecular structure of aspirin. J. Chem. Soc. 1964, 6036–6048. [Google Scholar] [CrossRef]
- Cabezas, C.; Alonso, J.L.; López, J.C.; Mata, S. Unveiling the shape of aspirin in the gas phase. Angew. Chem. Int. Ed. 2012, 51, 1375–1378. [Google Scholar] [CrossRef]
- Ouvrard, C.; Price, S.L. Toward crystal structure prediction for conformationally flexible molecules: The headaches illustrated by aspirin. Cryst. Growth Des. 2004, 4, 1119–1127. [Google Scholar] [CrossRef]
- Bond, A.D.; Boese, R.; Desiraju, G.R. On the polymorphism of aspirin. Angew. Chem. Int. Ed. 2007, 46, 615–617. [Google Scholar] [CrossRef]
- Bond, A.D.; Boese, R.; Desiraju, G.R. On the polymorphism of aspirin: Crystalline aspirin as intergrowths of two “polymorphic” domains. Angew. Chem. Int. Ed. 2007, 46, 618–622. [Google Scholar] [CrossRef]
- Wen, S.; Beran, G.J.O. Accidental degeneracy in crystalline aspirin: New insights from high-level ab initio calculations. Cryst. Growth Des. 2012, 12, 2169–2172. [Google Scholar] [CrossRef]
- Cui, X.; Rohl, A.L.; Shtukenberg, A.; Kahr, B. Twisted aspirin crystals. J. Am. Chem. Soc. 2013, 135, 3395–3398. [Google Scholar] [CrossRef]
- Palik, E.D. History of far-infrared research. I. The Rubens era. J. Opt. Soc. Am. 1977, 67, 857–865. [Google Scholar] [CrossRef]
- Grinsburg, N. History of far-infrared research. II. The grating era, 1925–1960. J. Opt. Soc. Am. 1977, 67, 865–871. [Google Scholar] [CrossRef]
- Smith, P.R.; Auston, D.H.; Nuss, M.C. Subpicosecond photoconducting dipole antennas. IEEE J. Quant. Electron. 1988, 24, 255–260. [Google Scholar] [CrossRef]
- Fattinger, C.; Grischkowsky, D. Terahertz beams. Appl. Phys. Lett. 1989, 54, 490–492. [Google Scholar] [CrossRef]
- Saykally, R.J.; Blake, G.A. Molecular interactions and hydrogen bond tunneling dynamics: Some new perspectives. Science 1993, 259, 1570–1575. [Google Scholar]
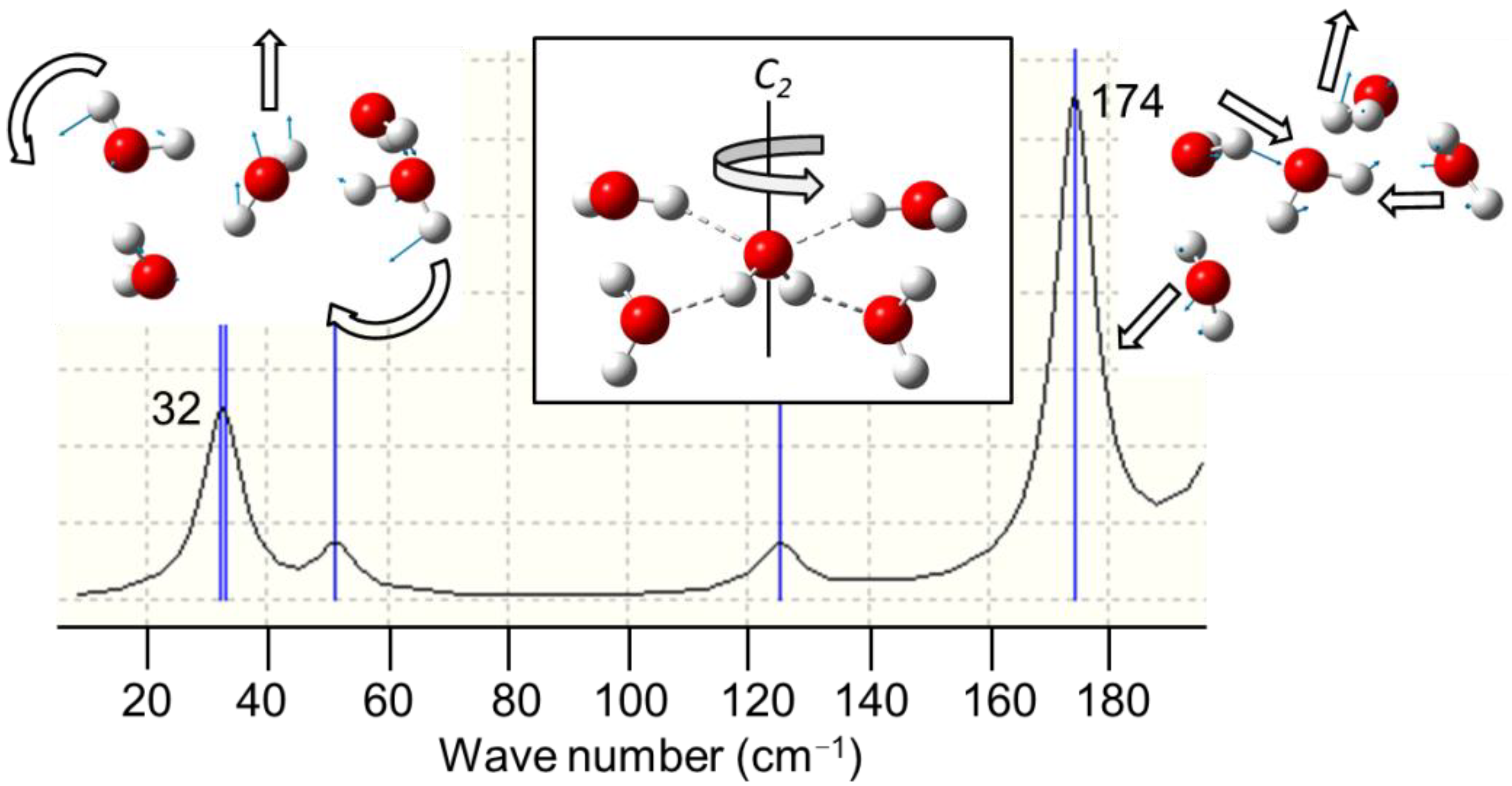
- Yu, B.L.; Yang, Y.; Zeng, F.; Xin, X.; Alfano, R.R. Reorientation of the H2O cage studied by terahertz time-domain spectroscopy. Appl. Phys. Lett. 2005, 86. [Google Scholar] [CrossRef]
- Walther, M.; Fischer, B.; Schall, M.; Helm, H.; Jepsen, P.U. Far-infrared vibrational spectra of all-trans, 9-cis and 13-cis retinal measured by THz time-domain spectroscopy. Chem. Phys. Lett. 2000, 332, 389–395. [Google Scholar] [CrossRef]
- Markelz, A.G.; Roitberg, A.; Heilweil, E.J. Pulsed terahertz spectroscopy of DNA, bovine serum albumin and collagen between 0.1 and 2.0 THz. Chem. Phys. Lett. 2000, 320, 42–48. [Google Scholar] [CrossRef]
