Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide

Abstract
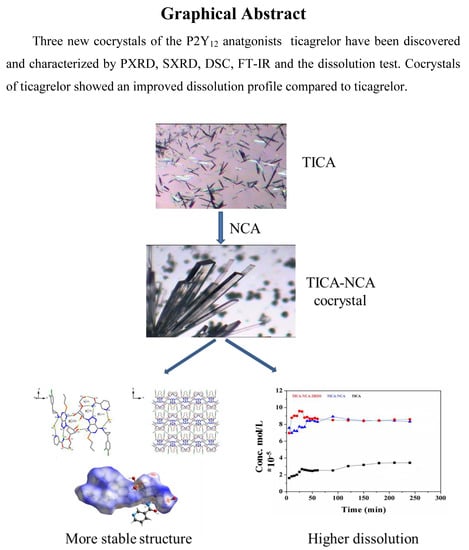
:
1. Introduction
2. Experimental Section
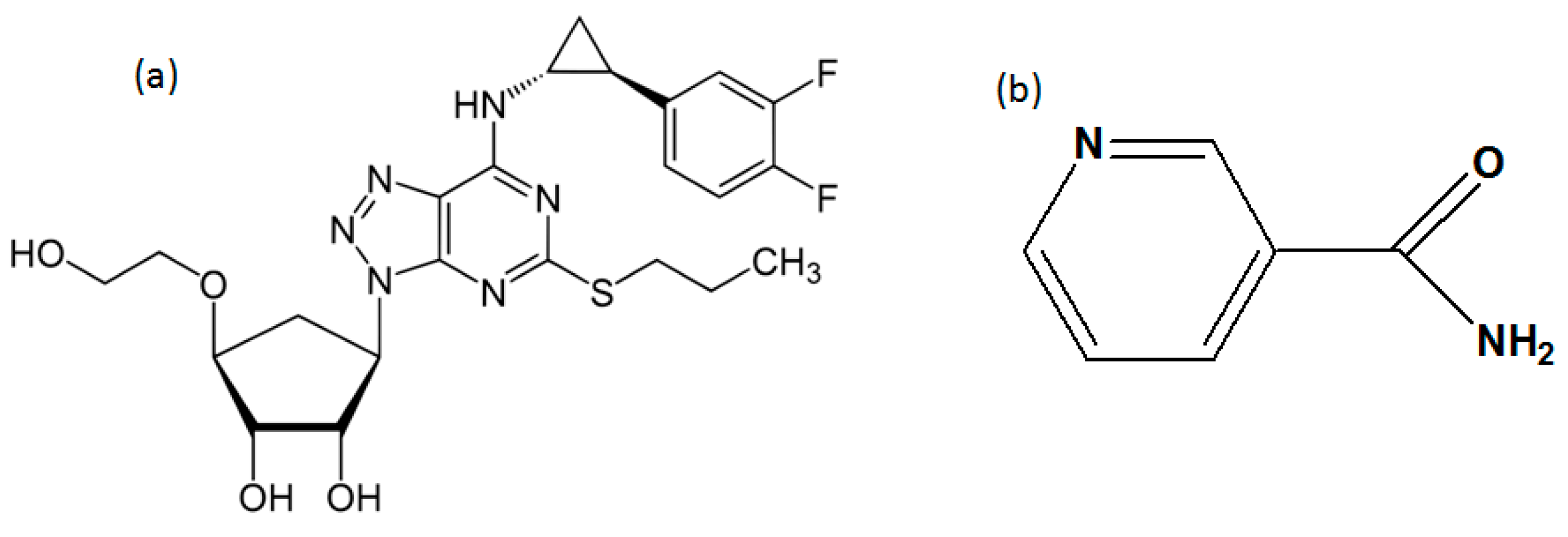
2.1. Materials
2.2. Preparation of New Solid State Forms
2.3. Powder X-ray Diffraction (PXRD) Study
2.4. Differential Scanning Calorimeter (DSC) Study
2.5. Fourier Transform-Infrared Spectroscopy (FT-IR)
2.6. Single Crystal X-ray Diffraction (SCXRD)
2.7. Transformation Study
2.8. Solubility and Dissolution Measurement
3. Results and Discussion
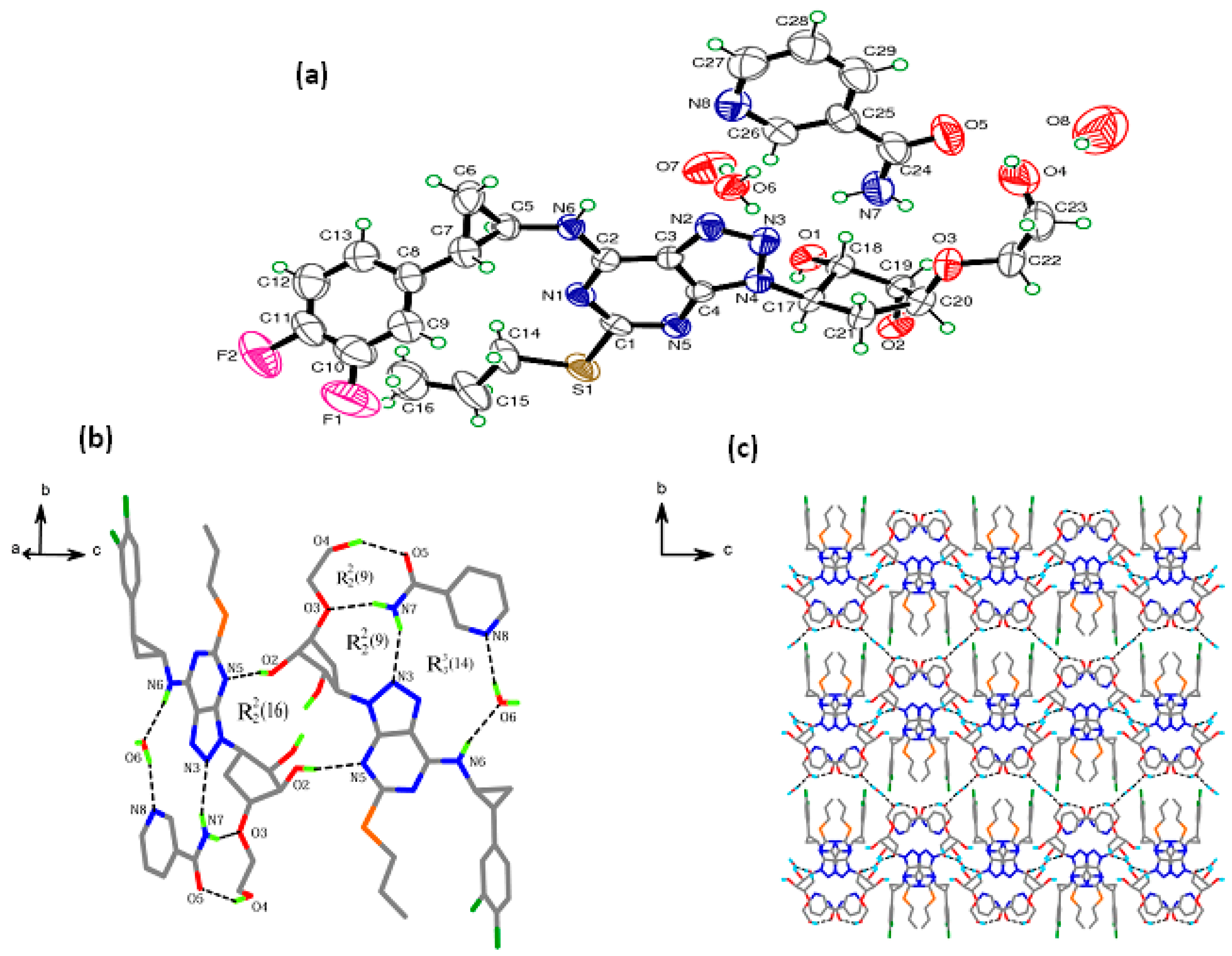
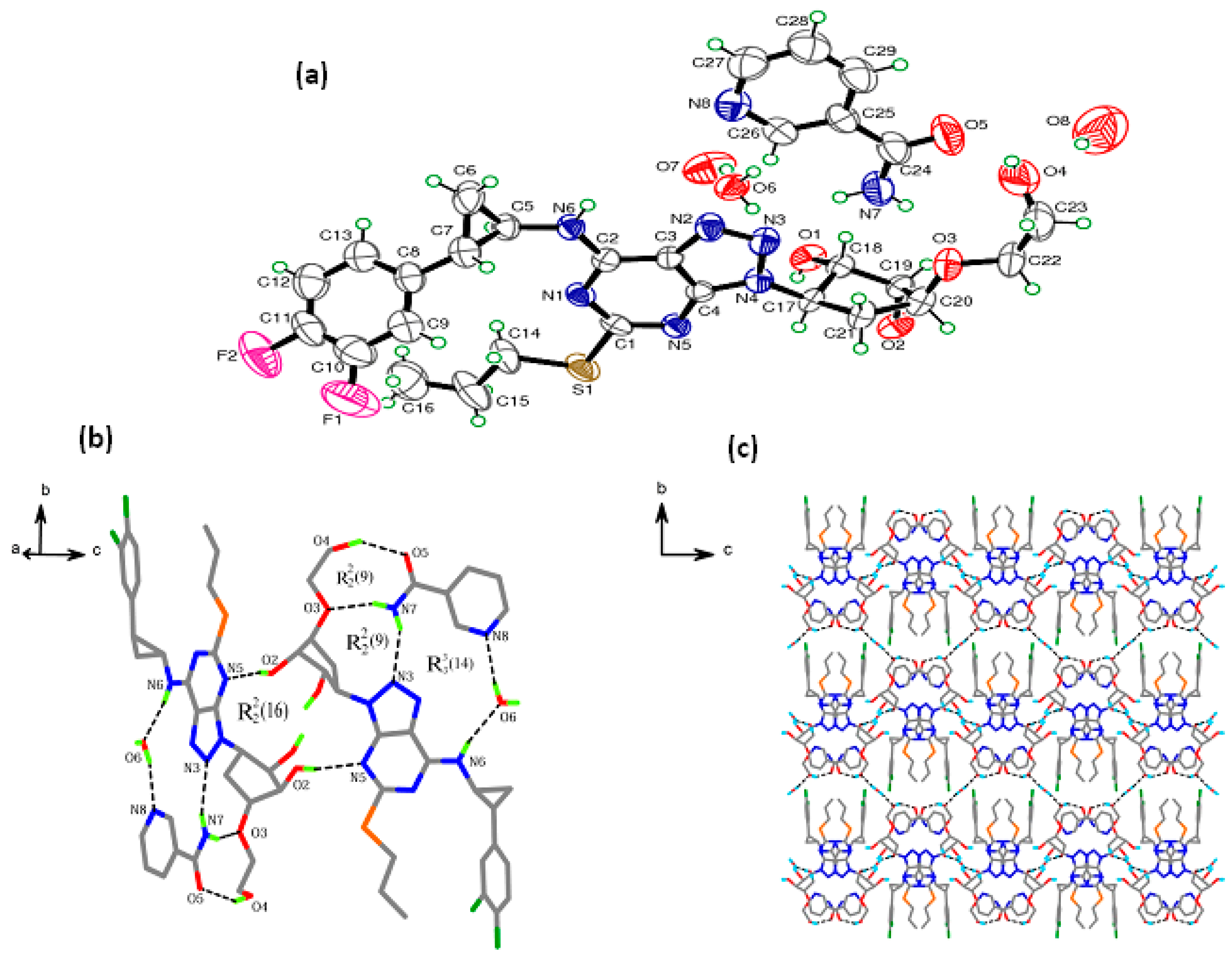
3.1. Crystal Structure Analyses
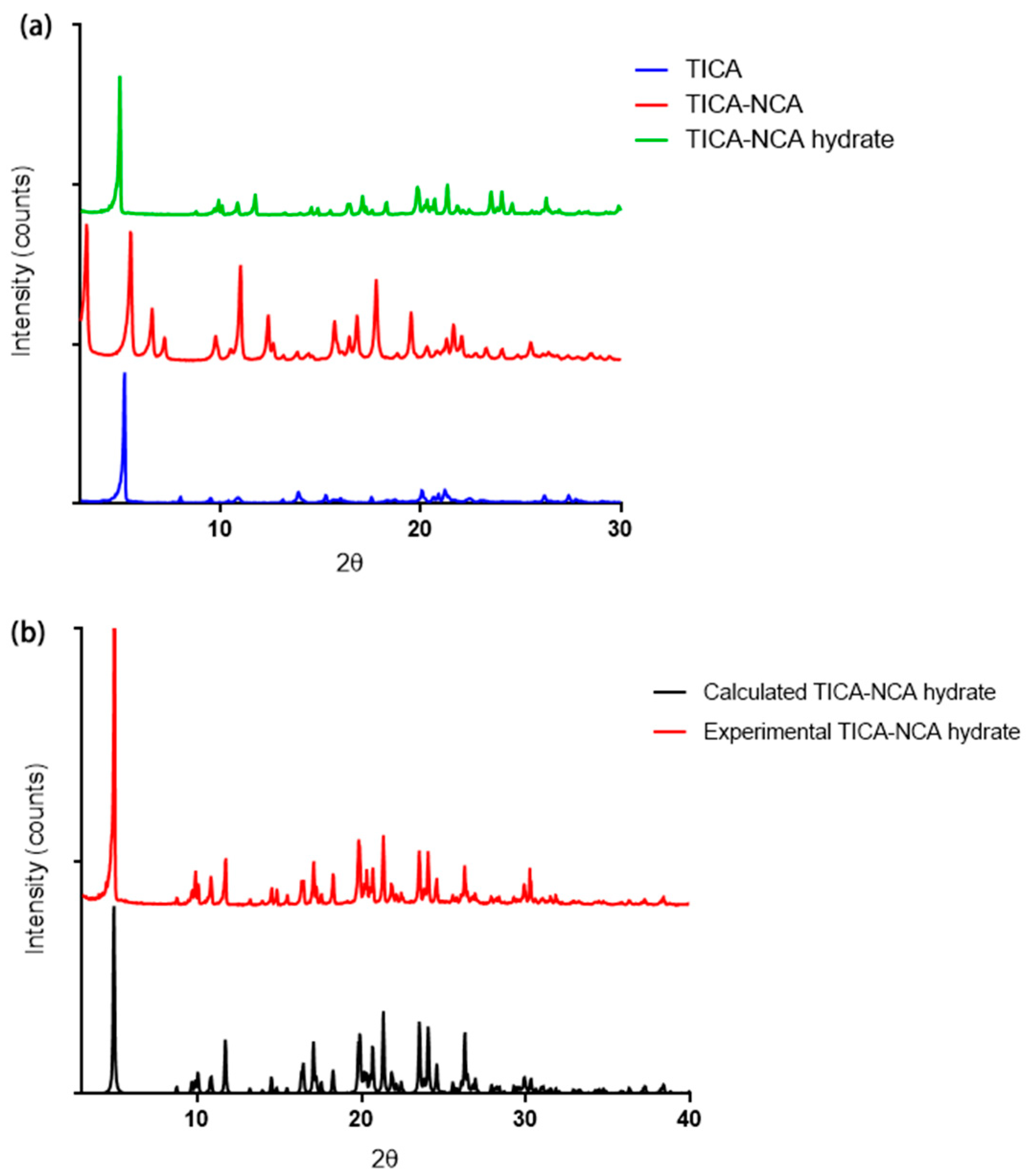
3.2. Powder X-ray Diffraction (PXRD)
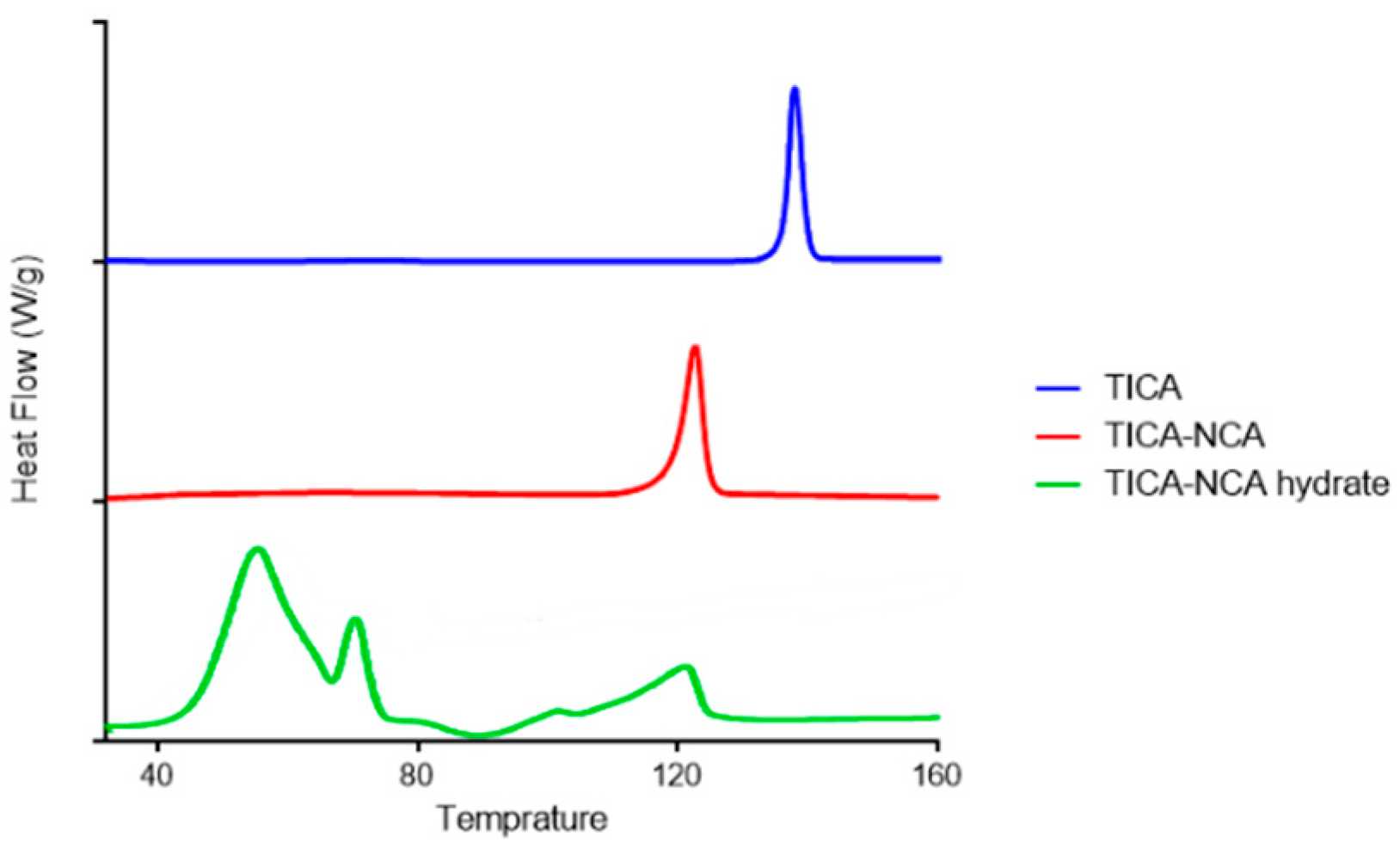
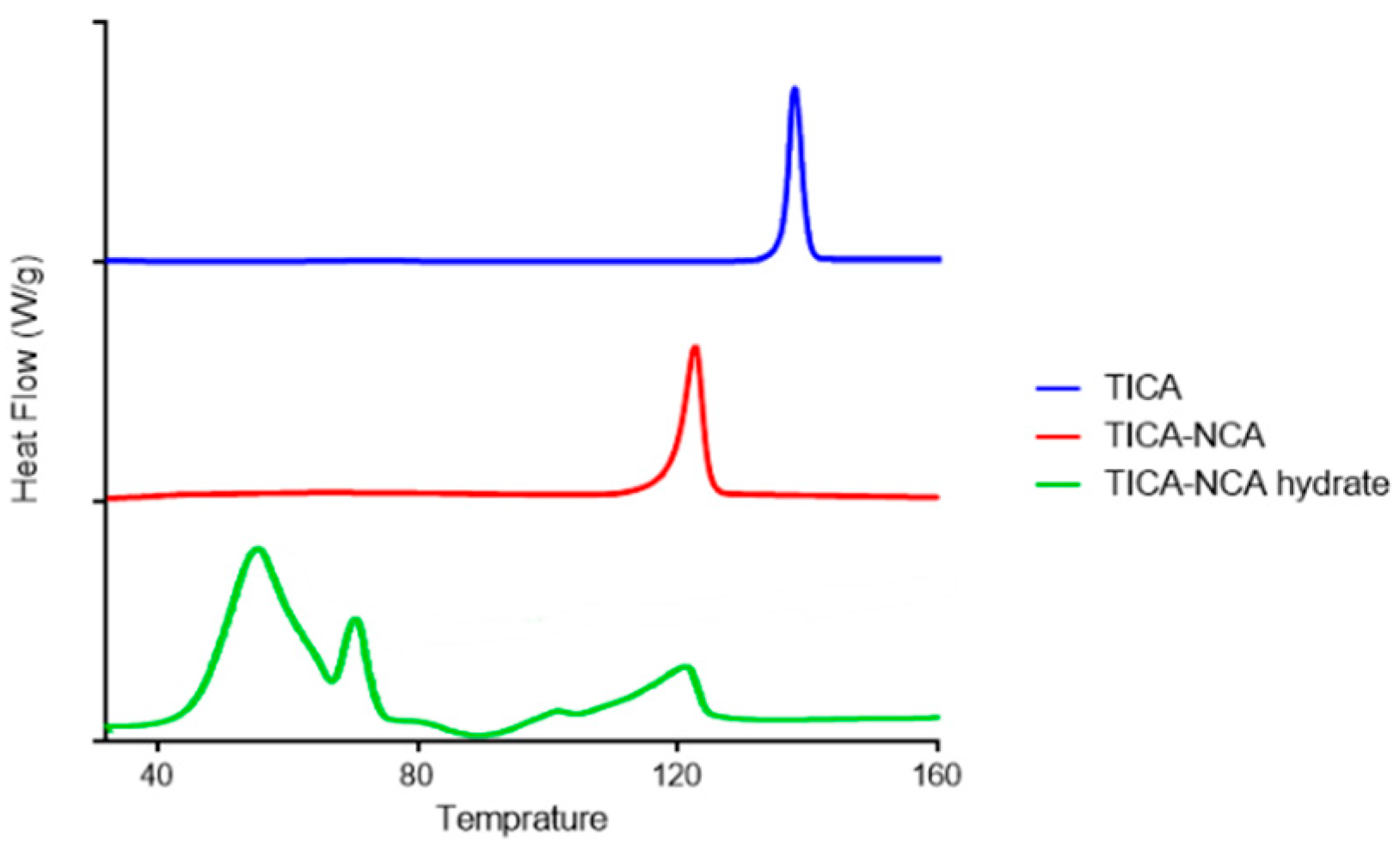
3.3. Differential Scanning Calorimeter (DSC) Analysis
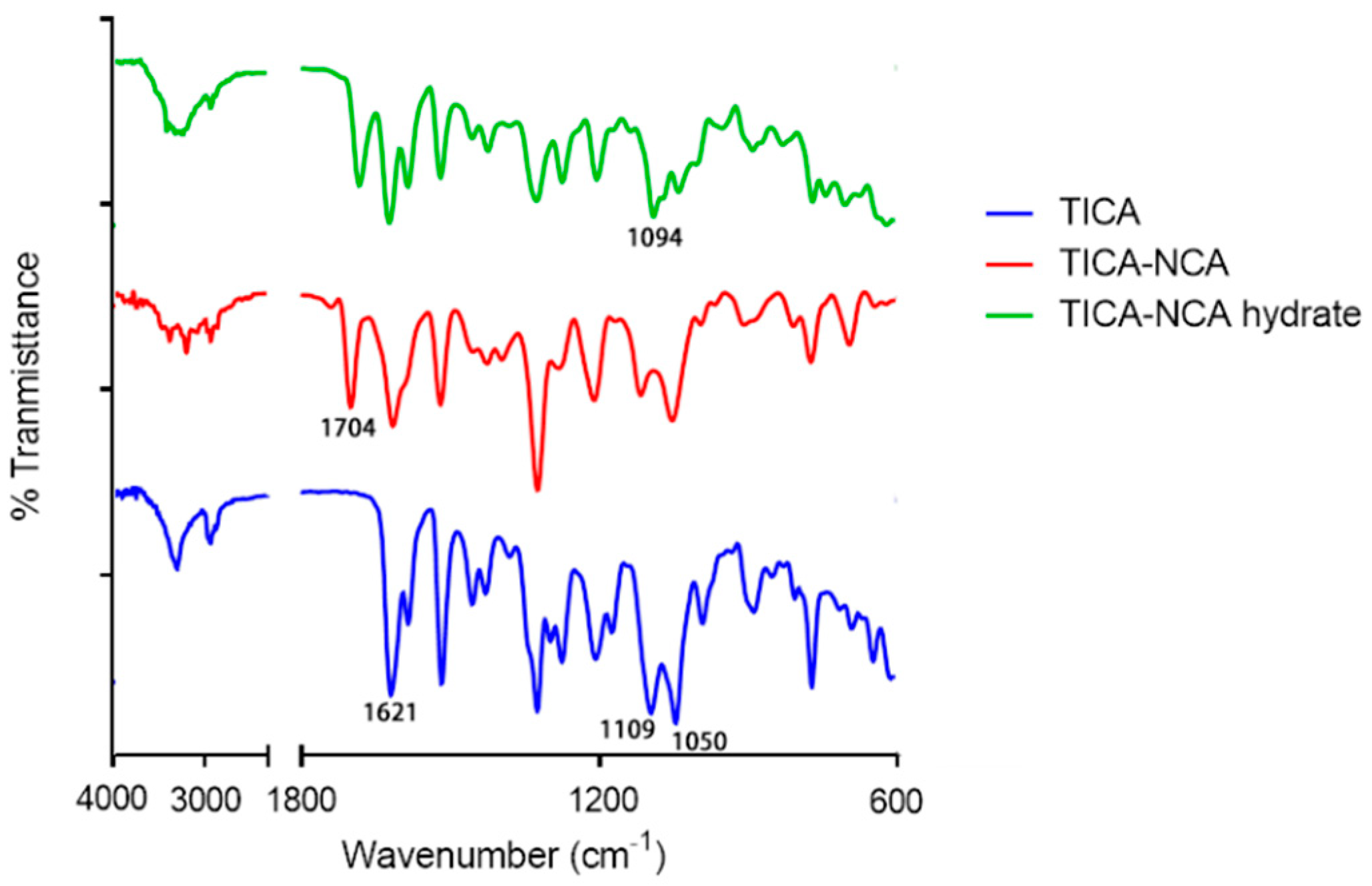
3.4. FT-IR Spectroscopy Analysis
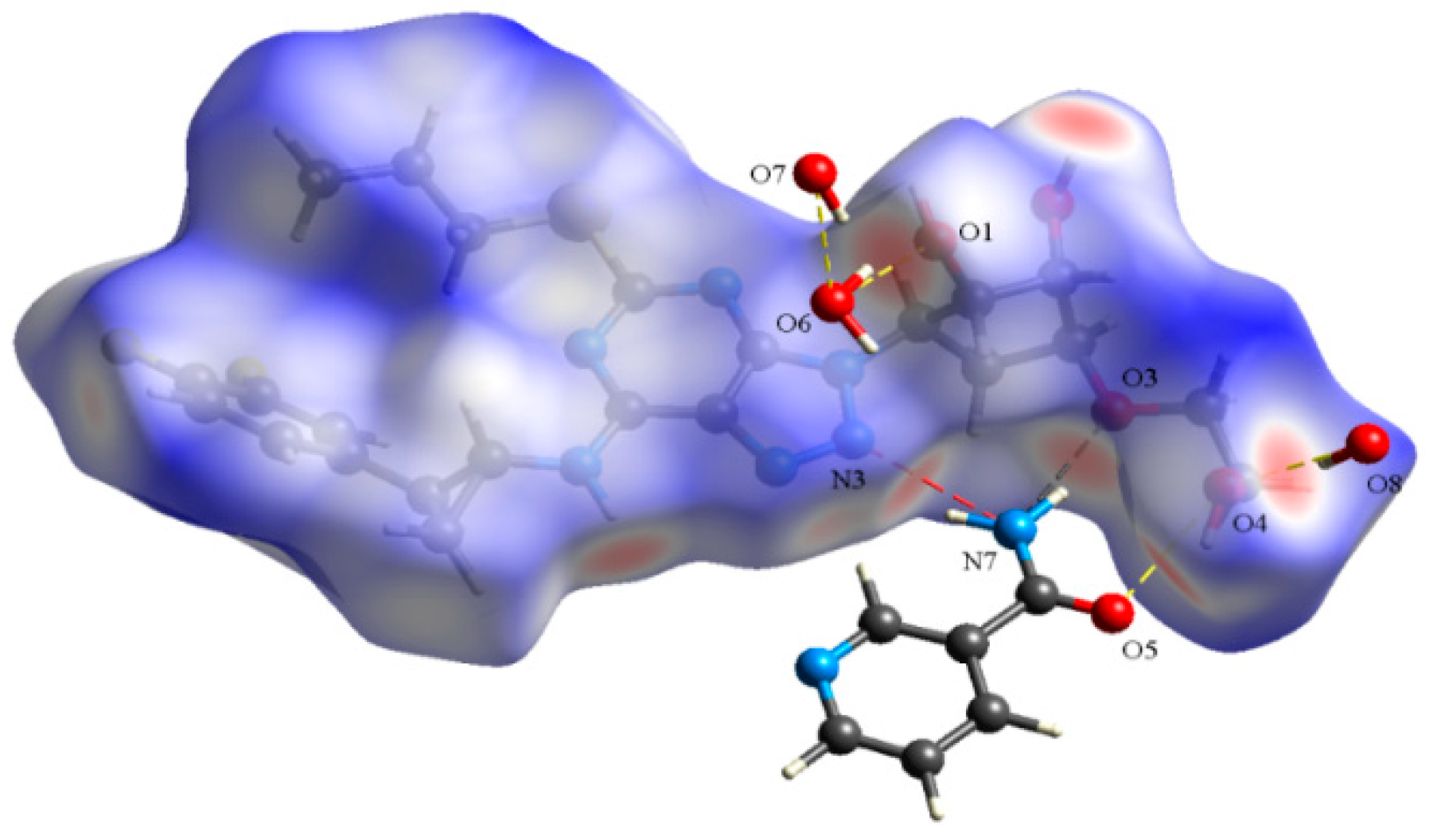
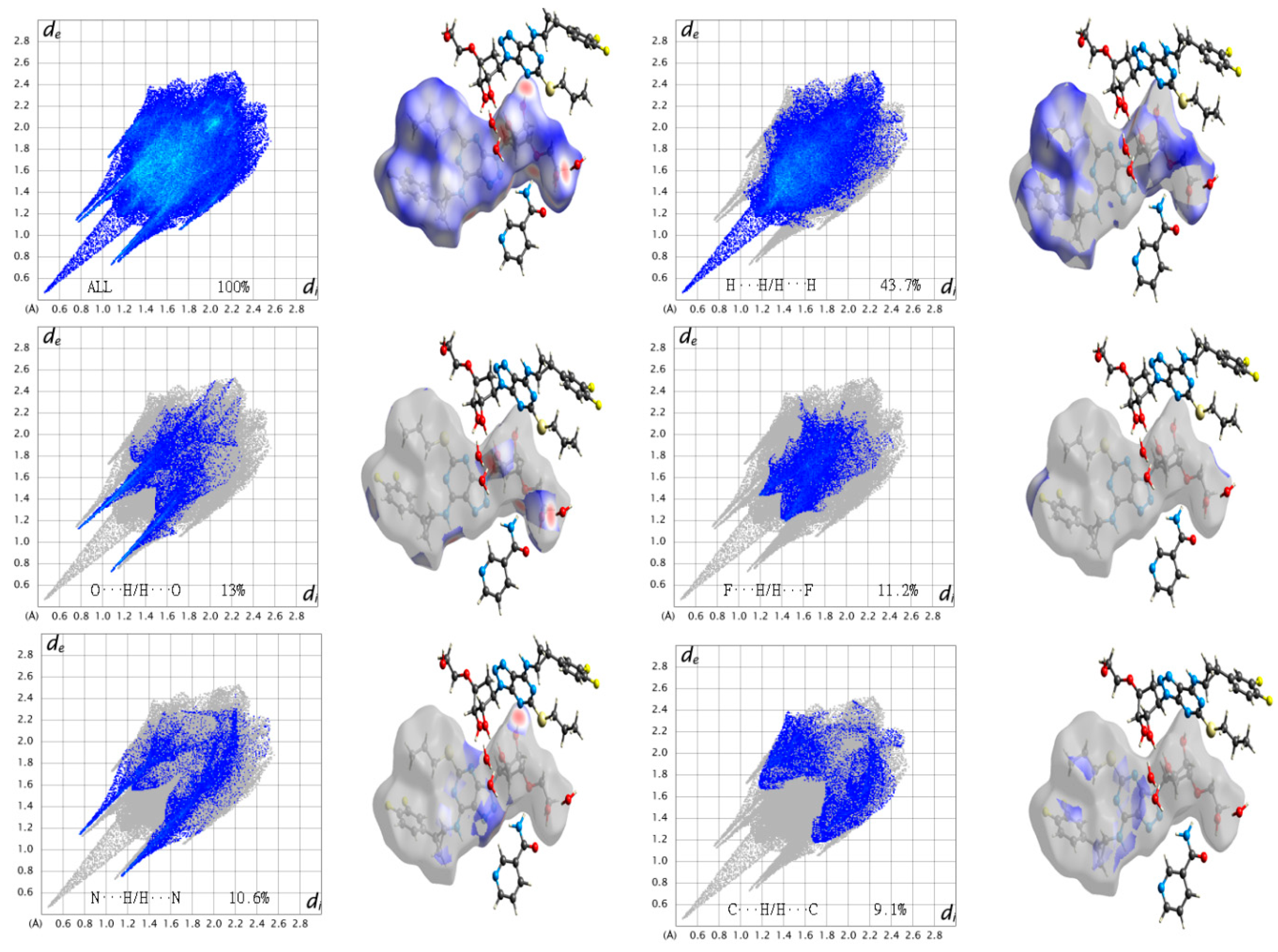
3.5. Hirshfeld Surface (HS) Analysis
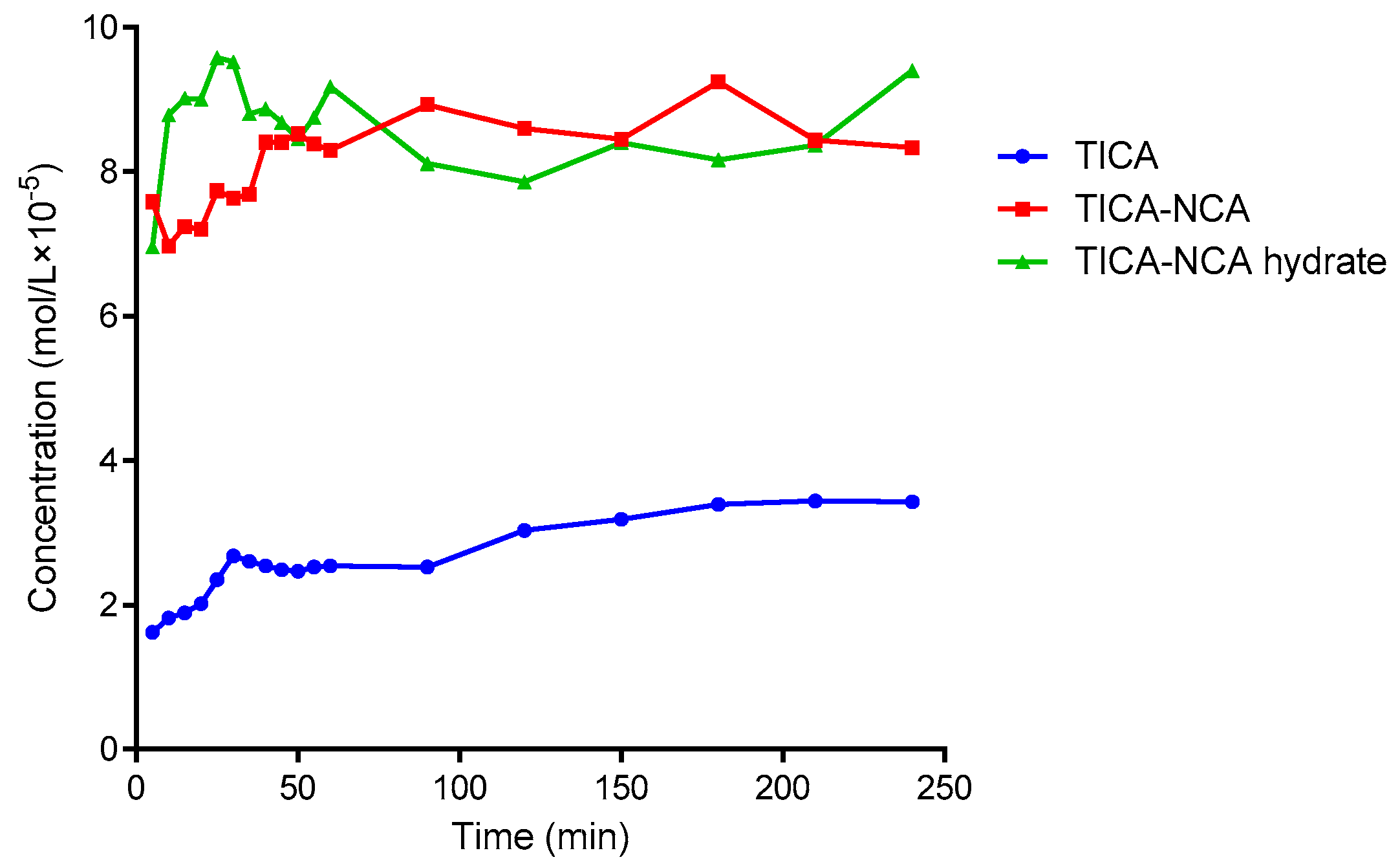
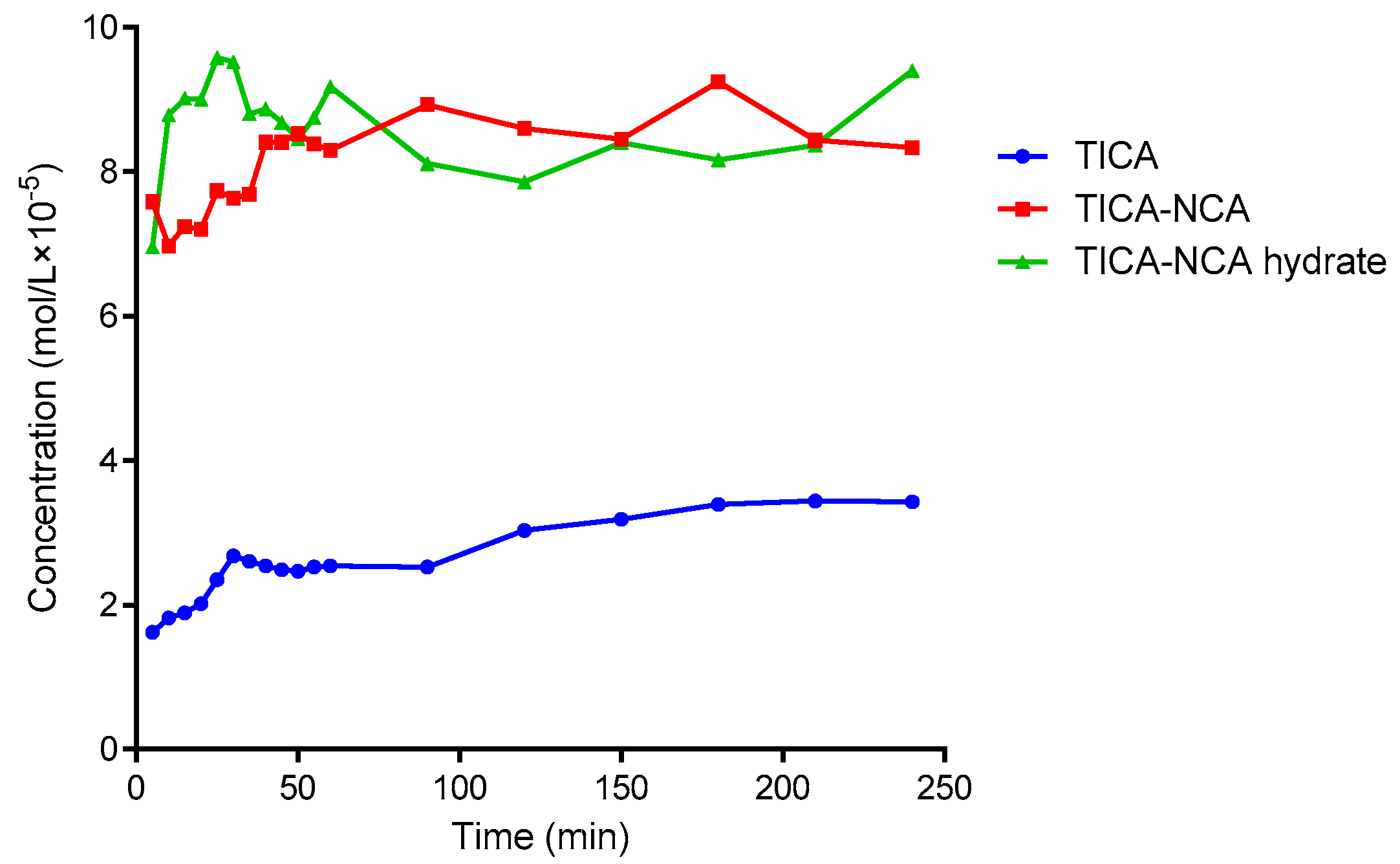
3.6. Solubility and Dissolution Rate
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Acknowledgments
Conflicts of Interest
References
