Crystal Structure of Shigella flexneri SF173 Reveals a Dimeric Helical Bundle Conformation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
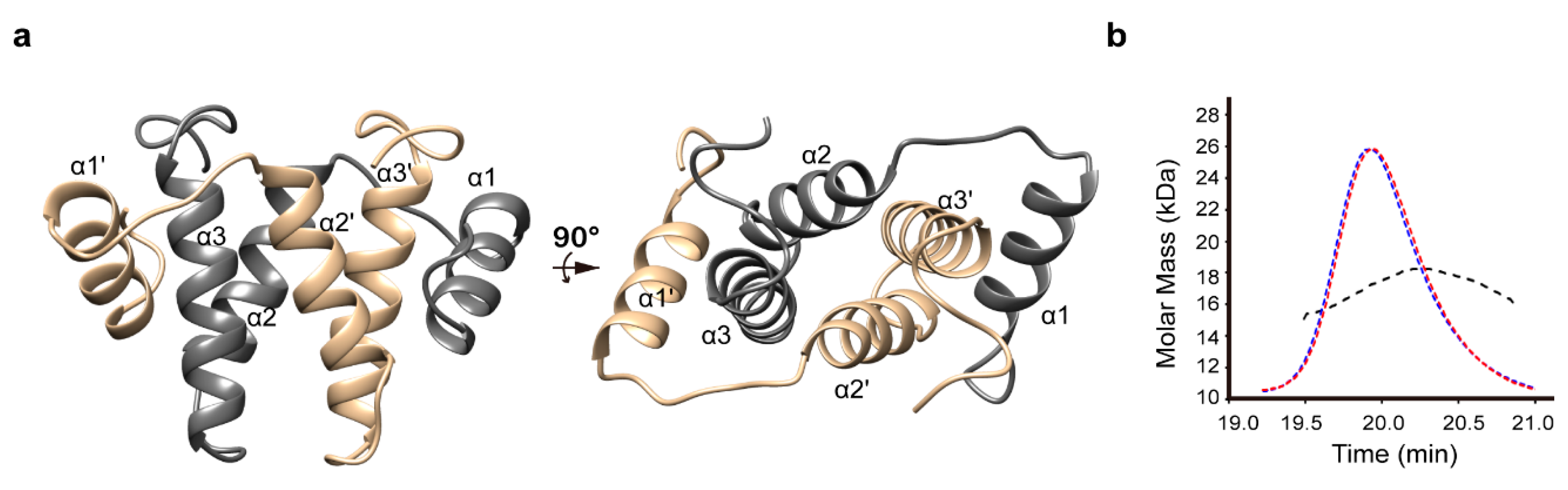
2.1. Protein Purification and SEC-MALS Analysis
2.2. Crystallization and Data Collection
2.3. Structure Determination and Refinement
3. Results and Discussion
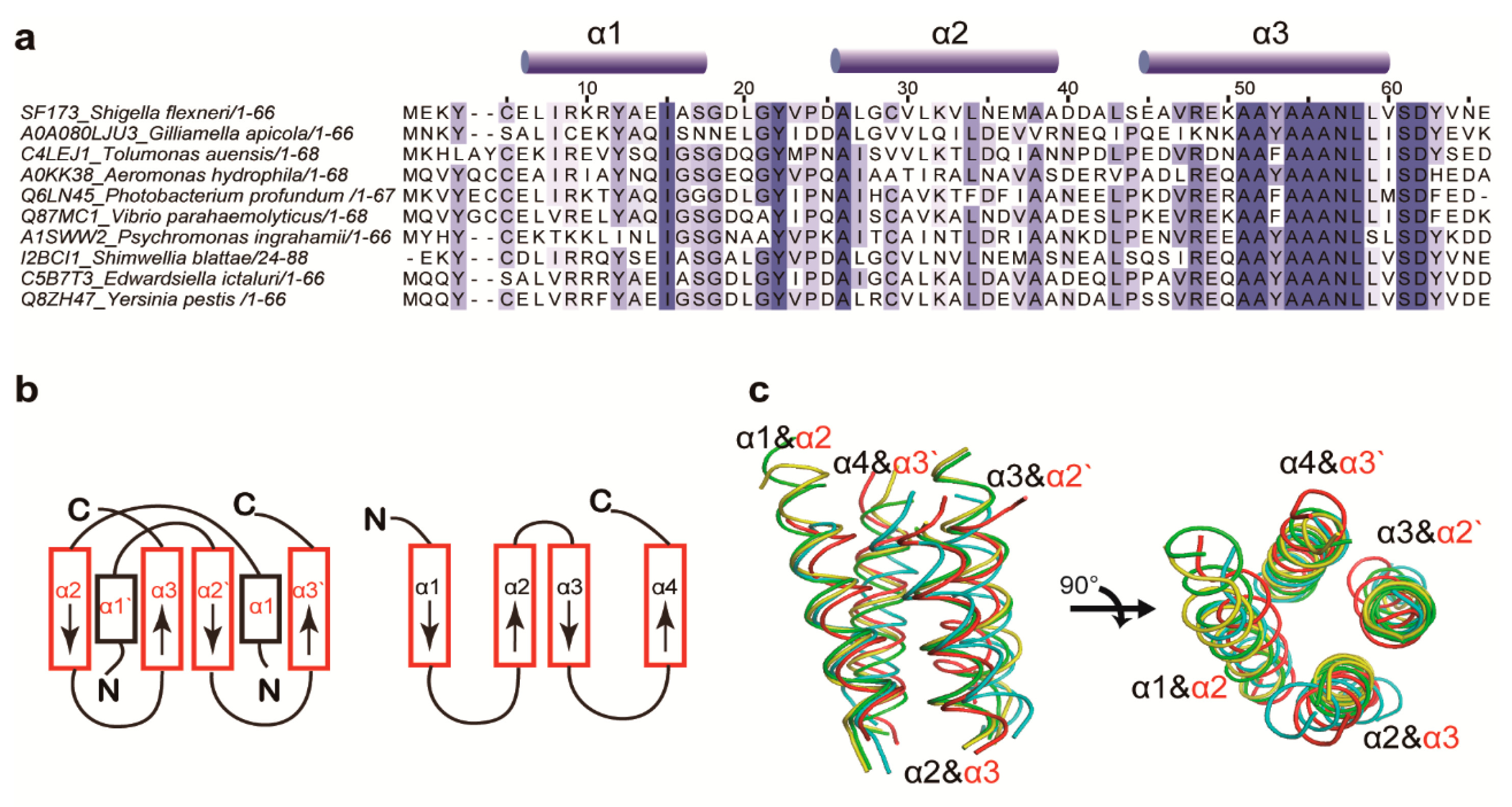
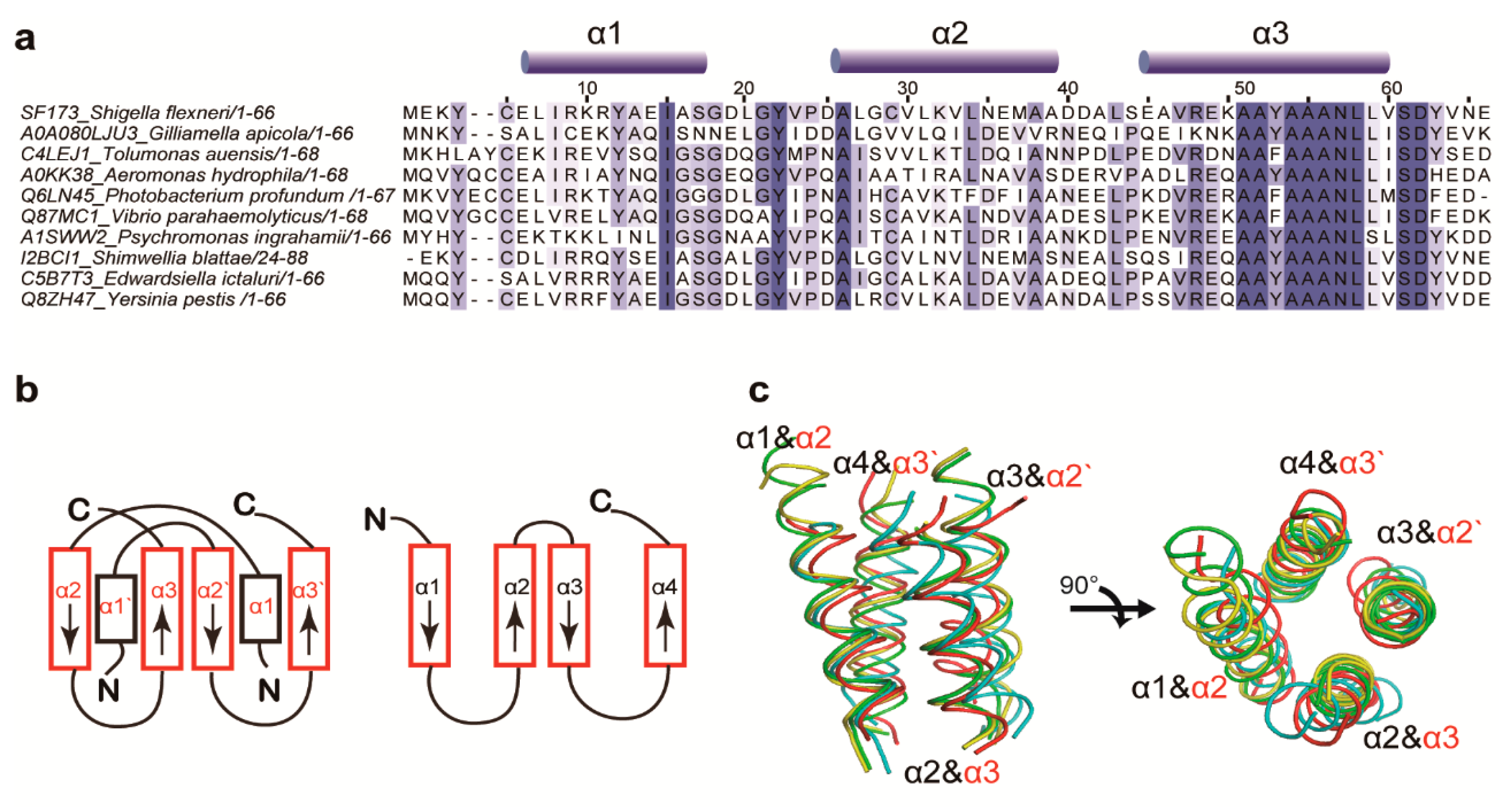
3.1. Structure Determination and Overall Fold
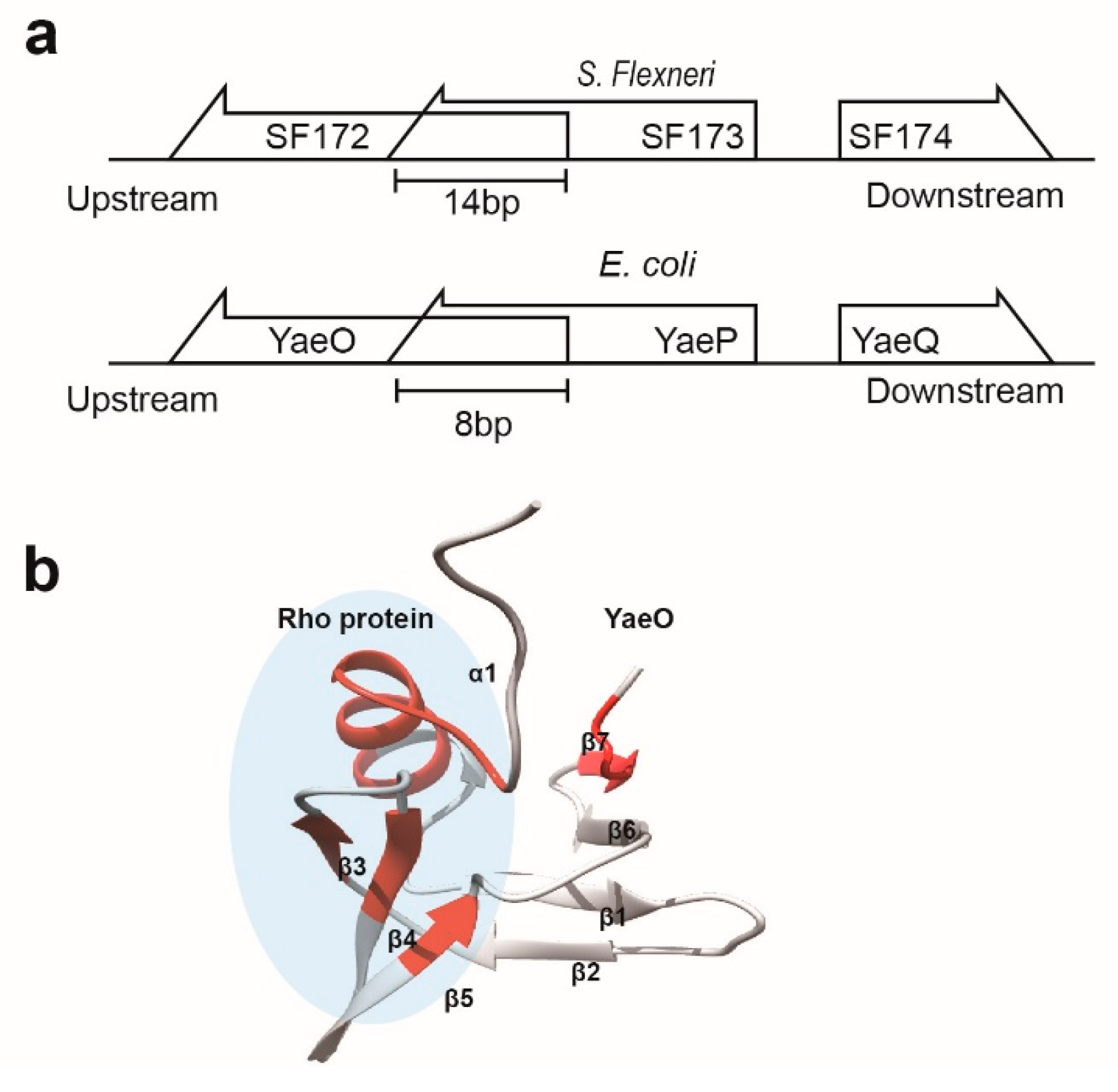
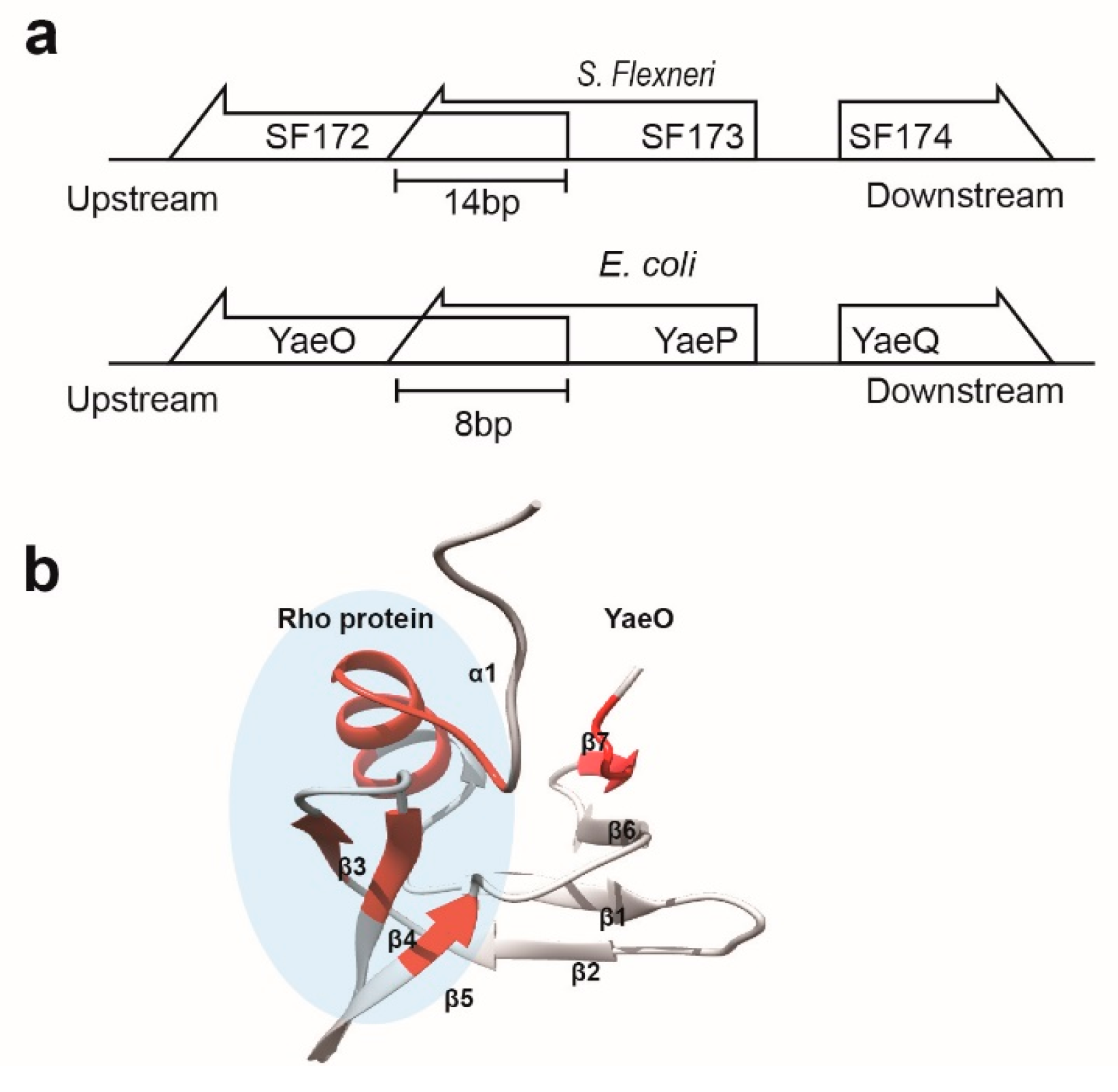
3.2. Structural Similarity to Colicin Immunity Proteins
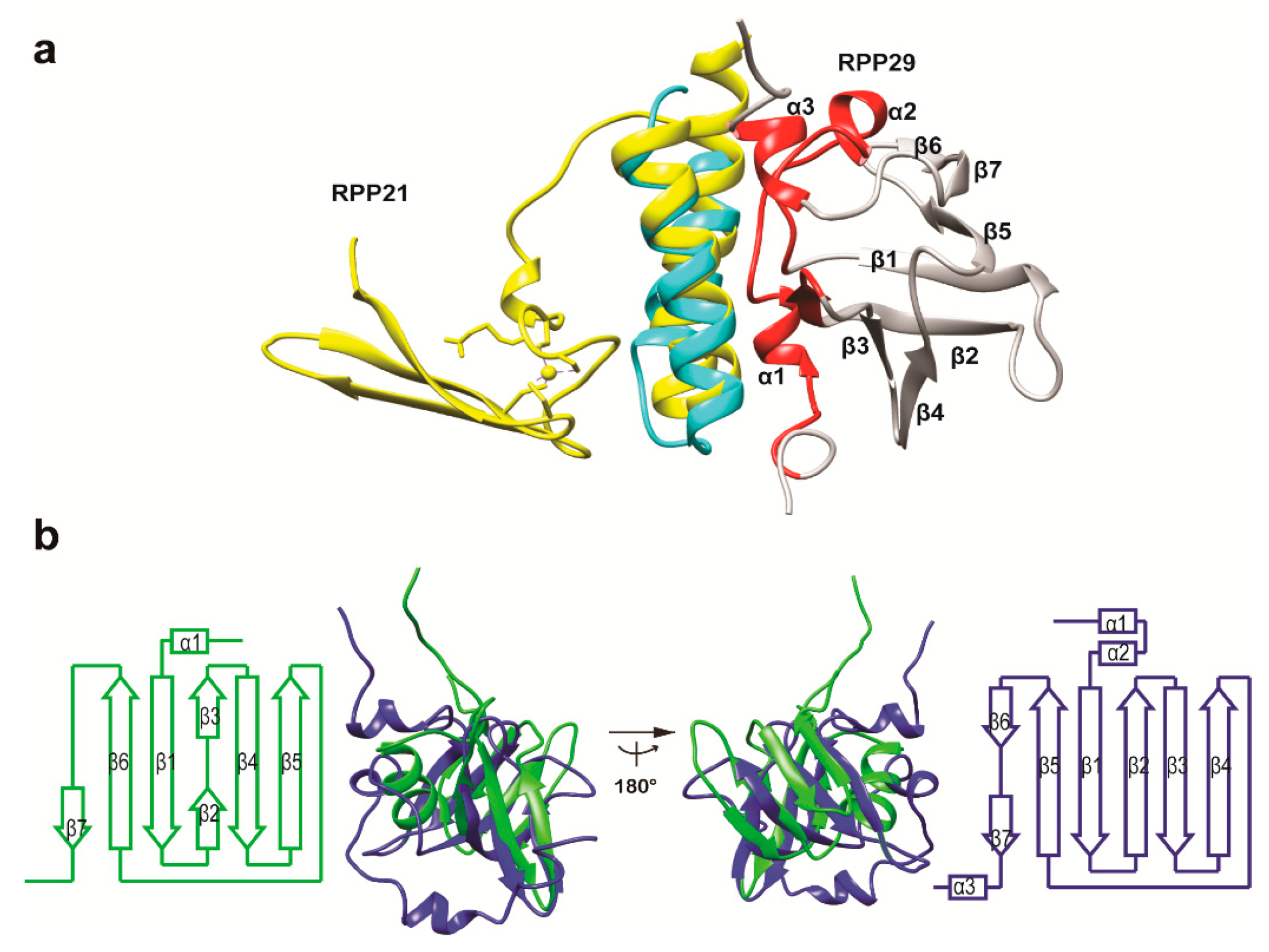
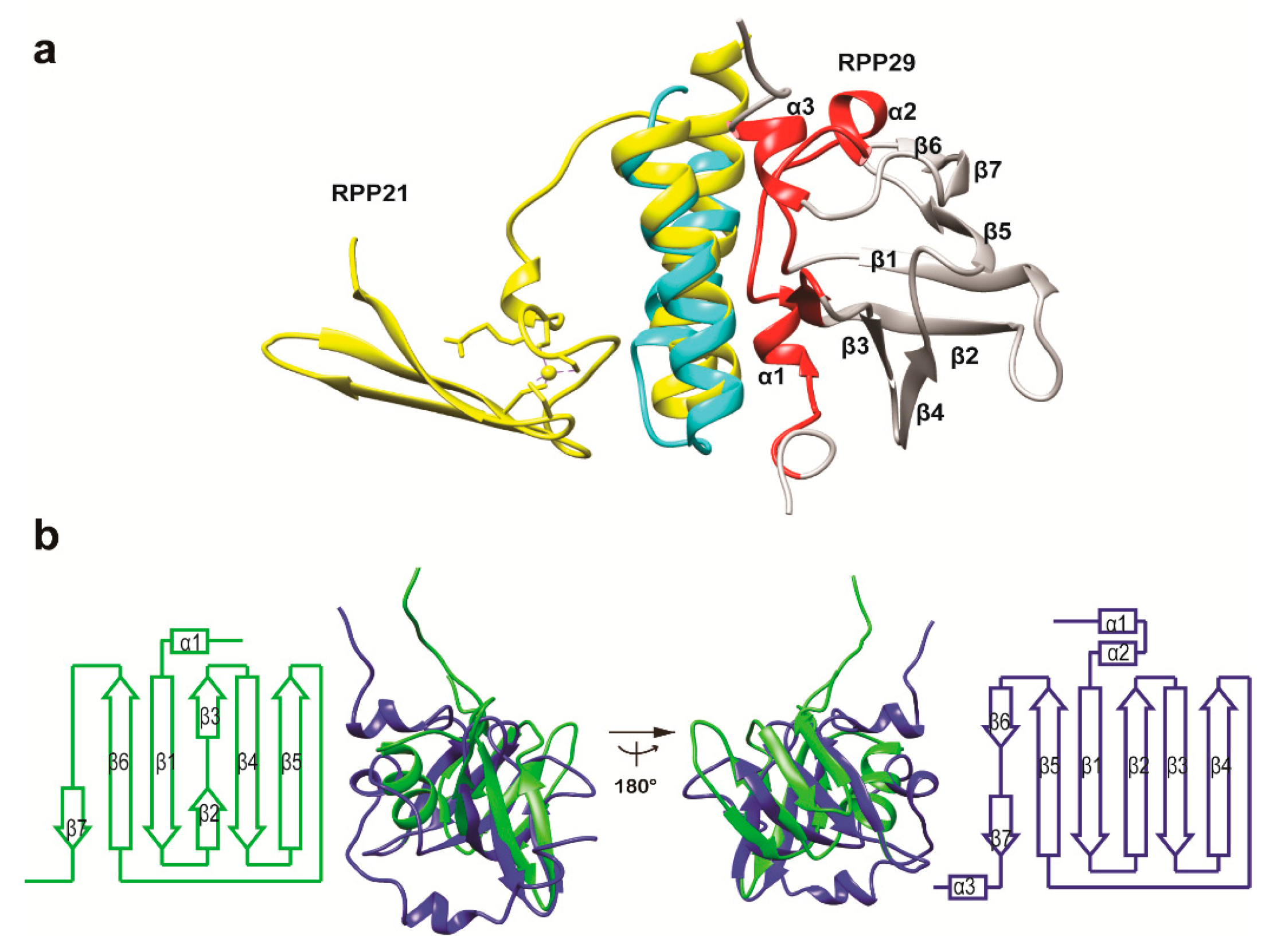
3.3. Structural Correlation with ROF/RNase P-Like Domain Proteins
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.; Chruszcz, M.; Zimmerman, M.D.; Kirillova, O.; Minor, W. Benefits of structural genomics for drug discovery