Theoretical Study on Electronic, Optical Properties and Hardness of Technetium Phosphides under High Pressure

1
The High Pressure Research Center of Science and Technology, Zhengzhou University of Light Industry, Zhengzhou 450002, China
2
Institute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, China
3
Luoyang Sunrui Sprcial Equipment CO., LTD, Luoyang 471003, China
*
Author to whom correspondence should be addressed.
Crystals 2017, 7(6), 176; https://doi.org/10.3390/cryst7060176
Submission received: 4 April 2017
/
Revised: 18 May 2017
/
Accepted: 15 June 2017
/
Published: 18 June 2017
(This article belongs to the Special Issue Crystal Indentation Hardness)
Abstract
:In this paper, the structural properties of technetium phosphides Tc3P and TcP4 are investigated by first principles at zero pressure and compared with the experimental values. In addition, the electronic properties of these two crystals in the pressure range of 0–40 GPa are investigated. Further, we discuss the change in the optical properties of technetium phosphides at high pressures. At the end of our study, we focus on the research of the hardness of TcP4 at different pressures by employing a semiempirical method, and the effect of pressure on the hardness is studied. Results show that the hardness of TcP4 increases with the increasing pressure, and the influence mechanism of pressure effect on the hardness of TcP4 is also discussed.
1. Introduction
Due to their excellent performance in cutting, abrasion, and drilling, exploring new types of ultrahard materials has become a subject of inherent interest in recent decades. Until now, the development of superhard materials has gone through stages of traditional superhard materials, second kinds of superhard materials, and novel superhard materials.
As the well-known hardest traditional ultrahard material, diamond is of great interest for application and basic research [1]. However, its limitations in cutting iron and other ferrous metals greatly limits its application [2]. The second types of ultrahard materials are compounds composed of light elements (B–C–N–O system) with strong and short covalent bonds. These compounds have made great progress in cutting iron and other ferrous metals, but the severe preparation conditions and high cost of these compounds limit their large-scale production and application. Novel superhard materials are transition metals combined with light elements—especially transition metal borides. These compounds have a high electron concentration due to the transition metals, and they contain short covalent bonds for the presence of boron atoms. The unique structural properties determine the high bulk moduli and hardness of transition-metal borides.
Transition-metal phosphides are a class of compounds similar to transition-metal borides, and have attracted much attention due to their wide applications in superconductivity, magnetocaloric behavior, catalytic activity, lithium intercalation capacity, and so on [3,4]. However, few investigations have been done on their hardness. Cadmium diphosphide is an important technical material as a wide-gap semiconductor. Due to its superior optical properties, CdP2 has a wide range of application prospects in the fabrication of solar cells [5]. In addition, its large thermo-optic coefficient leads to numerous applications in thermal sensors [6]. Further, its wide band gap and anisotropic electrical properties make CdP2 a promising material in electronic engineering [7]. In addition, many investigations [8,9,10] have also been done on the optical and electronic properties of α-ZnP2 to investigate its applications.
Neither CdP2 nor ZnP2 are ultrahard materials, but high pressure has an important effect on the hardness of materials. Studying the mechanism of the influence of high pressure on the hardness of materials is of great significance to improve their hardness characteristics. In our previous study, we have systematically explored the pressure effect on the elastic properties, mechanical stability, and hardness for transition-metal borides W2B5 [11]. Recently, we investigated the hardness properties of CdP2 at high pressure, and found that the hardness of CdP2 increases as the pressure is increased [12].
Technetium phosphides are a member of the transition-metal phosphides, and have seldom been studied. Using the X-ray diffraction method, Ruhl et al. [13] obtained the crystal structure of technetium phosphides for the first time in 1981. In their work, Ruhl et al. pointed out that Tc3P crystallizes in the tetragonal Mn3P (Fe3P type) structure, and the tetragonal Tc3P crystalizes in a unit cell with lattice parameters a = b = 9.568 ± 0.005 Å, c = 4.736 ± 0.003 Å, c/a = 0.4950 ± 0.0006, β = 90°, V = 433.6 ± 0.6 Å3, and the unit cell comprises eight structure units (32 atoms); meanwhile, TcP4 has an orthorhombic ReP4-type structure, and the orthorhombic cell of TcP4 crystalizes in a unit cell with lattice constants a = 6.238 ± 0.001 Å, b = 9.215 ± 0.003 Å, c = 10.837 ± 0.003 Å, V = 623.0 ± 0.1 Å3, with eight formula units (40 atoms) in the cell.
In this paper, we intend to systematically explore the structural, electronic, optical properties, and hardness of technetium phosphides on the basis of the former experimental results under high pressure. The purpose of our work is two-fold. First of all, it is to give a comprehensive and complementary investigation of the effect of pressure on the hardness of technetium phosphides. Second, it is to provide powerful guidelines for future experimental investigations; we hope that such an investigation might provide a way to explore novel ultrahard materials.
2. Computational Methods and Details
In this paper, by using the standard Kohn–Sham self-consistent density functional theory [14,15,16,17] based on SIESTA code, we study the structural, electronic, and elastic properties of two forms of technetium phosphides: Tc3P and TcP4 crystals. To get the calculated values, the conjugate gradient minimization method is used to relax the positions of atoms and lattice vectors of technetium phosphides at ambient and high pressures. In our MD simulation, the single-crystal diffractometer data obtained by Ruhl et al. are used as initial structure of calculations. The tetragonal Tc3P crystallizes in a unit cell with lattice parameters a = b = 9.568 ± 0.005 Å, c = 4.736 ± 0.003 Å, β = 90°, and the unit cell comprises eight structural units (32 atoms); while the orthorhombic crystallizes in a unit cell with lattice parameters a = 6.238 Å, b = 9.215 Å, and c = 10.837 Å, β = 90°, and the unit cell comprises eight structural units (40 atoms). For the exchange-correlation energy, the generalized gradient approximation (GGA) designed by Perdew, Burke, and Ernzerhof (PBE) [18,19,20] is adopted. Additionally, the local density approximation (LDA) [21] method is also used as auxiliary calculations. At the same time, we adopt norm-conserving pseudopotentials in the form of Kleinman and Bylander [22] using Troullier and Martins’ scheme to describe the valence electron interaction with the atomic core, and choose a split-valence double-ζ basis set plus polarization function (DZP) with an energy shift of 0.005 Ry as our atomic orbital basis set in all the computations.
The optical properties of a material can be described by the complex dielectric function as ε(ω) = ε1(ω) + iε2(ω). The imaginary part of the dielectric function ε2(ω) can be considered as detailing of the real transitions between the occupied and unoccupied states. The real part ε1(ω) can be obtained from the imaginary part by Kramers–Kronig relationship. In addition, other optical parameters (absorption coefficient, reflectance, refractive index, and energy loss spectroscopy) can be obtained from ε1(ω) and ε2(ω). Here we focus on the absorption coefficient of TcP4. It can be derived from the following formula [23].
