1. Introduction
Crystals of the potassium titanyl phosphate KTiOPO
4 family (KTP) are materials of a special type, namely, ferroelectrics-superionic conductors. They exhibit high nonlinear susceptibility and stability to external effects over a wide temperature range. KTP crystals are widely used in nonlinear optical devices, in particular, for doubling and tuning the frequency of laser radiation [
1,
2,
3]. They are employed as active elements in electrooptic modulators of solid-state lasers for parametric light generation and as waveguides in integrated optics [
4,
5,
6,
7].
For nonlinear optics applications, high-resistance (ρ ~ 10
7–10
11 Ω cm) crystals with a low defect concentration are suitable. Therefore, obtaining large, optically pure crystals of the KTP family is a highly important technological concern [
8]. Concurrently, the search for new compounds with nonlinear optical characteristics exceeding those of the pure (undoped) KTP crystals is ongoing. The search for compounds with improved nonlinear optical characteristics is carried out through the use of multiple isomorphous substitutions. As of this writing, the intensity of the second harmonic generation (SHG) signal is found to increase by a factor of 1.6 in KTiOAsO
4 (KTA) crystals, which remain isostructural to KTP crystals with complete substitution of arsenic for phosphorus [
9]. The SHG signal increases by 10% and 20% upon the partial (~5% and 4% respectively) substitution of niobium for titanium [
10,
11,
12], by a factor of approximately two upon the partial substitution of zirconium for titanium [
13,
14], and by 30–40% in KTP crystals doped with hafnium [
15].
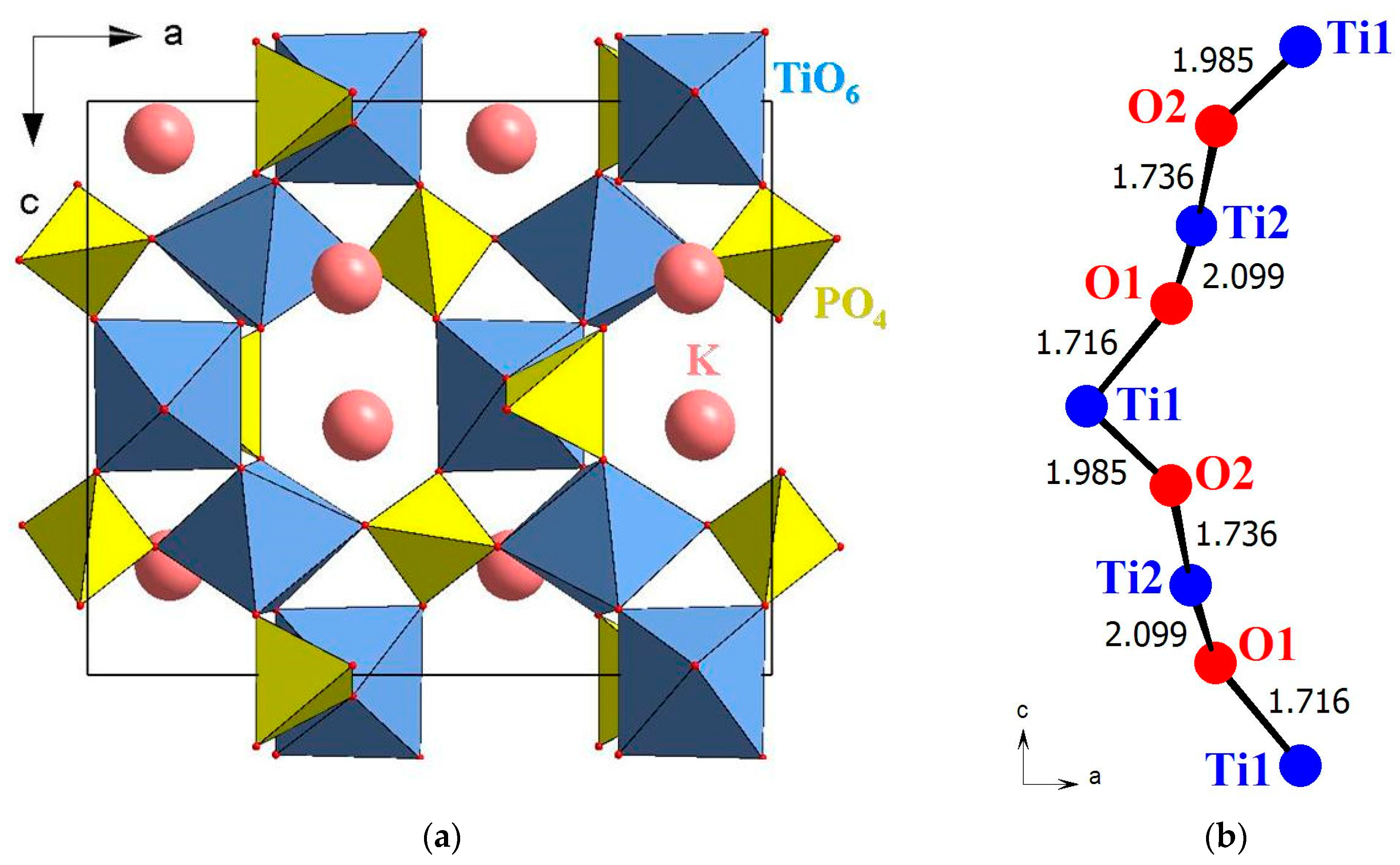
The unique combination of physical properties derives from the specific features of the crystal structure of KTP (polar space group
Pna2
1 [
16] in the ferroelectric phase existing below 934 °С [
17]). It is a rigid three-dimensional framework consisting of alternating vertex-sharing TiO
6 octahedra and PO
4 tetrahedra with channels running along the
c axis (
Figure 1a). The channels contain cations of potassium or other alkali elements. The high ionic conductivity of KTP crystals is caused by vacancies at alkali element positions and diffusion of cations over these vacancies. Relaxation of potassium atoms, which is connected to the formation of alkali cation-vacancy dipoles, appears on the temperature-permittivity plot as a broad anomaly in a range from 200 to 700 °С. The state of the cation sublattice has been shown to affect the ferroelectric properties of crystals of this family [
18].
Until recently, the nonlinear optical susceptibility of KTP crystals was associated with the alternation of long and short Ti–O bonds in the chains of TiO
6 octahedra (
