
Reversible Single-Crystal-to-Single-Crystal Structural Transformation in a Mixed-Ligand 2D Layered Metal-Organic Framework: Structural Characterization and Sorption Study


Abstract
:1. Introduction
2. Results and Discussion
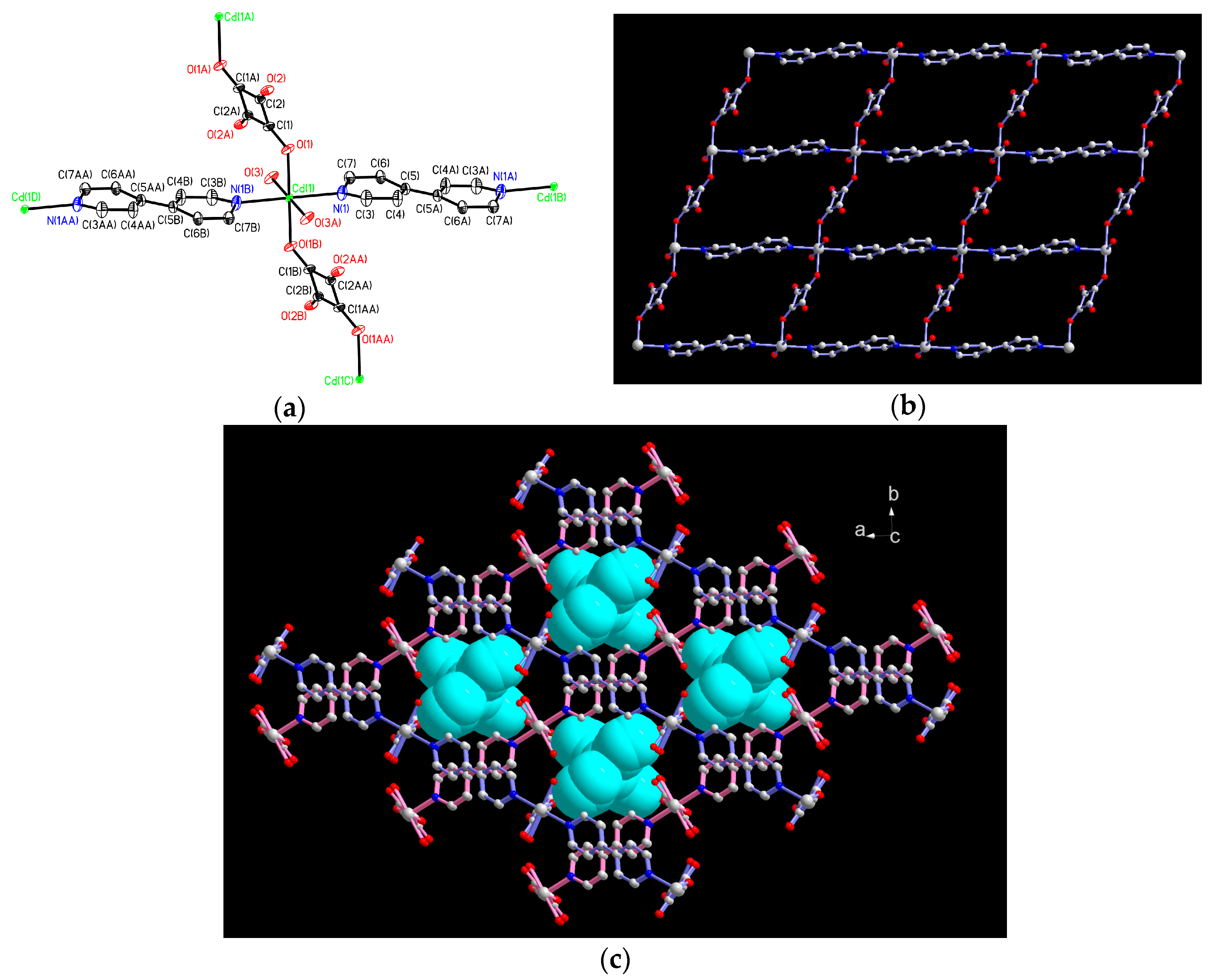
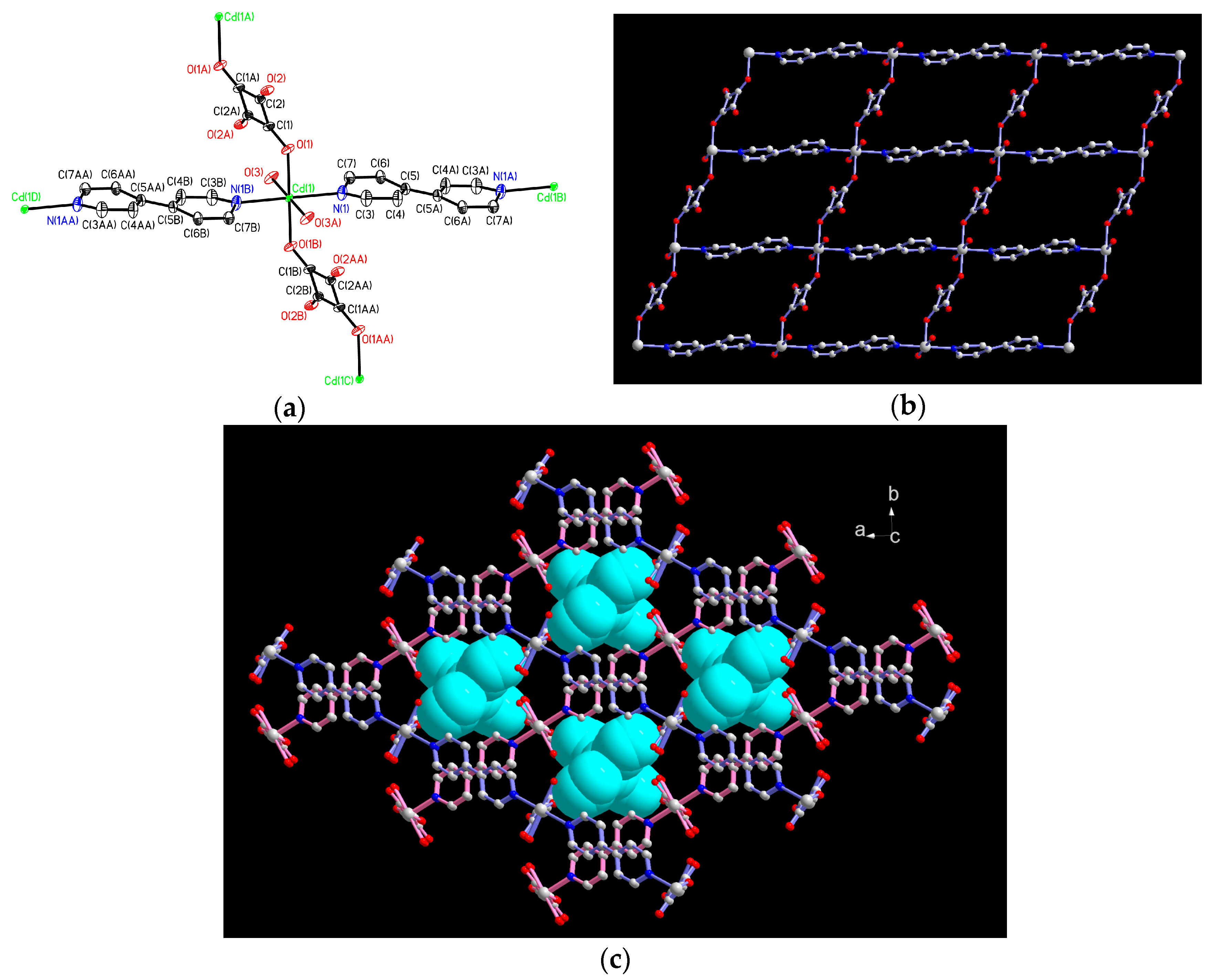
2.1. Structural Description of 1
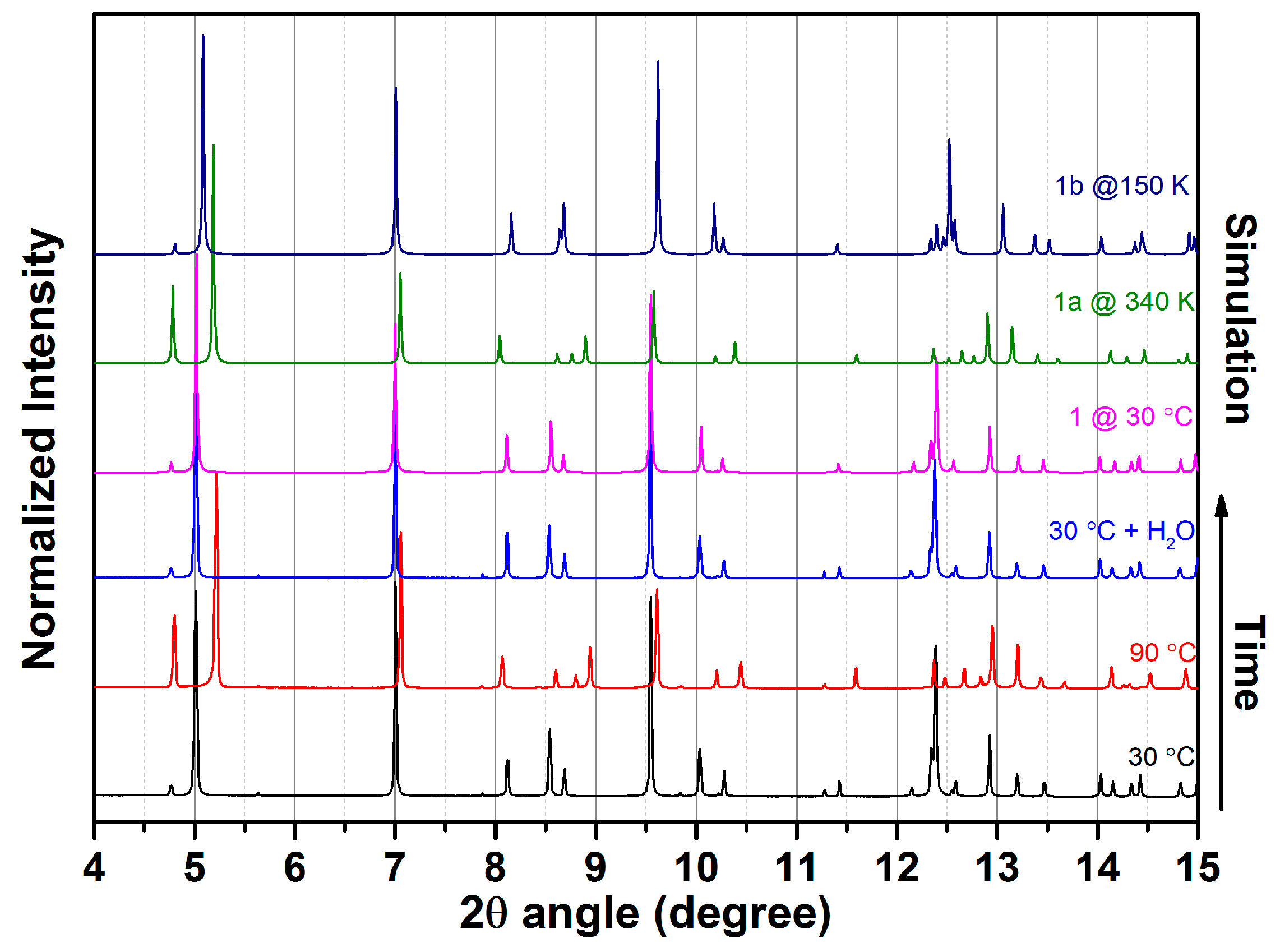
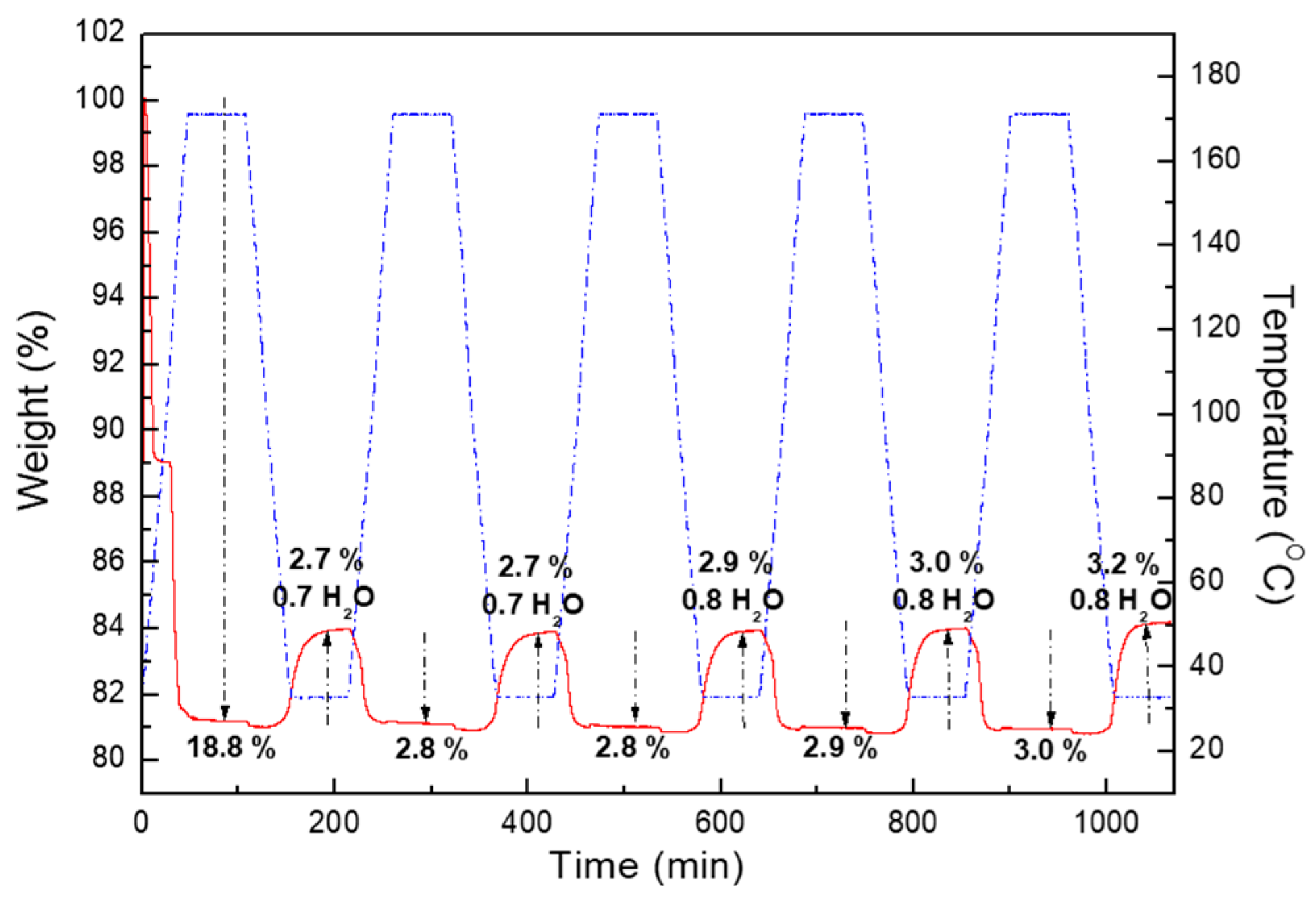
2.2. Water Adsorption Property of (1) by Cyclic TG Aanalsis and PXRD Measurements
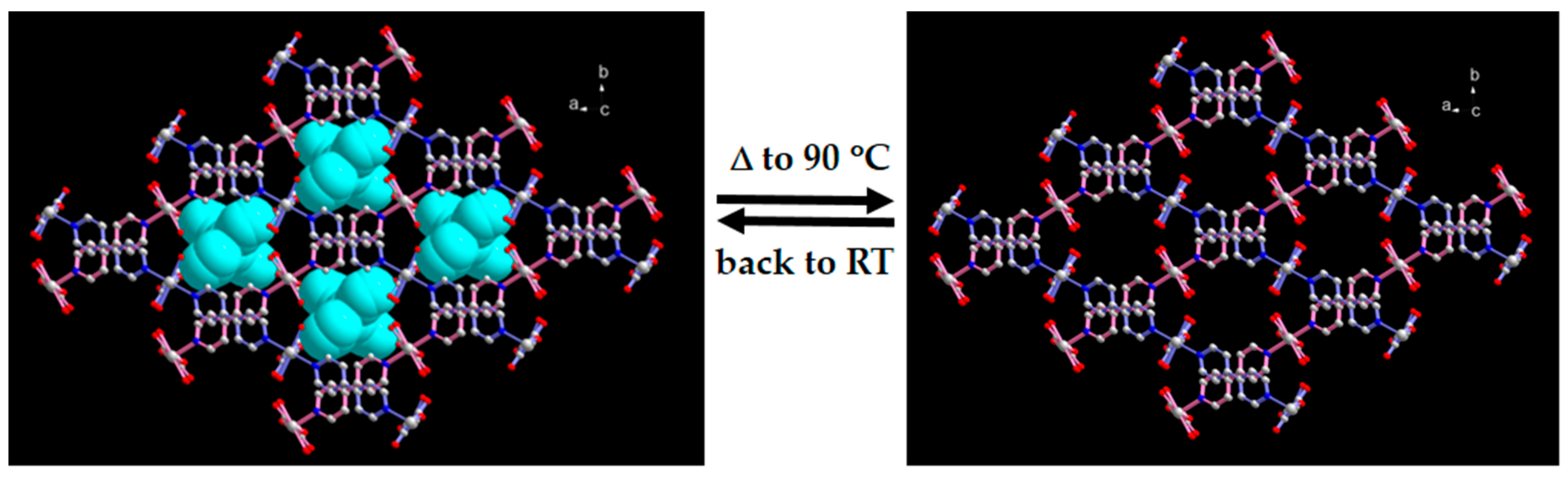
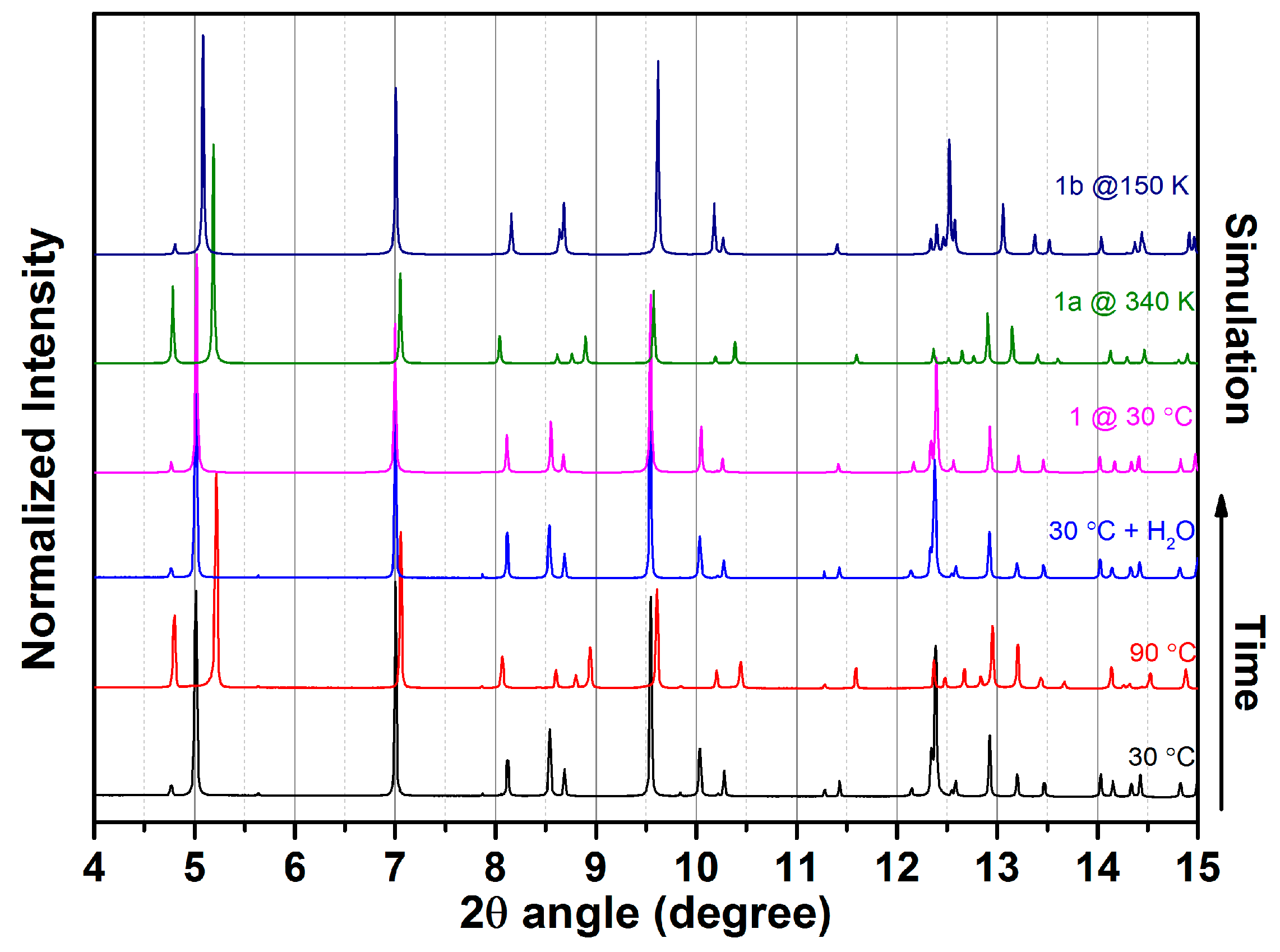
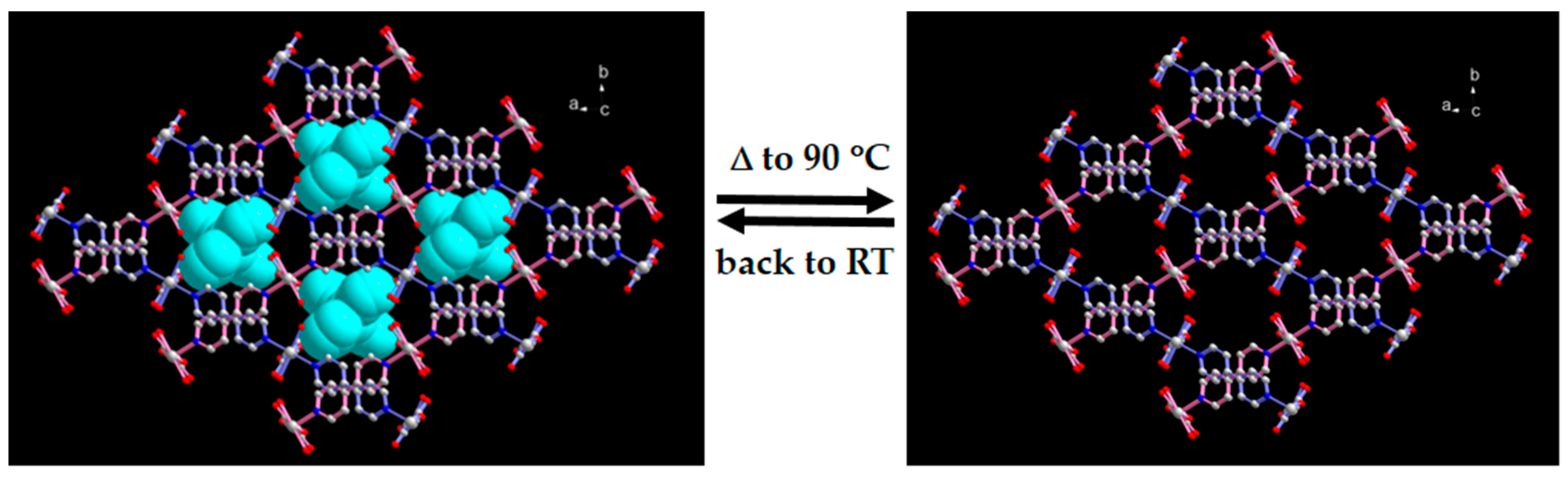
2.3. SCSC Transformation Associated with Guest Water De-/Rehydration
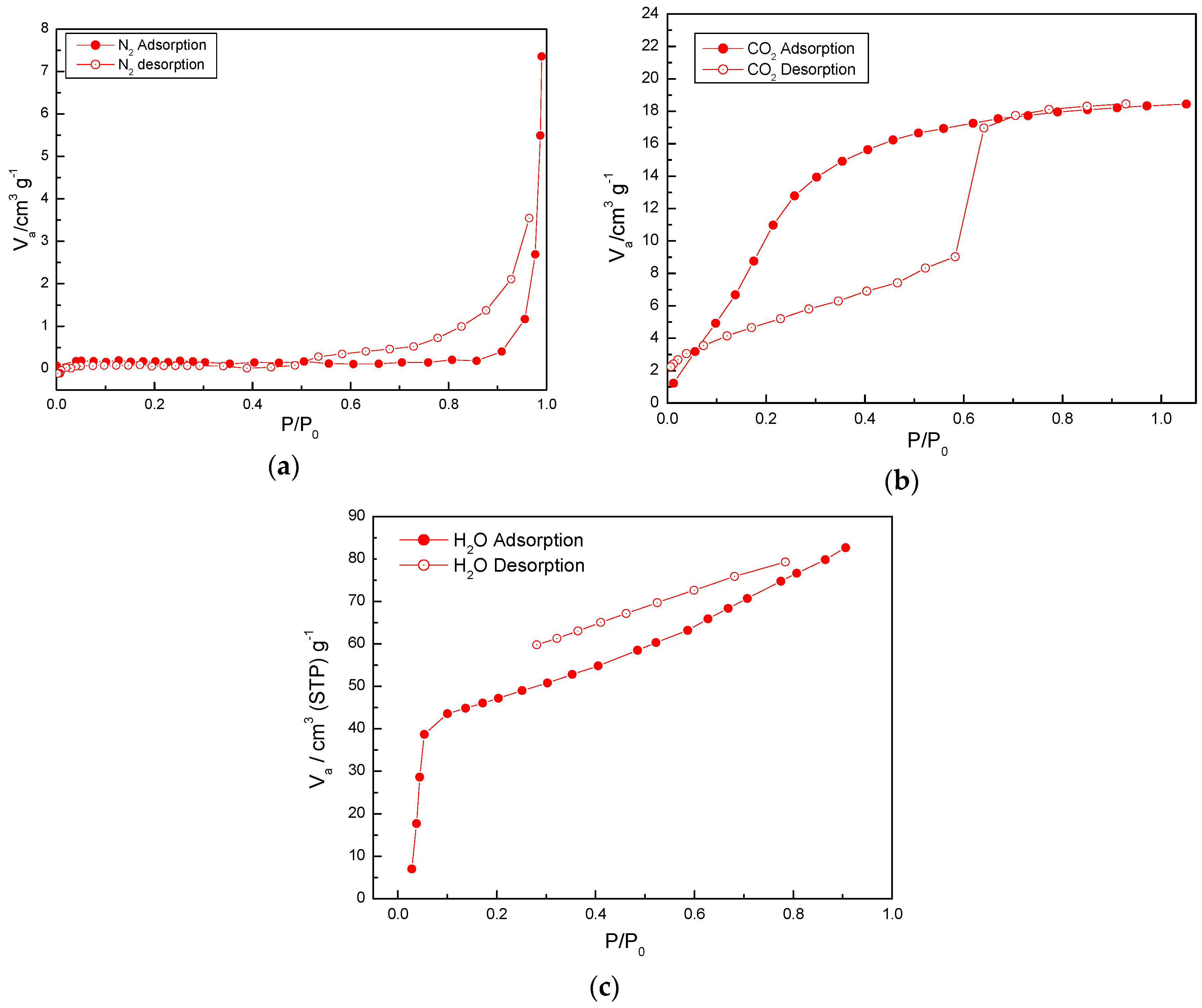
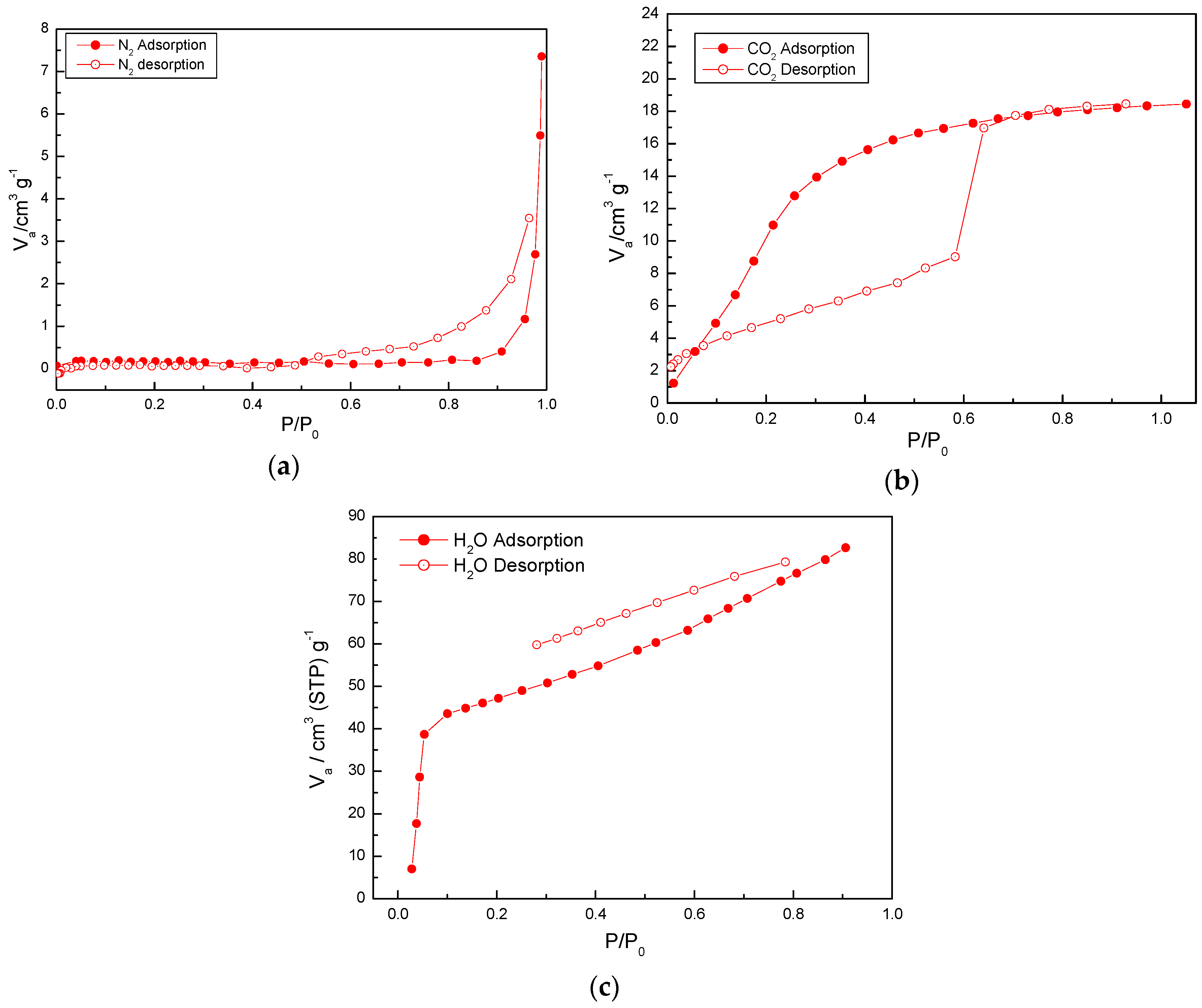
2.4. Adsorption Properties of (1)
3. Materials and Methods
3.1. Materials and Physical Techniques
3.2. Synthesis of {[Cd(C4O4)(bipy)(H2O)2]·3H2O}∞ (1), Dehydrated {[Cd(C4O4)(bipy)(H2O)2]}∞ (1a) and Rehydrated {[Cd(C4O4)(bipy)(H2O)2]·3H2O}∞ (1b)
3.3. Crystallographic Data Collection and Refinements
3.4. In Situ X-ray Powder Diffraction
3.5. Measurements of Gas Adsorption of Activated 1a
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Batten, S.R.; Champness, N.R.; Chen, X.M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Terminology of metal-organic frameworks and coordination polymers. Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Coordination polymers, metal-organic frameworks and the need for terminology guidelines. CrystEngComm 2012, 14, 3001–3004. [Google Scholar] [CrossRef]
- Li, B.; Chrzanowski, M.; Zhang, Y.; Ma, S. Applications of metal-organic frameworks featuring multi-functional sites. Coord. Chem. Rev. 2016, 307, 106–129. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Hu, Z.; Wang, G.; Uvdal, K. Coordination polymers for energy transfer: Preparations, properties, sensing applications, and perspectives. Coord. Chem. Rev. 2015, 284, 206–235. [Google Scholar] [CrossRef]
- Bradshaw, D.; Claridge, J.B.; Cussen, E.J.; Prior, T.J.; Rosseinsky, M.J. Design, Chirality, and Flexibility in Nanoporous Molecule-Based Materials. Acc. Chem. Res. 2015, 38, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.; Vilela, S.M.F.; Tome, J.P.C.; Paz, F.A.A. Multifunctional metal–organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Huo, F. Metal-organic framework composites: From fundamentals to applications. Nanoscale 2015, 7, 7482–7501. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C.; Vieth, J.K. MOFs, MILs and more: Concepts, properties and applications for porous coordination networks (PCNs). New J. Chem. 2010, 34, 2366–2388. [Google Scholar] [CrossRef]