In this study, we employ the model proposed by Gao et al. [24] to calculate the theoretical Vickers hardness of TcP4 at different pressures. Generally, the Vickers hardness value of a material is defined as the ratio of F/A in an experiment, where F is the pressed force applied to the diamond penetrator on the measured material in kilograms-force and A is the impression area of the resulting indentation in square millimeters. In this theoretical model, the Vickers hardness can be calculated by the following three formulae:
where Hv is the hardness of the calculated material, is the hardness of μ-type bond in material, Nμ, Pμ, are the total bond number, the Mulliken overlap population, and the bond volume of μ-type bond in the cell. fm and dμ are the metallicity and the bond length of μ-type bond. Ω is the cell volume.
The metallicity fm can be obtained by Formula (5),
where nm is the number of electrons that can be excited at the ambient temperature, ne is the total number of the valence electrons in a unit cell, and DF is the electron density of states at the Fermi level.
3. Results and Discussions
3.1. Structure Properties
Combined with both GGA and LDA approximations, the calculated lattice parameters and cell volume of the crystal Tc3P and TcP4 at ambient pressure are obtained by the conjugate gradient (CG) minimization method. The theoretical results at ambient pressure are shown in Table 1 and are compared with the experimental ones. From Table 1, it is noted that for the tetragonal Tc3P crystals, the deviations of GGA and LDA results are within 3.7% and 2.6% for the lattice parameters and cell volume, respectively. For the orthorhombic TcP4 crystals, the error of the results obtained by GGA and LDA are within 4.1% and 1.0% for the lattice parameters and cell volume, respectively. So, the LDA results match the experimental values better than the GGA results, and the LDA method is a better approximation than GGA to study technetium phosphides at zero pressure.
3.2. Electronic Properties
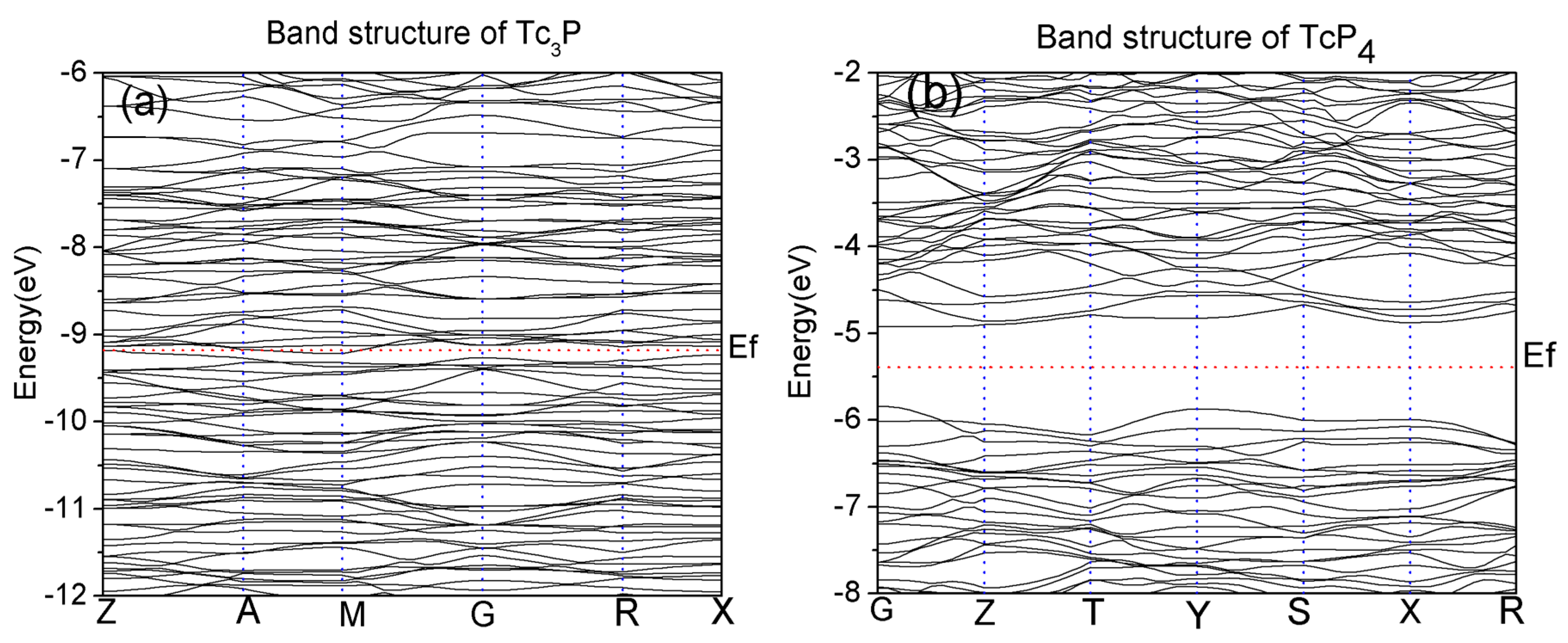
In this paper, the GGA approximation and LDA approximations are used to calculate the band structure of Tc3P and TcP4. The band structures calculated by these two methods are similar, so here we just present the GGA results in Figure 1 (only −12 to −6 eV for Tc3P, and −8 to −2 eV for TcP4). From Figure 1a, it can be seen that there are some points of intersection between the Fermi level and energy bands for the Tc3P crystal. Therefore, the Tc3P crystal can be considered as a conductor material. However, for the TcP4 crystal in Figure 1b, there is a direct band gap of 0.91 eV at the high symmetry point G. So, the TcP4 is a semiconductor material.