Figure 1b) resulting in a non-uniform distribution of the electron density among these structural units. Such distribution creates anharmonicity in induced vibrations of atoms in a crystal exposed to a harmonic electromagnetic wave, and thereby the appearance of the nonlinear optical properties. The correlation between the SHG signal intensity and the anharmonicity of effective displacements of atoms in the octahedron chains can be clearly traced in continuous series of solid solutions K(Ti
1−xGe
x)OPO
4 [
19] and K(Ti
1−xSn
x)OPO
4 [
20]. The stronger the distortions in the octahedra, the higher the intensity of the SHG signal.
Sometimes, the intensity of SHG signal decreases in compounds with strongly distorted octahedra [
2] (for example, in NaTiOPO
4 with complete substitution of sodium for potassium). On the other hand, the optical nonlinearity of KTP crystals doped with zirconium [
21] and of KTA crystals [
22] is larger than that of crystals of pure KTP, but their octahedra are less distorted. Analyzing AsO
4 tetrahedra, the authors of [
22] noticed a decrease in the Ti–O–As angles by 2–5° in comparison with the Ti–O–P angles in PO
4 tetrahedra, but the existence of chains of TiO
6 octahedra with alternating long and short Ti–O bonds in the structure was assumed to be the main reason for the high optical susceptibility of KTA crystals.
The study of the structure and distribution of the electron density in RTA crystals at
T = 9.6 K and room temperature [
23,
24] revealed asymmetric dipole-like peaks of electron density near the Ti and Rb atomic positions in the
c direction. The optical susceptibility of these crystals was attributed to the possible localization of the electron density, as well as to charge transfer along the Ti–O–Ti–O and Ti–O–As–O chains.
At the end of the 20th century, studies [
17,
25,
26] appeared in which the optical susceptibility of KTP crystals was associated with PO
4 tetrahedra and KO
8/KO
9 groups. The authors of [
25] calculated the polarizability of the bonds in each of the structural groups of many nonlinear optical compounds, taking into account the fact that each type of constituent chemical bond contributed to the total linearity and nonlinearity of the crystal. In both KTP and KTA compounds, the origin of nonlinearity was concluded to be KO
x (
x = 8, 9) groups and P(As)O
4 groups. The authors of [
26] believed that a strong distortion of octahedra was a necessary, but not sufficient, condition for high nonlinearity in crystals of the KTP family. In the opinion of the authors of [
27], one should not to confine oneself to analyzing only a part of the structure while neglecting the remaining atoms when studying the causes of nonlinear optical properties. The second harmonic generation was shown to be connected with the acentricity of the atomic structure as a whole in the course of theoretical study on the relation between the yield of the second harmonic of laser radiation and degree of centrosymmetricity of the electric potential function of the whole atomic structure of the KTP crystal [
27].