- Möller, K.D.; Rothschild, W.G. Far-Infrared Spectroscopy; Wiley-Interscience: New York, NY, USA, 1971. [Google Scholar]
- Frank, W.F.X.; Fiedler, H. On the problem of direct observation of H-bridge interactions in polymers by FIR spectroscopy. Infrared Phys. 1979, 19, 481–489. [Google Scholar] [CrossRef]
- Shen, D.Y.; Pollack, S.K.; Hsu, S.L. Far-infrared study of hydrogen bonding in semicrystalline polyurethane. Macromolecules 1989, 22, 2564–2569. [Google Scholar] [CrossRef]
- Kato, T.; Shirota, H. Intermolecular vibrational modes and orientational dynamics of cooperative hydrogen-bonding dimer of 7-azaindole in solution. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef]
- Morgan, J.; Warren, B.E. X-ray analysis of the structure of water. J. Chem. Phys. 1938, 6, 666–673. [Google Scholar] [CrossRef]
- Brady, G.W.; Romanow, W.J. Structure of water. J. Chem. Phys. 1960, 32. [Google Scholar] [CrossRef]
- Cartwright, C.H. Hindered rotation in liquid H2O and D2O. Phys. Rev. 1936, 49, 470–471. [Google Scholar] [CrossRef]
- Falk, M.; Giguère, P.A. Infrared spectrum of the H3O+ ion in aqueous solutions. Can. J. Chem. 1957, 35, 1195–1204. [Google Scholar] [CrossRef]
- Walrafen, G.E. Raman spectral studies of water structure. J. Chem. Phys. 1964, 40, 3249–3256. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Yokouchi, M.; Chatani, Y.; Tadokoro, H.; Teranishi, K.; Tani, H. Structural studies of polyesters: 5. Molecular and crystal structures of optically active and racemic poly(β-hydroxybutyrate). Polymer 1973, 14, 267–272. [Google Scholar] [CrossRef]
- Iwata, T.; Aoyagi, Y.; Tanaka, T.; Fujita, M.; Takeuchi, A.; Suzuki, Y.; Uesugi, K. Microbeam X-ray diffraction and enzymatic degradation of poly[(R)-3-hydroxybutyrate] fibers with two kinds of molecular conformations. Macromolecules 2006, 39, 5789–5795. [Google Scholar] [CrossRef]
- Yamamoto, S.; Morisawa, Y.; Sato, H.; Hoshina, H.; Ozaki, Y. Quantum mechanical interpretation of intermolecular vibrational modes of crystalline poly-(R)-3-hydroxybutyrate observed in low-frequency Raman and terahertz spectra. J. Phys. Chem. B 2013, 117, 2180–2187. [Google Scholar] [CrossRef]
- Takahashi, M.; Kawazoe, Y.; Ishikawa, Y.; Ito, H. Interpretation of temperature-dependent low frequency vibrational spectrum of solid-state benzoic acid dimer. Chem. Phys. Lett. 2009, 479, 211–217. [Google Scholar] [CrossRef]
- Fumino, K.; Reichert, E.; Wittler, K.; Hempelmann, R.; Ludwig, R. Low-frequency vibrational modes of protic molten salts and ionic liquids: Detecting and quantifying hydrogen bonds. Angew. Chem. Int. Ed. 2012, 51, 6236–6240. [Google Scholar] [CrossRef]