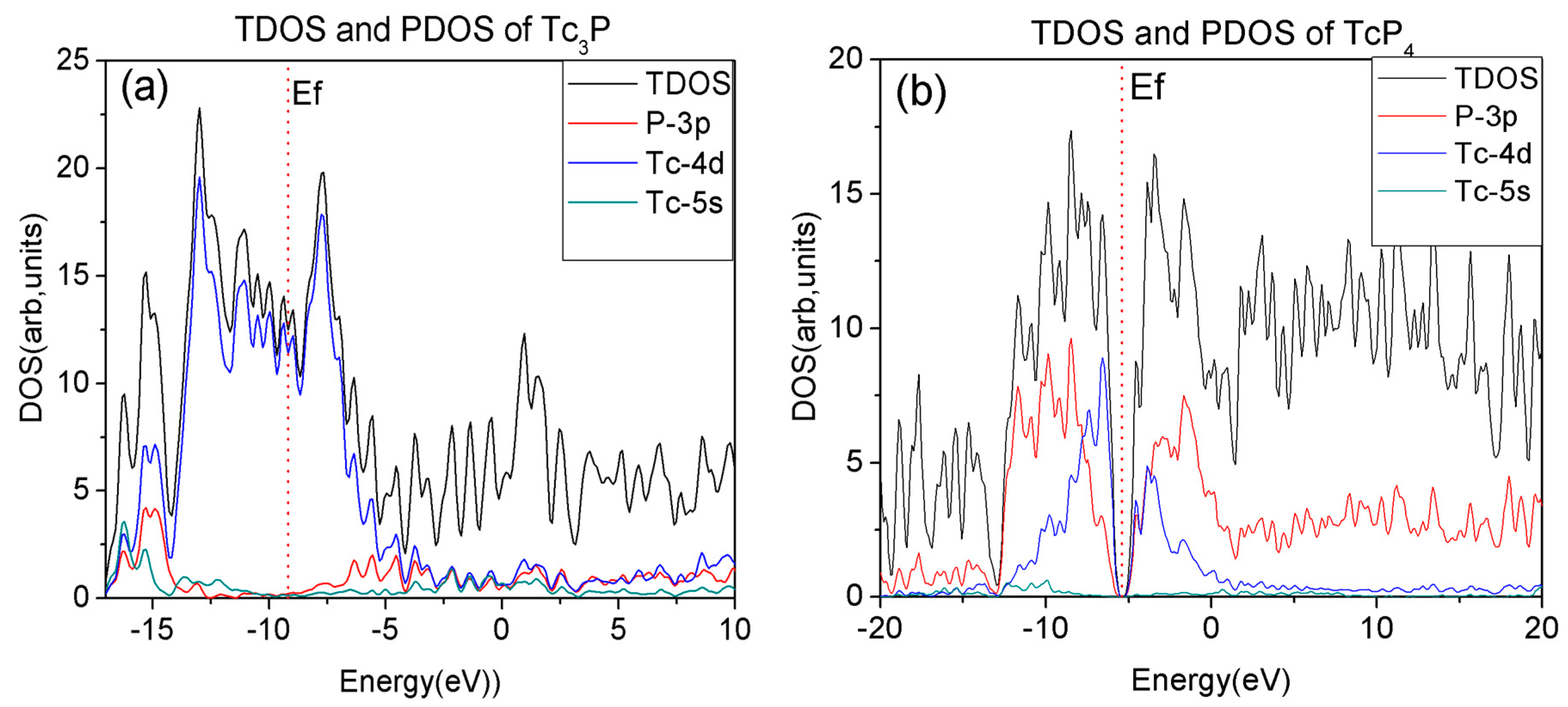
The properties of materials have much to do with the electron configuration of molecule. So, we further calculated the total and projected density of states (TDOS and PDOS) for Tc3P crystal to study the distribution of the electrons on different orbits to the conductibility of Tc3P crystal.
The total and projected electronic density of states of Tc3P with energy ranging from −16 to 0 eV is depicted in Figure 2a. At the same time, the projected density of states of some s, p, and d states of Tc and P atoms are calculated, respectively. From Figure 2a, the contribution of the Tc-4d electrons is predominant for the energy bands near the Fermi level of the Tc3P crystal. So, the Tc-4d electrons are important for the conductibility of this crystal.
In addition, Figure 2b shows us the TDOS and PDOS of the TcP4 crystal, and it can be seen that the whole band can be divided into two prominent valence-band regions and two conduction-band regions. The regions range from −20 to −13 eV, −13 to −6 eV, −6 to 1 eV, and 1 to 20 eV, respectively. The densities of states at these regions primarily consist of Tc-5s, Tc-4d, and P-3p states. From Figure 2b, we can see that the bonding peaks ranged from −20 to −13 eV and the bonding peaks between 1 and 20 eV are mainly dominated by the P-3p state. The peaks of the bonding ranged from −13 to −6 eV in the valence band and the peaks of the bonding ranged from −6 to 1 eV in the conduction band are not only predominated by the P-3p state, but also contributed from the Tc-4d state. In addition, because the electronic densities of states from −13 and −6 eV and the region range from −6 to 1 eV are located in the vicinity of the Fermi level, and the Tc-4d and P-3p states are principally for these two bands, the Tc-4d and P-3p electrons are very important for the energy bands of the TcP4 crystal.
According to above analysis, it is noted that the conductibility of Tc3P crystal is attributed to the Tc-4d electrons, while the distribution of the P-3p electrons weakens the conductivity of the TcP4 crystal. As a result, the Tc3P crystal is considered as a conductor material, while the TcP4 crystal is a semiconductor material.
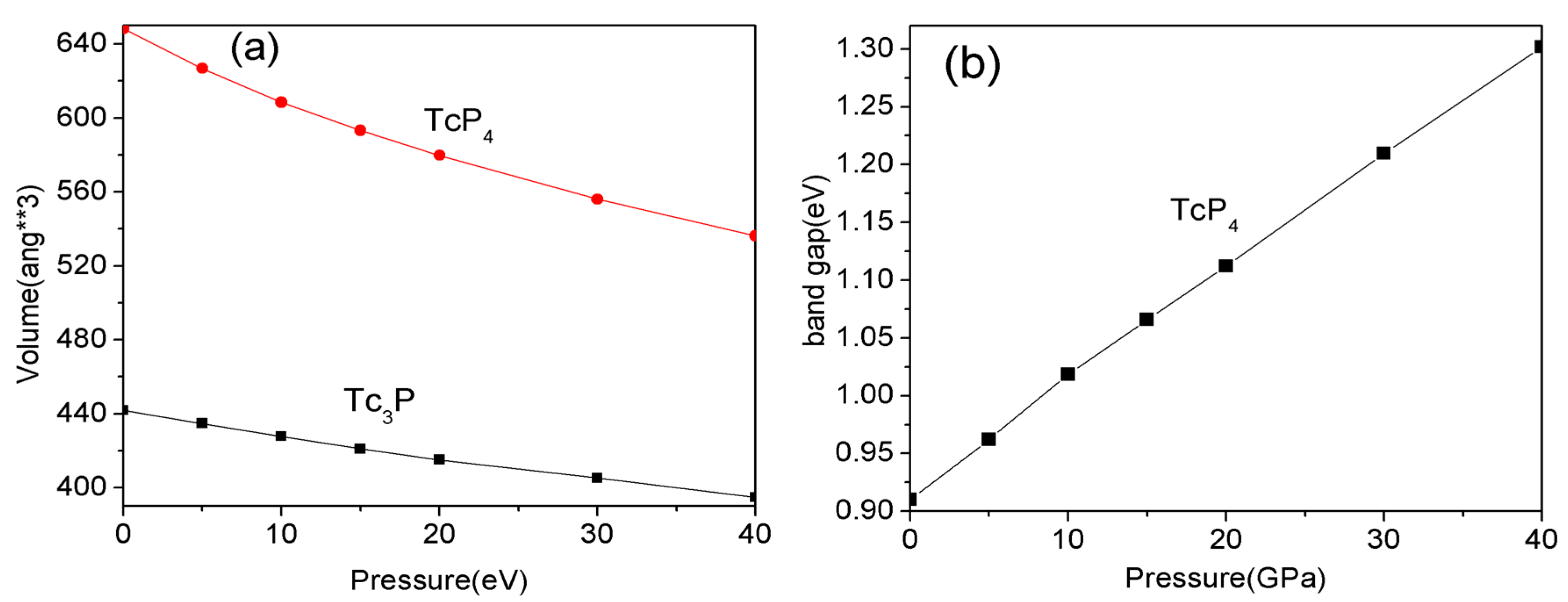
The structural and electronic properties of technetium phosphide crystals are also investigated with GGA and LDA approximations at the pressure range from 0 to 40 GPa. However, previous studies [25,26] show that the GGA method is better than LDA to study the properties of these types of crystal at a high pressure. Therefore, the following investigations are studied by GGA approximation. The cell volumes of Tc3P and TcP4 crystal changing with increasing pressure are shown in Figure 3a. In the pressure range between 0 and 40 GPa, it is noted that the cell volume of the Tc3P crystal decreases smoothly with the increasing pressure. In addition, Figure 3b shows that the band gap of the TcP4 crystal changed with the pressure. As is shown in Figure 3b, from 0 to 40 GPa, the band gap of the TcP4 crystal increases from 0.91 to 1.30 eV. Although the band gap increases with the increasing pressure, the TcP4 crystal is still a semiconductor under high pressure.