- Serre, C.; Millange, F.; Thouvenot, C.; Nogues, M.; Marsolier, G.; Louër, D.; Forey, G. Very Large Breathing Effect in the First Nanoporous Chromium(III)-Based Slids: MIL-53 or CrIII(OH)·{O2C–C6H4–CO2}·{HO2C–C6H4–CO2H}x·H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, R.; Seki, K.; Akiyama, G.; Kitagawa, S. Porous coordination-polymer crystals with gated channels specific for supercritical gases. Angew. Chem. Int. Ed. 2003, 42, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, T.; Serre, C.; Huguenard, C.; Fink, G.; Taulelle, F.; Henry, M.; Bataille, T.; Forey, G. A rationale for the large breathing of the porous aluminum terephthalate (MIL-53) upon hydration. Chem. Eur. J. 2004, 10, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Yagishita, S.; Tanaka, H.; Tohyama, K.; Adachi, K.; Kaizaki, S.; Kumagai, H.; Inoue, K.; Kitaura, R.; Chang, H.C.; et al. Metal-Complex Assemblies Constructed from the Flexible Hinge-Like Ligand H2bhnq: Structural Versatility and Dynamic Behavior in the Solid State. Chem. Eur. J. 2004, 10, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Nakata, E.; Saito, T.; Kojima, K. Structural determination of physisorbed sites for CO2 and Ar gases inside an organometallic framework. CrystEngComm 2003, 5, 411–413. [Google Scholar] [CrossRef]
- Takamizawa, S.; Nakata, E.; Yokoyama, H.; Mochizuki, K.; Mori, W. Carbon dioxide inclusion phases of a transformable 1D coordination polymer host [Rh2(O2CPh)4(pyz)]n. Angew. Chem. Int. Ed. 2003, 42, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Nakata, E.; Saito, T. Gas inclusion crystal between 1-D coordination polymer and nitrous oxide: Occurrence of crystal phase transition induced by a slight amount of fluid guests inside host. Inorg. Chem. Commun. 2003, 6, 1415–1418. [Google Scholar] [CrossRef]
- Takamizawa, S.; Nakata, E.; Saito, T. Single-crystal adsorbents: A new observation field for light aggregates. Angew. Chem. Int. Ed. 2004, 43, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Nakata, E.; Saito, T. Structural determination of copper(II) benzoate–pyrazine containing carbon dioxide molecules. Inorg. Chem. Commun. 2004, 7, 1–3. [Google Scholar] [CrossRef]