- Marzio, H.D.; Navarro, V.J. Hepatotoxicity of cardiovascular and antidiabetic drugs. In Drug-Induced Liver Disease, 3rd ed.; Elsevier: New York, NY, USA, 2013; Chapter 29; pp. 519–540. [Google Scholar]
- Bojarska, J.; Remko, M.; Fruziński, A.; Maniukiewicz, W. The experimental and theoretical landscape of a new antiplatelet drug ticagrelor: Insight into supramolecular architecture directed by C-H…F, π-π and C-H…π interactions. J. Mol. Struct. 2018, 1154, 290–300. [Google Scholar] [CrossRef]
- Weitz, J.I. Blood coagulation and anticoagulant, fibrinolytic, and antiplatelet drugs. In Goodman and Gilman’s The Pharmacological Basic of Therapeutics, 12th ed.; McGraw Hill: New York, NY, USA, 2011; Chapter 30; pp. 849–877. [Google Scholar]
- Husted, S.; Emanuelsson, H.; Heptinstall, S.; Sandset, P.M.; Wickens, M.; Peters, G. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible p2y (12) antagonist azd6140 with aspirin in patients with atherosclerosis: A double-blind comparison to clopidogrel with aspirin. Eur. Heart J. 2006, 27, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Emanuelsson, H. Safety, tolerability, and initial efficacy of azd6140, the first reversible oral adenosine diphosphate receptor antagonist, compared with clopidogrel, in patients with non–ST-segment elevation acute coronary syndrome: primary results of the DISPERSE-2 trial. J. Am. Coll. Cardiol. 2007, 50, 1844–1851. [Google Scholar]