- Xu, W.; Angell, C.A. Solvent-free electrolytes with aqueous solution-like conductivities. Science 2003, 302, 422–425. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Xu, W.; Angell, C.A. Ionic liquids by proton transfer: Vapor pressure, conductivity, and the relevance of ΔpKa from aqueous solutions. J. Am. Chem. Soc. 2003, 125, 15411–15419. [Google Scholar] [CrossRef]
- Angell, C.A.; Byrne, N.; Belieres, J.-P. Parallel developments in aprotic and protic ionic liquids: Physical chemistry and applications. Acc. Chem. Res. 2007, 40, 1228–1236. [Google Scholar] [CrossRef]
- Nuthakki, B.; Greaves, T.L.; Krodkiewska, I.; Weerawardena, A.; Burgar, M.I.; Mulder, R.J.; Drummond, C.J. Protic ionic liquids and iconicity. Aust. J. Chem. 2007, 60, 21–28. [Google Scholar] [CrossRef]
- Greaves, T.L.; Drummond, C.J. Protic ionic liquids: Properties and applications. Chem. Rev. 2008, 108, 206–237. [Google Scholar] [CrossRef]
- Fumino, K.; Wulf, A.; Ludwig, R. Hydrogen bonding in protic ionic liquids: Reminiscent of water. Angew. Chem. Int. Ed. 2009, 48, 3184–3186. [Google Scholar] [CrossRef]
- Stoimenovski, J.; Izgorodina, E.I.; MacFarlane, D.R. Ionicity and proton transfer in protic ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 10341–10347. [Google Scholar] [CrossRef]
- Bodo, E.; Postorino, P.; Mangialardo, S.; Piacente, G.; Ramondo, F.; Bosi, F.; Ballirano, P.; Caminiti, R. Structure of the molten salt methyl ammonium nitrate explored by experiments and theory. J. Phys. Chem. B 2011, 115, 13149–13161. [Google Scholar]
- Krüger, M.; Bründermann, E.; Funkner, S.; Weingärtner, H.; Havenith, M. Communications: Polarity fluctuations of the protic ionic liquid ethylammonium nitrate in the terahertz regime. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef]
- Abdul-Sada, A.K.; Greenway, A.M.; Hitchcock, P.B.; Mohammed, T.J.; Seddon, K.R.; Zora, J.A. Upon the structure of room temperature halogenoaluminate ionic liquids. J. Chem. Soc. Chem. Commun. 1986, 1753–1754. [Google Scholar]
- Wulf, A.; Fumino, K.; Ludwig, R. Spectroscopic evidence for an enhanced anion–cation interaction from hydrogen bonding in pure imidazolium ionic liquids. Angew. Chem. Int. Ed. 2010, 49, 449–453. [Google Scholar] [CrossRef]
- Fumino, K.; Wulf, A.; Ludwig, R. The cation–anion interaction in ionic liquids probed by far-infrared spectroscopy. Angew. Chem. Int. Ed. 2008, 47, 3830–3834. [Google Scholar] [CrossRef]
- Wulf, A.; Fumino, K.; Ludwig, R.; Taday, P.F. Combined THz, FIR and Raman spectroscopy studies of imidazolium-based ionic liquids covering the frequency range 2–300 cm−1. ChemPhysChem 2010, 11, 349–353. [Google Scholar] [CrossRef]