- Takamizawa, S.; Nakata, E.; Saito, T. Saito, Inclusion Formation between 1D Coordination Polymer Host and CS2 through Vapor Adsorption. Chem. Lett. 2004, 33, 538–539. [Google Scholar] [CrossRef]
- Takamizawa, S.; Nakata, E. Direct observation of H2 adsorbed state within a porous crystal by single crystal X-ray diffraction analysis. CrystEngComm 2005, 7, 476–479. [Google Scholar] [CrossRef]
- Takamizawa, S.; Nakata, E.; Saito, T.; Akatsuka, T. Generation of a Plastic Crystal Including Methane Rotator within Metal-Organic Cavity by Forcible Gas Adsorption. Inorg. Chem. 2005, 44, 1362–1366. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Nakata, E.; Akatsuka, T. Magnetic Behavior of a 1D Molecular-Oxygen System Included within a Transformable Single-Crystal Adsorbent. Angew. Chem. Int. Ed. 2006, 45, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Kobayashi, Y.; Ozeki, T.; Fujita, M. Direct Crystallographic Observation of a Coordinatively Unsaturated Transition-Metal Complex in situ Generated within a Self-Assembled Cage. J. Am. Chem. Soc. 2006, 128, 6558–6559. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Tamura, M.; Fujita, M. Diels-alder in aqueous molecular hosts: Unusual regioselectivity and efficient catalysis. Science 2006, 312, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Kachi-Terajima, C.; Akatsuka, T.; Kohbara, M.; Takamizawa, S. Structural and Magnetic Study of N2, NO, NO2, and SO2 Adsorbed within a Flexible Single-Crystal Adsorbent of [Rh2(bza)4(pyz)]n. Chem. Asian J. 2007, 2, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Kachi-Terajima, C.; Akatsuka, T.; Kohbara, M.; Jin, T. Alcohol-Vapor Inclusion in Single-Crystal Adsorbents [MII2(bza)4(pyz)]n (M = Rh, Cu): Structural Study and Application to Separation Membranes. Chem. Asian J. 2007, 2, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Akatsuka, T.; Ueda, T. Gas-Conforming Transformability of an Ionic Single-Crystal Host Consisting of Discrete Charged Components. Angew. Chem. Int. Ed. 2008, 47, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Kohbara, M.; Akatsuka, T.; Miyake, R. Gas-adsorbing ability of tris-ethylenediamine metal complexes (M = Co(III), Cr(III), Rh(III), Ir(III)) as transformable ionic single crystal hosts. New J. Chem. 2008, 32, 1782–1787. [Google Scholar] [CrossRef]
