Synthesis, Crystal Structure, DFT Studies, Docking Studies, and Fluorescent Properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile


Abstract
1. Introduction
2. Results and Discussion
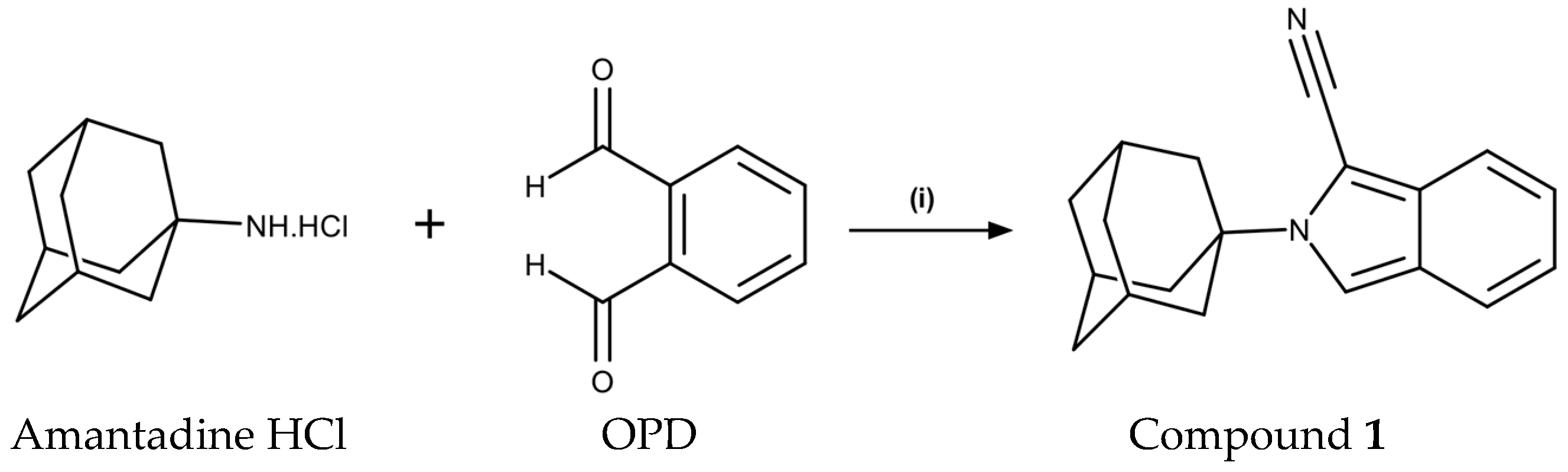
2.1. Chemistry
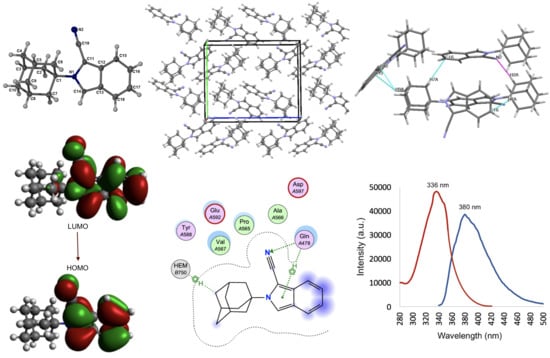
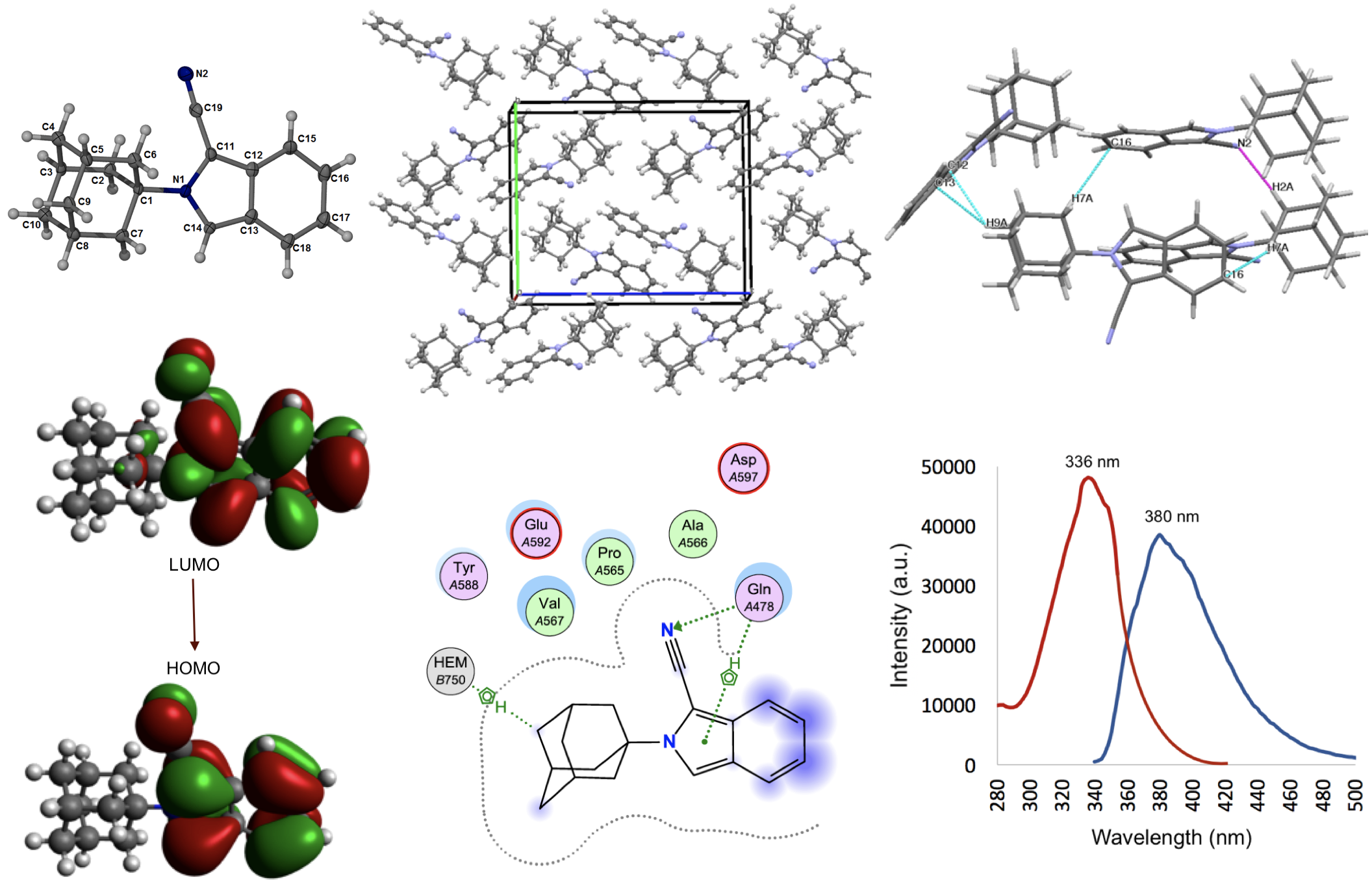
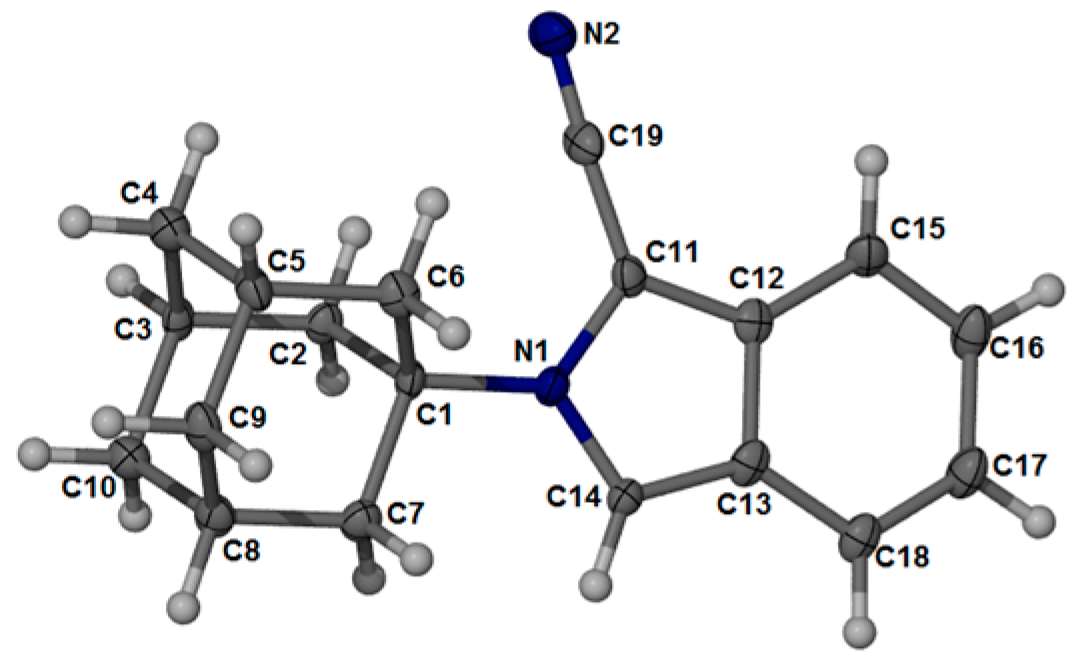
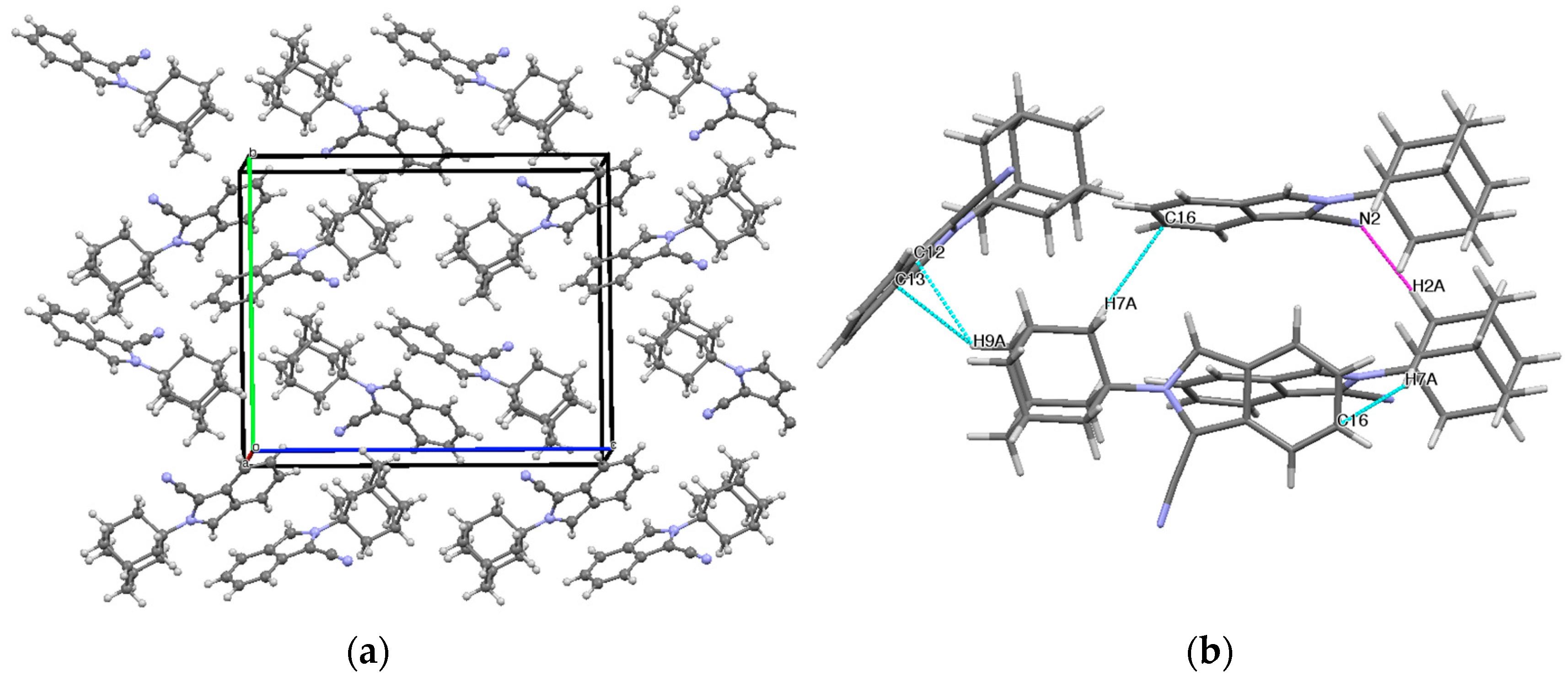
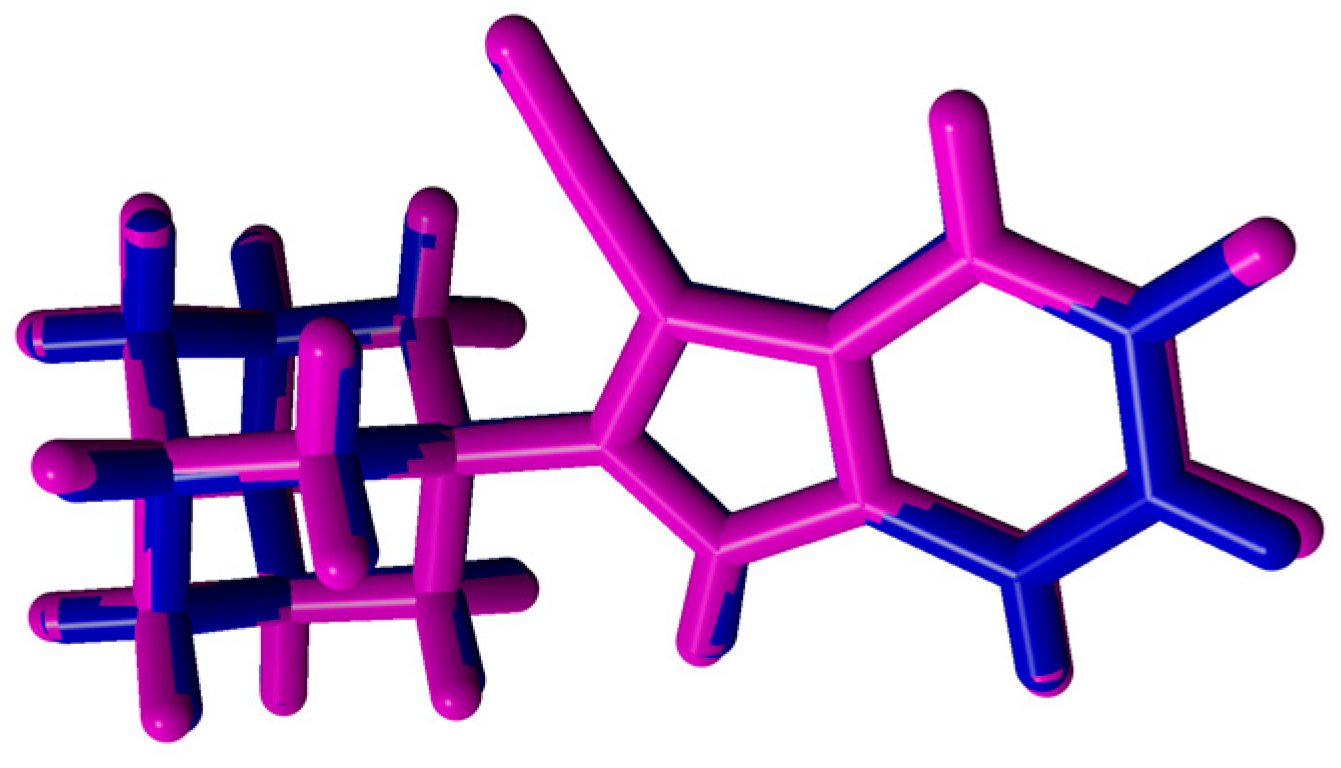
2.2. Crystal Structure and Geometry Optimization
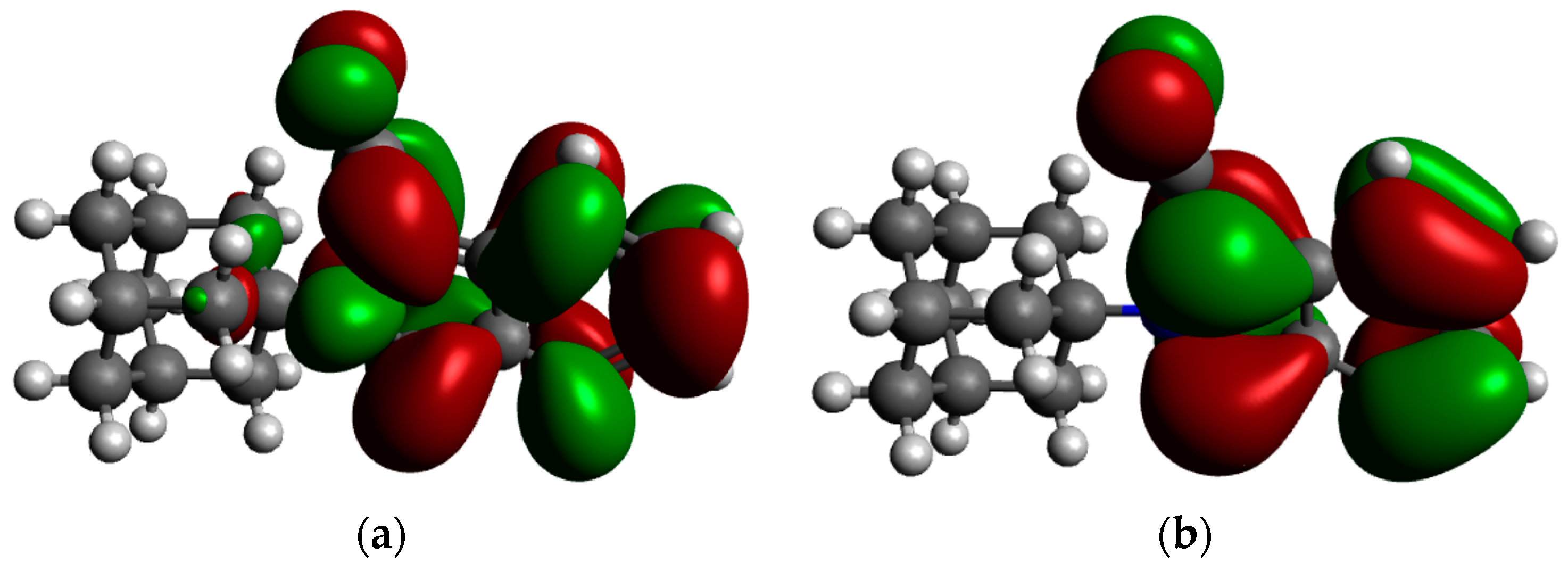
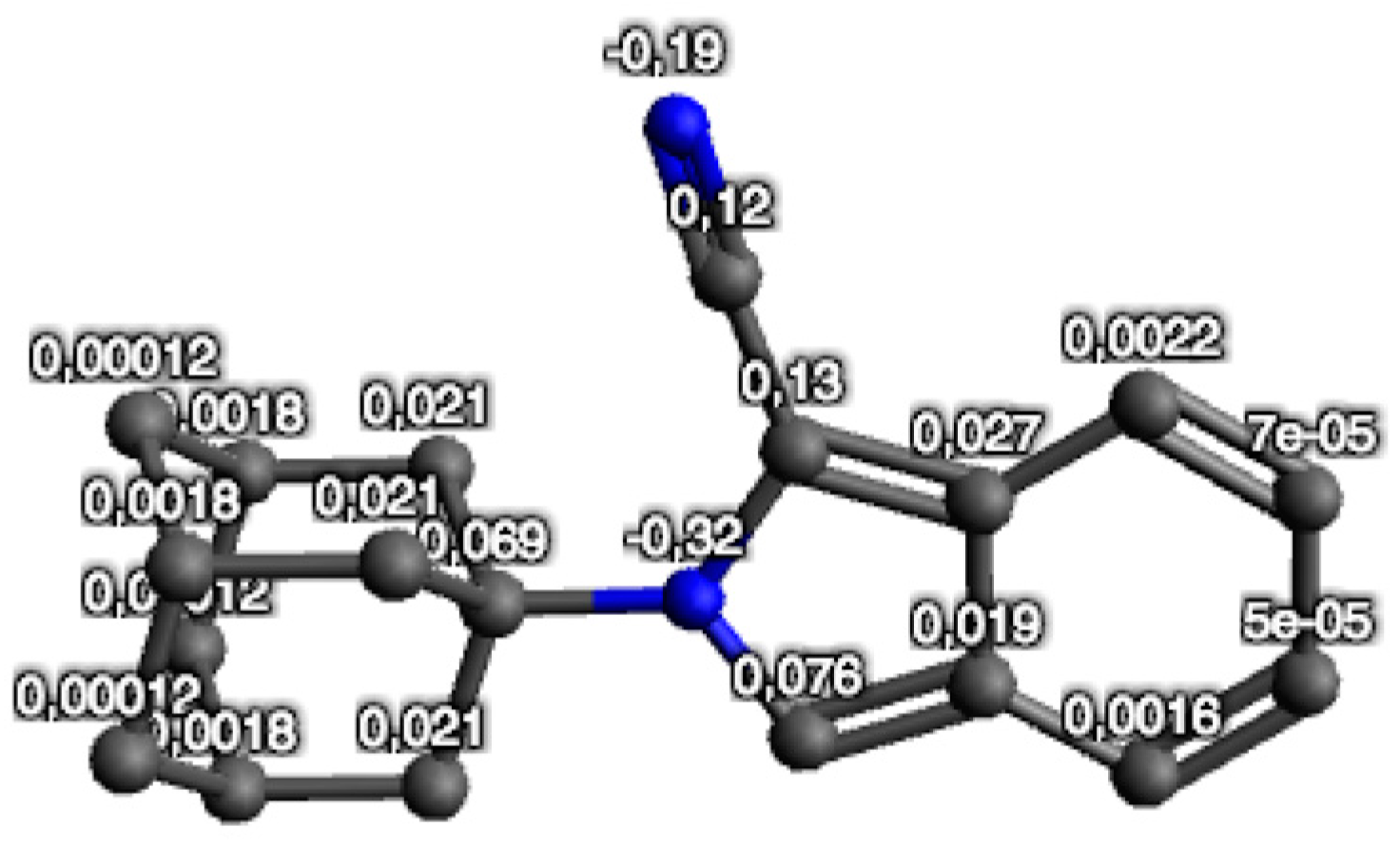
2.3. Frontier Molecular Orbitals and Atomic Net Charges
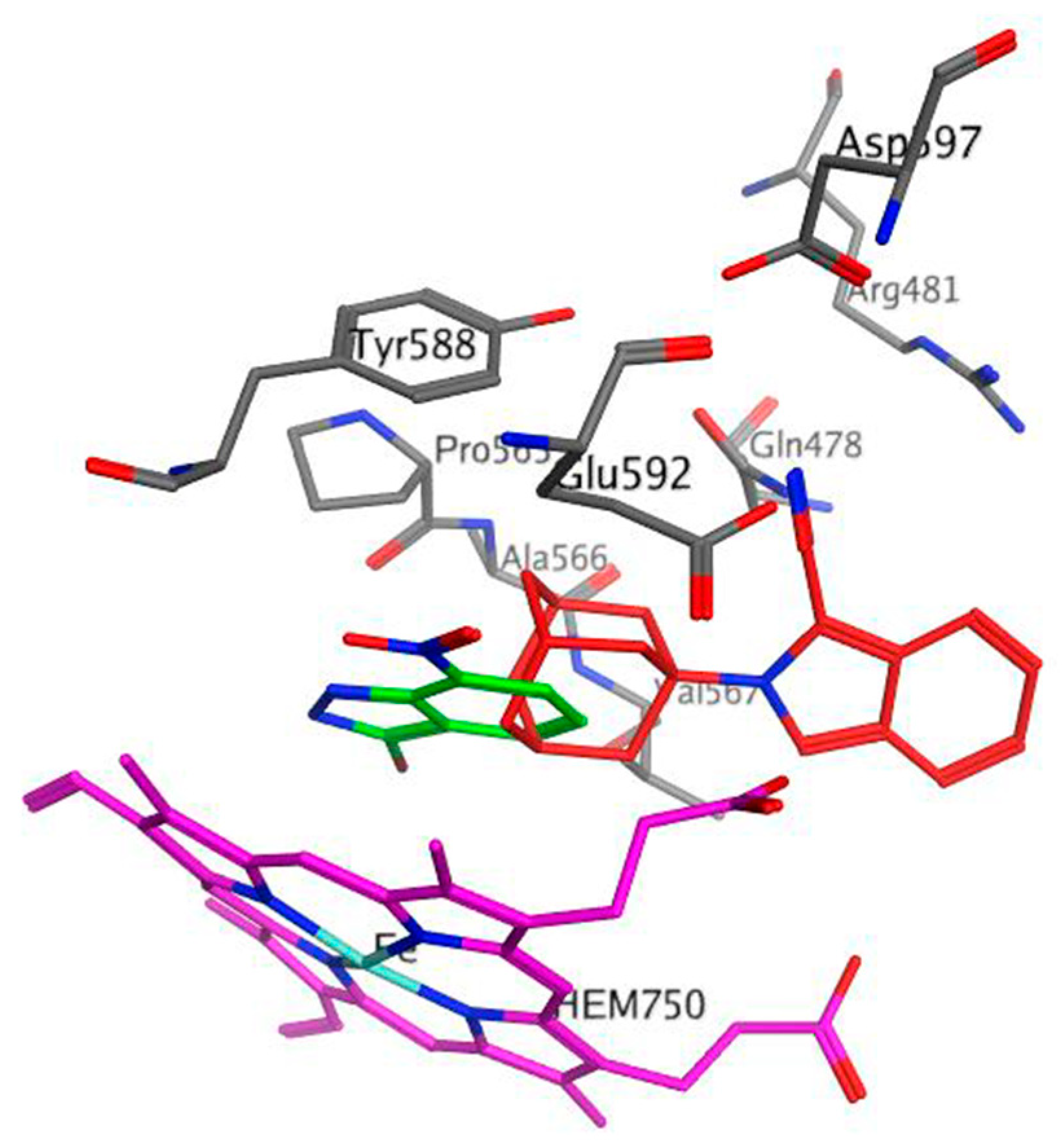
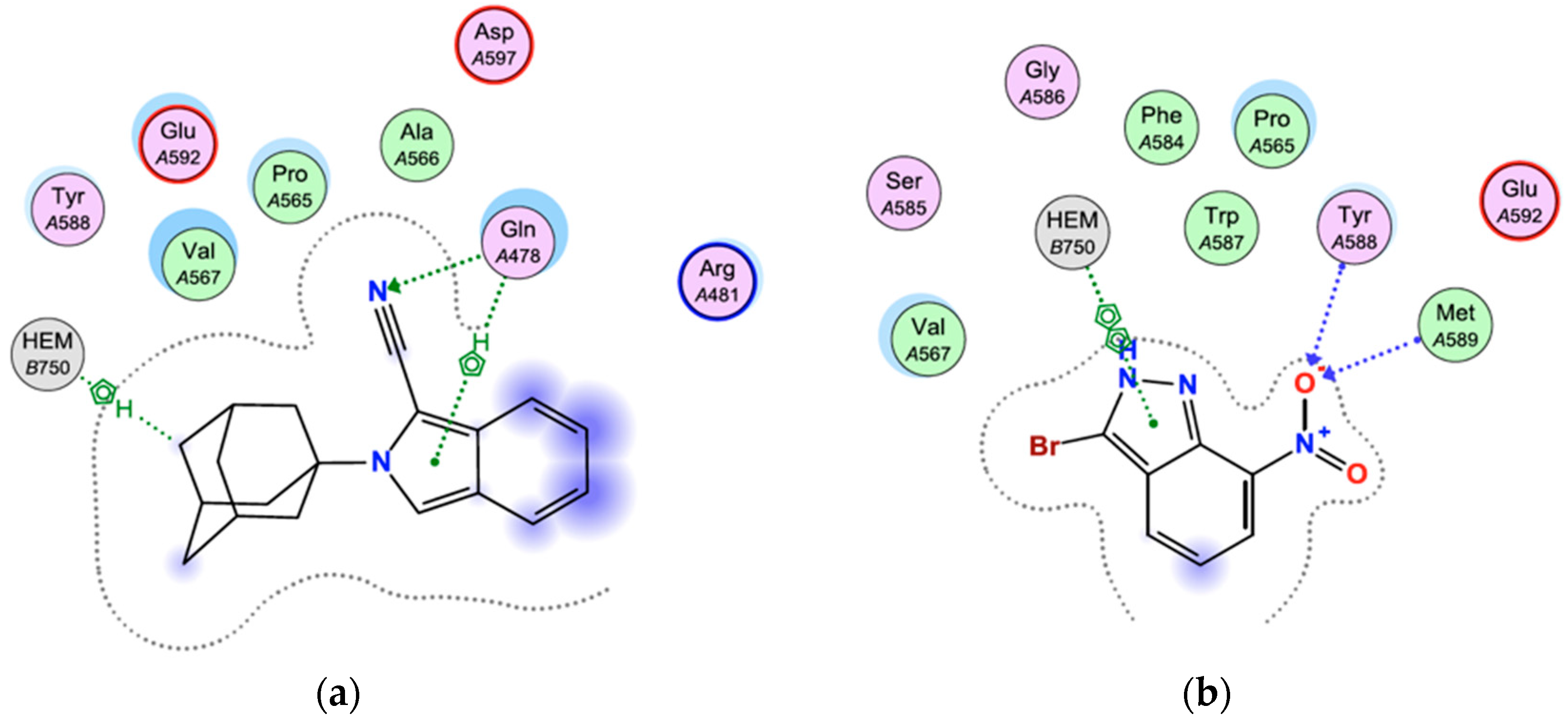
2.4. Docking Studies
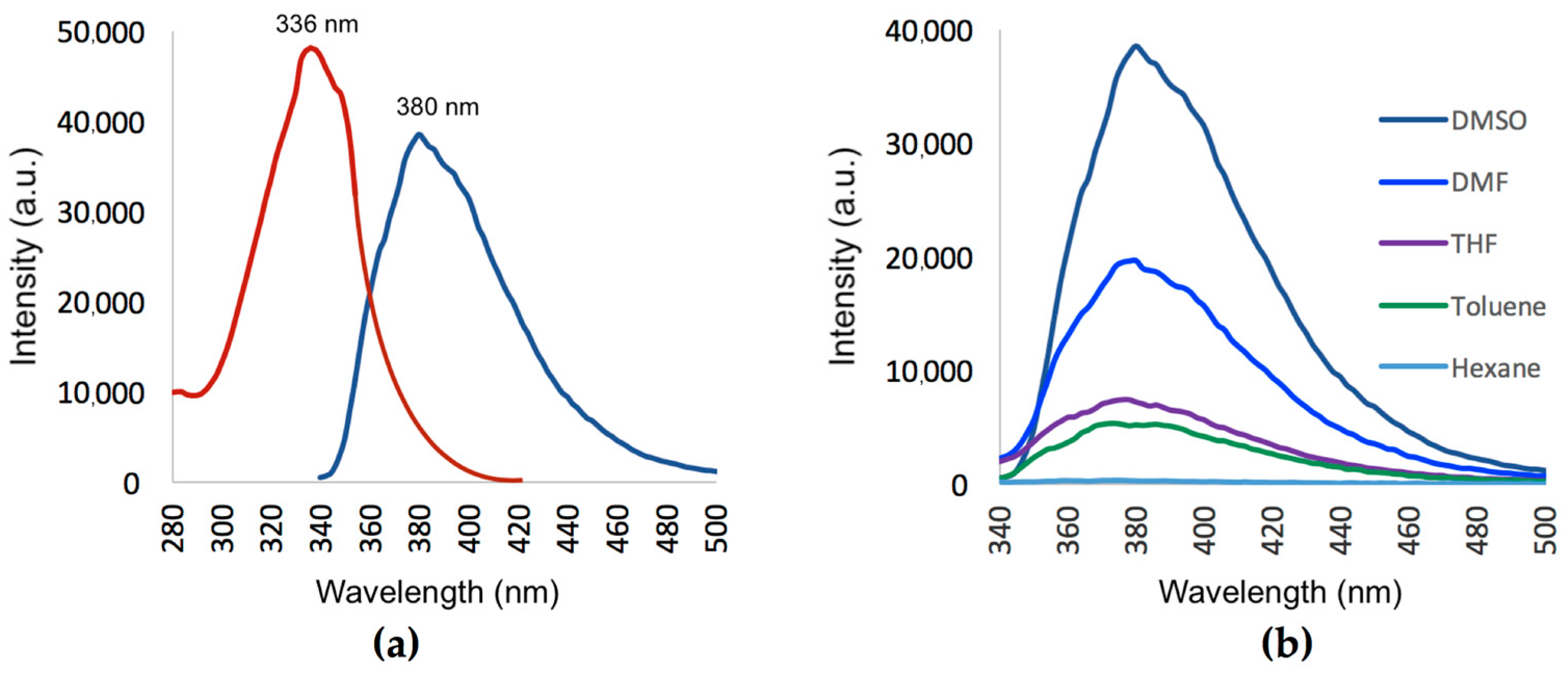
2.5. Fluorescent Properties
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of 2-(adamantan-1-yl)-2H-isoindole-1-carbonitrile (1)
3.2. X-Ray Crystallography
3.3. Theoretical Calculations
3.4. Docking Studies
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Laruelle, M. Imaging synaptic neurotransmission with in vivo binding competition techniques: A critical review. J. Cereb. Blood Flow Metab. 