
3D DNA Crystals and Nanotechnology

1
Department of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA
2
Department of Chemistry, New York University, New York, NY 10003, USA
*
Author to whom correspondence should be addressed.
Crystals 2016, 6(8), 97; https://doi.org/10.3390/cryst6080097
Submission received: 28 July 2016
/
Revised: 11 August 2016
/
Accepted: 12 August 2016
/
Published: 18 August 2016
(This article belongs to the Special Issue Nucleic Acid Crystallography)
Abstract
:DNA’s molecular recognition properties have made it one of the most widely used biomacromolecular construction materials. The programmed assembly of DNA oligonucleotides has been used to create complex 2D and 3D self-assembled architectures and to guide the assembly of other molecules. The origins of DNA nanotechnology are rooted in the goal of assembling DNA molecules into designed periodic arrays, i.e., crystals. Here, we highlight several DNA crystal structures, the progress made in designing DNA crystals, and look at the current prospects and future directions of DNA crystals in nanotechnology.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The DNA double helical structure has become an iconic symbol, both in science and in popular culture, due in part to the elegance and simplicity of the helical double arrangement of DNA strands. This simplicity is also reflected in the immediately recognizable relationship between the double helical structure and DNA’s role in genetic information storage. The original determination of the double helical DNA structure was itself deeply rooted in the exploration of another class of structures that have fascinated humans for millennia: crystals. This fascination comes in part because of the crystal shape, or habit, which is a consequence of the periodic nature of crystals as first suggested by René Just Haüy (1743–1822). From an analytical perspective, crystalline substances are attractive because their periodicity allows the structure of their component molecules to be established through X-ray diffraction. Though small molecule crystallography was already an established discipline in the 1950s, it was at the dawn of macromolecular crystallography that Franklin and Gosling collected their early diffraction images of microcrystalline DNA fibers [1]. These diffraction images enabled Crick and Watson to develop a model for the helical repeat structure of duplex DNA that ultimately initiated the development of molecular biology [2]. Not long after, the maturation of macromolecular crystallography began to reveal the structural and functional details of proteins, but nucleic acid crystal structures were largely limited to relatively simple dinucleoside phosphate structures [3], primarily owing to the expense of longer oligonucleotides of discrete sequences needed for crystallization. It was not until the development of low-cost chemical DNA synthesis by Caruthers and colleagues [4] that facile automated synthesis of longer oligonucleotides with designed sequences was possible. This in turn opened the door for the determination of DNA crystal structures [5,6,7]. DNA crystal structures are responsible for a great deal of our knowledge about the diversity of DNA structure, its sequence-dependent heterogeneity [8,9], and how it interacts with other biological molecules to express the information stored in its molecular structure/sequence [10,11].
Much as the discovery of the DNA duplex structure was linked with the exploration of crystals, DNA crystals are inextricably linked with the field of DNA nanotechnology. Even before the term “nanotechnology” had been coined, it was suggested that DNA could be used as a material to organize matter. It was originally proposed that DNA could be used as a programmable construction material for rationally designed branched junctions that could be assembled to form periodic three-dimensional crystals [12]. The crystals could serve as porous scaffolds to orient and position guest molecules, such as proteins, at specific positions (Figure 1), effectively making the guest an integral part of the crystal lattice. A scaffold for crystallizing proteins could provide a unique method for overcoming the macromolecular crystallization bottleneck and enable rapid structure determination, thereby enhancing and accelerating the structure-based drug design pipeline. The ability to position guest molecules in 3D would enable many applications beyond the original notion of a crystallization scaffold. 3D DNA crystals have also been envisioned as information storage devices [13], as zeolite-like materials capable of macromolecular separations [14], and/or catalysis with appropriate functionalization [15].
Any number of materials could be suitable for constructing molecular scaffolding materials. In one example, crystalline metal complexes have been effectively used as “crystal sponges” to determine small molecule crystal structures [16] and metal-organic frameworks have been used to house protein enzymes [17]. However, DNA has a number of features that make it an ideal material for designing scaffolds that could be applied in many different circumstances. First and most uniquely, DNA is a programmable molecule. Base pairing provides specificity to inter- or intramolecular interactions that allows single strands of DNA to form structurally well-designed higher order assemblies. Second, the interaction between these single stranded sticky ends leads to a predictable local structure—the Watson-Crick duplex—when programmed via complementary base pairs [18]. Sequence-specific structural variations that can lead to deviations in the Watson-Crick duplex [19,20], and a number of non-canonical DNA base pairing motifs that can compete with Watson-Crick base pairing [21,22,23,24], can be mitigated by careful sequence selection. Third, the association between DNA strands is a self-assembly process. The ability for individual strands, programmed by their sequences, to find complementary regions in other strands when part of a mixture allows for significant complexity in design. There is also a code external to the double helix, enabling the binding of DNA triplexes, as well as their recognition by other macromolecules [25]. Finally, the cost of DNA synthesis continues to drop. In 1981, “Only a highly trained and skilled chemist could produce a single 12-unit DNA sequence in less than 5 months.” [26] Thus, in the earliest days of DNA nanotechnology, the synthesis cost per DNA nucleotide was over $300, whereas today this cost is below $0.20 a base on a useful scale [27]. These decreased synthesis costs have allowed the construction of DNA assemblies from hundreds to thousands of individual strands [28,29].
One of the inherent design principles of DNA nanotechnology is that on short length-scales (well below the persistence length), the DNA duplex behaves as a linear, fairly-rigid rod [30]. Constructing branching structures of sufficient rigidity and uniformity to connect these duplexes is the central challenge of DNA crystal design. The need to understand more thoroughly how DNA behaved in multi-arm junctions was directly responsible for the broadening of the DNA nanotechnology landscape. The first such attempts were the construction of geometric objects, with DNA duplexes as edges [31,32]. While successful, these constructs highlighted the “floppiness” of the junctions that connected the duplex edge as one of the major challenges in using DNA as a building material. Robust motifs, like the DX and TX motifs [33,34] based on multiple reciprocal exchanges of strands between antiparallel DNA duplexes, were found to enhance DNA rigidity [35,36], and led to the construction of DNA tiled arrays [37] and a host of 2D periodic and discrete assemblies, DNA nanotubes [38], the use of DNA tiles for logical computation [39], and to the development of dynamic DNA machines and circuits [40,41,42]. The field saw an explosion in 2005 with the development of DNA origami [43] leading to a new and powerful method for creating diverse 2D and 3D DNA nanostructures with over 7000 nt pairs. More recently, DNA brick assemblies [28,44] have been utilized to create many types of arrangements from a starting set of numerous short DNA strands. Both origami and brick structures are largely used without strand purification, making them attractive to a large audience of non-chemists (physicists, computer scientists, engineers and materials-scientists), but this feature renders them unlikely as components for 3D crystal formation. Finally, the versatility of DNA as a construction material has also allowed it to become an important player in different areas of material science. DNA has been used as the connective material for the construction of crystalline nanoparticle arrays [45,46]. DNA hydrogels offer a number of remarkable properties with wide-ranging application [47].
The wonderful diversity of discrete DNA structures created through planar tile motifs, DNA origami, and DNA brick methods have clearly demonstrated the power of DNA as a self-assembling construction material, but they have also shifted emphasis from the initial motivations of the field. Further, there are several significant differences between these types of nanoscale assemblies of large, single objects and the assembly of robust 3D DNA crystals. It is the programmed cooperative interactions between many DNA strands with distinct sequences that facilitates nucleation and stabilizes these nanoassemblies [48]. For 3D DNA crystals, the crystal contacts are by their nature isomorphous, meaning there is much less room for subtle structural heterogeneity. In addition, the key feature of a crystal to be used for diffraction purposes is not its stability, but its resolution, i.e., the extent of its scattering. Resolution and scattering power are not direct functions of the thermodynamic stability of the crystalline contacts as a whole, but on the homogeneity of the crystal components and other features that are not well understood. Thus, the notion of constructing porous 3D DNA crystals with sufficient uniformity to study their structures by X-ray diffraction has proved to be quite distinct from its progeny.