- Roth, C.; Peppel, T.; Fumino, K.; Köckerling, M.; Ludwig, R. The importance of hydrogen bonds for the structure of ionic liquids: Single-crystal X-ray diffraction and transmission and attenuated total reflection spectroscopy in the terahertz region. Angew. Chem. Int. Ed. 2010, 49, 10221–10224. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. How well can new-generation density functional methods describe stacking interactions in biological systems? Phys. Chem. Chem. Phys. 2005, 7, 2701–2705. [Google Scholar] [CrossRef]
- Hazra, M.K. Inter-molecular vibrational frequencies of doubly hydrogen-bonded complexes involving 2-pyridone: Reliability of few selected theoretical level of calculations. Chem. Phys. Lett. 2009, 473, 10–16. [Google Scholar] [CrossRef]
- Dkhissi, A.; Blossey, R. Performance of DFT/MPWB1K for stacking and H-bonding interactions. Chem. Phys. Lett. 2007, 439, 35–39. [Google Scholar] [CrossRef]
- Antony, J.; Helden, G.; Meijer, G.; Schmidt, B. Anharmonic midinfrared vibrational spectra of benzoic acid monomer and dimer. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef]
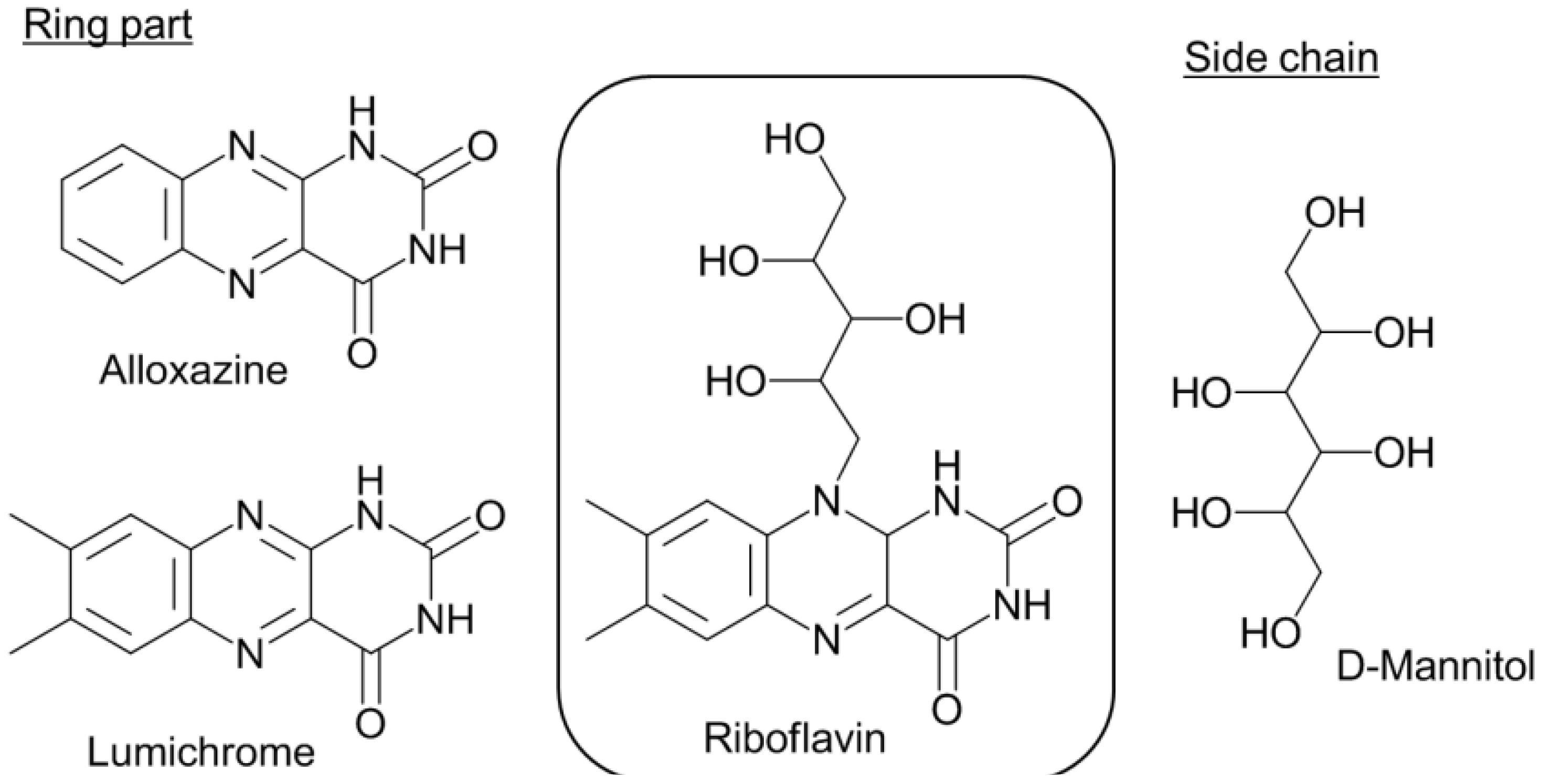
- Takahashi, M.; Ishikawa, Y.; Nishizawa, J.; Ito, H. Low-frequency vibrational modes of riboflavin and related compounds. Chem. Phys. Lett. 2005, 401, 475–482. [Google Scholar] [CrossRef]
- Hakey, P.M.; Allis, D.G.; Hudson, M.R.; Ouellette, W.; Korter, T.M. Investigation of (1R,2S)-(–)-ephedrine by cryogenic terahertz spectroscopy and solid-state density functional theory. ChemPhysChem 2009, 10, 2434–2444. [Google Scholar] [CrossRef]
- Mourik, T.; Duijneveldt, F.B. Ab initio calculations on the C–H···O hydrogen-bonded systems CH4–H2O, CH3NH2–H2O, CH3NH3+–H2O. J. Mol. Struct. THEOCHEM 1995, 341, 63–73. [Google Scholar] [CrossRef]
- Jurečka, P.; Šponer, J.; Černý, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef]
- Cybulski, S.M.; Lytle, M.L. The origin of deficiency of the supermolecule second-order Møller-Plesset approach for evaluating interaction energies. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef]
- Zhao, Y.; Schulz, N.E.; Truhlar, D.G. Exchange-correlation functional with broad accuracy for metallic and nonmetallic compounds, kinetics and noncovalent interactions. J. Chem. Phys. 2005, 123, 161103:1–161103:4. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Takahashi, K. Theoretical study on the effect of intramolecular hydrogen bonding on OH stretching overtone decay lifetime of ethylene glycol, 1,3-propanediol, and 1,4-butanediol. Phys. Chem. Chem. Phys. 2010, 12, 13950–13961. [Google Scholar] [CrossRef]