2000, 20, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W.C.; Lyoo, C.H.; McGwier, M.; Snow, J.; Jenko, K.J.; Kimura, N.; Corona, W.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136, 2228–2238. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Terre’Blanche, G.; Van der Schyf, C.J.; Malan, S.F. Screening of novel pentacyclo-undecylamines for neuroprotective activity. Eur. J. Pharmacol. 2003, 458, 73–79. [Google Scholar] [CrossRef]
- Mayer, B.; Klatt, P.; Werner, E.R.; Schmidt, K. Identification of imidazole as l-arginine-competitive inhibitor of porcine brain nitric oxide synthase. FEBS Lett. 1994, 350, 199–202. [Google Scholar] [CrossRef]
- Zheng, W. Characterization of aclcium channel binding. Curr. Prot. Pharmacol. 2001, 14, 1–12. [Google Scholar]
- Auer, M.; Moore, K.J.; Myers-Almes, F.J.; Guenther, R.; Pope, A.J.; Stoekli, K.A. Fluorescence correlation spectroscopy: Lead discovery by miniaturized HTS. Drug Discov. Today 1998, 3, 457–465. [Google Scholar] [CrossRef]
- McGrath, J.C.; Arribas, S.; Daily, C.J. Fluorescent ligands for the study of receptors. Trends Pharmacol. Sci. 1996, 17, 393–399. [Google Scholar] [CrossRef]
- McCabe, R.T.; Newman, A.H.; Skolnick, P. AHN 683: A fluorescent ligand for peripheral-type benzodiazepine receptors. J. Pharmacol. Exp. Ther. 1992, 262, 734–740. [Google Scholar]