There are over 1000 examples of “naked” DNA crystal structures deposited in the Nucleic Acid Database (NDB) [49], representing a wide array of DNA sequences and structures. In this review, we restrict our view to those DNA crystal structures with features that lend themselves toward nanotechnology applications based on several specific criteria. First, these DNA lattices show continuous hydrogen bonding. In this case, “continuous” refers to crystal packing interactions between strands that result in any DNA molecule in any unit cell of the crystal being reachable from any starting point by tracing through the sugar-phosphate backbone or through nucleobase hydrogen bonding interactions. Surprisingly, very few DNA crystal structures in the NDB meet this criterion, with a majority relying on end-to-end base stacking interactions between duplex segments, or groove-to-groove interactions to facilitate crystal packing. Non-hydrogen-bonding contacts certainly have the potential to be another parameter used in designing and optimizing DNA crystals, but as of now their programmability is not well understood in most cases. Second, the one thing in common with nearly all of the proposed applications of DNA crystals is the positioning of guest molecules. Therefore, the crystals must contain sufficient solvent space in the form of channels or pockets to allow guest incorporation. The geometry and volume of the solvent space dictate what type of guest molecules (e.g., small molecules or macromolecules) can be incorporated, but densely packed DNA would make the inclusion of guest molecules—either before or after crystallization—significantly more difficult. There are two different classes of DNA lattices present in the NDB that meet these criteria.
2. Tensegrity Triangle Crystals
The first fully designed 3D DNA crystal lattice was based on a three-fold symmetric structural motif known as the tensegrity triangle [50,51]. The motif is characterized by the helix axes of each of the triangle edges extending in linearly independent directions (Figure 2a). This provides an inherent 3D geometry that can be extended into a periodic structure by end-to-end translation of the motif along each linear direction. To replicate the full motif structure with DNA, seven strands are required (Figure 2b,c), although they are often three-fold symmetrized using three strands in a 3:3:1 ratio. First, a central strand (black) containing three repeating 7 nt segments base paired with three identical edge strands (green) to form the central three-fold symmetric triangle with a single nick site. These pairing interactions occur between the central 7 nt of the 21 nt edge strands, leaving 7 free nt on either side. These 7 nt extensions occur where two edges cross, and they are held together and oriented by the addition of three cross-over strands (blue) that form four-arm branched junctions at the triangle vertices. While this motif could be robustly assembled in solution, the programmability for these motifs to assemble together and form a larger periodic structure is dependent on 2 nt “sticky ends” present at the 5' ends of both cross-over and duplex strands (Figure 2b, red outline). These nucleotides direct the assembly of the independently folding triangle motifs into a higher order periodic structure and provide continuous connectivity within the lattice.
As described above, nearly all of the proposed applications of DNA crystals involve placement of guest molecules relative to the DNA, and this requires the crystals to have adequate solvent space for the guests to occupy. As part of their design, the tensegrity triangle crystals contain rhombohedral solvent pockets of ~200 nm3 (Figure 3). These solvent spaces are connected, effectively generating a series of solvent channels that run through the crystal along each of the triangle edges (Figure 4a). When considered as a macroscopic object, the lattice arrangement creates a highly porous crystal. Figure 4a–c shows several different lattice views that create discernible solvent channels (when viewed in projection) running through the entire crystal.
The robust design of the tensegrity triangle crystals has allowed the construction of several other types of DNA lattices. One of the advantages of this construct is the ability to increase the overall complexity of the components, while maintaining programmability. This was demonstrated by the construction of crystals with two different triangles in the crystal’s asymmetric unit [52]. The duplex regions of the two triangles contained different sequences, and the sticky ends were programmed to enable the two motifs to assemble through an alternating pattern. As designed, the cell constants of these crystals now contained 1/3 of each of the two triangles in the asymmetric unit, and two complete tensegrity triangles in the unit cell. Significantly, the increased complexity of the unit cell contents allows increased diversity of the placement of guest molecules (see below). The tensegrity triangle described in Figure 2, Figure 3 and Figure 4 is a two-turn variety, with each triangle edge containing the 21 nucleotides of two turns of B-form duplex. However, the ability to program turn length based on the number of base pairs per arm provides another method for altering the unit cell dimensions and tuning the crystal properties. Both three- and four-turn tensegrity triangle crystals have been assembled into crystals [51]. While these crystals only diffract to 5.5 Å (modified from [51]) and 10–14 Å resolution, respectively, they clearly influence the properties of the unit cell, most notably the solvent content. The three-turn would contain solvent channels with ~568 nm3, while the four-turn crystals have channels of 850–1100 nm3.
An interesting rule of thumb arose in another context, but the tensegrity triangle (which precedes it) obeys the rule. At one juncture, attempts were made to make 2D crystals of DNA origami. The first attempts used the same strategy as employed in the first 2D DX crystals [37], wherein all the helix axes were parallel. The origami crystals that were formed were extremely narrow (<10 tiles), although quite long. Wenyan Liu, working on the problem, recognized that the long direction was the direction of the helix axes: He designed a cross-shaped origami tile that yielded roughly isotropic crystals in 2D; hence, the notion that DNA crystals seem better if their helix axes span the dimensions of the crystal is known as Wenyan Liu’s Rule [53]. It is worth pointing out that one of us (NCS) has tried numerous motifs that ought to form 3D crystals, but which contain parallel helix axes. These included TX motifs with non-integral twists between two TXs, as well as 6-helix bundles. 3D crystals were often obtained, but no diffraction was observed, thus vindicating Wenyan Liu’s rule. The rule is an empirical observation and the basis of this design principle remains unknown.
3. DNA 13-Mer Crystals with Non-Canonical Base Pairs
The tensegrity triangle crystals described above were the result of many trials and years of dedicated work to identify an assembly motif that gave crystals that diffracted usable resolution. However, the first description of a continuous 3D DNA crystal came several years earlier [54]. In this case, the crystals were not initially designed, but were instead the serendipitous product of an attempt to construct a lattice from both DNA and protein components. The DNA 13-mer, GGACAGATGGGAG was used as one strand in a two member, four-way junction motif that was based on the RNA strand in a DNA-RNA hybrid four-way junction [55]. The longer partner strand of this motif (originally described as an in vitro evolved DNA enzyme) was biotinylated, with the goal of assembling a periodic DNA/protein lattice built from DNA four-way junctions and tetrameric streptavidin. Despite having regions of complete complementarity to its partner strand, the 13-mer crystallized independently from the mixture through self-pairing interactions. Remarkably, the only crystallization requirement was the addition of Mg2+.
In this lattice, every DNA strand is identical, with the crystallographic symmetry resulting in each strand forming base pairs with three other strands to form two distinct structural regions (Figure 5a). The first structural region is composed of self-pairing between nucleotides 4 and 9 of two 13-mers to form an anti-parallel B-form double helix containing two central G-A non-canonical pairs. The second structural region is formed through base pairing between G1-A3 of one strand and G10-A12 of another to form a parallel-stranded GGA-GGA motif. This region, denoted the homopurine junction, forms two distinct G-G base pairs (G1(syn)-G10(anti)), G2(anti)-G11(anti)), and one A-A base pair (A3-A12) (Figure 5b). G13 is disordered in the structure, and is not necessary for crystallization. It is the homopurine junction region of this crystal that is critical for connecting the short B-form duplex regions into a continuously hydrogen-bonded DNA network.
The organization of the crystal lattice can be viewed as layers of coaxially stacked duplexes separated by 20 Å of solvent space. Each of these parallel arrays is rotated by 120° with respect to the flanking layers down the 64 screw axis. Each layer is continuously hydrogen bonded to the flanking layers through the homopurine junctions which simultaneously serve to bridge the coaxially stacked duplex regions within each layer. Similar to the tensegrity triangle crystal, this geometric layout also results in the lattice having extensive solvent pockets. The overall solvent content of nearly 70% of the unit cell volume (59,483 Å3 of solvent in a 88,334 Å3 cell) is a result of solvent channels that run both down the six-fold axis, and along each of the helical layers perpendicular to the six-fold axis (Figure 6a,b)
This crystal structure provides an example of DNA’s structural plasticity in the absence of Watson-Crick interactions. Alternate DNA structures are now beginning to be a useful tool for DNA nanotechnology applications [54,55,56,57,58,59,60]. However, these have been primarily limited to well-understood motifs, such as G-quadruplex and DNA i-motifs, where the structure can be predicted from a combination of sequence and environmental conditions. It is the lack of predictability that significantly limits the use of non-canonical motifs as junctions for DNA assembly. There is mounting evidence that the GGA-GGA parallel motif is such a predictable motif. It has been observed in solution [61], and it is structurally related to another well-studied parallel-stranded motif known as π-DNA [62,63]. Finally, though this initial DNA crystal was not the product of deliberate design, the resulting structure enabled the construction of a number of derivative DNA lattices using the interactions of the homopurine junction.