- Chalasani, N.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Davern, T.; Serrano, J.; Yang, H.; Rochon, J. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008, 135, 1924. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.A.; Goldberg, T.; Dorsch, M.P.; Cheng, J.W.M. Antiplatelet therapy after placement of a drug-eluting stent: A review of efficacy and safety studies. Clin. Ther. 2010, 32, 2265–2281. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M. Drug-induced acute liver failure: Results of a US. Multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Salmon, D.J. Building co-crystals with molecular sense and supramolecular sensibility. CrystEngComm 2005, 7, 439–448. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Mcmahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Zaworotko, M.J. The role of cocrystals in pharmaceutical science. Drug Discovery Today 2008, 13, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular synthons in crystal engineering & mdash; a new organic synthesis. Angew. Chem. Int. Ed. 2010, 34, 2311–2327. [Google Scholar]
- Brittain, H.G. Cocrystal systems of pharmaceutical interest: 2010. Cryst. Growth Des. 2012, 12, 1046–1054. [Google Scholar] [CrossRef]
- Editor, N.R.G. Cocrystals: Molecular design of pharmaceutical materials. Mol. Pharm. 2007, 4, 299–300. [Google Scholar]
- Babu, N.J.; Nangia, A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst. Growth Des. 2011, 11, 2662–2679. [Google Scholar] [CrossRef]
- Aitipamula, S.; Chow, P.S.; Tan, R.B.H. Dimorphs of a 1:1 cocrystal of ethenzamide and saccharin: Solid-state grinding methods result in metastable polymorph. CrystEngComm 2009, 11, 889–895. [Google Scholar] [CrossRef]
- Childs, S.L.; Chyall, L.J.; Dunlap, J.T.; Smolenskaya, V.N.; Stahly, B.C.; Stahly, G.P. Crystal engineering approach to forming cocrystals of amine hydrochlorides with organic acids. Molecular complexes of fluoxetine hydrochloride with benzoic, succinic, and fumaric acids. J. Am. Chem. Soc. 2004, 126, 13335–13342. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zu, H.; Zhang, J. Enhanced dissolution and stability of adefovir dipivoxil by cocrystal formation. J. Pharm. Pharmacol. 2011, 63, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Maddileti, D.; Jayabun, S.K.; Nangia, A. Soluble cocrystals of the xanthine oxidase inhibitor febuxostat. Cryst. Growth Des. 2013, 13, 3188–3196. [Google Scholar] [CrossRef]
- Aitipamula, S.; Wong, A.B.H.; Chow, P.S.; Tan, R.B.H. Novel solid forms of the anti-tuberculosis drug, isoniazid: Ternary and polymorphic cocrystals. CrystEngComm 2013, 15, 5877–5887. [Google Scholar] [CrossRef]