- Joubert, J.; van Dyk, S.; Malan, S.F. Small molecule fluorescent ligands as central nervous system imaging probes. Mini-Rev. Med. Chem. 2013, 13, 682–696. [Google Scholar] [CrossRef]
- McCarron, S.T.; Chambers, J.J. Modular chemical probes for visualizing and tracking endogenous ion channels. Neuropharmacology 2015, 98, 41–47. [Google Scholar] [CrossRef]
- Joubert, J.; van Dyk, S.; Malan, S.F. Fluorescent polycyclic ligands for nitric oxide synthase (NOS) inhibition. Bioorg. Med. Chem. 2008, 16, 8952–8958. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Dyk, S.V.; Green, I.R.; Malan, S.F. Synthesis and evaluation of fluorescent heterocyclic aminoadamantanes as multifunctional neuroprotective agents. Bioorg. Med. Chem. 2011, 19, 3935–3944. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Dyk, S.V.; Green, I.R.; Malan, S.F. Synthesis, evaluation and application of polycyclic fluorescent analogues as N-methyl-d-aspartate receptor and voltage gated calcium channel ligands. Eur. J. Med. Chem. 2011, 46, 5010–5020. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Samsodien, H.; Baber, Q.R.; Cruickshank, D.L.; Caira, M.R.; Malan, S.F. Synthesis and structural analysis of novel neuroprotective pentacyclo[5.4.1.02,6.03,10.05,9]undecane- and adamantane-derived propargylamines. J. Chem. Cryst. 2014, 44, 194–204. [Google Scholar] [CrossRef]