The first derivative of the 13-mer lattice was the creation of a fully self-complementary duplex region by removing the central G-A base pairs (Figure 5a). Converting these to G-C base pairs resulted in crystals with isomorphous unit cell parameters and overall crystal structure [64]. Testing all 64 possible self-complementary sequences in the duplex region showed that the combination of the GGA-GGA parallel motif and self-complementary duplex segments was a robust design, with over 20 different sequences yielding crystals. Thirteen of these were solved to resolutions better than 2.3 Å. This study allowed the examination of the role of the sequence in crystal self-assembly. There was a clear selection against the G4-C9 base pair, and a strong selection for the A5-T8 base pair in the duplex region, suggesting that base pairing identities can have a large impact on crystallizability in certain contexts. Because these crystals were isomorphous, it was also possible to construct “mixed” single crystals containing more than one oligonucleotide sequence [64].
The 13-mer DNA crystals has also served as a template for the design of a 3D DNA crystal with expanded solvent channels. The addition of 10 or 11 nucleotides between the duplex region and the 3' GGA motif was designed to extend the duplex region by one turn on either side (Figure 7a). In this design, a second strand was required to pair with the expansion region [54]. This construct crystallized under the same conditions as the DNA 13-mer (only requiring the addition of Mg2+), but only diffracted to ~5 Å resolution. This diffraction was sufficient to confirm the predicted unit cell symmetry and constants, and a model of the lattice structure showed the solvent channels down the six-fold symmetry axis expanded substantially, going from channels of ~2 nm in diameter (Figure 6a) to channels of ~9 nm in diameter (Figure 7b). The channels perpendicular to the six-fold axis were also expanded (Figure 7c). Though the structure could not be definitively solved at low-resolution, atomic force microscopy of the crystal surface reinforced both the cell dimensions and the presence of solvent channels [65] (Figure 7b,c).
4. Lessons for DNA Crystal Design
The two types of DNA crystal lattice described above arise from very different types of interactions and geometries, but there are observed commonalities between the structures and their assembly. First, and not surprisingly, both structures require multi-cations for crystallization. These cation concentrations, including the relative amount and identity of mono- and divalent cations, is one of the main environmental conditions that requires optimization of nucleic acid crystallization [66,67]. Appropriate cation concentrations are necessary to shield the charge of the polyanionic DNA backbone to promote crystal assembly, while sub-optimal concentrations will not crystallize or result in aggregation. In both of these cases, metallic cations are apparently necessary for both crystal formation and stability, and this is likely an inherent property of most DNA crystals that might require crosslinking or otherwise affect their utility in applications where high salt concentrations may not be desirable.
A second common feature of the two structures is that the motifs that provide continuity to the base pairing interactions between crystallizing units (the sticky ends of the tensegrity triangle and the homopurine junction of the 13-mer lattice) are relatively short. This may be a significant feature that facilitates crystal assembly into macroscale objects, and supports the idea that not all interactions that could “pre-organize” the components are beneficial for crystal assembly. For the tensegrity triangle motif, many different sticky end lengths were assessed [68], but those crystals of two-turn triangles with the 1 and 2 nt sticky ends diffracted to the highest resolution. This is contrary to the simple interpretation that longer sticky ends would provide more cohesive connectivity between triangle units through more favorable thermodynamics. The importance of the shortness of the sticky ends is that they facilitate reversibility; the useful presence of phosphates [69] is likely to have a similar origin. For the 13-mer lattice, the homopurine junction length could not be altered, but in the final structure G1 is rotated about the glycosidic bond to adopt the less common syn orientation (Figure 5b). Attempts to pre-organize G1 into the syn orientation using an 8-amino dG substitution resulted in 13-mers that crystallized extremely rapidly (in minutes) at typical DNA concentrations upon the addition of Mg2+, but they formed primarily microcrystal clusters that diffracted poorly (PJP, unpublished). These results suggest that the desire to design components that pre-organize stable lattice contacts can in some cases be detrimental to crystal assembly. There may be a variety of different reasons for this in both cases. The argument can be made that the short sticky ends in the tensegrity triangle allow reversibility that can help avoid kinetic traps during crystal nucleation or growth. In the 13-mer case, it appears the G1 syn pre-organization alters crystal nucleation, perhaps by reducing the activation free energy of nucleation at the expense of crystal growth [70].
Another feature common between the two types of lattice is the impact expansion has on the diffraction limit. The three- and four-turn tensegrity triangle crystals (~5.5 Å with LNA additions, and ~10 Å, respectively) and the expanded non-canonical crystals diffract poorly relative to the shorter versions. There are likely a number of factors that contribute to the decreased resolution, but one thing these constructs all have in common is an increase in solvent content. This phenomenon is emphasized in the 20 Å resolution found in the 3D tensegrity triangle crystals when the edges are 8-turn DX molecules (NCS, unpublished). The longer helices used in these constructs result in greater separation between strands and ultimately larger solvent spaces. While this type of expansion may be desirable, for example, as a scaffold for proteins, there is a strong negative correlation between solvent content and diffraction limits in macromolecular crystals [71]. For some applications, these decreased resolution limits may not be an important issue, but for use as a macromolecular scaffold for structure determination, it is significant. Indeed, one of the significant challenges for future applications that rely on x-ray diffraction and structure determination will be to mitigate the effects of increased solvent.
5. DNA Crystal Applications
Though the goal of using DNA crystals as a scaffold for protein crystallization has yet to be achieved, the DNA crystal lattices described here have already begun to push toward this goal, while revealing other potential applications. The first example of integrating proteins, albeit non-specifically, within a 3D DNA crystal was the demonstration that the expanded non-canonical lattice could function as a macromolecule sieve [14]. The 9 nm diameter solvent channels running down the six-fold symmetry axis of these crystals were capable of absorbing proteins up to ~45 kDa, with the amount of absorbed protein inversely proportional to its molecular weight. Selective absorption was demonstrated using a fluorescent protein mixture. 28 kDa green-fluorescent protein could be visualized throughout the interior of the crystal, while a 280 kDa red-fluorescent protein was excluded from the crystal.
The ability to incorporate proteins into the expanded non-canonical lattice suggested the possibility of constructing solid-state biocatalysts by encapsulation of protein enzymes. As a proof-of-concept, RNase A was encapsulated within the DNA crystal and used to cleave a fluorescently-quenched dinucleotide substrate [15]. RNase A provided a convenient model system, as a potent protein inhibitor of RNase A could be used in large excess due to its being too large to enter the crystal. Thus, any observed enzyme activity occurred in the crystal solvent channels. These enzyme-infused crystals were capable of multiple rounds of catalysis following washing to remove substrate and product.
Several examples of specific guest molecule placement have been demonstrated with the tensegrity triangle crystals. As one way of assessing that the crystals with two triangles in the asymmetric unit contained both constructs, different color fluorescent dyes were linked covalently to each construct [52]. Crystals grown with one cyan labeled triangle and the other unlabeled triangle produced distinctly cyan crystals, and the opposite combination using a red dye produced red crystals. Strikingly, crystals grown from a mixture of the two labeled constructs yielded purple crystals, consistent with the co-incorporation of the dyes. The relatively low diffraction limit (5 Å), the three-fold averaging of the dyes within the crystal, and the flexible organic linker prevented their observation in the electron density. However, the site-specific placement of small molecules into DNA crystals is a first step toward the reaching the goal of using DNA crystals as molecular scaffolds.
6. Future Directions and Applications
There has been a push to expand the types, and thus the applications of DNA crystals by incorporating different guest molecules. One potential application that has already been explored with DNA origami constructs is as a drug delivery vehicle. Anti-cancer drugs, such as doxorubicin, can be intercalated into DNA origami designs and used to improve drug efficacy in cell culture models [72,73]. DNA crystals may be suitable for similar use, but with the potential of drugs beyond just small molecule intercalators. The ability to house proteins, including antibody-based drugs [74], would provide a new type of use for DNA crystals. Shielding these types of drugs in a biomolecular scaffold could enhance biological compatibility and improve drug efficacy. However, macroscopic DNA crystals have yet to be tested in delivery applications. Future work in this area will require understanding how DNA crystals (either macroscopic or nanoscale) interact with cells, similar to the developing body of work with other DNA constructs [75].