- Pan, Q.H.; Li, J.Y.; Chen, Q.; Han, Y.D.; Chang, Z.; Song, W.C.; Bu, X.H. [Co(en)3]1/3[In(ox)2]·3.5H2O: A zeolitic metal-organic framework templated by Co(en)3Cl3. Microporous Mesoporous Mater. 2010, 132, 453–457. [Google Scholar] [CrossRef]
- Pan, Q.H.; Chen, Q.; Song, W.C.; Hu, T.L.; Bu, X.H. Template-directed synthesis of three new open-framework metal(II) oxalates using Co(III) complex as template. CrystEngComm 2010, 12, 4198–4204. [Google Scholar] [CrossRef]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen bonding in metal-organic-supramolecular networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Ingleson, M.J.; Bacsa, J.; Rosseninsky, M.J. Homochiral H-bonded proline based metal organic frameworks. Chem. Commun. 2007, 3036–3038. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Yang, C.C.; Yeh, C.T.; Ceng, K.Y.; Chang, P.C.; Ho, M.L.; Lee, G.H.; Shih, W.J.; Sheu, H.S. Reversible Solid-State Structural Transformation of a 1D–2D Coordination Polymer by Thermal De/Rehydration Processes. Inorg. Chem. 2011, 50, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. C–H···O and other weak hydrogen bonds. From crystal engineering to virtual screening. Chem. Commun. 2005, 2995–3001. [Google Scholar] [CrossRef] [PubMed]
- García-Báez, E.V.; Martínez-Martínez, F.J.; Höpfl, H.; Padilla-Martínez, I.I. π-Stacking Interactions and CH···X (X = O, Aryl) Hydrogen Bonding as Directing Features of the Supramolecular Self-Association in 3-Carboxy and 3-Amido Coumarin Derivatives. Cryst. Growth Des. 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Jabiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Claessens, C.G.; Stoddart, J.F. π–π Interaction in self-assembly. J. Phys. Org. Chem. 1997, 10, 254–272. [Google Scholar] [CrossRef]
- Guo, H.; Guo, X.; Batten, S.R.; Song, J.; Song, S.; Dang, S.; Zhang, G.; Tang, J.; Zhang, H. Hydrothermal Synthesis, Structures, and Luminescent Properties of Seven d10 Metal-Organic Frameworks Based on 9,9-Dipropylfluorene-2,7-Dicarboxylic Acid (H2DFDA). Cryst. Growth Des. 2009, 9, 1394–1401. [Google Scholar] [CrossRef]