- Tao, Q.; Chen, J.-M.; Ma, L.; Lu, T.-B. Phenazopyridine cocrystal and salts that exhibit enhanced solubility and stability. Cryst. Growth Des. 2012, 12, 3144–3152. [Google Scholar] [CrossRef]
- Sun, C.C.; Hou, H. Improving mechanical properties of caffeine and methyl gallate crystals by cocrystallization. Cryst. Growth Des. 2008, 8, 1575–1579. [Google Scholar] [CrossRef]
- Hickey, M.B.; Peterson, M.L.; Scoppettuolo, L.A.; Morrisette, S.L.; Vetter, A.; Guzman, H.; Remenar, J.F.; Zhang, Z.; Tawa, M.D.; Haley, S.; et al. Performance comparison of a co-crystal of carbamazepine with marketed product. Eur. J. Pharm. Biopharm. 2007, 67, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, S.D.; Jonaitis, D.T.; Sutch, J.C.D. Novel Ticagrelor Co-Crystal. U.S. Patent 20,160,176,912A1, 23 June 2016. [Google Scholar]
- Rigaku. Process-Auto and Crystalstructure; Rigaku Corporation: Tokyo, Japan, 2006. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A-Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. Wingx and ortep for windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND, version 3.1; Crystal Impact GbR: Bonn, Germany, 2005. [Google Scholar]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; Academic Press: Washington, DC, USA, 1982. [Google Scholar]
- Vangala, V.R.; Chow, P.S.; Tan, R.B.H. Co-crystals and co-crystal hydrates of the antibiotic nitrofurantoin: Structural studies and physicochemical properties. Cryst. Growth Des. 2012, 12, 5925–5938. [Google Scholar] [CrossRef]