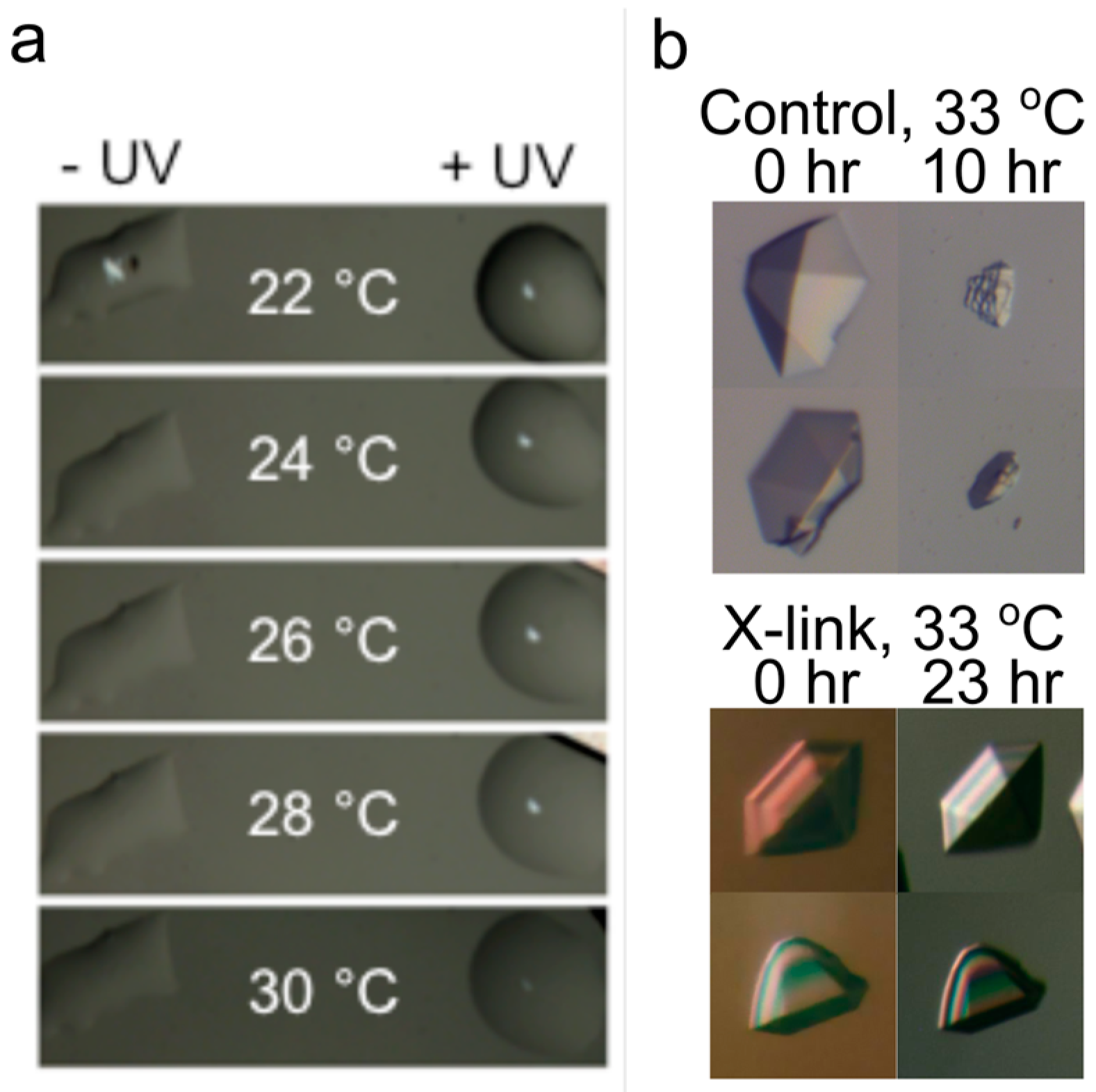
One challenge for any nucleic acid construct in these types of applications is longevity in biological environments. In some cases, DNA nanoconstructs seem to have an inherent resistance to nucleases [76,77], though others show extreme sensitivity in tissue culture mediums [78]. Several different methods for enhancing the durability of nanoscale DNA constructs have been explored [79,80]. Studies on DNA crystals have been limited, but both intrinsic design and extrinsic approaches have been explored. The salt stability of the triangle crystals has been enhanced through the conversion of an interconnecting duplex arm into a triplex by the addition of appropriately designed single strands [81]. The addition of the triplex-forming strand enhances the stability of crystals in low salt, and provides an alternative method for the introduction of guest molecules, for example, by the incorporation of fluorescent dyes [82]. Triplex formation has also been used to introduce photo-crosslinkers site-specifically to the triangle crystals. When activated by UV exposure, these crosslinks enhance crystal thermostability (Figure 8a) [83]. Non-specific chemical crosslinking has been shown to be an effective way of enhancing 13-mer crystal durability under a number of conditions. The bis-alkylating reagent nornitrogen mustard formed multiple interstrand crosslinks within the crystals, leading to significantly enhanced thermostability (Figure 8b), tolerance in low cation environments, and decreased nuclease sensitivity [84].
The last decade has seen significant advances in the design, characterization, and application of DNA crystals. It is now routine to produce robust DNA crystals from specific motifs, and there continue to be advances in RNA crystal design [85]. Both the conventional Watson-Crick paired tensegrity triangle and the novel parallel GGA-GGA motif can be used to produce crystalline arrangements, and new designs and motifs will further expand the exploration of DNA crystals. There are at least two key routes to pursue now: improving the diffracting capability of large-solvent-content variants of these crystals, and putting interesting guests inside them. It should be clear that larger units of these motifs can also be used (e.g., the whole rhombohedron in the case of the tensegrity triangle), so that complex guests and diverse guests can be included in the crystals. As DNA nanotechnology goes back to its roots, we expect the next decade to provide even more excitement in this area than was provided by the last one.
Acknowledgments
NCS has been supported by grants EFRI-1332411, and CCF-1526650 from the NSF, MURI W911NF-11-1-0024 from ARO, MURI N000140911118 from ONR, DE-SC0007991 from DOE for partial salary support, and grant GBMF3849 from the Gordon and Betty Moore Foundation. PJP has been supported by NSF CAREER Award (DMR11496650).
