Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases


1
Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Leninskii pr. 31, 119991 Moscow, Russia
2
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, 119991 Moscow, Russia
*
Author to whom correspondence should be addressed.
Crystals 2019, 9(7), 330; https://doi.org/10.3390/cryst9070330
Submission received: 24 May 2019
/
Revised: 18 June 2019
/
Accepted: 27 June 2019
/
Published: 28 June 2019
(This article belongs to the Special Issue Chemical Bonding in Crystals and Their Properties)
Abstract
:Dihydrogen bonds attract much attention as unconventional hydrogen bonds between strong donors of H-bonding and polyhedral (car)borane cages with delocalized charge density. Salts of closo-borate anions [B10H10]2− and [B12H12]2− with protonated organic ligands 2,2’-dipyridylamine (BPA), 1,10-phenanthroline (Phen), and rhodamine 6G (Rh6G) were selectively synthesized to investigate N−H...H−B intermolecular bonding. It was found that the salts contain monoprotonated and/or diprotonated N-containing cations at different ratios. Protonation of the ligands can be implemented in an acidic medium or in water because of hydrolysis of metal cations resulting in the release of H3O+ cations into the reaction solution. Six novel compounds were characterized by X-ray diffraction and FT-IR spectroscopy. It was found that strong dihydrogen bonds manifest themselves in FT-IR spectra that allows one to use this technique even in the absence of crystallographic data.
1. Introduction
The term “secondary bonds” was introduced by Alcock almost 50 years ago [1] when he described weak bonds found in crystals of inorganic compounds which are shorter than the sum of the wan der Waals radii of the atoms involved in the interaction but longer than the covalent bonds. These bonds are also called non-bonding or non-valent specific bonds [2,3,4,5]. Among all types of non-bonding bonds, hydrogen bonds were the only ones to be considered in detail for a long time, as this type of bond is the most important among intermolecular interactions [6,7,8]. Hydrogen bonds are attractions arising between a hydrogen bond bound with a more electronegative atom (a hydrogen bond donor) and another atom bearing a long pair of electrons (a hydrogen bond acceptor).
There is no doubt that these interactions play a critical role in the organization of the structure of solid compounds and packing, which directly affects their properties. A great number of studies are devoted to this problem, and this topic remains of current interest.
We focused on so-called dihydrogen bonds (DHB) that arise between B–H groups of boron clusters and a protic hydrogen moiety (H–X). Boron clusters (boranes [9,10,11,12,13,14], carboranes [15], metalloboranes [16,17], and their derivatives) belong to boron hydrides and tend to form numerous DHB with the H–X groups (X = C, O, N) of cations, ligands or solvent molecules found in crystals of their compounds; some of them have been highlighted in a number of studies [18,19,20,21,22,23,24] and generalized in reviews [25,26,27,28]. The indirect evidence of DHB formed is provided by IR spectroscopy when analyzing the region of the stretching vibrations of the BH groups (2500–2100 cm–1). It should be noted that the perhalogenated boron clusters also participate in non-bonding interactions and can be identified by spectroscopic methods. In particular, numerous B–Cl…H–X and B–Cl…X interactions were found in salts and complexes of the [B10Cl10]2− anion by analyzing the 35Cl NQR spectroscopy and X-ray diffraction data of the products obtained [29,30,31].
The most common type of DHB found in compounds of the [BnHn]2− boron clusters (n = 10, 12) are the B–H…H–N bonds realized between the BH groups of the boron clusters and N-containing cations or molecules. The closest to the present study’s detailed discussion of this type of bonds was reported for salts of the closo-decaborate anion with monoprotonated and diprotonated bipyridine [HBipy]2[B10H10] and [H2Bipy][B10H10] [32,33], respectively, which were studied by X-ray diffraction; atomic charges were determined by the Hirshfeld’s method. The authors reported the difference density maps and discussed the electron distribution of the electron density and the electron transfer between the boron cluster and the cation.
Here, we consider the B–H…H–N DHB found in salts of the [BnHn]2− boron clusters (n = 10, 12) and N-containing monoprotonated and diprotonated organic bases (2,2’-dipyridylamine (BPA), 1,10-phenanthroline (Phen), and rhodamine 6G (Rh6G)); the compounds have been studied by X-ray diffraction techniques and IR spectroscopy.