- Aitipamula, S.; Vangala, V.R.; Chow, P.S.; Tan, R.B.H. Cocrystal hydrate of an antifungal drug, griseofulvin, with promising physicochemical properties. Cryst. Growth Des. 2012, 12, 5858–5863. [Google Scholar] [CrossRef]
- Ueto, T.; Takata, N.; Muroyama, N.; Nedu, A.; Sasaki, A.; Tanida, S.; Terada, K. Polymorphs and a hydrate of furosemide-nicotinamide 1:1 cocrystal. Cryst. Growth Des. 2012, 12, 485–494. [Google Scholar] [CrossRef]
- Eddleston, M.D.; Sivachelvam, S.; Jones, W. Screening for polymorphs of cocrystals: A case study. CrystEngComm 2012, 15, 175–181. [Google Scholar] [CrossRef]
- Abourahma, H.; Cocuzza, D.S.; Melendez, J.; Urban, J.M. Pyrazinamide cocrystals and the search for polymorphs. CrystEngComm 2011, 13, 6442–6450. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic-compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Newman, A.W.; Byrn, S.R. Solid-state analysis of the active pharmaceutical ingredient in drug products. Drug Discovery Today 2003, 8, 898–905. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Watson, D.G.; Taylor, R. International Tables for Crystallography; D. Reidel: Dordrecht, The Netherlands, 1983. [Google Scholar]
