1. Introduction
The recent awareness of the threat to human health and to the environment of air and water pollutants has led scientists to search for a new technology able to reduce the contaminants’ concentrations. In the last two decades, and after the discovery of the properties of photocatalytic metal oxide nanoparticles (NPs) in the degradation of pollutants, the interest in the development of photocatalytic self-cleaning paints and coatings has increased enormously in academia and industry [
1,
2,
3]. The mechanism of self-cleaning paints is based on the presence of a photocatalyst (commonly composed of metal oxide NPs) that, under UV light exposure, exceeds its band gap energy and generates electron hole pairs. Although, in most cases, the electron hole pairs recombine to generate heat, in some cases the photocatalytic phenomena takes place and electrons and holes reach the surface of the photocatalyst where they can participate in redox reactions with the present oxygen and water molecules, creating highly active radicals. These radicals are able to decompose organic compounds and bacteria and, thus, they are able to clean surfaces [
4,
5,
6]. Furthermore, since the surface of the photocatalyst becomes superhydrophilic under UV irradiation, it is also claimed that photocatalytic materials might also have self-cleaning properties [
7].
Among the different photocatalysts, titanium dioxide (TiO
2) is the most thoroughly used because of its low toxicity, chemical stability, low cost, abundance as a raw material, and its high photocatalytic efficiency [
8]. Nonetheless, self-cleaning paints with other metal oxides, like ZnO, have also been reported [
9,
10].
A conventional waterborne paint is prepared in two steps. In the first step, the dispersion of the pigment in water, stabilizer, thickener, and auxiliary solvents, so-called mill base, is prepared. The pigment can be composed by non-photocatalytic TiO2 (rutile rich) and other inorganic materials, like calcium carbonate, depending on the final application and price sought for the paint (e.g., indoor or outdoor use). In the second step, the mill base is blended with the binder (polymer latex) and the paint is obtained. Waterborne photocatalytic paints differ from conventional waterborne paints in the addition of the photocatalyst (anatase phase rich TiO2 NPs) in the formulation. There is little information in the scientific literature about the preparation of photocatalytic paints, the way the photocatalyst is incorporated, and its final distribution on the paint film.
Commercial photocatalytic paints are mostly implemented in exterior (as façade paints) to avoid soiling and to keep the surface of the buildings clean [
11]. More recently, indoor photocatalytic paints have also been implemented with the aim to control the odour, degrade volatile organic compounds (VOC) and NO
X compounds, and maintain surfaces free of bacteria and fungi [
9,
12,
13,
14,
15,
16], among other applications.
Hochmannova and Vytrasova [
9] found that the photocatalytic activity of coatings was influenced by the type of acrylic dispersion used as a binder, the total pigment volume concentration (PVC), the type of the photocatalytic nanoparticles, and by the morphology of the surfaces.
Nevertheless, one of the most controversial aspects of the photocatalytic paints is related with the potential degradation of the organic binder constituents during long exposure to UV irradiation. Thus, there are works that show that under UV light irradiation the organic binder in the photocatalytic paint degrades forming other stable compounds, like acetaldehydes and ketones, which do not proceed to further mineralization [
17], or carbonyls [
18]. Other authors [
19] also reported leaching of the TiO
2 NPs from the paint matrix to the environment during very long periods of UV exposure (one year) although the measured values were considered low (1.5 µg/dm
3 in a climate chamber, and between 0.5 and 14 µg/dm
3 during leaching tests). The detachment of the NPs was observed in photocatalytic paints that contained TiO
2 NPs, but this was not observed in paints that only contained the usual TiO
2 pigment. This behaviour was attributed to a weaker binding of the TiO
2 NPs to the polymer matrix, which was observed in the scanning electron microscope (SEM) images of the surface of the paints.
In this context, Baudys et al. [
10] interestingly pointed out that the key point in the development of stable paints is the creation of better barriers to the photocatalytic mineralization process, so that the polymer binder remains intact longer and the paint exhibits an appreciable lifetime as a coated film. They noted that in the preparation of photocatalytic paints it is important to find a compromise between maintaining a reasonable degree of photocatalytic activity whilst retaining a good paint stability. Clearly, the higher the photocatalytic activity of the paint, the lower the paint lifetime due to the light-induced weathering and photo-chalking.