research. Infect. Disord. Drug Targets 2009, 9, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J. Microbes and microbial toxins: Paradigms for microbial-mucosal interactions III. Shigellosis: From symptoms to molecular pathogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G319–G323. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, O.J.; Cavaillon, J.M.; Huerre, M.; Ohayon, H.; Gounon, P.; Sansonetti, P.J. Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J. Exp. Med. 1994, 180, 1307–1319. [Google Scholar] [CrossRef] [PubMed]
- Kotloff, K.L.; Winickoff, J.P.; Ivanoff, B.; Clemens, J.D.; Swerdlow, D.L.; Sansonetti, P.J.; Adak, G.K.; Levine, M.M. Global burden of Shigella infections: Implications for vaccine development and implementation of control strategies. Bull. World Health Organ. 1999, 77, 651–666. [Google Scholar] [PubMed]
- Hale, T.L. Genetic basis of virulence in Shigella species. Microbiol. Rev. 1991, 55, 206–224. [Google Scholar] [PubMed]
- Jin, Q.; Yuan, Z.; Xu, J.; Wang, Y.; Shen, Y.; Lu, W.; Wang, J.; Liu, H.; Yang, J.; Yang, F.; et al. Genome sequence of Shigella flexneri 2a: Insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 2002, 30, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Onodera, N.T.; Ryu, J.; Durbic, T.; Nislow, C.; Archibald, J.M.; Rohde, J.R. Genome sequence of Shigella flexneri serotype 5a strain M90T Sm. J. Bacteriol. 2012, 194, 3022. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, S.; Levy, I.; Kazaronovski, V.; Samra, Z. Growing antimicrobial resistance of Shigella isolates. J. Antimicrob. Chemother. 2003, 51, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J. Shigellosis: An old disease in new clothes? PLoS Med. 2006, 3, e354. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.J.; Chopra, I.; Fishwick, C.W. Structure-based discovery of antibacterial drugs. Nat. Rev. Microbiol. 