3.3. Optical Properties
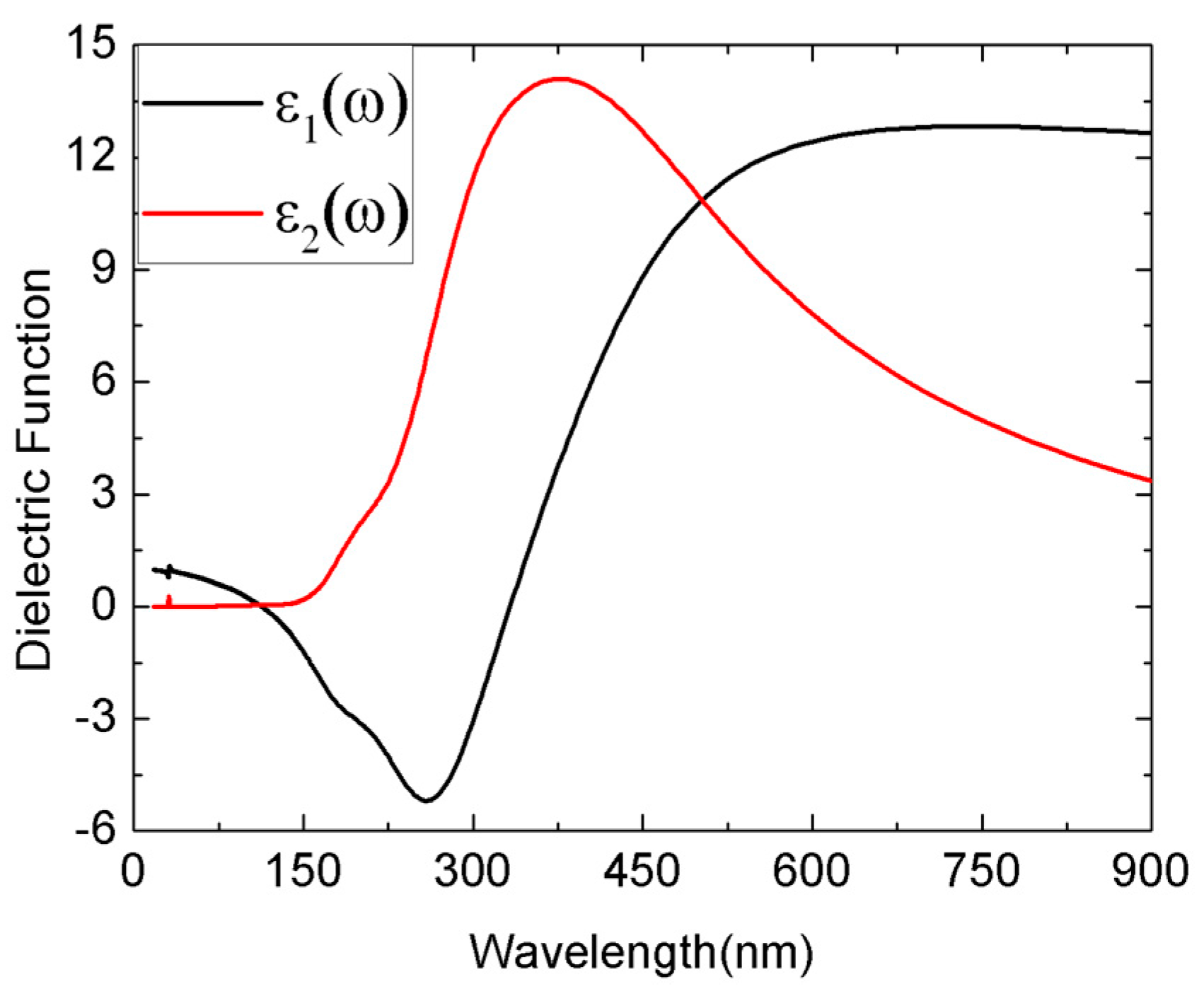
Commonly, transition-metal phosphides have good optical properties. The Tc3P crystal is a conductor material, while the TcP4 is a semiconductor material with a wide direct band gap. Therefore, here we only investigate the optical properties of TcP4. The complex dielectric function of TcP4 at zero pressure is presented in Figure 4. In order to discuss the absorption coefficient of TcP4 under high pressure, we computed the dielectric function of this crystal at different pressures. The imaginary part of the dielectric function and the absorption coefficient of TcP4 crystal at different pressures are shown in Figure 5.
From Figure 5b, it is noted that a primary peak of the absorption coefficient of TcP4 occurred in the wavelength region of the ultraviolet band. In addition, TcP4 has a large absorption coefficient in the visible-light region, while the imaginary part of the dielectric function has a close connection with the absorption coefficient. From Figure 5a,b, we can see the change of optical properties of TcP4 at high pressure. The absorption of visible and ultraviolet light for TcP4 enhances as the pressure is increased. Combined with the study of the change in band gap with pressure in Section 3.2, it can be seen that the pressure effect has an important influence on the photo-catalytic properties of TcP4. In the future, we can consider increasing the photo-catalytic performance of TcP4 by a high-pressure method.