In situ neutron powder diffraction analysis from 297 to 1358 K [
28] indicated the largest average displacements of K
+ and P
5+ cations during the transition from paraelectric (space group
Pnna in the authors’ set) to ferroelectric (space group
Pna2
1) phase. The average values of the displacements at 297 K were 0.16, −0.59, 0.04 and 0.14 Å for O
2−, K
+, Ti
4+, and P
5+ ions, respectively. Total spontaneous polarization was estimated to be −0.19 μC cm
−2. Contributions of O
2−, K
+, Ti
4+, and P
5+ ions to the total spontaneous polarization were −0.23, −0.087, 0.016 and 0.105 μC cm
−2, respectively. The intensity of SHG decreases with increasing temperature [
17], and the decrease in the spontaneous polarization with increasing temperature corresponds to the dependence of the SHG intensity on the temperature ~(1 −
T/
TC)
0.5. Thus, the results of studying the paraelectric phase of KTP crystals also suggest that the K
+ and P
5+ ions significantly affect the bond polarizability and consequently, the PO
4 tetrahedra and the KO
8/KO
9 groups contribute to the nonlinear susceptibility of crystals of the KTP family. The results of a study of neutron total (Bragg and diffusion) scattering in KTP crystals [
29] from room temperature to 900 °C additionally suggested that changes in the local arrangement of oxygen atoms around Ti
4+ and the displacement of K
+ were the reasons for the SHG signal decrease with increasing temperature, and therefore, a significant part of the SHG effect came from potassium cations.
Recent publications with
ab initio computations for the electronic structure and nonlinear optical characteristics of KTP-type crystals [
30,
31,
32] emphasized the distortion of TiO
6 octahedra, originating the superior nonlinear optical properties of these crystals. The distorted octahedron allows the dipolar excited states to mix with the bonding electronic states producing a strong hyperpolarizability on the Ti–O bonds [
30]. In [
31], it was found that the band structure of KTP-type phosphate crystals was dominated by Ti and O states and weakly dependent on the nature of the alkali-site element. Thus, the nonlinear optical properties of KTP-type solid solutions should be weakly influenced by ion substitution at the alkali-site positions. The observed anisotropy in the linear optical susceptibilities [
32] was shown to provide enhanced phase matching conditions for the second harmonic generation. The strongest contributions to optical dielectric constant peaks were found to be due to the inter-band transitions between the valence-band maximum and conduction-band minimum. These transitions are often originated from O-2
p to Ti-3
d states.
To date, a large number of studies of the atomic structure of crystals of the KTP family have been conducted, some of them at the Institute of Crystallography [
33]. These studies were quite high level, but in order to analyze the defect structure of single crystals and structural reasons for their physical properties, a new, accurate approach to data collection was needed. This approach was used in the study of the atomic structure of crystals of pure KTP [
34], specially selected single crystals (KTA [
35], KTP, doped with zirconium (KTP:Zr) [
36], hafnium (KTP:Hf) [
37], and niobium (KTP:Nb) [
38]) whose nonlinear susceptibility was higher than that of KTP crystals. This review summarizes the results of those studies.
3. Single Crystals of Pure KTP
The purpose of the accurate X-ray diffraction study of pure KTP single crystals grown by one method (crystallization from the solution in the melt) in two ways (the top-seeded solution growth and spontaneous flux crystallization) [
34] was to obtain the most complete and precise data on their structure in order to first, evaluate how the growth conditions affect the structure of the KTP crystals and second, to use the obtained data in the further investigations of the structure of KTP crystals doped with isovalent or heterovalent impurities. An analysis of the results of the refinement of the structure of KTP single crystals grown by one method—but in different ways—made it possible to reveal similarities and differences in their structure. Similarities in the unit cell parameters and the average interatomic distances in the structures were found. Furthermore, the potassium sublattices in the crystals under investigation were characterized by a similar disordering (existence of additional K positions). Despite the similarity in crystal-chemical parameters, the distribution of the electron density in the crystals was different due to different numbers of defects in the crystal structures. On the whole, the analysis of the difference distribution of the electron density suggested that, compared to the crystals grown by the top-seeded solution method, the crystals grown through the spontaneous flux crystallization contained a larger number of defects. This was indicated by higher peaks of the residual electron density near cation positions and by the presence of a larger number of uninterpreted residual peaks in the difference electron density maps.