- Jones, G.B.; Chapman, B.J. 2.01—Pyrroles and their Benzo Derivatives: Structure. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Pergamon: Oxford, UK, 1996; pp. 1–38. ISBN 9780080965185. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Honarparvar, B.; Govender, T.; Maguire, G.E.; Soliman, M.E.; Kruger, H.G. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2013, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. A survey of carbon-carbon bond lengths. Tetrahedron 1962, 17, 125–134. [Google Scholar] [CrossRef]
- Becker, H. Jan Fleming, Frontier Orbitals and Organic Chemical Reactions., John Wiley u. Sons LTD. J. Praktische Chem. 1978, 320, 879–880. [Google Scholar] [CrossRef]
- Contreras, R.; Domingo, L.R.; Andrés, J.; Pérez, P.; Tapia, O. Nonlocal (pair site) reactivity from second-order static density response function: Gas-and solution-phase reactivity of the acetaldehyde enolate as a test case. J. Phys. Chem. A 1999, 103, 1367–1375. [Google Scholar] [CrossRef]
- Clare, B.W. Frontier orbital energies in quantitative structure-activity relationships: A comparison of quantum chemical methods. Theor. Chim. Acta 1994, 87, 415–430. [Google Scholar] [CrossRef]
- Clare, B.W. The relationship of charge transfer complexes to frontier orbital energies in QSAR. J. Mol. Struct. Theochem. 1995, 331, 63–78. [Google Scholar] [CrossRef]
- Clare, B.W. Charge transfer complexes and frontier orbital energies in QSAR: A congeneric series of electron acceptors. J. Mol. Struct. Theochem. 1995, 337, 139–150. [Google Scholar] [CrossRef]
- Heaton, C.; Miller, A.; Powell, R. Predicting the reactivity of fluorinated compounds with copper using semi-empirical calculations. J. Fluor. Chem. 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, R.M.; López, C.; Pérez-Medina, C.; Pérez-Torralba, M.; Elguero, J.; Escames, G.; Acuña-Castroviejo, D. Fluorinated indazoles as novel selective inhibitors of nitric oxide synthase (NOS): Synthesis and biological evaluation. Bioorg. Med. Chem. 2006, 17, 6180–6189. [Google Scholar] [CrossRef] [PubMed]
- Callis, P.R.; Burgess, B.K. Tryptophan Fluorescence Shifts in Proteins from Hybrid Simulations: An Electrostatic Approach. J. Phys. Chem. B 1997, 101, 9429–9432. [Google Scholar] [CrossRef]