- Mohamed, M.N.A.; Watts, H.D.; Guo, J.; Catchmark, J.M.; Kubicki, J.D. MP2, density functional theory, and molecular mechanical calculations of C–H···π and hydrogen bond interactions in a cellulose-binding module-cellulose model system. Carbohydrate Res. 2010, 345, 1741–1751. [Google Scholar] [CrossRef]
- Cockett, M.C.R.; Miyazaki, M.; Tanabe, K.; Fujii, M. Isomer selective IR-UV depletion spectroscopy of 4-fluorotoluene-NH3: Evidence for π-proton-acceptor and linear hydrogen-bonded complexes. Phys. Chem. Chem. Phys. 2011, 13, 15633–15638. [Google Scholar]
- Schmies, M.; Patzer, A.; Fujii, M.; Dopfer, O. Structures and IR/UV spectra of neutral and ionic phenol–Arn cluster isomers (n ≤ 4): Competition between hydrogen bonding and stacking. Phys. Chem. Chem. Phys. 2011, 13, 13926–13941. [Google Scholar] [CrossRef]
- Kumar, S.; Biswas, P.; Kaul, I.; Das, A. Competition between hydrogen bonding and dispersion interactions in the indole···pyridine dimer and (Indole)2···pyridine trimer studied in a supersonic jet. J. Phys. Chem. A 2011, 115, 7461–7472. [Google Scholar] [CrossRef]
- Ivanova, B.B. Solid-state Raman spectra of non-centrosymmetric crystals-theoretical vs. experimental study towards an application in THz-regime. J. Mol. Struct. 2012, 1016, 47–54. [Google Scholar] [CrossRef]
- Vianello, R.; Mavri, J. Microsolvation of the histamine monocation in aqueous solution: The effect on structure, hydrogen bonding ability and vibrational spectrum. New J. Chem. 2012, 36, 954–962. [Google Scholar] [CrossRef]
- Lippert, K.M.; Hof, K.; Gerbig, D.; Ley, D.; Hausmann, H.; Guenther, S.; Schreiner, P.R. Hydrogen-bonding thiourea organocatalysts: The privileged 3,5-bis(trifluoromethy)phenyl group. Eur. J. Org. Chem. 2012, 2012, 5919–5927. [Google Scholar]
- León, I.; Cocinero, E.J.; Rijs, A.M.; Millán, J.; Alonso, E.; Lesarri, A.; Fernández, J.A. Formation of water polyhedrons in propofol-water clusters. Phys. Chem. Chem. Phys. 2013, 15, 568–575. [Google Scholar] [CrossRef]
- Blaser, S.; Ottiger, P.; Frey, H.-M.; Leutwyler, S. NH3 as a strong H-bond donor in singly- and doubly-bridged ammonia solvent clusters: 2-Pyridone·(NH3)n, n = 1–3. J. Phys. Chem. A 2013, 117, 7523–7534. [Google Scholar] [CrossRef]
- Jaeqx, S.; Du, W.; Meijer, E.J.; Oomens, J.; Rijs, A.M. Conformational study of Z-Glu-OH and Z-Arg-OH: Dispersion interactions versus conventional hydrogen bonding. J. Phys. Chem. A 2013, 117, 1216–1227. [Google Scholar] [CrossRef]