3.4. Hardness
Based on previous work [27,28], it is known that the metallic bond limits the hardness of transition metal phosphides. Compared Tc3P and TcP4, it is not difficult to find that there are many more Tc-Tc bonds in Tc3P than that of TcP4, so we focus our study on TcP4 in the present work. There are many theoretical models to calculate theoretical Vickers hardness. For example, Tian et al. [29] proposed the formulation of hardness for crystals, Hv = 0.92 k1.137G0.708, where k = G/B, B is the bulk modulus, and G is the shear modulus. Both bulk and shear moduli are macroscopic concepts. Liu et al. [30] employed this model to calculate the hardness in pyrite-type transition-metal pernitrides. In this study, we employ the model proposed by Gao et al. [24] to calculate the theoretical Vickers hardness. This model considers the effect of metal bonds on the hardness, and presents the relationship between hardness and microscopic parameters. According to this model, the bond parameters and Vickers hardness of TcP4 at ambient pressure are calculated and presented in Table 2. Compared with the experimental values that can be obtained, theoretical hardness obtained by Tian’s and Gao’s model are in better agreement with the experimental results. However, the latter can explain the origin of hardness at atomic level.
In addition, to study the effect of pressure on the hardness property of TcP4, we computed its Vickers hardness from 0 to 40 GPa. The results are presented in Table 3. It is not difficult to see that the hardness of TcP4 increases with the increasing pressure. By analyzing the bond parameter and bond component of TcP4 at different pressures, we found that the hardness is most closely related with the metallicity, the Mulliken overlap population of the bonds in the crystal. Further analysis showed that there are two reasons explaining the increasing hardness at high pressure: (1) the Mulliken overlap populations of the covalent bonds (P-P bonds) and ionic bonds (Cd-P bonds) in TcP4 increase as the pressure is increased, leading to both the hardness of P-P bonds Cd-P bonds increasing at a higher pressure; (2) the metallicity of metallic bonds in TcP4 are weakened as the pressure increases, leading to the limit to the hardness in the crystal being weakened. In a word, due to the enhancement of covalent bonds and the weakening of metal bonds, the hardness of TcP4 is increased at a higher pressure.
4. Conclusions
In this paper, density functional theory is employed to investigate the structure, electronic properties, optical properties, and hardness of technetium phosphides in the pressure range of 0 to 40 GPa. The lattice parameters and the final atomic positions obtained theoretically are in excellent agreement with the experimental values at ambient pressure. In addition, the electronic properties of Tc3P and TcP4 crystal are also studied and presented. The results show that the Tc3P crystal is a conductor material. However, for the TcP4 crystal, there is a direct band gap of 0.91 eV at the high symmetry point G. Additionally, the band gap of the TcP4 crystal increased from 0.91 to 1.30 eV as the pressure increased from 0 to 40 GPa. The calculated total electronic density of states and projected densities of states for the Tc3P and TcP4 are presented. Results show that the predominant distribution of Tc-4d to the DOS near the Fermi level leads to the classification of Tc3P crystal as a conductor material, while the distribution of both Tc-4d and P-3p to the DOS near the Fermi level leads to the conclusion that TcP4 crystal is a semiconductor material. What is more, the optical properties of TcP4 are discussed, and we find the absorption of visible and ultraviolet light for TcP4 enhances as the pressure is increased. This indicates that the high pressure method is an effective way to regulate the photo-catalytic performance of TcP4.
At the end of our study, we research the effect of pressure on the hardness of TcP4. Results show that both the Mulliken overlap populations of P-P bonds and Cd-P bonds in TcP4 increase as the pressure is increased, which leads to the hardness of covalent bonds and ionic bonds increasing as the pressure is increased. Meanwhile, as the pressure increases, the metallicity weakens. This leads to the hardness of TcP4 increasing as the pressure is increased. As the pressure rises from 0 to 40 GPa, the Vickers hardness of TcP4 increases from 20.14 to 25.74 GPa. So, the effect of pressure plays an important role on the hardness of Tc-P crystals; it changes the hardness of the material by affecting its chemical bond parameters. This study would provide a theoretical basis for improving the hardness of transition metal phosphides and a theoretical guidance for the synthesis of novel ultrahard materials for experiment.
Acknowledgments
Supported by the National Natural Science Foundation of China (NSFC. Grant No. 11374217), the Key Scientific Research Projects of Henan Province (No. 17A140030) and the Doctoral Fund of Zhengzhou University of Light Industry (No. 2014BSJJ088 and 2015XJJZ022)
Author Contributions
Shiquan Feng conceived and designed the work, he supervised the whole work, and wrote the paper. Xuerui Cheng calculated the data, and revised part of the paper. The optical and electronic properties were analyzed by Xinlu Cheng. The hardness properties analysis was performed by Jinsheng Yue. Junyu Li revised the paper and did the work of document inquire.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sumiya, H.; Toda, N.; Satoh, S. Mechanical Properties of Synthetic Type IIa Diamond Crystal. Diam. Relat. Mater. 1997, 6, 1841–1846. [Google Scholar] [CrossRef]