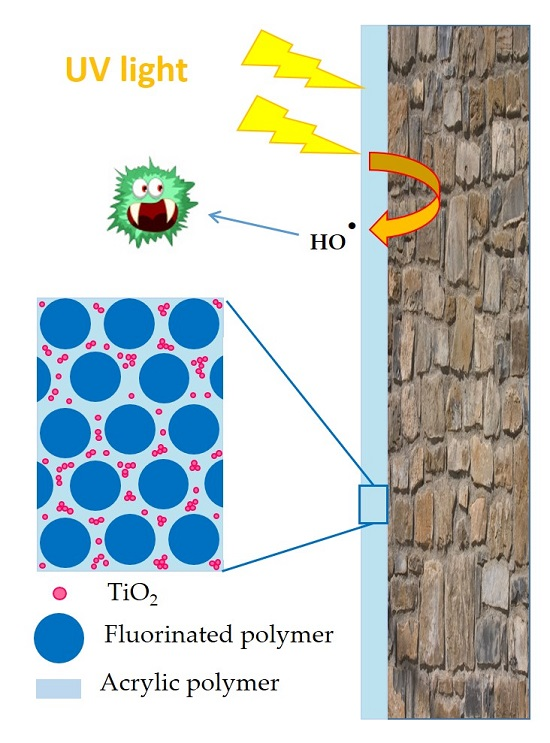
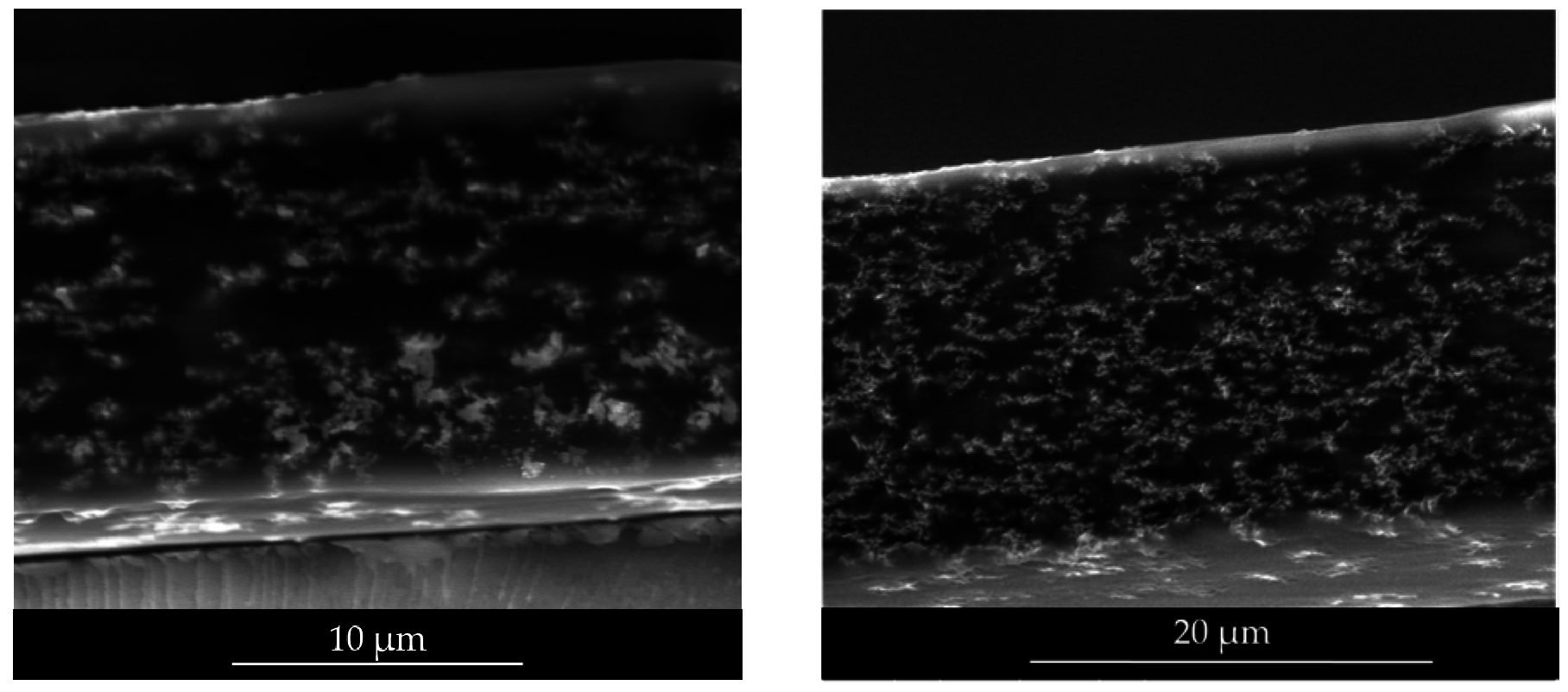
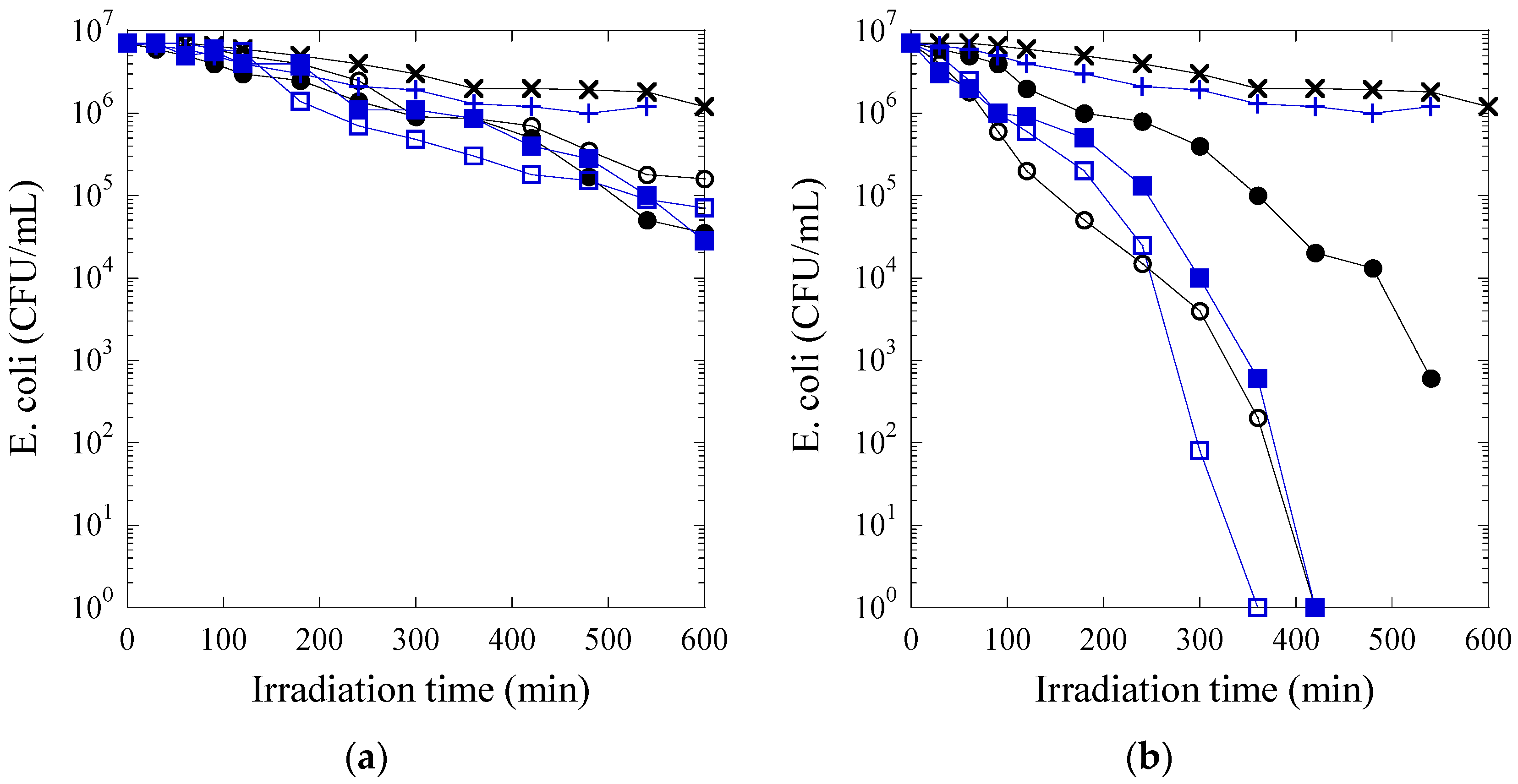
In this work, photocatalytic paints were formulated using two types of polymeric binders (a pure (meth)acrylic latex (LM1) and a fluorinated/(meth)acrylic latex (CS2) with core-shell morphology) and two types of photocatalytic TiO2 NPs (Degussa P25 and a sample from Johnson and Matthey (JM)). First, hybrid dispersions were prepared by blending the photocatalytic TiO2 aqueous dispersions and the latexes. Polymer films were prepared and their photocatalytic activity was assessed by analyzing the inactivation of Escherichia coli bacteria. The photoactivity was found superior in the substrate-film interface due to the higher concentration of the TiO2 in the bottom of the film. Then, the stability of the photocatalytic paints formulated with a hybrid binder composed by the fluorinated/(meth)acrylic core-shell latex and JM TiO2 was analyzed by measuring the evolution of gloss retention and color change during the exposure of coated aluminum panels to accelerated weathering tests (QUV-B) for 5000 hours. Both gloss and color decreased during the first 1000 hours of exposure and then remained constant at acceptable values.