- Chen, P.; Zhang, H.B.; Lin, G.D.; Hong, Q.; Tsai, K.R.; Alvarado, V.; Davis, H.T.; Scriven, L.E.; Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, K.; Shekar, B.C.; Khadgapathi, P.; Bhikshapathi, D.; Renuka, K. Development, characterization and in vivo evaluation of tolvaptan solid dispersions via solvent evaporation technique. Int. J. Drug Deliv. 2015, 7, 32–43. [Google Scholar]
- Sugano, K.; Kataoka, M.; Mathews, C.C.S.; Yamashita, S. Prediction of food effect by bile micelles on oral drug absorption considering free fraction in intestinal fluid. Eur. J. Pharm. Sci. 2010, 40, 118–124. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Reference | TICA-NCA Hydrate |
|---|---|
| CCDC No | 1497021 |
| Chemical formula | C29H38F2N8O7S |
| Mr | 608.73 |
| Crystal system | Orthorhombic |
| Space group | P2221 |
| a/Å | 9.1347(5) |
| b/Å | 17.9018(10) |
| c/Å | 20.1929(11) |
| α/° | 90 |
| B/° | 90 |
| γ/° | 90 |
| Unit cell volume/Å | 3302.1(3) |
| No of formula units per unit cell, Z | 4 |
| Radiation type | MoKα |
| μ (mm−1) | 0.167 |
| Temperature/K | 296(2) |
| Crystal size (mm) | 0.43 × 0.38 × 0.07 |
| Dcalc/g cm−3 | 1.369 |
| Rint | 0.1262 |
| Reflections collected | 32621 |
| Unique reflections | 7530 |
| Observed reflections | 3600 |
| Final R1 values (I > 2δ(I)) | 0.0794 |
| Final WR(F2) values (all date) | 0.1609 |
| Goodness-of-fit | 1.006 |
| Atom D,H,A | D–H…Aa | H…A/Å | D…A/Å | D–H…A/° |
|---|---|---|---|---|