Author Contributions
Paul J. Paukstelis and Nadrian C. Seeman wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Franklin, R.E.; Gosling, R.G. Molecular Configuration in Sodium Thymonucleate. Nature 1953, 171, 740–741. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C.; Sussman, J.L.; Berman, H.M.; Kim, S.H. Nucleic Acid Conformation: Crystal Structure of a Naturally Occurring Dinucleoside Phosphate (UpA). Nature 1971, 233, 90–92. [Google Scholar] [CrossRef]
- Caruthers, M.H. Gene synthesis machines: DNA chemistry and its uses. Science 1985, 230, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.H.-J.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; van Boom, J.H.; van der Marel, G.; Rich, A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Drew, H.R.; Wing, R.M.; Takano, T.; Broka, C.; Tanaka, S.; Itakura, K.; Dickerson, R.E. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. USA 1981, 78, 2179–2183. [Google Scholar] [PubMed]
- Brown, T.; Hunter, W.N.; Kneale, G.; Kennard, O. Molecular structure of the G.A base pair in DNA and its implications for the mechanism of transversion mutations. Proc. Natl. Acad. Sci. USA 1986, 83, 2402–2406. [Google Scholar] [CrossRef] [PubMed]
- Sines, C.C.; McFail-Isom, L.; Howerton, S.B.; VanDerveer, D.; Williams, L.D. Cations Mediate B-DNA Conformational Heterogeneity. J. Am. Chem. Soc. 2000, 122, 11048–11056. [Google Scholar] [CrossRef]
- Hays, F.A.; Teegarden, A.; Jones, Z.J.R.; Harms, M.; Raup, D.; Watson, J.; Cavaliere, E.; Ho, P.S. How sequence defines structure: A crystallographic map of DNA structure and conformation. Proc. Natl. Acad. Sci. USA 2004, 102, 7157–7162. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Geiger, J.H.; Hahn, S.; Sigler, P.B. Crystal structure of a yeast TBP/TATA-box complex. Nature 1993, 365, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [PubMed]
- Seeman, N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Robinson, B.H.; Seeman, N.C. The design of a biochip: A self-assembling molecular-scale memory device. Protein Eng. 1987, 1, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Paukstelis, P.J. Three-dimensional DNA crystals as molecular sieves. J. Am. Chem. Soc. 2006, 128, 6794–6795. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Paukstelis, P.J. DNA Crystals as Vehicles for Biocatalysis. J. Am. Chem. Soc. 2014, 136, 7817–7820. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, Y.; Yoshioka, S.; Ariyoshi, J.; Arai, T.; Hitora, Y.; Takada, K.; Matsunaga, S.; Rissanen, K.; Fujita, M. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature 2013, 495, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Moon, S.-Y.; Guelta, M.A.; Harvey, S.P.; Hupp, J.T.; Farha, O.K. Encapsulation of a Nerve Agent Detoxifying Enzyme by a Mesoporous Zirconium Metal-Organic Framework Engenders Thermal and Long-Term Stability. J. Am. Chem. Soc. 2016, 138, 8052–8055. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Dewan, J.C.; Seeman, N.C. A DNA decamer with a sticky end: The crystal structure of d-CGACGATCGT. J. Mol. Biol. 1997, 267, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.-S.; Wu, H.-M.; Crothers, D.M. DNA bending at adenine-thymine tracts. Nature 1986, 320, 501–506. [Google Scholar] [CrossRef] [PubMed]
- DiGabriele, A.D.; Steitz, T.A. A DNA dodecamer containing an adenine tract crystallizes in a unique lattice and exhibits a new bend. J. Mol. Biol. 1993, 231, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.H.; Wartell, R.M. The thermal stability of DNA fragments with tandem mismatches at a d(CXYG).d(CY’X’G) site. Nucleic Acids Res. 1996, 24, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, P.; Ohmichi, T.; Sugimoto, N. Characterization and thermodynamic properties of quadruplex/duplex competition. FEBS Lett. 2002, 526, 77–81. [Google Scholar] [CrossRef]
- Mendoza, O.; Elezgaray, J.; Mergny, J.-L. Kinetics of quadruplex to duplex conversion. Biochimie 2015, 118, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Thirugnanasambandam, A.; Karthik, S.; Artheswari, G.; Gautham, N. DNA polymorphism in crystals: Three stable conformations for the decadeoxynucleotide d(GCATGCATGC). Acta Crystallogr. Sect. Struct. Biol. 2016, 72, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C.; Rosenberg, J.M.; Rich, A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA 1976, 73, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Urbina, G.; Sathe, G.M.; Liu, W.C.; Gillen, M.F.; Duck, P.D.; Bender, R.; Ogilvie, K.K. Automated synthesis of gene fragments. Science 1981, 214, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Structural DNA Nanotechnology; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Ke, Y.; Ong, L.L.; Shih, W.M.; Yin, P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Marchi, A.N.; Saaem, I.; Vogen, B.N.; Brown, S.; LaBean, T.H. Toward Larger DNA Origami. Nano Lett. 2014, 14, 5740–5747. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P.J. Flexibility of DNA. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Seeman, N.C. Synthesis from DNA of a molecule with the connectivity of a cube. Nature 1991, 350, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Seeman, N.C. Symmetric Holliday junction crossover isomers. J. Mol. Biol. 1994, 238, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.J.; Seeman, N.C. DNA double-crossover molecules. Biochemistry 1993, 32, 3211–3220. [Google Scholar] [CrossRef] [PubMed]
- LaBean, T.H.; Yan, H.; Kopatsch, J.; Liu, F.; Winfree, E.; Reif, J.H.; Seeman, N.C. Construction, Analysis, Ligation, and Self-Assembly of DNA Triple Crossover Complexes. J. Am. Chem. Soc. 2000, 122, 1848–1860. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Seeman, N.C. Direct evidence for Holliday junction crossover isomerization. Biochemistry 1997, 36, 4240–4247. [Google Scholar] [CrossRef] [PubMed]
- Sa-Ardyen, P.; Vologodskii, A.V.; Seeman, N.C. The flexibility of DNA double crossover molecules. Biophys. J. 2003, 84, 3829–3837. [Google Scholar] [CrossRef]
- Winfree, E.; Liu, F.; Wenzler, L.A.; Seeman, N.C. Design and self-assembly of two-dimensional DNA crystals. Nature 1998, 394, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, F.; Liao, S.; Kopatsch, J.; Wang, T.; Mao, C.; Seeman, N.C. Six-helix bundles designed from DNA. Nano Lett. 2005, 5, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; LaBean, T.H.; Reif, J.H.; Seeman, N.C. Logical computation using algorithmic self-assembly of DNA triple-crossover molecules. Nature 2000, 407, 493–496. [Google Scholar] [PubMed]
- Mao, C.; Sun, W.; Shen, Z.; Seeman, N.C. A nanomechanical device based on the B-Z transition of DNA. Nature 1999, 397, 144–146. [Google Scholar] [PubMed]
- Yan, H.; Zhang, X.; Shen, Z.; Seeman, N.C. A robust DNA mechanical device controlled by hybridization topology. Nature 2002, 415, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Seeman, N.C. Circuits and programmable self-assembling DNA structures. Proc. Natl. Acad. Sci. USA 2002, 99, 12577–12582. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ong, L.L.; Sun, W.; Song, J.; Dong, M.; Shih, W.M.; Yin, P. DNA brick crystals with prescribed depths. Nat. Chem. 2014, 6, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lytton-Jean, A.K.R.; Lee, B.; Weigand, S.; Schatz, G.C.; Mirkin, C.A. DNA-programmable nanoparticle crystallization. Nature 2008, 451, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Nykypanchuk, D.; Maye, M.M.; van der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 2008, 451, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Um, S.H.; Lee, J.B.; Park, N.; Kwon, S.Y.; Umbach, C.C.; Luo, D. Enzyme-catalysed assembly of DNA hydrogel. Nat. Mater. 2006, 5, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, A.; Frenkel, D. Numerical Evidence for Nucleated Self-Assembly of DNA Brick Structures. Phys. Rev. Lett. 2014, 112, 238103. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, B.C.; Westbrook, J.; Ghosh, S.; Petrov, A.I.; Sweeney, B.; Zirbel, C.L.; Leontis, N.B.; Berman, H.M. The Nucleic Acid Database: New features and capabilities. Nucleic Acids Res. 2014, 42, D114–D122. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, M.; Deng, Z.; Walulu, R.; Mao, C. Tensegrity: Construction of Rigid DNA Triangles with Flexible Four-Arm DNA Junctions. J. Am. Chem. Soc. 2004, 126, 2324–2325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Birktoft, J.J.; Chen, Y.; Wang, T.; Sha, R.; Constantinou, P.E.; Ginell, S.L.; Mao, C.; Seeman, N.C. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 2009, 461, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sha, R.; Birktoft, J.; Zheng, J.; Mao, C.; Seeman, N.C. A DNA Crystal Designed to Contain Two Molecules per Asymmetric Unit. J. Am. Chem. Soc. 2010, 132, 15471–15473. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhong, H.; Wang, R.; Seeman, N.C. Crystalline Two-Dimensional DNA Origami Arrays. Angew. Chem. Int. Ed. Engl. 2011, 50, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Paukstelis, P.J.; Nowakowski, J.; Birktoft, J.J.; Seeman, N.C. Crystal Structure of a Continuous Three-Dimensional DNA Lattice. Chem. Biol. 2004, 11, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, J.; Shim, P.J.; Prasad, G.S.; Stout, C.D.; Joyce, G.F. Crystal structure of an 82-nucleotide RNA-DNA complex formed by the 10–23 DNA enzyme. Nat. Struct. Mol. Biol. 1999, 6, 151–156. [Google Scholar]
- Chakraborty, S.; Sharma, S.; Maiti, P.K.; Krishnan, Y. The poly dA helix: A new structural motif for high performance DNA-based molecular switches. Nucleic Acids Res. 2009, 37, 2810–2817. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Chakraborty, K.; Krishnan, Y. Tunable, colorimetric DNA-based pH sensors mediated by A-motif formation. Chem. Commun. 2012, 48, 2513–2515. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Nizak, C.; Surana, S.; Halder, S.; Krishnan, Y. Two DNA nanomachines map pH changes along intersecting endocytic pathways inside the same cell. Nat. Nanotechnol. 2013, 8, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.C.; Henderson, E. G-Wires: Self-Assembly of a Telomeric Oligonucleotide, d(GGGGTTGGGG), into Large Superstructures. Biochemistry 1994, 33, 10718–10724. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yang, Z.; Liu, D. DNA nanotechnology based on i-motif structures. Acc. Chem. Res. 2014, 47, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Kettani, A.; Bouaziz, S.; Skripkin, E.; Majumdar, A.; Wang, W.; Jones, R.A.; Patel, D.J. Interlocked mismatch-aligned arrowhead DNA motifs. Struct. Lond. Engl. 1999, 7, 803–815. [Google Scholar] [CrossRef]
- Robinson, H.; van der Marel, G.A.; van Boom, J.H.; Wang, A.H. Unusual DNA conformation at low pH revealed by NMR: Parallel-stranded DNA duplex with homo base pairs. Biochemistry 1992, 31, 10510–10517. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.; Wang, A.H. 5’-CGA sequence is a strong motif for homo base-paired parallel-stranded DNA duplex as revealed by NMR analysis. Proc. Natl. Acad. Sci. USA 1993, 90, 5224–5228. [Google Scholar] [CrossRef] [PubMed]
- Saoji, M.; Zhang, D.; Paukstelis, P.J. Probing the role of sequence in the assembly of three-dimensional DNA crystals. Biopolymers 2015, 103, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Seeman, N.C.; Paukstelis, P.J. Surface AFM of a 3D DNA crystal. University of Maryland: College Park, MD, USA, Unpublished work. 2010. [Google Scholar]
- Berger, I.; Kang, C.; Sinha, N.; Wolters, M.; Rich, A. A Highly Efficient 24-Condition Matrix for the Crystallization of Nucleic Acid Fragments. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Viladoms, J.; Parkinson, G.N. HELIX: A new modular nucleic acid crystallization screen. J. Appl. Crystallogr. 2014, 47, 948–955. [Google Scholar] [CrossRef]
- Ohayon, Y.P.; Chandrasekaran, A.R.; Hernandez, C.; Jong, M.A.; Abdallah, H.O.; Fisher, N.; Mohsen, M.; Shah, C.; Demirel, E.; Tan, A.; et al. Designing Higher Resolution Self-Assembled 3D DNA Crystals via Strand Terminus Modifications. New York University: New York, NY, USA, Unpublished work. 2016. [Google Scholar]
- Sha, R.; Birktoft, J.J.; Nguyen, N.; Chandrasekaran, A.R.; Zheng, J.; Zhao, X.; Mao, C.; Seeman, N.C. Self-Assembled DNA Crystals: The Impact on Resolution of 5′-Phosphates and the DNA Source. Nano Lett. 2013, 13, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Boistelle, R.; Astier, J.P. Crystallization mechanisms in solution. J. Cryst. Growth 1988, 90, 14–30. [Google Scholar] [CrossRef]
- Kantardjieff, K.A.; Rupp, B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein–nucleic acid complex crystals. Protein Sci. 2003, 12, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Song, C.; Nangreave, J.; Liu, X.; Lin, L.; Qiu, D.; Wang, Z.-G.; Zou, G.; Liang, X.; Yan, H.; et al. DNA Origami as a Carrier for Circumvention of Drug Resistance. J. Am. Chem. Soc. 2012, 134, 13396–13403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, Q.; Li, N.; Dai, L.; Liu, Q.; Song, L.; Wang, J.; Li, Y.; Tian, J.; Ding, B.; et al. DNA Origami as an In Vivo Drug Delivery Vehicle for Cancer Therapy. ACS Nano 2014, 8, 6633–6643. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.L.; Mrsny, R.J. Formulation and delivery issues for monoclonal antibody therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Qian, H.; Tay, C.Y.; Leong, D.T. Cellular processing and destinies of artificial DNA nanostructures. Chem. Soc. Rev. 2016, 45, 4199–4225. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.S.; Yin, H.; Erben, C.M.; Wood, M.J.A.; Turberfield, A.J. DNA Cage Delivery to Mammalian Cells. ACS Nano 2011, 5, 5427–5432. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.; Mohammed, A.; Gardell, J.; Masich, S.; Czeizler, E.; Orponen, P.; Högberg, B. DNA rendering of polyhedral meshes at the nanoscale. Nature 2015, 523, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Wickham, S.F.J.; Shih, W.M.; Perrault, S.D. Addressing the Instability of DNA Nanostructures in Tissue Culture. ACS Nano 2014, 8, 8765–8775. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, A.; Endo, M.; Katsuda, Y.; Hidaka, K.; Sugiyama, H. Photo-Cross-Linking-Assisted Thermal Stability of DNA Origami Structures and Its Application for Higher-Temperature Self-Assembly. J. Am. Chem. Soc. 2011, 133, 14488–14491. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, V.; Oberleitner, B.; Sobotta, J.; Nickels, P.; Grossi, G.; Kempter, S.; Frischmuth, T.; Liedl, T.; Manetto, A. One-Step Formation of “Chain-Armor”-Stabilized DNA Nanostructures. Angew. Chem. Int. Ed. Engl. 2015, 54, 7795–7798. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chandrasekaran, A.R.; Li, Q.; Li, X.; Sha, R.; Seeman, N.C.; Mao, C. Post-Assembly Stabilization of Rationally Designed DNA Crystals. Angew. Chem. Int. Ed. 2015, 54, 9936–9939. [Google Scholar] [CrossRef] [PubMed]
- Rusling, D.A.; Chandrasekaran, A.R.; Ohayon, Y.P.; Brown, T.; Fox, K.R.; Sha, R.; Mao, C.; Seeman, N.C. Functionalizing Designer DNA Crystals with a Triple-Helical Veneer. Angew. Chem. Int. Ed. Engl. 2014, 53, 3979–3982. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.O.; Ohayon, Y.P.; Chandrasekaran, A.R.; Sha, R.; Fox, K.R.; Brown, T.; Rusling, D.A.; Mao, C.; Seeman, N.C. Stabilisation of self-assembled DNA crystals by triplex-directed photo-cross-linking. Chem. Commun. 2016, 52, 8014–8017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Paukstelis, P.J. Enhancing DNA Crystal Durability through Chemical Crosslinking. ChemBioChem 2016, 17, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Boerneke, M.A.; Dibrov, S.M.; Hermann, T. Crystal-Structure-Guided Design of Self-Assembling RNA Nanotriangles. Angew. Chem. Int. Ed. Engl. 2016, 55, 4097–4100. [Google Scholar] [CrossRef] [PubMed]
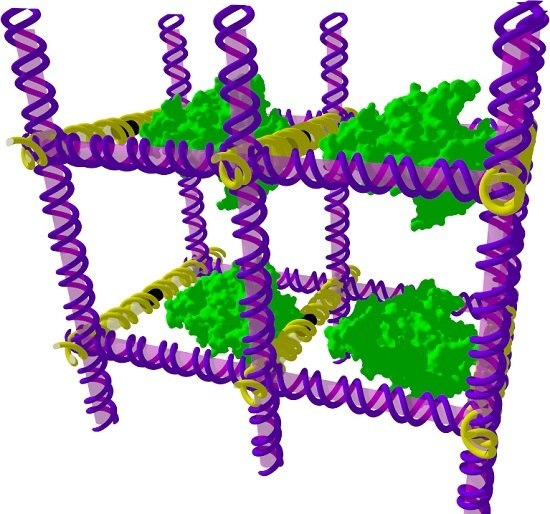
Figure 1.
A periodic DNA scaffold. This schematic imagines that DNA (purple/yellow) could be assembled into 3D periodic arrays, or crystals, to which guest molecules such as proteins (green) could be tethered. The guest molecule would then be part of the crystal lattice, allowing its molecular structure to be determined by X-ray diffraction. This idea sparked the field of DNA nanotechnology.