3. Materials and Methods
3.1. Materials
Methyl methacrylate (MMA, Quimidroga, Spain), n-butyl acrylate (BA, Quimidroga, Spain), and methacrylic acid (MAA, Aldrich, Spain) monomers were used as received. The P25 and JM TiO2 NPs were kindly provided by Degussa and Johnson and Matthey, respectively. Potassium persulfate (KPS, Sigma, Spain), tert-butyl hydroperoxide (TBHP, Sigma-Aldrich, Spain), and ascorbic acid (AsAc, Sigma-Aldrich, Spain) were used as initiators. Sodium lauryl sulfate emulsifier (SLS, Sigma, Spain) was used as received. Ammonium hydroxide (NH4OH, Sigma, Spain) was employed as a base. Doubly-deionized (DDI, MilliQ quality) water was used throughout the work.
The P(VDF-co-HFP) (88.2/11.8 wt%) seed latex employed for the synthesis of the core-shell NPs was provided by Solvay Specialty Polymers (Italy). The solids content of the latex was 31 wt% and the average diameter of particles was 102 nm.
3.2. Synthesis of the Acrylic Latex LM1
The acrylic latex was synthesized by a seeded semi-batch emulsion polymerization reaction carried out in a 1 L jacketed reactor equipped with a reflux condenser, nitrogen inlet, a sampling device, and a stainless steel anchor stirrer rotating at 250 rpm. The formulation used to prepare the seed is given in
Table 2. The organic phase containing the monomers was added to the aqueous phase composed of the emulsifier dissolved in water and the resulting dispersion was charged into the reactor. The temperature was increased to 70 °C and once the temperature was reached, the initiator (potassium persulfate, KPS) was added in a shot. The final solids content of the seed was 20 wt% and the average diameter of particles was 74 nm. The seeded semi-batch emulsion polymerization was prepared following the recipe given in
Table 3. The seed was diluted with water to reach a solids content of 1.3 wt% and then a pre-emulsion containing the mixture of monomers, water, and emulsifier, an aqueous phase containing the oxidant (tert-butyl hydroperoxide, TBHP), and another one containing the reductant (ascorbic acid, AsAc), were fed using three different streams to the reactor for 4 h. After the feed period had finished, the system was left to react for one more hour and the temperature was decreased. The final solids content of the LM1 latex was 50 wt% and the final diameter of particles was 332 nm.