- Komanduri, R.; Shaw, M.C. Wear of synthetic diamond when grinding ferrous materials. Nature 1975, 255, 211–213. [Google Scholar] [CrossRef]
- Yang, S.; Liang, C.; Prins, R. Preparation and hydrotreating activity of unsupported nickel phosphide with high surface area. J. Catal. 2006, 241, 465–469. [Google Scholar] [CrossRef]
- Muetterties, E.L.; Sauer, J.C. Catalytic Properties of Metal Phosphides: I. Qualitative Assay of Catalytic Properties. J. Am. Chem. Soc. 1974, 96, 3410–3415. [Google Scholar] [CrossRef]
- Dmitruk, N.L.; Zuev, V.A.; Stepanova, M.A. Spectral distribution of the photoconductivity of cadmium diphosphide. Russ. Phys. J. 1991, 34, 642–644. [Google Scholar]
- Kushnir, O.S.; Bevz, O.A.; Polovinko, I.I.; Sveleba, S.A. Temperature dependence of optical activity and circular dichroism in α-ZnP2 crystals. Phys. Status Solidi B 2003, 238, 92–101. [Google Scholar] [CrossRef]
- Lazarev, V.B.; Shevchenko, V.Y.; Grinberg, L. K.; Sobolev, V.V. Semiconducting II-V Compounds; Nauka: Moscow, Russia, 1976. [Google Scholar]
- Morozova, V.A.; Marenkin, S.F.; Koshelev, O.G.; Trukhan, V.M. Optical absorption in monoclinic zinc diphosphide. Inorg. Mater. 2006, 42, 221. [Google Scholar] [CrossRef]
- Sheleg, A.U.; Zaretskii, V.V. X-ray study of the commensurate—Incommensurate phase transitions in α-ZnP2. Phys. Status Solidi A 1984, 86, 517–523. [Google Scholar] [CrossRef]
- Slobodyanyuk, A.V.; Schaack, G. Measurement of Raman scattered intensities in media with natural or field-induced optical activity. J. Raman Spectrosc. 1987, 18, 561–568. [Google Scholar] [CrossRef]
- Feng, S.Q.; Wang, L.L.; Jiang, X.X.; Li, H.N.; Cheng, X.L.; Su, L. High-pressure dynamic, thermodynamic properties, and hardness of CdP2. Chin. Phys. B 2017, 4, 046301. [Google Scholar] [CrossRef]
- Feng, S.Q.; Yang, Y.; Li, J.Y.; Jiang, X.X.; Li, H.N.; Cheng, X.L. Pressure effect on the hardness of diamond and W2B5: First-principle. Mod. Phys. Lett. B 2017, 31, 1750137. [Google Scholar] [CrossRef]
- Ruehl, R.; Jeitschko, W.; Schwochau, K. Preparation and Crystal Structures of Technetium Phosphides. J. Solid. State. Chem. 1982, 44, 134–140. [Google Scholar] [CrossRef]
- Ordejón, P.; Artacho, E.; Soler, J.M. Self-consistent order-N density-functional calculations for very large systems. Phys. Rev. B 1996, 53, R10441. [Google Scholar] [CrossRef]
- Barth, U.; Von Hedin, L. A local exchange-correlation potential for the spin polarized case: I. J. Phys. C. Solid State Phys. 1972, 5, 1629. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Strobel, R.; Maciejewski, M.; Pratsinis, S.E.; Baiker, A. Unprecedented formation of metastable monoclinic BaCO3 nanoparticles. Therm. Acta 2006, 445, 23–26. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E. High-density limit of the Perdew-Burke-Ernzerhof generalized gradient approximation and related density functionals. Phys. Rev. A 2006, 74, 044501. [Google Scholar] [CrossRef]
- Wu, Z.G.; Cohen, R.E. More accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5075. [Google Scholar] [CrossRef]
- Kleinman, L.; Bylander, D.M. Efficacious Form for Model Pseudopotentials. Phys. Rev. Lett. 1982, 48, 1425. [Google Scholar] [CrossRef]
- Okoye, C.M.I. Theoretical study of the electronic structure, chemical bonding and optical properties of KNbO3 in the paraelectric cubic phase. J. Phys. Condens. Matter. 2003, 15, 5945–5958. [Google Scholar] [CrossRef]
- Gao, F. Theoretical model of intrinsic hardness. Phys. Rev. B 2006, 73, 132104. [Google Scholar] [CrossRef]
- Fan, C.L.; Cheng, X.L.; Zhang, H. First-principles study of the structural and electronic properties of the alpha modification of zinc diphosphide. Phys. Status Solidi B 2009, 246, 77–81. [Google Scholar] [CrossRef]
- Feng, S.Q.; Cheng, X.L. Theoretical study on electronic properties and pressure-induced phase transition in β-CdP2. Comput. Theor. Chem. 2011, 966, 149–153. [Google Scholar] [CrossRef]
- Feng, S.Q.; Li, X.D.; Su, L.; Li, H.N.; Yang, H.Y.; Cheng, X.L. Ab initio Study on Structural, Electronic Properties, and Hardness of Re-doped W2B5. Solid. Stat. Commun. 2016, 245, 60–64. [Google Scholar] [CrossRef]
- Feng, S.Q.; Guo, F.; Li, J.Y.; Wang, Y.Q.; Zhang, L.M.; Cheng, X.L. Theoretical investigations of physical stability, electronic properties and hardness of transition-metal tungsten borides WBx (x = 2.5, 3). Chem. Phys. Lett. 2015, 635, 205–209. [Google Scholar] [CrossRef]
- Liu, Z.T.; Gall, Y.; Khare, S.V.D. Electronic and bonding analysis of hardness in pyrite-type transition-metal pernitrides. Phys. Rev. B 2014, 90, 134102. [Google Scholar] [CrossRef]
- Tian, Y.J.; Xu, B.; Zhao, Z.S. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. H. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Zhong, M.M.; Kuang, X.Y.; Wang, Z.H.; Shao, P.; Ding, L.P.; Huang, X.F. Phase Stability, Physical Properties, and Hardness of Transition-Metal Diborides MB2 (M = Tc, W, Re, and Os): First-Principles Investigations. J. Phys. Chem. C 2013, 117, 10643. [Google Scholar] [CrossRef]
- Gu, Q.F.; Krauss, G.; Steurer, W. Transition metal borides: Superhard versus ultra-incompressihle. Adv. Mater. 2008, 20, 3620–3626. [Google Scholar] [CrossRef]
Figure 1.
Electronic band structures of the (a) Tc3P and (b) TcP4 crystals in the vicinity of the Fermi level. The red horizontal dotted lines correspond to the Fermi level Ef.
Figure 1.
Electronic band structures of the (a) Tc3P and (b) TcP4 crystals in the vicinity of the Fermi level. The red horizontal dotted lines correspond to the Fermi level Ef.
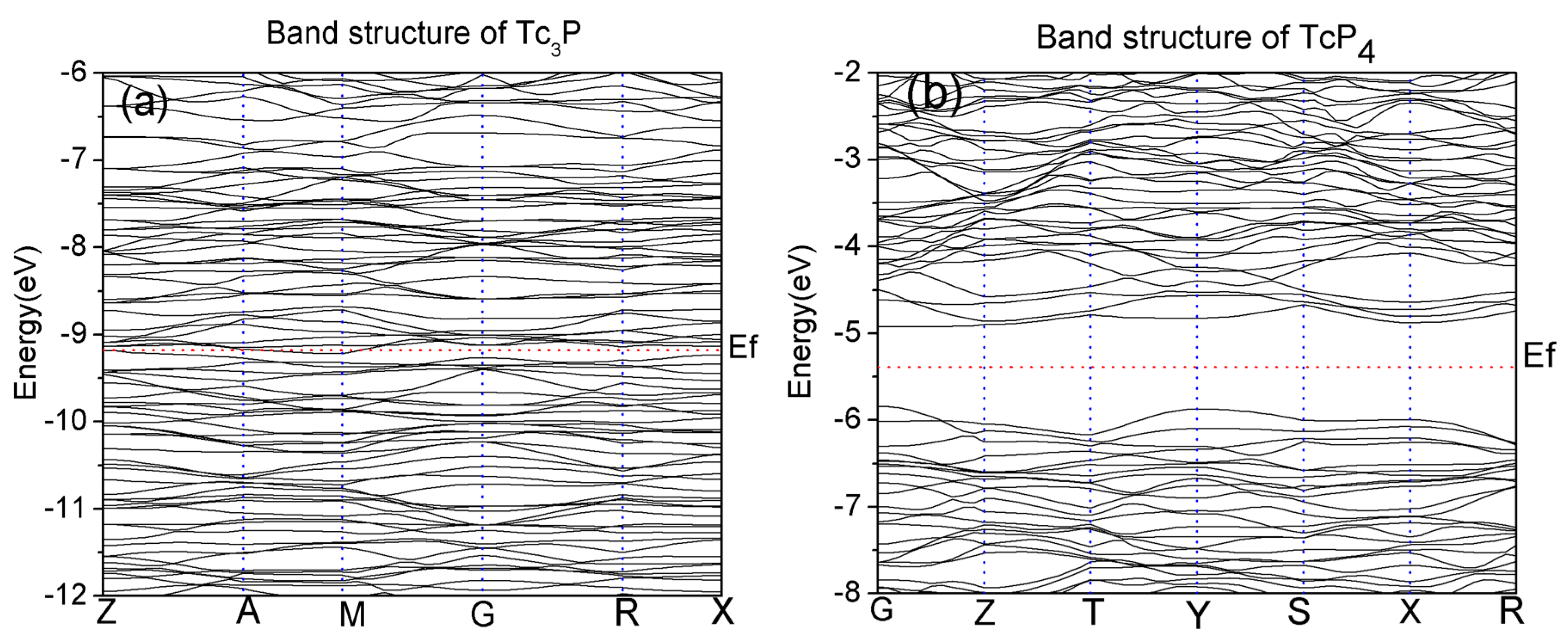
Figure 2.