| N6–H6–O6i | 0.8600 | 2.1400 | 2.982(4) | 168.000 |
| O6–H62–N8i | 0.8200 | 2.0300 | 2.839(5) | 167.000 |
| O1–H1–O1ii | 0.8200 | 1.9400 | 2.714(6) | 155.000 |
| O2–H2–N5ii | 0.8200 | 2.0600 | 2.868(4) | 170.000 |
| N7–H7A–O3 | 0.8600 | 2.1200 | 2.946(5) | 160.000 |
| N7–H7B–N3 | 0.8600 | 2.2100 | 3.005(5) | 154.000 |
| O4–H4–O5 | 0.8200 | 2.0400 | 2.770(6) | 148.000 |
| O6–H61–O1 | 0.8200 | 1.9600 | 2.768(4) | 165.000 |
| O7–H71–O6 | 0.8300 | 2.1000 | 2.859(5) | 152.000 |
| O8–H81–O4 | 0.8200 | 2.1000 | 2.906(6) | 169.000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inam, M.; Wu, J.; Shen, J.; Phan, C.U.; Tang, G.; Hu, X. Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide. Crystals 2018, 8, 336. https://doi.org/10.3390/cryst8090336
Inam M, Wu J, Shen J, Phan CU, Tang G, Hu X. Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide. Crystals. 2018; 8(9):336. https://doi.org/10.3390/cryst8090336
Chicago/Turabian StyleInam, Muhammad, Jiajia Wu, Jie Shen, Chi Uyen Phan, Guping Tang, and Xiurong Hu. 2018. "Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide" Crystals 8, no. 9: 336. https://doi.org/10.3390/cryst8090336
APA StyleInam, M., Wu, J., Shen, J., Phan, C. U., Tang, G., & Hu, X. (2018). Preparation and Characterization of Novel Pharmaceutical Co-Crystals: Ticagrelor with Nicotinamide. Crystals, 8(9), 336. https://doi.org/10.3390/cryst8090336