3.3. Synthesis of The Core-Shell Latex Cs2
The core-shell latex CS2 was synthesized by seeded semi-batch emulsion polymerization following the recipe presented in
Table 4. First, the P(VDF-
co-HFP) seed latex provided by Solvay Specialty Polymers (Italy) was loaded in a 1 L jacketed reactor equipped with a reflux condenser, nitrogen inlet, a sampling device, and a stainless steel anchor stirrer rotating at 250 rpm, and the temperature was increased to 70 ºC. Once the temperature was reached, a pre-emulsion containing the mixture of acrylic monomers, water, and emulsifier, an aqueous phase containing the oxidant (TBHP), and another one containing the reductant (AsAc), were fed using three different streams to the reactor for 3 h. After the feed period had finished, the system was left to react for one more hour and the temperature was decreased. The final solids content of the core-shell latex was 40 wt% and the final average diameter of particles was 140 nm.
3.4. Preparation of The Hybrid Blends
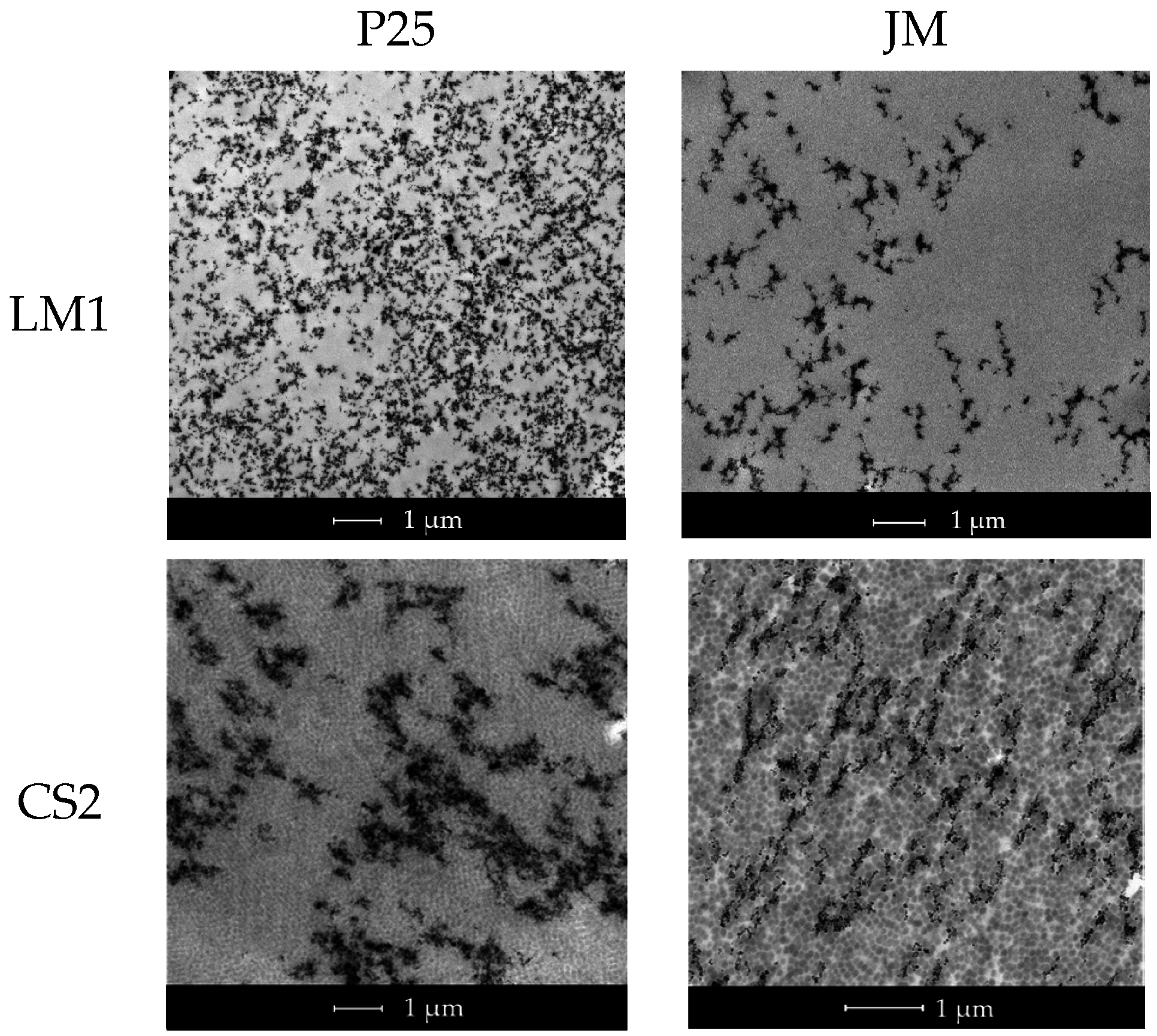
Two different latexes were used in order to prepare the hybrid blends. On the one hand, a conventional (meth)acrylic latex (poly(methyl methacrylate-
co-butyl acrylate-
co-methacrylic acid), P(MMA-
co-BA-
co-MMA), 49.5 wt% MMA, 49.5 wt% BA, and 1% wt MAA) commonly used for coating applications was employed (labeled as LM1). As acrylic polymers may suffer degradation under UV exposure [
17,
18,
23], a core-shell latex containing perfluorinated polymer in the core was also used in order to improve the UV resistance of the final blends. P(VDF) is a high molecular weight polymer used in architectural applications due to its exceptional weathering resistance: indeed, the presence of strong C-F bonds allows the polymer to resist against the photo-oxidation initiated by UV light [
24,
25]. The core-shell latex was produced by seeded semi-batch polymerization using as seed a poly(vinylidene fluoride-
co-hexafluoro propene) (P(VDF-
co-HFP) latex (provided by Solvay Specialty Polymers, Italy) and copolymerizing a mixture of MMA/BA/MAA (49.5/49.5/1 wt%) on the shell to produce a film forming latex (labeled as CS2). It is worth pointing out that the P(VDF-
co-HFP) latex is not film-forming by itself due to its high Tg (well above room temperature) and partial crystallinity. Thus, a shell of a film-forming acrylic polymer is produced to render the composite latex the ability to form a film at room temperature (see