The total density of states (DOS) and projected DOS (PDOS) of P(3p), Tc(4d) and Tc(5s)(d) for (a) Tc3P crystal in the range from −16 to 10 eV and (b) TcP4 crystal in the range from −20 to 20 eV. The red vertical dotted lines correspond to the Fermi level Ef.
Figure 2.
The total density of states (DOS) and projected DOS (PDOS) of P(3p), Tc(4d) and Tc(5s)(d) for (a) Tc3P crystal in the range from −16 to 10 eV and (b) TcP4 crystal in the range from −20 to 20 eV. The red vertical dotted lines correspond to the Fermi level Ef.
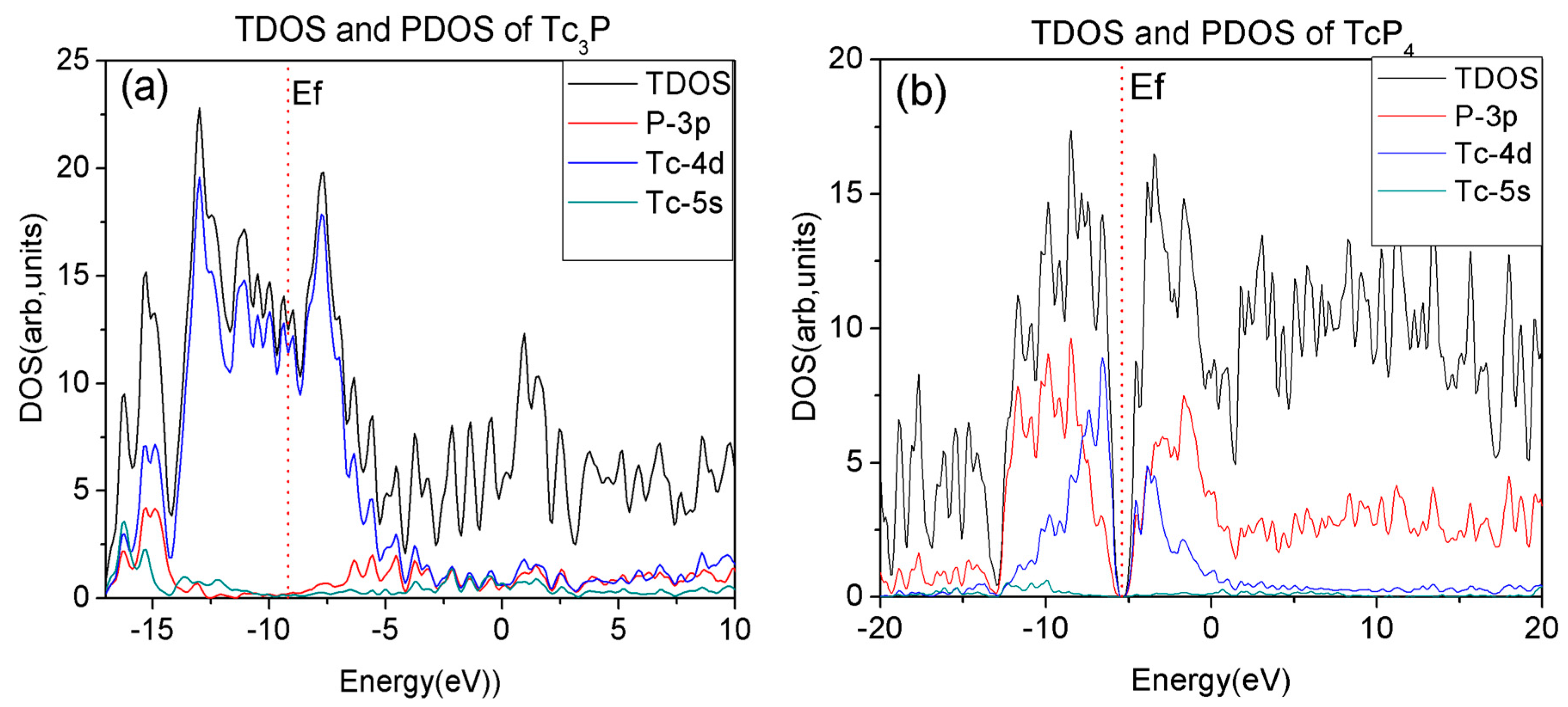
Figure 3.
(a) The dependence of the cell volume on pressure for the Tc3P and TcP4 crystal; (b) The dependence of the band gap on pressure for the TcP4 crystal.
Figure 3.
(a) The dependence of the cell volume on pressure for the Tc3P and TcP4 crystal; (b) The dependence of the band gap on pressure for the TcP4 crystal.