- Reger, D.L.; Horger, J.J.; Smith, M.D.; Long, G.J.; Grandjean, F. Homochiral, Helical Supramolecular Metal-Organic Frameworks Organized by Strong π···π Stacking Interactions: Single-Crystal to Single-Crystal Transformations in Closely Packed Solids. Inorg. Chem. 2011, 50, 686–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Yang, C.C.; Chung, W.C.; Lee, G.H.; Ho, M.L.; Yu, Y.C.; Chung, M.W.; Sheu, H.S.; Shih, C.H.; Cheng, K.Y.; et al. A New Coordination Polymer Exhibiting Unique 2D Hydrogen-Bonded (H2O)16 Ring Formation and Water-Dependent Luminescence Properties. Chem. Eur. J. 2011, 17, 9232–9241. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, A.; Greenwood, A.R.; Filatov, A.S.; Gallagher, A.T.; Galli, G.; Anderson, J.S. Incorporation of Pyrazine and Bipyridine Linkers with High-Spin Fe(II) and Co(II) in a Metal-Organic Framework. Inorg. Chem. 2017, 56, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Satomi, T.; Azuma, K.; Takeda, R.; Maeda, K. New layered copper 1,3,5-benzenetriphosphonates pillared with N-donor ligands: Their synthesis, crystal structures, and adsorption properties. Dalton Trans. 2015, 44, 12717–12725. [Google Scholar] [CrossRef] [PubMed]
- Botezat, O.; van Leusen, J.; Kravtsov, V.C.; Filippova, I.G.; Hauser, J.; Speldrich, M.; Hermann, R.P.; Kramer, K.W.; Liu, S.X.; Decurtins, S. Interpenetrated (8,3)-c and (10,3)-b Metal-Organic Frameworks Based on {Fe-3(III)} and ((Fe2CoII)-Co-III} Pivalate Spin Clusters. Cryst. Growth Des. 2014, 14, 4721–4728. [Google Scholar] [CrossRef]
- Wang, F.; Jing, X.M.; Zheng, B.; Li, G.H.; Zeng, G.; Huo, Q.S.; Liu, Y.L. Four Cd-Based Metal-Organic Frameworks with Structural Varieties Derived from the Replacement of Organic Linkers. Cryst. Growth Des. 2013, 13, 3522–3527. [Google Scholar] [CrossRef]
- Thuery, P. Sulfonate Complexes of Actinide Ions: Structural Diversity in Uranyl Complexes with 2-Sulfobenzoate. Inorg. Chem. 2013, 52, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.Y.; Liu, S.X.; Xie, L.H.; Sun, C.Y.; Cao, J.F.; Ren, Y.H.; Feng, D.; Su, Z.M. Rational design microporous pillared-layer frameworks: Syntheses, structures and gas sorption properties. CrystEngComm 2009, 11, 177–182. [Google Scholar] [CrossRef]
- Mao, H.Y.; Zhang, C.H.; Li, G.; Zhang, H.Y.; Hou, H.W.; Li, L.K.; Wu, Q.G.; Zhu, Y.; Wang, E.B. New types of the flexible self-assembled Metal-Organic coordination polymers constructed by aliphatic dicarboxylates and rigid bidentate nitrogen ligands. Dalton Trans. 2004, 22, 3918–3925. [Google Scholar] [CrossRef] [PubMed]