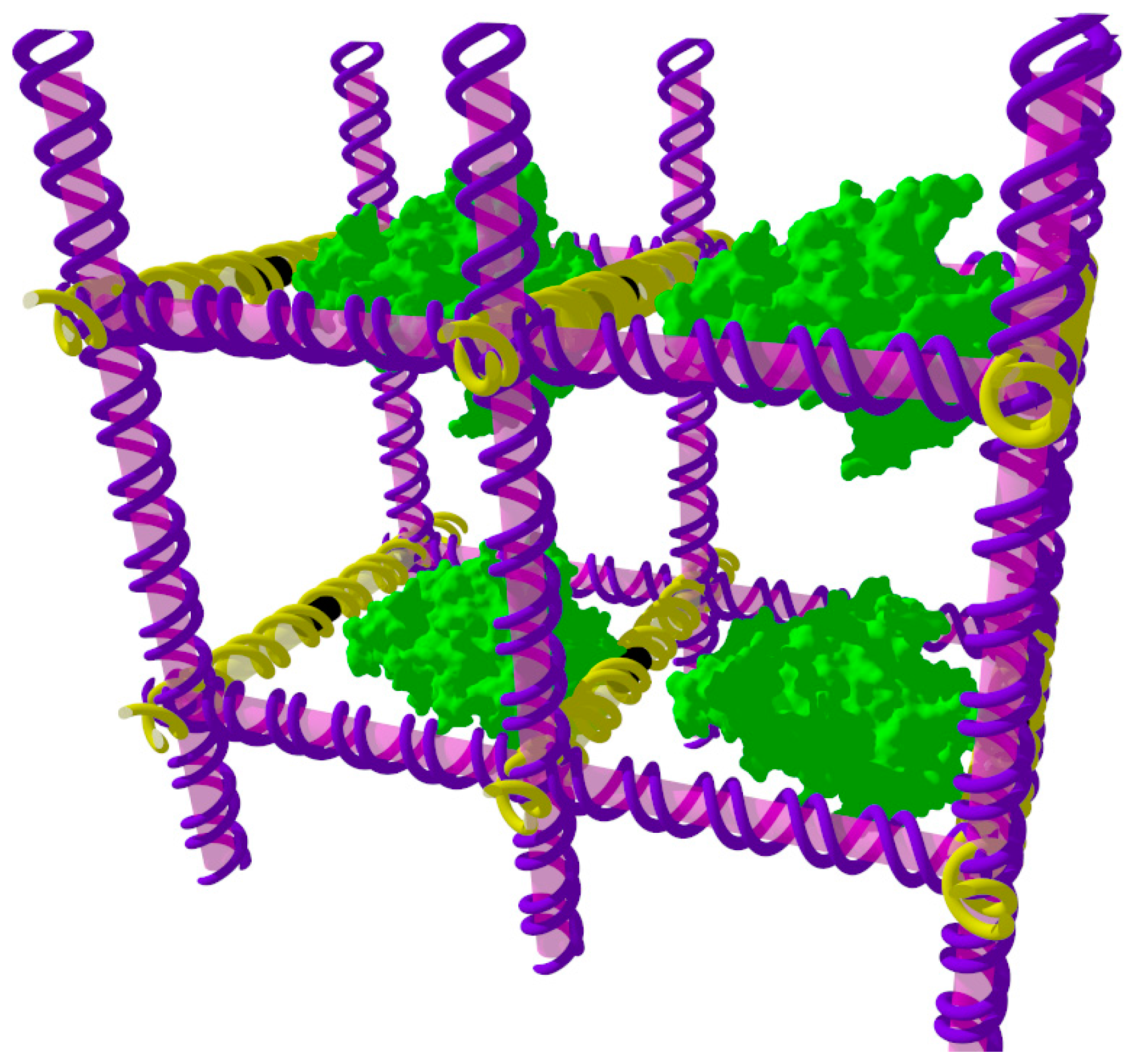
Figure 1.
A periodic DNA scaffold. This schematic imagines that DNA (purple/yellow) could be assembled into 3D periodic arrays, or crystals, to which guest molecules such as proteins (green) could be tethered. The guest molecule would then be part of the crystal lattice, allowing its molecular structure to be determined by X-ray diffraction. This idea sparked the field of DNA nanotechnology.

Figure 2.
The tensegrity triangle motif. (a) Cylindrical representation of a tensegrity triangle, highlighting the three independent linear directions of the cylinders; (b) One example of three-fold symmetrized DNA tensegrity triangle. The central strand is in black, the edge strands are in green, and the cross-over strands in blue. 2 nt sticky ends are outlined in red; (c) The single DNA tensegrity triangle motif described by the crystal structure (PDBID: 3GBI). Strands are colored as in (b) and yellow cylinders connect the helix ends to show the three independent linear directions that multiple motifs extend into a 3D lattice.
Figure 2.
The tensegrity triangle motif. (a) Cylindrical representation of a tensegrity triangle, highlighting the three independent linear directions of the cylinders; (b) One example of three-fold symmetrized DNA tensegrity triangle. The central strand is in black, the edge strands are in green, and the cross-over strands in blue. 2 nt sticky ends are outlined in red; (c) The single DNA tensegrity triangle motif described by the crystal structure (PDBID: 3GBI). Strands are colored as in (b) and yellow cylinders connect the helix ends to show the three independent linear directions that multiple motifs extend into a 3D lattice.

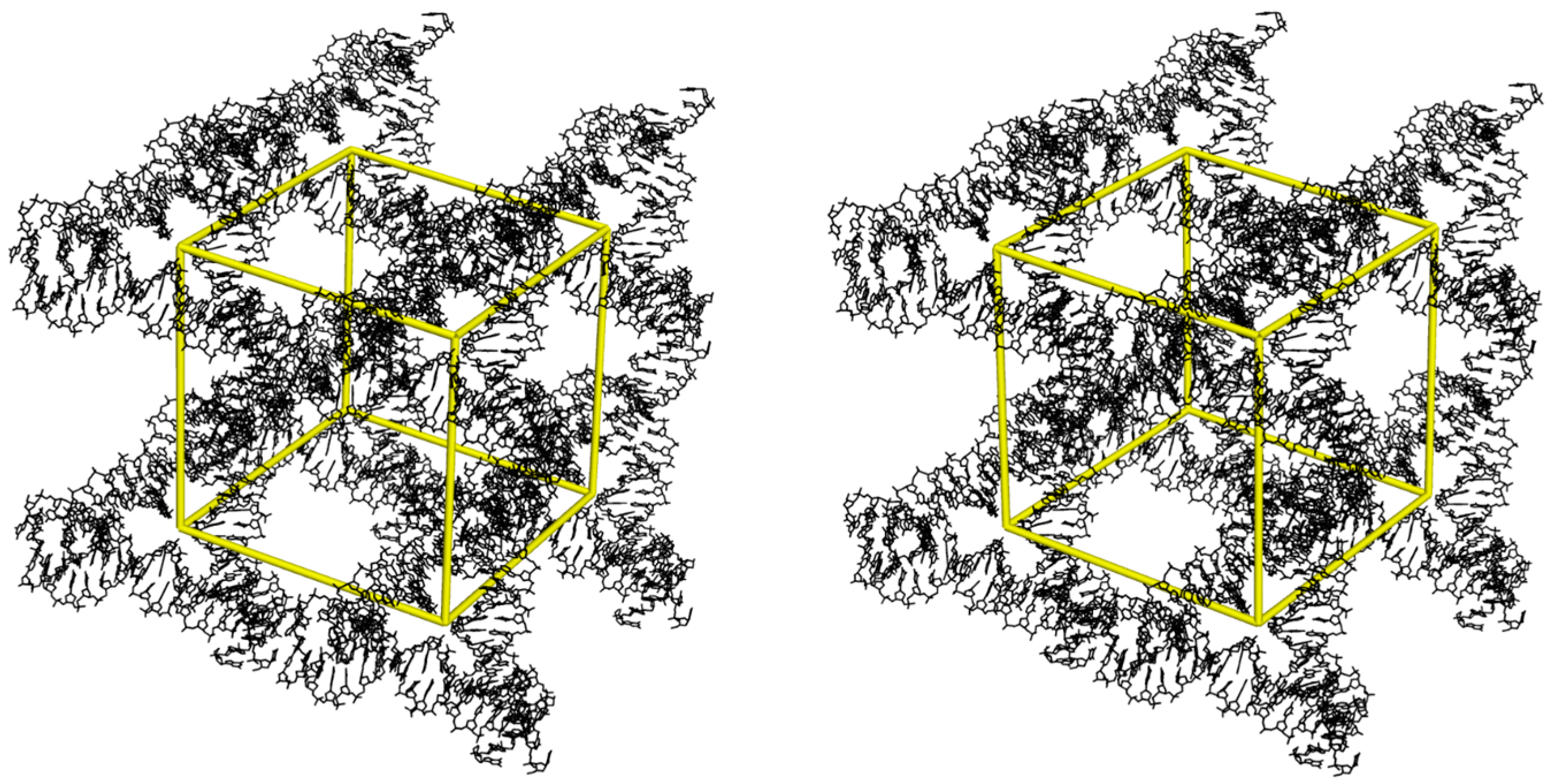
Figure 3.