2. Materials and Methods
Elemental analysis for carbon, hydrogen, and nitrogen content was performed using a CHNS-3 FA 1108 automated elemental analyzer (Carlo Erba Instruments, Milan, Italy). Boron content was determined on an iCAP 6300 Duo ICP emission spectrometer (Thermo Scientific, Waltham, USA) with inductively coupled plasma. The samples were dried in a vacuum at room temperature to a constant weight, thus obtaining solvent-free forms.
IR spectra of the crystals obtained were recorded on an Infralum FT-02 Fourier-transform spectrophotometer (Lumex, St. Petersburg, Russia) in the range of 4000–600 cm−1 at a resolution of 1 cm−1. Samples were prepared as Nujol mulls or were recorded in thin layer; NaCl pellets were used.
2.1. Synthesis
DMF (HPLC), acetonitrile (HPLC), CF3COOH as well as solid BPA, Phen, rhodamine Rh6G · HCl, and CuSO4·5 H2O were purchased from Sigma-Aldrich and used without additional purification. [Et3NH]2[B10H10] and [Et3NH]2[B12H12] were prepared from decaborane-14 using the known synthetic procedures [34,35].
(HBPA)2(H2BPA)[B10H10]2 (1)
A solution of [Et3NH]2[B10H10] (0.4 mmol) in water (10 mL) was added to a solution of BPA (0.8 mmol) in acetonitrile (10 mL). Glacial trifluoroacetic acid CF3COOH (5 mL) was added dropwise to the resulting mixture when stirring. The reaction mixture was allowed to stand at room temperature in a beaker covered with a watch glass. Yellow crystals 1·2H2O precipitated after 48 h from the corresponding reaction mixture, which were filtered off, washed with water (2 × 5 mL), and dried in air. Yield, 82%. 1: C30H51N9B20: Calculated (%): C, 47.79; H, 6.82; N, 16.72; B, 28.7. Found: C, 47.71; H, 6.83; N, 16.69; B, 28.5. IR, cm−1 (Nujol mull): 3286s, 3236, 3204, 3136, 2543, 2492, 2444, 2420, 1641s, 1591s, 1557, 1528, 1468s, 1435, 1378s, 1237, 1162, 1108, 1061, 1013, 904, 839, 787s, 781, 773s.
[PhenH]2[B12H12] (2)
A solution of [Et3NH]2[B12H12] (0.4 mmol) in water (10 mL) was added to a solution of Phen (0.8 mmol), respectively, in acetonitrile (10 mL). Glacial trifluoroacetic acid CF3COOH (5 mL) was added dropwise to the resulting mixtures. The reaction mixture was allowed to stand at room temperature in a beaker covered with a watch glass. Yellow crystals 2·2H2O precipitated after 48 h, which were filtered off, washed with acetonitrile (2 × 5 mL), and dried in air. Yield, 77%. 2: C24H30N4B12: Calculated (%): C, 57.17; H, 6.00; N, 11.11; B, 25.7. Found: C, 57.15; H, 6.02; N, 11.09; B, 25.6. IR, cm−1 (Nujol mull): 3098, 3063, 2487, 2443, 1629, 1608, 1586, 1522s, 1461s, 1431s, 1416s, 1226, 1056s, 875, 851s, 783, 738s, 719.
(NHEt3)(HBPA)[B10H10] (3)
A solution of [Et3NH]2[B10H10] (0.4 mmol) in acetonitrile (10 mL) was added to a solution of BPA (0.4 mmol) in the same solvent (10 mL). Glacial trifluoroacetic acid CF3COOH (5 mL) was added dropwise when stirring the reaction mixture. The reaction mixture was allowed to stand at room temperature. Yellow crystals 3·2CH3CN started to precipitate after 30 min; after 2 h, they were filtered off, washed with acetonitrile (2 × 5 mL), and dried in air. Yield, 55%. 3: C16H36N4B10: Calculated (%): C, 48.95; H, 9.24; N, 14.27; B, 27.5. Found: C, 48.96; H, 9.22; N, 14.25; B, 27.2. IR, cm−1 (Nujol mull): 3467, 3316, 3255, 3212, 3145, 3111, 2499, 2466, 2448, 2404, 1648s, 1612, 1591s, 1528s, 1488s, 1466s, 1435, 1378, 1273, 1165s, 1018, 773s, 721.