- Batten, S.R.; Murray, K.S. Structure and magnetism of coordination polymers containing dicyanamide and tricyanomethanide. Coord. Chem. Rev. 2003, 246, 103–130. [Google Scholar] [CrossRef]
- Tong, M.L.; Ye, B.H.; Cai, J.W.; Chen, X.M.; Ng, S.W. Clathration of Two-Dimensional Coordination Polymers: Synthesis and Structures of [M(4,4′-bpy)2(H2O)2](ClO4)2·(2,4′-bpy)2·H2O and [Cu(4,4′-bpy)2(H2O)2](ClO4)4·(4,4′-H2Bpy) (M = CdII, ZnII and bpy = Bipyridine). Inorg. Chem. 1998, 37, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Näther, C.; Greve, J.; Jeβ, I. New Coordination Polymer Changing Its Color upon Reversible Deintercalation and Reintercalation of Water: Synthesis, Structure, and Properties of Poly[Diaqua-(μ2-Squarato-O,O′)-(μ2–4,4′-Bipyridine-N,N′)- Manganese(II)] Trihydrate. Chem. Mater. 2002, 14, 4536–4542. [Google Scholar] [CrossRef]
- Greve, J.; Jeβ, I.; Näther, C.J. Synthesis, crystal structures and investigations on the dehydration reaction of the new coordination polymers poly[diaqua-(μ2-squarato-O,O′)-(μ2–4,4′-bipyridine-N,N′)Me(II)] hydrate (Me = Co, Ni, Fe). Solid State Chem. 2003, 175, 328–340. [Google Scholar] [CrossRef]
- Konar, S.; Corbella, M.; Zangrando, E.; Ribas, J.N.; Chaudhuri, R. The first unequivocally ferromagnetically coupled squarato complex: Origin of the ferromagnetism in an interlocked 3D Fe(II) system. Chem. Commun. 2003, 1424–1425. [Google Scholar] [CrossRef]
- Wang, C.C.; Yang, C.H.; Tseng, S.M.; Lee, G.H.; Sheu, H.S.; Phyu, K.W. A New Moisture-Sensitive Metal-Coordination Solids, {[Cd(C4O4)(bipy)(H2O)2]·3H2O}∞ (bipy = 4,4’-bipyridine). Inorg. Chim. Acta 2004, 357, 3759–3764. [Google Scholar]
- Ito, M.; West, R. New Aromatic Anions. IV. Vibrational Spectra and Force Constants for C4O4−2 and C5O5−2. J. Am. Chem. Soc. 1963, 85, 2580–2584. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Serezhkin, V.N. TOPOS3.2: A new version of the program package for multipurpose crystal-chemical analysis. J. Appl. Crystallogr. 2000, 33, 1193. [Google Scholar] [CrossRef]
- Blatov, V.A.; Carlucci, L.; Ciani, G.; Proserpio, D.M. Interpenetrating metal–organic and inorganic 3D networks: A computer-aided systematic investigation. Part I. Analysis of the Cambridge structural database. CrystEngComm 2004, 6, 377–395. [Google Scholar] [CrossRef]