Tensegrity crystal solvent pockets. A stereoscopic view of eight tensegrity triangle motifs in the crystal lattice that generate a rhombohedral solvent space (yellow outline).
Figure 3.
Tensegrity crystal solvent pockets. A stereoscopic view of eight tensegrity triangle motifs in the crystal lattice that generate a rhombohedral solvent space (yellow outline).

Figure 4.
Solvent channels. The tensegrity triangle crystals form multiple solvent channels that run through the crystal in multiple directions. (a) Solvent channels that run along each helical axis of the motif; (b,c) Two other channels that run through the crystal.
Figure 4.
Solvent channels. The tensegrity triangle crystals form multiple solvent channels that run through the crystal in multiple directions. (a) Solvent channels that run along each helical axis of the motif; (b,c) Two other channels that run through the crystal.

Figure 5.
The DNA 13-mer lattice with non-canonical base pairs. (a) Secondary structure of base pairing interactions. Four identical strands are colored differently. Dashes indicate Watson-Crick base pairs, and circles indicate non-canonical base pairs. G13 is disordered in the crystal structure and is not shown; (b) The three unique non-canonical base pairs of the homopurine junction. Black dashes indicate hydrogen bonding.
Figure 5.
The DNA 13-mer lattice with non-canonical base pairs. (a) Secondary structure of base pairing interactions. Four identical strands are colored differently. Dashes indicate Watson-Crick base pairs, and circles indicate non-canonical base pairs. G13 is disordered in the crystal structure and is not shown; (b) The three unique non-canonical base pairs of the homopurine junction. Black dashes indicate hydrogen bonding.

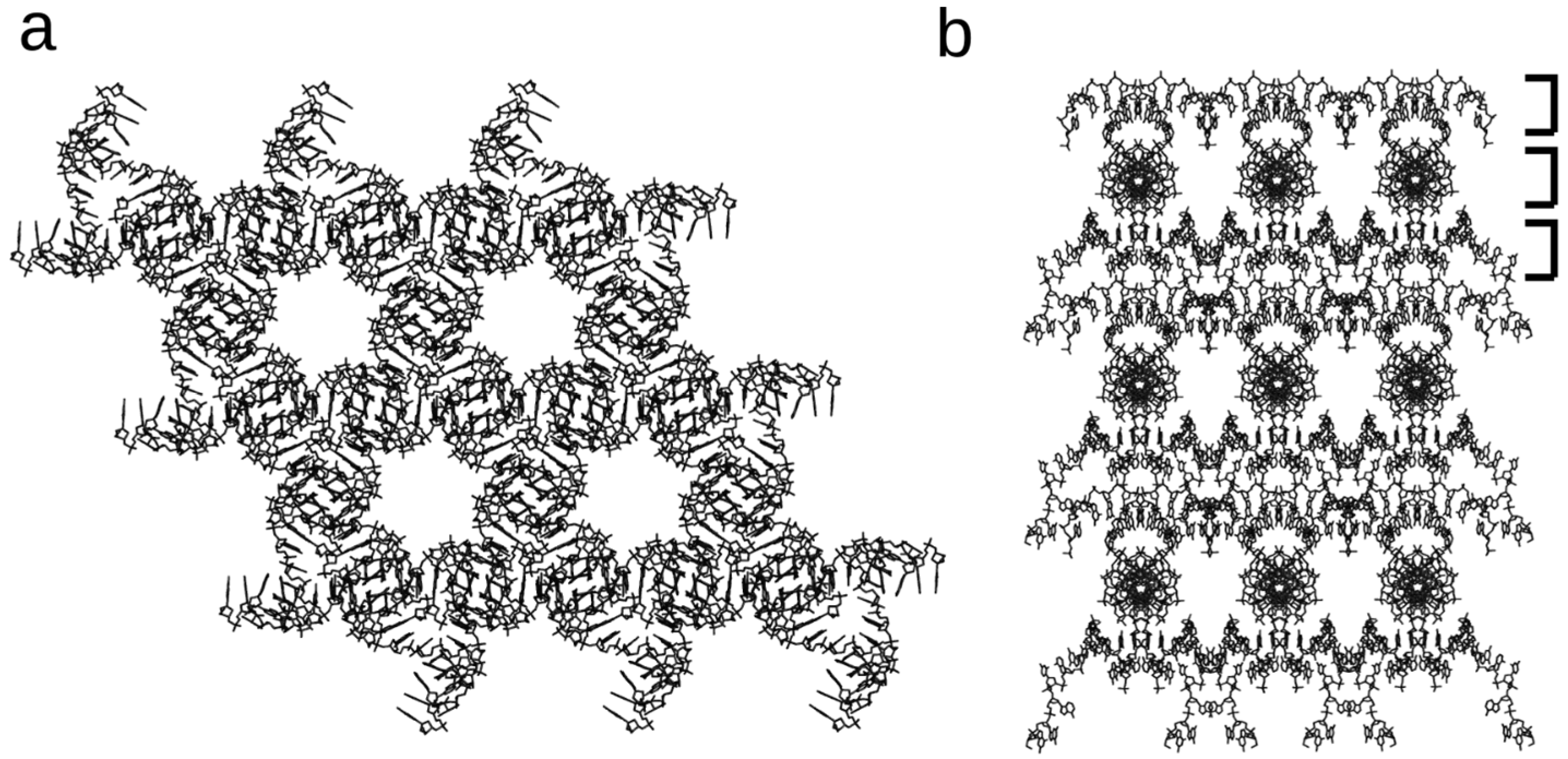
Figure 6.
Solvent channels of the 13-mer lattice. Solvent channels run through the 13-mer crystals in multiple directions. (a) Solvent channels running down the six-fold axis are ~2 nm in diameter; (b) Solvent channels run along each of the three helical layers (shown by brackets) that repeat down the six-fold axis.
Figure 6.
Solvent channels of the 13-mer lattice. Solvent channels run through the 13-mer crystals in multiple directions. (a) Solvent channels running down the six-fold axis are ~2 nm in diameter; (b) Solvent channels run along each of the three helical layers (shown by brackets) that repeat down the six-fold axis.

Figure 7.
The expanded non-canonical lattice. (a) Secondary structure of the expanded lattice. Green “spacer” strands are required to pair with the 10 or 11 nucleotide expansion; (b,c) Models of the expanded solvent channels parallel and perpendicular to the six-fold symmetry axis, respectively. AFM images confirmed the presence of the solvent channels and unit cell dimensions, along with low resolution diffraction [65]. Scale bars are 20 nm.
Figure 7.
The expanded non-canonical lattice. (a) Secondary structure of the expanded lattice. Green “spacer” strands are required to pair with the 10 or 11 nucleotide expansion; (b,c) Models of the expanded solvent channels parallel and perpendicular to the six-fold symmetry axis, respectively. AFM images confirmed the presence of the solvent channels and unit cell dimensions, along with low resolution diffraction [65]. Scale bars are 20 nm.

Figure 8.
Crosslinking enhances DNA crystal thermostability. (a) Site-specific introduction of photo crosslinking groups to the tensegrity triangle crystals through triplex formation enhances crystal stability in a UV-dependent manner; (b) Comparison of 13-mer DNA crystals with and without nornitrogen mustard crosslinking following incubation at 33 °C at the specified times.
Figure 8.
Crosslinking enhances DNA crystal thermostability. (a) Site-specific introduction of photo crosslinking groups to the tensegrity triangle crystals through triplex formation enhances crystal stability in a UV-dependent manner; (b) Comparison of 13-mer DNA crystals with and without nornitrogen mustard crosslinking following incubation at 33 °C at the specified times.

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Paukstelis, P.J.; Seeman, N.C. 3D DNA Crystals and Nanotechnology. Crystals 2016, 6, 97. https://doi.org/10.3390/cryst6080097
AMA Style
Paukstelis PJ, Seeman NC. 3D DNA Crystals and Nanotechnology. Crystals. 2016; 6(8):97. https://doi.org/10.3390/cryst6080097
Chicago/Turabian StylePaukstelis, Paul J., and Nadrian C. Seeman. 2016. "3D DNA Crystals and Nanotechnology" Crystals 6, no. 8: 97. https://doi.org/10.3390/cryst6080097
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.