- Vivian, J.T.; Callis, P.R. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef]
- Hilaire, M.R.; Mukherjee, D.; Troxler, T.; Gai, F. Solvent Dependence of Cyanoindole Fluorescence Lifetime. Chem. Phys. Lett. 2017, 685, 133–138. [Google Scholar] [CrossRef]
- APEX3, SAINT, and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2018.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Atwood, J.L.; Barbour, L.J. Molecular Graphics: From Science to Art. Cryst. Growth Des. 2003, 3, 3–8. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A Software Tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. Version 1.2.0. Available online: http://avogadro.cc/ (accessed on 13 October 2018).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Foxen, E.B.; Malan, S.F. Microwave Optimized Synthesis of N-(adamantan-1-yl)-4-[(adamantan-1-yl)-sulfamoyl]benzamide and Its Derivatives for Anti-Dengue Virus Activity. Molecules 2018, 23, 1678. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), Version 2018.01. Available online: http://www. chemcomp.com (accessed on 8 November 2018).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths (Å) | X-ray Crystal | DFT a | Bond Angles (°) | X-ray Crystal | DFT a |
|---|---|---|---|---|---|
| N1-C14 | 1.352 | 1.360 | C14-N1-C11 | 108.77 | 109.13 |
| N1-C1 | 1.498 | 1.499 | C1-N1-C14 | 126.57 | 125.31 |
| N1-C11 | 1.389 | 1.395 | C2-C3-C10 | 110.53 | 109.52 |
| N2-C19 | 1.136 | 1.161 | C10-C3-C4 | 110.10 | 109.74 |
| C3-C10 | 1.520 | 1.539 | N1-C1-C2 | 109.54 | 109.72 |
| C1-C2 | 1.531 | 1.548 | C2-C1-C6 | 110.85 | 110.21 |
| C11-C12 | 1.397 | 1.408 | C6-C1-C7 | 108.39 | 108.39 |
| C12-C13 | 1.433 | 1.433 | C11-C12-C13 | 106.98 | 106.78 |
| C13-C18 | 1.412 | 1.417 | C11-C12-C15 | 132.27 | 132.92 |
| C5-C6 | 1.526 | 1.542 | C13-C12-C15 | 120.67 | 120.30 |
| C15-C16 | 1.375 | 1.375 | N1-C11-C12 | 107.88 | 107.88 |
| C16-C17 | 1.408 | 1.425 | N1-C11-C19 | 125.49 | 125.69 |
| C13-C18-C17 | 118.66 | 118.44 | |||
| N2-C19-C11 | 176.20 | 176.79 | |||
| C12-C15-C16 | 117.31 | 118.21 | |||
| C15-C16-C17 | 122.20 | 121.68 |
| Crystal Data | 1 |
|---|---|
| Chemical formula | C19H20N2 |
| Mr | 276.37 |
| Crystal system, space group | Orthorhombic, P212121 |
| Temperature (K) | 100 |
| a, b, c (Å) | 6.4487 (12), 13.648 (3), 16.571 (3) |
| V (Å3) | 1458.4 (5) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.07 |
| Crystal size (mm) | 0.59 × 0.05 × 0.05 |
| Diffractometer | Bruker APEX II DUO CCD |
| Tmin, Tmax | 0.931, 1.000 |