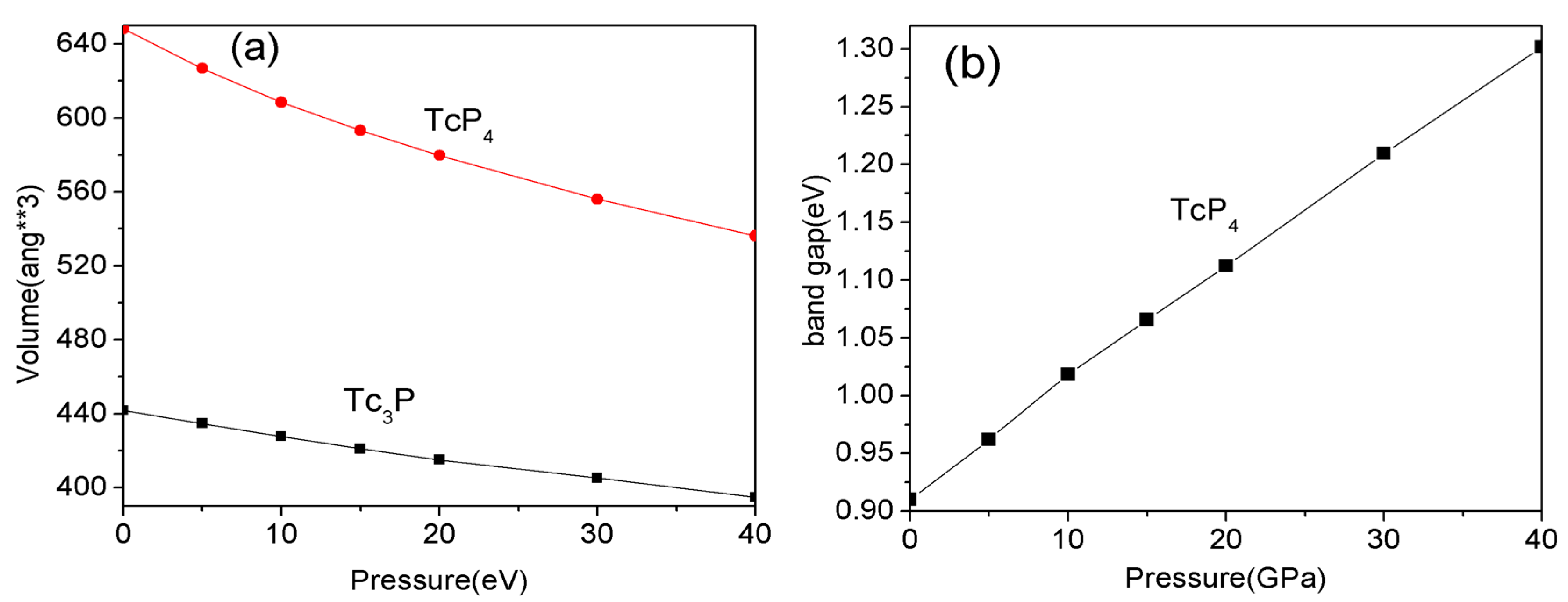
Figure 4.
The complex dielectric function for the TcP4 crystal; the black solid line corresponds to the real part, and the red solid line corresponds to the imaginary part.
Figure 4.
The complex dielectric function for the TcP4 crystal; the black solid line corresponds to the real part, and the red solid line corresponds to the imaginary part.
Figure 5.
(a) The imaginary part of the dielectric function for the TcP4 crystal at different pressures. (b) The absorption coefficient of TcP4 crystal at different pressures.
Figure 5.
(a) The imaginary part of the dielectric function for the TcP4 crystal at different pressures. (b) The absorption coefficient of TcP4 crystal at different pressures.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated and experimental lattice parameters of the crystal Tc3P and TcP4 at zero pressure. GGA: generalized gradient approximation; LDA: local density approximation.
Table 1.
Calculated and experimental lattice parameters of the crystal Tc3P and TcP4 at zero pressure. GGA: generalized gradient approximation; LDA: local density approximation.
| Lattice Parameter | Tc3P | TcP4 | ||||
|---|---|---|---|---|---|---|
| Theoretical Results | Experimental Values [13] | Theoretical Results | Experimental Values [13] | |||
| GGA | LDA | GGA | LDA | |||
| a0 (Å) | 9.490 | 9.376 | 9.568 | 6.341 | 6.280 | 6.238 |
| b0 (Å) | 9.490 | 9.376 | 9.568 | 9.339 | 9.241 | 9.215 |
| c0 (Å) | 4.907 | 4.856 | 4.736 | 10.949 | 10.827 | 10.837 |
| V0 (Å3) | 441.9 | 426.9 | 433.6 | 648.3 | 628.3 | 623.0 |
Table 2.
Calculated bond parameters and theoretical hardness obtained by the models of Tian et al. and Gao et al. at ambient pressure. Experimental data are presented for comparison.
Table 2.
Calculated bond parameters and theoretical hardness obtained by the models of Tian et al. and Gao et al. at ambient pressure. Experimental data are presented for comparison.
| Compounds | Bond | dμ (Å) | Pμ | Ω (Å3) | vbμ (Å3) | fm (10−3) | Hvμ (GPa) | Hv Tian | Hv Gao | Hv exp |
|---|---|---|---|---|---|---|---|---|---|---|
| WB2-WB2 | B-B (1) | 1.727 | 0.76 | 2.521 | 0 | 28.8 | 25.6 [31] | 27.7 [32] | ||
| B-B (2) | 1.838 | 0.65 | 3.038 | 0 | ||||||
| W-B (1) | 2.335 | 0.26 | 6.229 | 1.787 | ||||||
| ReB2-ReB2 | B-B | 1.807 | 0.64 | 1.846 | 0 | 43.4 | 39.1 [31] | 39.3 [32] | ||
| Re-B | 2.240 | 0.25 | 3.515 | 1.311 | ||||||
| TcP4 | P-P | 2.178 | 0.58 | 621.7 | 5.362 | 0 | 24.68 | 19.7 | 20.1 | |
| P-P | 2.190 | 0.56 | 5.451 | 0 | 23.19 | |||||
| P-P | 2.191 | 0.51 | 5.458 | 0 | 21.07 | |||||
| P-P | 2.203 | 0.60 | 5.549 | 0 | 24.12 | |||||
| P-P | 2.252 | 0.51 | 5.927 | 0 | 18.36 | |||||
| Tc-P | 2.342 | 0.52 | 6.667 | 0.929 | 14.39 | |||||
| Tc-P | 2.356 | 0.33 | 6.787 | 0.929 | 8.86 | |||||
| Tc-P | 2.358 | 0.54 | 6.804 | 0.929 | 14.44 | |||||
| Tc-P | 2.377 | 0.53 | 6.970 | 0.929 | 13.62 | |||||
| Tc-P | 2.425 | 0.57 | 7.401 | 0.929 | 13.25 | |||||
| Tc-P | 2.523 | 0.30 | 8.335 | 0.929 | 5.72 | |||||
| Tc-Tc | 3.000 | 0.63 | 14.012 | 0.929 | 5.05 |
Table 3.
Calculated Vickers hardness of TcP4 at different pressures.
| Compounds | 0 GPa | 10 GPa | 20 GPa | 40 GPa |
|---|---|---|---|---|
| Hardness (GPa) | 20.14 | 22.35 | 24.88 | 25.74 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Feng, S.; Cheng, X.; Cheng, X.; Yue, J.; Li, J. Theoretical Study on Electronic, Optical Properties and Hardness of Technetium Phosphides under High Pressure. Crystals 2017, 7, 176. https://doi.org/10.3390/cryst7060176
AMA Style
Feng S, Cheng X, Cheng X, Yue J, Li J. Theoretical Study on Electronic, Optical Properties and Hardness of Technetium Phosphides under High Pressure. Crystals. 2017; 7(6):176. https://doi.org/10.3390/cryst7060176
Chicago/Turabian StyleFeng, Shiquan, Xuerui Cheng, Xinlu Cheng, Jinsheng Yue, and Junyu Li. 2017. "Theoretical Study on Electronic, Optical Properties and Hardness of Technetium Phosphides under High Pressure" Crystals 7, no. 6: 176. https://doi.org/10.3390/cryst7060176
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.