- León, I.; Millán, J.; Cocinero, E.J.; Lesarri, A.; Fernández, J.A. Transition from planar to nonplanar hydrogen bond networks in the solvation of aromatic dimers: Propofol2-(H2O)2–4. J. Phys. Chem. A 2013, 117, 3396–3404. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef]
- Lilienfeld, O.A.; Tkatchenko, A. Two- and three-body interatomic dispersion energy contributions to binding in molecules and solids. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [Green Version]
- Al-Saidi, W.A.; Voora, V.K.; Jordan, K.D. An assessment of the vdW-TS method for extended systems. J. Chem. Theory Comput. 2012, 8, 1503–1513. [Google Scholar] [CrossRef]
- Hujo, W.; Grimme, S. Comparison of the performance of dispersion-corrected density functional theory for weak hydrogen bonds. Phys. Chem. Chem. Phys. 2011, 13, 13942–13950. [Google Scholar] [CrossRef]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 1693–1694. [Google Scholar] [CrossRef]
- King, M.D.; Buchanan, W.D.; Korter, T.M. Application of London-type dispersion corrections to the solid-state density functional theory simulation of the terahertz spectra of crystalline pharmaceuticals. Phys. Chem. Chem. Phys. 2011, 13, 4250–4259. [Google Scholar] [CrossRef]
- King, M.D.; Korter, T.M. Application of London-type dispersion corrections in solid-state density functional theory for predicting the temperature-dependence of crystal structures and terahertz spectra. Cryst. Growth Des. 2011, 11, 2006–2010. [Google Scholar] [CrossRef]
- King, M.D.; Ouellette, W.; Korter, T.M. Noncovalent interactions in paired DNA nucleobases investigated by terahertz spectroscopy and solid-state density functional theory. J. Phys. Chem. A 2011, 115, 9467–9478. [Google Scholar] [CrossRef]
- Takahashi, M.; Ishikawa, Y.; Ito, H. The dispersion correction and weak-hydrogen-bond network in low-frequency vibration of solid-state salicylic acid. Chem. Phys. Lett. 2012, 531, 98–104. [Google Scholar]
- Beyer, T.; Price, S.L. The errors in lattice energy minimisation studies: Sensitivity to experimental variations in the molecular structure of paracetamol. CrystEngComm 2000, 34, 1–8. [Google Scholar]
- Pulay, P. Ab initio calculation of force constants and equilibrium geometries in polyatomic molecules. I. Theory. Mol. Phys. 1969, 17, 197–204. [Google Scholar] [CrossRef]
