
Synthesis and Crystallographic Characterization of a Maleimide Derivative of Tryptamine


Abstract
:
1. Introduction
2. Results and Discussion
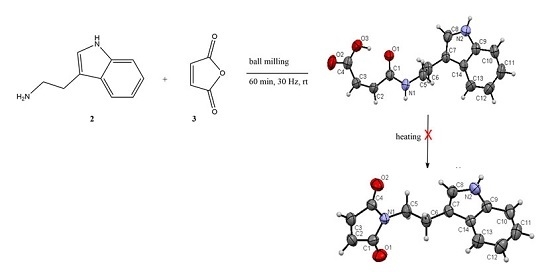
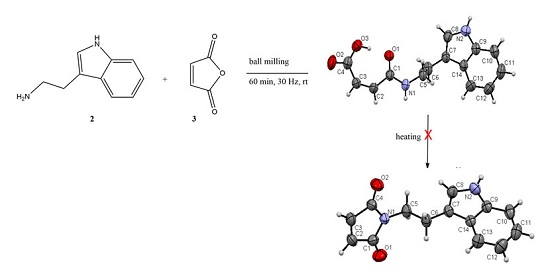
2.1. Synthesis and Crystallization
2.1.1. Mechanosynthesis
2.1.2. Synthesis Using Activated Maleic Acid
2.2. Structural Commentary
3. Materials and Methods
3.1. NMR Data
3.2. Crystal Data Collection and Refinement
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rondelet, G.; Dal Maso, T.; Willems, L.; Wouters, J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 2016, 194, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.; Vandermeers, F.; de Brogniez, A.; Florins, A.; Nigro, A.; François, C.; Bouzar, A.-B.; Verlaeten, O.; Stern, E.; Lambert, D.; et al. Chemoresistance to valproate treatment of bovine leukemia virus-infected sheep; identification of improved HDAC inhibitors. Pathogens 2012, 1, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanaka, R.; Hamada, S.; Nakagawa, H.; Miyata, N. Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Colaço, M.O.; Dubois, J.; Wouters, J. Mechanochemical synthesis of phthalimides with crystal structures of intermediates and products. CystEngComm 2015, 17, 2523–2528. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, A.G.; Neudorfl, J.; de Kiff, A. Photoinduced electron-transfer chemistry of the bielectrophoric N-phthaloyl derivatives of the amino acids tyrosine, histidine and tryptophan. Beilstein J. Org. Chem. 2011, 7, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Boittiaux, I.; Tilborg, A.; Lambert, D.; Wouters, J. The dicyclohexylamine salt of RG108 (N-phthalyl-l-tryptophan), a potential epigenetic modulator. Acta Cryst. 2010, 66, o3175–o3176. [Google Scholar] [CrossRef] [PubMed]
- Tilborg, A.; Boittiaux, I.; Norberg, B.; Lambert, D.; Wouters, J. 4-Nitro-N-phthalyl-l-tryptophan. Acta Cryst. 2011, 67, o2116. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Chhatwal, A.R.; Williams, J.M. Direct amide formation from unactivated carboxylic acids and amines. Chem. Commun. 2012, 48, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef] [PubMed]
- Casimir, J.R.; Guichard, G.; Briand, J.-P. Methyl 2-((succinimidooxy)carbonyl)benzoate (MSB): A new, efficient reagent for N-phthaloylation of amino acid and peptide derivatives. J. Org. Chem. 2002, 67, 3764–3768. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Caro, L.D.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (1) | (1o) | |
|---|---|---|
| Chemical formula | C14H12N2O2 | C14H14N2O3 |
| Mr | 240.26 | 258.27 |
| Crystal system, space group | Orthorhombic, P212121 | Monoclinic, P21/n |
| a, b, c (Å) | 6.2720 (3), | 8.9037 (6), |
| 12.8304 (6), | 12.0034 (10), | |
| 15.0252 (6) | 12.1047 (8) | |
| α, β, γ (°) | 90, 90, 90 | 90, 92.357 (6), 90 |
| V (Å3) | 1209.11 (9) | 1292.59 (16) |
| Z | 4 | 4 |
| Radiation type | Mo Kα | Cu Kα |
| µ (mm−1) | 0.09 | 0.78 |
| Crystal size (mm) | 1.0 × 0.8 × 0.5 | 0.13 × 0.07 × 0.05 |
| Tmin, Tmax | 0.917, 0.956 | 0.930, 0.960 |
| No. of ref. measured, independent, and observed | 4460, 2144, 1789 | 5570, 2181, 1524 |
| Rint | 0.036 | 0.069 |
| θmax (°) | 25.0 | 64.0 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.041, 0.097, 1.05 | 0.060, 0.185, 1.02 |
| No. of reflections | 2144 | 2011 |
| No. of parameters | 163 | 172 |
| Δρmax, Δρmin (e Å−3) | 0.11, −0.16 | 0.23, −23 |
| Compound 1 | |||
| N1–C4 | 1.377 (3) | C1–C2 | 1.482 (4) |
| N1–C1 | 1.380 (4) | C2–C3 | 1.307 (4) |
| O1–C1 | 1.205 (3) | C3–C4 | 1.480 (4) |
| O2–C4 | 1.208 (3) | ||
| C1–N1–C5–C6 | −88.3 (3) | C5–C6–C7–C8 | 15.9 (4) |
| N1–C5–C6–C7 | −177.1 (2) | ||
| Compound 1o | |||
| N1–C1 | 1.323 (3) | C1–C2 | 1.471 (4) |
| O2–C4 | 1.220 (3) | C2–C3 | 1.330 (4) |
| O3–C4 | 1.290 (4) | C3–C4 | 1.479 (4) |
| C1–N1–C5–C6 | 88.6 (3) | C5–C6–C7–C8 | 93.4 (3) |
| N1–C5–C6–C7 | −178.3 (2) |
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| Compound 1 | ||||
| N2–H2···O2 i | 0.86 | 2.30 | 2.952 (3) | 133 |
| Compound 1o | ||||
| O3–H3O···O1 | 0.82 | 1.68 | 2.490 (3) | 171 |
| N2–H2···O1 ii | 0.86 | 2.09 | 2.938 (3) | 169 |
| N1–H1···O2 iii | 0.86 | 2.18 | 2.941 (3) | 147 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois, J.; Colaço, M.; Rondelet, G.; Wouters, J. Synthesis and Crystallographic Characterization of a Maleimide Derivative of Tryptamine. Crystals 2016, 6, 153. https://doi.org/10.3390/cryst6110153
Dubois J, Colaço M, Rondelet G, Wouters J. Synthesis and Crystallographic Characterization of a Maleimide Derivative of Tryptamine. Crystals. 2016; 6(11):153. https://doi.org/10.3390/cryst6110153
Chicago/Turabian StyleDubois, Jean, Melwin Colaço, Grégoire Rondelet, and Johan Wouters. 2016. "Synthesis and Crystallographic Characterization of a Maleimide Derivative of Tryptamine" Crystals 6, no. 11: 153. https://doi.org/10.3390/cryst6110153