| No. of measured, independent, and observed [I > 2σ(I)] reflections | 14040, 3372, 2528 |
| Rint | 0.072 |
| (sin θ/λ)max (Å−1) | 0.650 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.074, 0.199, 1.04 |
| No. of reflections | 3372 |
| No. of parameters | 190 |
| Δρmax, Δρmin (e Å−3) | 0.69, −0.28 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joubert, J. Synthesis, Crystal Structure, DFT Studies, Docking Studies, and Fluorescent Properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile. Crystals 2019, 9, 24. https://doi.org/10.3390/cryst9010024
Joubert J. Synthesis, Crystal Structure, DFT Studies, Docking Studies, and Fluorescent Properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile. Crystals. 2019; 9(1):24. https://doi.org/10.3390/cryst9010024
Chicago/Turabian StyleJoubert, Jacques. 2019. "Synthesis, Crystal Structure, DFT Studies, Docking Studies, and Fluorescent Properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile" Crystals 9, no. 1: 24. https://doi.org/10.3390/cryst9010024
APA StyleJoubert, J. (2019). Synthesis, Crystal Structure, DFT Studies, Docking Studies, and Fluorescent Properties of 2-(Adamantan-1-yl)-2H-isoindole-1-carbonitrile. Crystals, 9(1), 24. https://doi.org/10.3390/cryst9010024