- Smart, V. 4.043 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, USA, 1995. [Google Scholar]
- Saint, V. 4.035 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, USA, 1995. [Google Scholar]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- Sheldrick, G.M. SHELXTL 5.03 (PC-Version), Program Liberary for Structure Solution and Molecular Graphics; Siemens Analytical Instruments Division: Madison, WI, USA, 1995. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 1a | 1b |
|---|---|---|---|
| Cd(1)–O(3) | 2.264(3) | 2.303(3) | 2.265(2) |
| Cd(1)–O(3)i | 2.264(3) | 2.303(3) | 2.265(2) |
| Cd(1)–O(1) | 2.294(3) | 2.273(2) | 2.296(2) |
| Cd(1)–O(1)i | 2.294(3) | 2.273(2) | 2.296(2) |
| Cd(1)–N(1) | 2.341(4) | 2.339(3) | 2.340(2) |
| Cd(1)–N(1)i | 2.341(4) | 2.339(3) | 2.340(2) |
| O(3)–Cd(1)–O(3)i | 180 | 180.0 | 180.0 |
| O(3)i–Cd(1)–O(1)i | 92.77(12) | 93.61(11) | 92.76(7) |
| O(3)–Cd(1)–O(1)i | 87.23(12) | 86.39(11) | 87.24(7) |
| O(3)i–Cd(1)–O(1) | 87.23(12) | 86.39(11) | 87.24(7) |
| O(3)–Cd(1)–O(1) | 92.77(12) | 93.61(11) | 92.76(7) |
| O(1)–Cd(1)–O(1)i | 180 | 180.0 | 180.0 |
| O(3)i–Cd(1)–N(1) | 87.82(13) | 91.88(11) | 87.93(8) |
| O(3)–Cd(1)–N(1) | 92.18(13) | 88.12(11) | 92.07(8) |
| O(1)i–Cd(1)–N(1) | 85.78(14) | 86.46(11) | 85.60(8) |
| O(1)–Cd(1)–N(1) | 94.22(14) | 93.54(11) | 94.40(8) |
| O(3)i–Cd(1)–N(1)i | 92.18(13) | 88.12(11) | 92.07(8) |
| O(3)–Cd(1)–N(1)i | 87.82(13) | 91.88(11) | 87.93(8) |
| O(1)i–Cd(1)–N(1)i | 94.22(14) | 93.54(11) | 94.40(8) |
| O(1)–Cd(1)–N(1)i | 85.78(14) | 86.46(11) | 85.60(8) |
| N(1)–Cd(1)–N(1)i | 180.0 | 180.0 | 180.0 |
| Compound 1 | ||||
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | ∠ D–H⋯A (°) |
| O(3)–H(3A)⋯O(2)i | 0.81(7) | 1.88(7) | 2.664(5) | 162(6) |
| O(3)–H(3B)⋯O(2)ii | 0.87(7) | 1.85(7) | 2.698(5) | 163(6) |
| O(4)–H(4A)⋯O(5) | 0.83(5) | 1.96(5) | 2.762(5) | 162(6) |
| O(5)–H(5A)⋯O(1) | 0.71(8) | 2.11(8) | 2.807(5) | 171(8) |
| O(5)–H(5B)⋯O(4)iii | 0.79(7) | 2.00(7) | 2.782(5) | 168(7) |
| Compound 1b | ||||
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | ∠ D–H⋯A (°) |
| O(3)–H(3A)⋯O(2)i | 0.79(4) | 1.89(4) | 2.663(3) | 166(4) |
| O(3)–H(3B)⋯O(2)ii | 0.81(4) | 1.91(4) | 2.698(3) | 165(3) |
| O(4)–H(4A)⋯O(5) | 0.82(3) | 1.95(3) | 2.766(3) | 169(4) |