2010, 8, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; An, J.G.; Lee, Y.S.; Seok, S.H.; Yoo, H.S.; Seo, M.D. Overexpression, purification, crystallization and preliminary X-ray crystallographic analysis of SF173 from Shigella flexneri. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Jancarik, J.; Scott, W.G.; Milligan, D.L.; Koshland, D.E., Jr.; Kim, S.H. Crystallization and preliminary X-ray diffraction study of the ligand-binding domain of the bacterial chemotaxis-mediating aspartate receptor of Salmonella typhimurium. J. Mol. Biol. 1991, 221, 31–34. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. Part A 1997, 276, 307–326. [Google Scholar]
- Hendrickson, W.A.; Teeter, M.M. Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulphur. Nature 1981, 290, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Adams, P.D.; Read, R.J.; McCoy, A.J.; Moriarty, N.W.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Zwart, P.H.; Hung, L.-W. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 582–601. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.; Eisenhaber, F.; Argos, P. Principles of helix-helix packing in proteins: The helical lattice superposition model. J. Mol. Biol. 1996, 255, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Park, J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.; Chang, H.Y.; Daugherty, L.; Fraser, M.; Hunter, S.; Lopez, R.; McAnulla, C.; McMenamin, C.; Nuka, G.; Pesseat, S.; et al. The InterPro protein families database: The classification resource after 15 years. Nucleic Acids Res. 2015, 43, D213–D221. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Chang, C.; Coggill, P.; Bateman, A.; Finn, R.D.; Cymborowski, M.; Otwinowski, Z.; Minor, W.; Volkart, L.; Joachimiak, A. The structure of pyogenecin immunity protein, a novel bacteriocin-like immunity