[PhenH)]2[B10H10] (4)
A solution of [Et3NH]2[B10H10] (0.4 mmol) in acetonitrile (10 mL) was added to a solution of Phen (0.8 mmol) in the same solvent (10 mL). Glacial trifluoroacetic acid CF3COOH (5 mL) was added dropwise. The reaction mixture was allowed to stand at room temperature in a beaker covered with a watch glass. Yellow solvent-free crystals 4 precipitated after 24 h, which were filtered off, washed with acetonitrile (2 × 5 mL), and dried in air. Yield, 68%. C24H28N4B10: Calculated (%): C, 59.98; H, 5.87; N, 11.66; B, 22.5. Found: C, 59.99; H, 5.81; N, 11.59; B, 22.4. IR, cm−1 (thin layer): 3095, 3071, 30545, 3029, 2510, 2477, 2451, 2421, 1658, 1631, 1616s, 1595s, 1540s, 1468s, 1418, 1378s, 1316, 1285, 1214s, 1190, 1159, 1010s, 855, 844, 769, 719s, 715, 665, 622s.
[Rh6GH]2[B12H12] (5)
A solution of R6G·HCl (0.4 mmol) in water (10 mL) was added to a solution of [Et3NH]2[B12H12] (0.8 mmol) in acetonitrile (10 mL). The reaction mixture was allowed to stand at room temperature in a beaker covered with a watch glass. A red precipitate was formed after 24 h, which was filtered off, washed with acetonitrile (2 × 5 mL), and dried in air. Red crystals 5·2CH3CN were recrystallized from acetonitrile. Yield, 77%. 5: C56H74N4O6B12: Calculated (%): C, 65.37; H, 7.25; N, 5.45; B, 12.6. Found: C, 65.41; H, 7.03; N, 5.39; B, 12.5. IR, cm−1 (Nujol mull): 3410, 3361, 2484, 2461, 2447, 2422, 2251w, 1727s, 1648, 1607s, 1571, 1549, 1531, 1500, 1467, 1366, 1305, 1255, 1191, 1146, 1079, 1056, 1026, 900, 847, 812, 785, 738.
[PhenH]2(Phen)2.5[B10H10] (6) and [Co(Phen)3][B10H10] (7)
Compound 6 was obtained in the course of cobalt(II) complexation reaction proceeded in water. We used the procedure similar to those indicated in [36] but the reaction was carried out in water instead of acetonitrile or DMF as follows. A solution of [Et3NH]2[B10H10] (0.4 mmol) in water (10 mL) was added to a solution containing CoCl2·6 H2O (0.4 mmol) and Phen (1.2 mmol) in water (10 mL). Orange precipitate [Co(Phen)3][B10H10] (7) was formed in good yield (85%), which was filtered off and dried. After 48 h, light-yellow crystals 6·0.5H2O precipitated from the mother solution; they were filtered off and dried in air. Yield of 6, < 10%. 6: IR, cm−1 (Nujol mull): 3164, 3152, 3027, 2495, 2476, 2414, 1656, 1651, 1618, 1610, 1543, 824s, 761s. C54H48N9B10: Calculated (%): C, 34.72; H, 2.59; N, 33.75; B, 28.9. Found: C, 34.70; H, 2.57; N, 33.71; B, 28.7. 7: C36H34N6B10Co: Calculated (%): C, 60.24; H, 4.77; N, 11.71; B, 15.0. Found: C, 60.21; H, 4.71; N, 11, 73; B, 14.9. IR, cm−1 (Nujol mull): 3052w; 2469, 2434; 1625, 1582w, 1519s, 1463s, 1427s, 1379, 1342w, 1225w, 1143w, 1103, 861s; 1009; 844s, 727s.