| O(5)–H(5A)⋯O(1) | 0.81(4) | 2.01(4) | 2.808(3) | 171(4) |
| O(5)–H(5B)⋯O(4)iii | 0.76(4) | 2.04(4) | 2.783(3) | 168(4) |
| Compound | 1 | 1a | 1b |
|---|---|---|---|
| empirical formula | C14H18Cd1N2O9 | C14H12Cd1N2O6 | C14H18Cd1N2O9 |
| formula mass (g mol−1) | 470.70 | 416.66 | 470.70 |
| crystal system | Monoclinic | Monoclinic | Monoclinic |
| space group | C 2/c | C 2/c | C 2/c |
| a (Å) | 20.3421(8) | 19.7226(10) | 20.3589(12) |
| b (Å) | 11.6164(5) | 11.7865(5) | 11.6195(6) |
| c (Å) | 8.3198(3) | 8.1877(3) | 8.3196(4) |
| α (deg) | 90 | 90 | 90 |
| β (deg) | 113.7456(15) | 112.1639(15) | 113.7461(18) |
| γ (deg) | 90 | 90 | 90 |
| V (Å3) | 1799.55(12) | 1762.68(13) | 1801.47(17) |
| Z | 4 | 4 | 4 |
| T (K) | 150(2) | 340(2) | 150(2) |
| Dcalcd (g cm−3) | 1.737 | 1.570 | 1.736 |
| μ (mm−1) | 1.263 | 1.267 | 1.262 |
| θ range (deg) | 2.066–27.474 | 2.230–27.484 | 3.011–27.491 |
| total no. of data collected | 5780 | 5760 | 6113 |
| no. of unique data | 2070 | 2028 | 2066 |
| no. of obsd data (I > 2σ(I)) | 1681 | 1594 | 1649 |
| Rint | 0.0334 | 0.0318 | 0.0274 |
| refine params | 145 | 114 | 145 |
| R1, wR2 (I > 2σ(I)) 1 | 0.0360, 0.0924 | 0.0389, 0.0733 | 0.0250, 0.0482 |
| R1, wR2 (all data) 1 | 0.0464, 0.1003 | 0.0551, 0.0800 | 0.0376, 0.0537 |
| GOF 2 | 1.085 | 1.191 | 1.095 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-C.; Ke, S.-Y.; Chen, K.-T.; Hsieh, Y.-F.; Wang, T.-H.; Lee, G.-H.; Chuang, Y.-C. Reversible Single-Crystal-to-Single-Crystal Structural Transformation in a Mixed-Ligand 2D Layered Metal-Organic Framework: Structural Characterization and Sorption Study. Crystals 2017, 7, 364. https://doi.org/10.3390/cryst7120364
Wang C-C, Ke S-Y, Chen K-T, Hsieh Y-F, Wang T-H, Lee G-H, Chuang Y-C. Reversible Single-Crystal-to-Single-Crystal Structural Transformation in a Mixed-Ligand 2D Layered Metal-Organic Framework: Structural Characterization and Sorption Study. Crystals. 2017; 7(12):364. https://doi.org/10.3390/cryst7120364
Chicago/Turabian StyleWang, Chih-Chieh, Szu-Yu Ke, Kuan-Ting Chen, Yi-Fang Hsieh, Tzu-Heng Wang, Gene-Hsiang Lee, and Yu-Chun Chuang. 2017. "Reversible Single-Crystal-to-Single-Crystal Structural Transformation in a Mixed-Ligand 2D Layered Metal-Organic Framework: Structural Characterization and Sorption Study" Crystals 7, no. 12: 364. https://doi.org/10.3390/cryst7120364