- Hagen, K.; Cross, V.R.; Hedberg, K. The molecular structure of selenonyl fluoride, SeO2,F2,, and sulfuryl fluoride, SO2F2, as determined by gas-phase electron diffraction. J. Mol. Struct. 1978, 44, 187–193. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Schaefer, H.F., III. A systematic theoretical study of harmonic vibrational frequencies: The ammonium ion NH4+ and other simple molecules. J. Chem. Phys. 1980, 73, 2310–2318. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Frisch, M.; Gaw, J.; Schaefer, H.F., III.; Binkley, J.S. Analytic evaluation and basis set dependence of intensities of infrared spectra. J. Chem. Phys. 1986, 84, 2262–2278. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.R.; Pople, J. Ab Initio Molecular Orbital Theory, 1st ed.; Wiley-Interscience: New York, NY, USA, 1986. [Google Scholar]
- King, M.D.; Buchanan, W.D.; Korter, T.M. Investigating the anharmonicity of lattice vibrations in water-containing molecular crystals through the terahertz spectroscopy of L-serine monohydrate. J. Phys. Chem. A 2010, 114, 9570–9578. [Google Scholar]
- Brittain, H.G. Polymorphism in Pharmaceutical Solids; Brittain, H.G., Ed.; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Kowal, A.T. First-principles computation of the anharmonic vibrational spectra of sulfuryl halides SO2X2 (X = F, Cl, Br). Chem. Phys. 2006, 324, 359–366. [Google Scholar] [CrossRef]
- Chaban, G.M.; Jung, J.O.; Gerber, R.B. Anharmonic vibrational spectroscopy of hydrogen-bonded systems directly computed from ab initio potential surfaces: (H2O)n, n = 2, 3; Cl− (H2O)n, n = 1, 2; H+(H2O)n, n = 1, 2; H2O–CH3OH. J. Phys. Chem. A 2000, 104, 2772–2779. [Google Scholar] [CrossRef]
- Petković, M. Infrared spectroscopy of ClONO2 and BrONO2 investigated by means of anharmonic force fields. Chem. Phys. 2007, 331, 438–446. [Google Scholar] [CrossRef]
- Garrett, R.H.; Grisham, C.M. Biochemistry, 2nd ed.; Sanders College Publishing: Fort Worth, TX, USA, 1999. [Google Scholar]
- Shen, Y.C.; Upadhya, P.C.; Linfield, E.H.; Davies, A.G. Temperature-dependent low-frequency vibrational spectra of purine and adenine. Appl. Phys. Lett. 2003, 82, 2350–2352. [Google Scholar] [CrossRef]
- Plazanet, M.; Fukushima, N.; Johnson, M.R. Modelling molecular vibrations in extended hydrogen-bonded networks—Crystalline bases of RNA and DNA and the nucleosides. Chem. Phys. 2002, 280, 53–70. [Google Scholar] [CrossRef]
- Giese, B.; McNaughton, D. Surface-enhanced Raman spectroscopic and density functional theory study of adenine adsorption to silver surfaces. J. Phys. Chem. B 2002, 106, 101–112. [Google Scholar] [CrossRef]
- Nowak, M.J.; Lapinski, L.; Kwiatkowski, J.S.; Leszczyński, J. Molecular structure and infrared spectra of adenine. Experimental matrix isolation and density functional theory study of adenine 15N isotopomers. J. Phys. Chem. 1996, 100, 3527–3534. [Google Scholar] [CrossRef]