Figure S4, supporting information). Note that both latexes (LM1 and CS2) contain 1 wt% of MAA in the formulation to obtain carboxylic acid surface functionalized polymer particles. The pH of both latexes was adjusted to 10 by the addition of NH
4OH, in order to deprotonate the carboxylic acid groups at the surface of the particles and to obtain negatively charged polymer particles (pKa
COOH ≈ 4.15). The deprotonated carboxylic acid groups play a key role in the interaction with the TiO
2 surface to obtain stable blends.
For the preparation of the blends, different amounts of TiO
2 NP aqueous dispersions at pH = 2 were added to the latexes at pH = 10, and left overnight under stirring. The final pH of the blends was close to 9. At this pH, both the polymer particles and the TiO
2 NPs were negatively charged and the final blends obtained were stable and form good films (see
Figure 7). The amounts of TiO
2 in the final blends were 1, 5, and 10 wbp%.
3.5. Characterization of the TiO2 NPs
In this work, two different aqueous dispersions of TiO2 NPs containing 2.5 wt% TiO2 were employed for the preparation of the blends. TiO2 NPs were named P25 (Degussa P25 TiO2) and JM (Johnson and Matthey TiO2). Although the theoretical particle diameters were 20 and 30 nm for P25 and JM, respectively, the diameter measured experimentally by dynamic light scattering (DLS) in the aqueous dispersions after 20 min of sonication using a Branson 450 w (operating at 8-output control and 60% duty cycle) were 150 and 70 nm, respectively. These results indicate that there was some aggregation of the TiO2 NPs in the aqueous dispersions. The isoelectric point (IEP) of the TiO2 NPs (measured by zeta potential measurements using DLS) were 7 and 6 for P25 and JM, respectively. The pH of the TiO2 NPs dispersions was around 2 in both cases, so the TiO2 NPs were positively charged.
3.6. Latex and Film Characterization.
Dynamic light scattering (Zetasizer Nano ZS, Malvern Instruments, Worcestershire, UK) was used to measure the z-average diameter of the TiO2 NP dispersion, mini-emulsion droplets, and final polymer particles.
The morphology of the films obtained by casting the latexes onto silicon molds was analyzed by transmission electron microscopy (TEM) (FEI, Eindhoven, The Netherlands) in a TECNAI G2 20 TWIN (FEI) operating at an accelerating voltage of 200 kV in a bright field image mode. The films cast at room temperature were trimmed at −40 °C using an ultramicrotome device (Leica EMFC6, Viena, Austria) equipped with a diamond knife. The ultrathin sections (100 nm) were placed on 300 mesh copper grids and were observed without further staining.