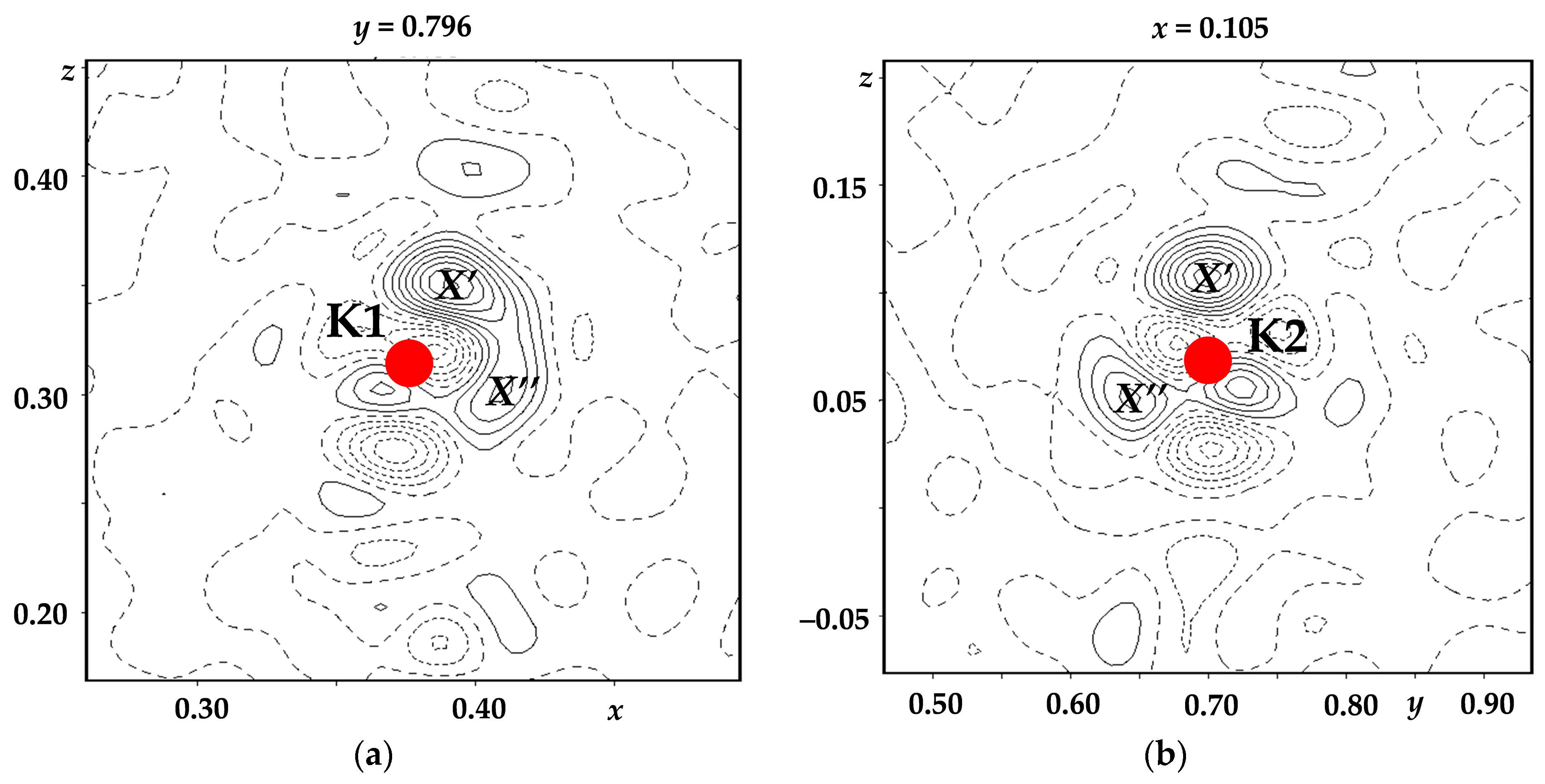
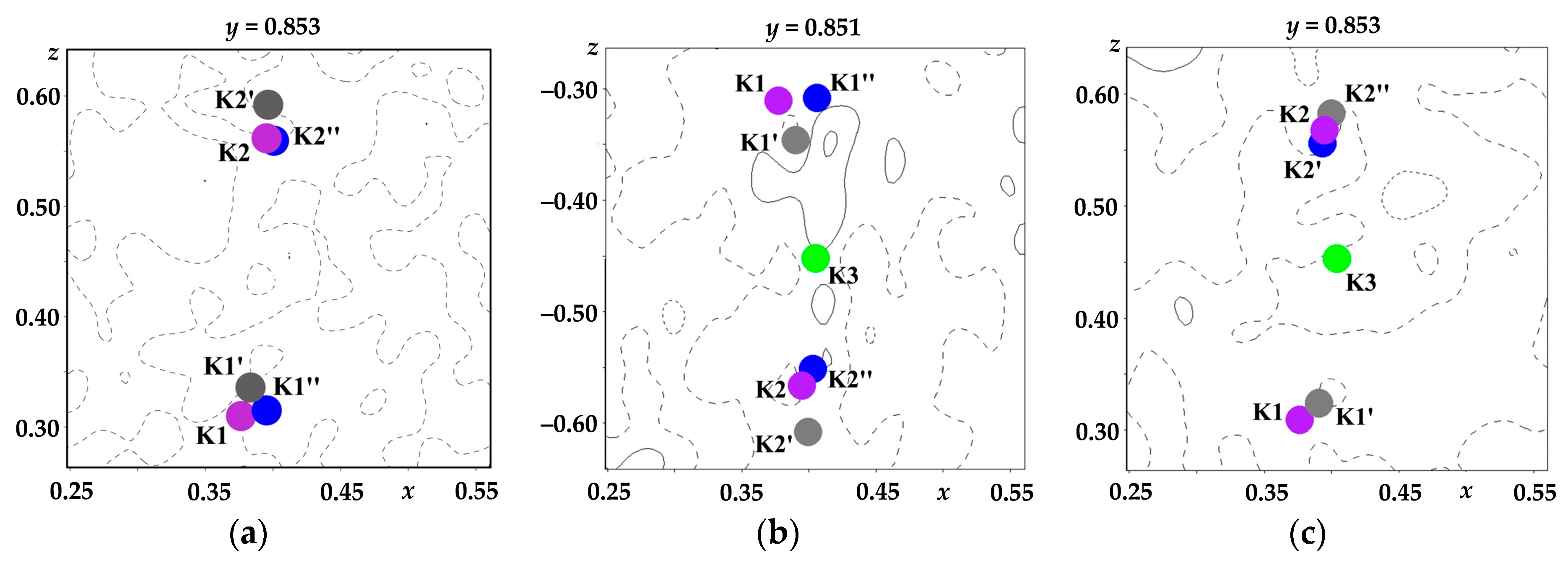
It should be noted that it was possible to perfectly analyze the distribution of potassium ions over the structure channels only due to the high quality of the experiments. A more detailed, in compared to [
34], analysis of the distribution of the residual electron density near the potassium atom positions was carried out in [
46]. In the case of top-seeded crystallization, the peaks
X′ and
X″ (
Figure 2) of the residual electron density Δρ
max = 0.83, 0.41 and 0.76, 0.47 e Å
−3, respectively, were located at distances of ~0.45 and 0.43 Å from the K1 (KO
8 groups) and K2 (KO
9 groups) atoms, respectively. In the case of spontaneous crystallization, analogous peaks were at distances of ~0.45 and 0.44 Å and their heights were Δρ
max = 0.88, 0.46 and 0.77, 0.41 e Å
−3, respectively.
In accordance with these peaks, the atoms K1′, K1″, K2′, and K2″ were localized. The occupancies of the main and additional potassium positions were refined by the step-scan technique [
47], which makes it possible to avoid a strong correlation between the atomic displacement parameters and the position occupancies. The final occupancies were assumed to be equal to the values corresponding to the midpoint of the confidence interval within which the discrepancy factor retained its minimum value. The error in the refinement was registered as equal to half the confidence interval. The occupancies of the main and additional positions of potassium atoms, as well as the distances between them, are listed in
Table 1.







{kind=link}
{kind=link}
{kind=link}
{kind=link}