- Gaigeot, M.-P.; Leulliot, N.; Ghomi, M.; Jobic, H.; Coulombeau, C.; Bouloussa, O. Analysis of the structural and vibrational properties of RNA building blocks by means of neutron inelastic scattering and density functional theory calculations. Chem. Phys. 2000, 261, 217–237. [Google Scholar] [CrossRef]
- Hielscher, R.; Hellwig, P. The temperature-dependent hydrogen-bonding signature of lipids monitored in the far-infrared domain. ChemPhysChem 2010, 11, 435–441. [Google Scholar]
- Takahashi, M.; Kawazoe, Y.; Ishikawa, Y.; Ito, H. Low-frequency vibrations of crystalline α,α-trehalose dihydrate. Chem. Phys. Lett. 2006, 429, 371–377. [Google Scholar] [CrossRef]
- Rungsawang, R.; Ueno, Y.; Tomita, I.; Ajito, K. Angle-dependent terahertz time-domain spectroscopy of amino acid single crystals. J. Phys. Chem. B 2006, 110, 21259–21263. [Google Scholar] [CrossRef]
- Whitley, V.H.; Hooks, D.E.; Ramos, K.J.; O’Hara, J.F.; Azad, A.K.; Taylor, A.J.; Barber, J.; Averitt, R.D. Polarization orientation dependence of the far infrared spectra of oriented single crystals of 1,3,5-trinitro-S-triazine (RDX) using terahertz time-domain spectroscopy. Anal. Bioanal. Chem. 2009, 395, 315–322. [Google Scholar] [CrossRef]
- Hoshina, H.; Morisawa, Y.; Sato, H.; Minamide, H.; Noda, I.; Ozaki, Y.; Otani, C. Polarization and temperature dependent spectra of poly(3-hydroxyalkanoate)s measured at terahertz frequencies. Phys. Chem. Chem. Phys. 2011, 13, 9173–9179. [Google Scholar] [CrossRef]
- Melinger, J.S.; Laman, N.; Harsha, S.S.; Cheng, S.; Grischkowsky, D. High-resolution waveguide terahertz spectroscopy of partially oriented organic polycrystalline films. J. Phys. Chem. A 2007, 111, 10977–10987. [Google Scholar] [CrossRef]
- Laman, N.; Harsha, S.S.; Grischkowsky, D. Narrow-line waveguide terahertz time-domain spectroscopy of aspirin and aspirin precursors. Appl. Spectrosc. 2008, 62, 319–326. [Google Scholar] [CrossRef]
- Melinger, J.S.; Laman, N.; Grischkowsky, D. The underlying terahertz vibrational spectrum of explosive solids. Appl. Phys. Lett. 2008, 93. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takahashi, M. Terahertz Vibrations and Hydrogen-Bonded Networks in Crystals. Crystals 2014, 4, 74-103. https://doi.org/10.3390/cryst4020074
Takahashi M. Terahertz Vibrations and Hydrogen-Bonded Networks in Crystals. Crystals. 2014; 4(2):74-103. https://doi.org/10.3390/cryst4020074
Chicago/Turabian StyleTakahashi, Masae. 2014. "Terahertz Vibrations and Hydrogen-Bonded Networks in Crystals" Crystals 4, no. 2: 74-103. https://doi.org/10.3390/cryst4020074