protein from Streptococcus pyogenes. BMC Struct. Biol. 2009, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Chavan, M.A.; Riley, M.A. Molecular evolution of bacteriocins in Gram-negative bacteria. In Bacteriocins; Springer: Berlin, Germany, 2007; pp. 19–43. [Google Scholar]
- Han, K.D.; Matsuura, A.; Ahn, H.C.; Kwon, A.R.; Min, Y.H.; Park, H.J.; Won, H.S.; Park, S.J.; Kim, D.Y.; Lee, B.J. Functional identification of toxin-antitoxin molecules from Helicobacter pylori 26695 and structural elucidation of the molecular interactions. J. Biol. Chem. 2011, 286, 4842–4853. [Google Scholar] [CrossRef] [PubMed]
- Kwon, A.R.; Kim, J.H.; Park, S.J.; Lee, K.Y.; Min, Y.H.; Im, H.; Lee, I.; Lee, B.J. Structural and biochemical characterization of HP0315 from Helicobacter pylori as a VapD protein with an endoribonuclease activity. Nucleic Acids Res. 2012, 40, 4216–4228. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, P.; Kozlov, G.; Gabrielli, L.; Elias, D.; Osborne, M.J.; Gallouzi, I.E.; Gehring, K. Solution structure of YaeO, a Rho-specific inhibitor of transcription termination. J. Biol. Chem. 2007, 282, 23348–23353. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, E.; Hartmann, R.K. The enigma of ribonuclease P evolution. Trends Genet. TIG 2003, 19, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Graille, M.; Mora, L.; Buckingham, R.H.; van Tilbeurgh, H.; de Zamaroczy, M. Structural inhibition of the colicin D tRNase by the tRNA-mimicking immunity protein. EMBO J. 2004, 23, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, J.; Frank, P.; Loffler, E.; Bennett, K.L.; Gerner, C.; Rossmanith, W. RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008, 135, 462–474. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Won, H.-S.; Yoon, W.-S.; Seok, S.-H.; Jung, B.-J.; Lee, S.-N.; Sim, D.-W.; Seo, M.-D. Crystal Structure of Shigella flexneri SF173 Reveals a Dimeric Helical Bundle Conformation. Crystals 2018, 8, 97. https://doi.org/10.3390/cryst8020097
Kim J-H, Won H-S, Yoon W-S, Seok S-H, Jung B-J, Lee S-N, Sim D-W, Seo M-D. Crystal Structure of Shigella flexneri SF173 Reveals a Dimeric Helical Bundle Conformation. Crystals. 2018; 8(2):97. https://doi.org/10.3390/cryst8020097
Chicago/Turabian StyleKim, Ji-Hun, Hyung-Sik Won, Won-Su Yoon, Seung-Hyeon Seok, Bong-Jun Jung, Seu-Na Lee, Dae-Won Sim, and Min-Duk Seo. 2018. "Crystal Structure of Shigella flexneri SF173 Reveals a Dimeric Helical Bundle Conformation" Crystals 8, no. 2: 97. https://doi.org/10.3390/cryst8020097
APA StyleKim, J.-H., Won, H.-S., Yoon, W.-S., Seok, S.-H., Jung, B.-J., Lee, S.-N., Sim, D.-W., & Seo, M.-D. (2018). Crystal Structure of Shigella flexneri SF173 Reveals a Dimeric Helical Bundle Conformation. Crystals, 8(2), 97. https://doi.org/10.3390/cryst8020097