2.2. X-ray Diffraction
Experimental data for 1·2H2O, 2·2H2O, 3·2CH3CN, 4, 5·CH3CN, and 6·0.5H2O were collected at low temperatures on Bruker Apex II CCD diffractometer using graphite monochromatted MoKα (1, 3, 5) radiation or CuKα (2) radiation with multilayer optics. Intensities of reflections for 4 and 6 were obtained on Bruker Smart Apex CCD diffractometer. The structures were solved by direct method and refined by full-matrix least squares against F2. Non-hydrogen atoms were refined anisotropically except some disordered carbon and boron atoms. A BPA cation in 3 and a boron cage in solid 6 are disordered over two sites, and were refined isotropically. A number of EADP and ISOR instructions were applied to refine some moieties. Positions of H(C) and H(B) atoms were calculated, and those of H(O) and H(N) atoms were located on difference Fourier maps, and then fixed at 0.87 and 0.88 Å. All hydrogen atoms were included in the refinement by the riding model with Uiso(H) = 1.5Ueq(X) for methyl groups and water molecules, and 1.2Ueq(X) for the other atoms. All calculations were made using the SHELXL2014 [37] and OLEX2 [38] program packages. The crystallographic data and experimental details are listed in Table S1 (see Supporting Information).
The crystallographic data for 1·2H, 2O2·2H, 2O3·2CH3CN, 4, 5·CH3CN, and 6·0.5H2O have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications under the CCDC numbers 1917700–1917705. This information may be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures. Hirshfeld surfaces were depicted with CrystalExplorer2.0 [39].
The X-ray powder diffraction patterns of compounds 4, 5·2CH3CN, and 7 were measured on a Bruker D8 Advance diffractometer (CuKα1) at RT with LynxEye detector and Ge(111) monochromator, θ/2θ scan from 5° to 80°, step size 0.01125° at the Shared Equipment Center of the Kurnakov Institute. The measurements were performed in transmission mode with the sample deposited in the single-crystal oriented silicon cuvette. The crystals were thoroughly triturated before measurements. The X-ray powder diffraction patterns for 4, 5·2CH3CN, and 7 are shown in Figures S11–S13 (see Supplementary Materials).
3. Results and Discussion
3.1. Synthesis and Structures of Salts of the Boron Clusters with Monoprotonated and Diprotonated Organic Bases
The target salts of the boron cluster anions and N-containing cations were synthesized by the reaction between salts [Et3NH]2[B10H10] or [Et3NH]2[B12H12] and neutral organic bases Phen, BPA in the presence of CF3COOH in CH3CN and CH3CN/water. The reactions proceeded according to the general reaction scheme:
[BnHn]2− + 2 L + CF3COOH → LH+ + LH2+ + [BnHn]2−
(L = Phen, BPA; n = 10, 12)
For Rh6G, the reaction proceeded between Rh6G·HCl and [Et3NH]2[B12H12] in acetonitrile.
It was found that the protonation of the N-containing organic bases L proceeded non-selectively; monoprotonated and diprotonated cations were present in the reaction solution simultaneously and the composition of the final product varied. The majority of crystals of the final products contained the corresponding solvent molecules.
When the reaction proceeded in CH3CN/water in the acidic medium, salts (HBPA)2(H2BPA)[B10H10]2·2H2O (1·2H2O) and [PhenH]2[B12H12]·2H2O (2·2H2O) were isolated which contained diprotonated and/or monoprotonated ligands.