SEM (FEI, Eindhoven, Netherlands) analysis was performed in a Quanta 250 (FEI) in the low vacuum mode at 10 kV and using a long field detector. Samples were dipped into liquid N2 to get fractures and they were adhered to stubs for cross-section analysis.
X-ray photoelectron spectroscopy (XPS) composition data and spectra were acquired on a SPECS (SPECS, Berlin, Germany) instrument equipped with a Phoibos 150 1D-DLD analyzer (SPECS, Berlin, Germany) and a monochromatic Al Kα X-ray source (SPECS, Berlin, Germany). Compositional survey and detail scans were acquired using a pass energy of 80 eV. High resolution spectra were acquired using a pass energy of 30 eV. The above data were taken at 90° takeoff angle. Data analysis was performed with Casa XPS 2.3.16 software (Casa Software Ltd, Cheshire, UK) to fit the signals to Gauss-Lorentzian curves, after removing the background (Shirley).
3.7. Formulation of the Photocatalytic Paints
For the formulation of the paints, first the pigment grind was prepared in a high speed mixer with glass beads following the recipe given in
Table 5, and then it was mixed with the other ingredients employed for the formulation of the paints (see
Table 6). Once the formulated paints were obtained, they were applied on aluminum panels with a drawdown bar and let to dry at room temperature.
3.8. Evaluation of the Bacterial Inactivation on Prepared Polymers under Solar Light Irradiation
Escherichia coli (E. coli K12) was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ) ATCC23716 (Braunschweig, Germany). The 200 µL culture aliquots with an initial concentration of ~106 CFU∙mL-1 in NaCl/KCl (pH 7) were placed either at the air-film or at the film-substrate interface. The polymeric sample area was 9 cm2. The polymer permits a homogeneous distribution of the inoculum evenly without needing an adsorption stage. A well-dispersed non-heterogeneous contact was established between the film and the bacterial solution. The 200 µL of the Escherichia coli solution was exposed at room temperature (25–28 °C). The films were then placed on Petri dishes provided with a lid to prevent evaporation. The used Petri dishes were selected according to their absorption edge. In our case they do not absorb the emitted light during our experiment.
After each pre-defined irradiation time, the films were transferred into sterile 10 mL tubes containing 5 mL autoclaved NaCl/KCl saline solution. This solution was subsequently mixed thoroughly using a vortex for 3 min. Serial dilutions were made in NaCl/KCl solution. A 100 µL sample of each dilution was pipetted onto a nutrient agar plate and then spread over the surface of the plate using the standard plate method. Agar plates were incubated lid down, at 37 °C, for 24 h before colonies were counted. To verify that no bacteria remained adsorbed to the polymeric surface, films were incubated on agar Petri dishes overnight. Almost no bacterial re-growth was observed as adhered bacteria to the polymeric surface. Samples were irradiated in the cavity of a Suntest solar simulator light CPS (Atlas GmbH, Hanau, Germany) at a light dose of 55 mW/cm2. The washing solution was a NaCl/KCl saline solution (8 g/L NaCl and 0.8 g/L KCl) followed with a MQ-water wash. Three independent assays were done for each sample. The statistical analysis of the results was performed for the colony-forming unit (CFU) values calculating the standard deviation values (n = 5%). The average values were compared by one-way analysis of variance and with the value of statistical significance. The one-way analysis of variance (one-way ANOVA) was used to compare the mean of the samples using the Fisher distribution.
3.9. Accelerated Weathering Test
QUV-B weathering tests were performed on the formulated paints coated on aluminum panels. Although the results produced by UV-A are closer to that of sunlight, the UV-B lamp is the most widely used fluorescent light source when durable materials are studied because the degradation takes place at faster rates [
26]. The tests lasted 5000 hours in total and cycles were as follow: eight hours of UVB-313 at 70 °C and four hours of humidity in the absence of irradiation (ASTM D 4329-84). Among the different properties that could be checked to determine the coating durability, it was decided to focus on color and gloss. Every 500 hours panels were taken out of QUV and the coating properties were measured. Moreover, a visual inspection of the film was made in order to check if chalking or other types of degradation forms were present.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}