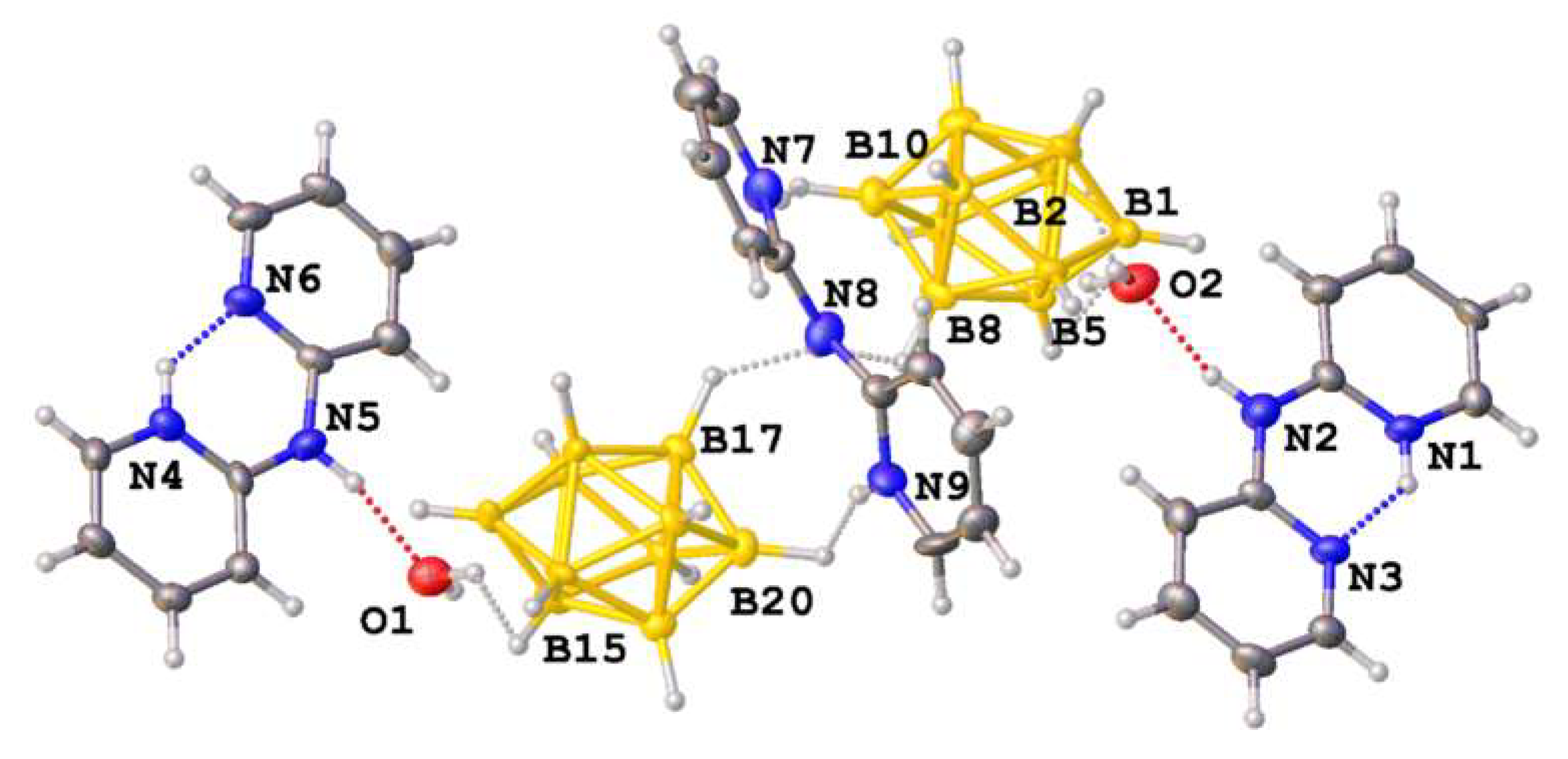
Asymmetric unit of 1·2H2O contains two monoprotonated cations HBPA+, a diprotonated cation H2BPA2+, two [B10H10]2- anions and two water molecules (Figure 1a). Cations HBPA+ are flat as their pyridyl fragments take part in intramolecular hydrogen bonding N–H...N (r(N...N) = 2.593(9) and 2.586(9) Å, NHN 133.6º and 133.4°). Both HBPA+ are also connected with water molecules via the N–H...O hydrogen bonds with bridge amino groups (r(N...O) = 2.727(6) and 2.751(6) Å, NHO = 158.7 and 162.0°). All OH groups of water molecules are directed to equatorial atoms of the boron cluster anions (H...H 1.94–2.26, H...B 2.45–2.57 Å). The H2BPA2+ cation is non-planar; the dihedral angle between planes of the pyridine cycles is 44.0(3)°. The NH groups of the pyridine cycles form very short contacts with the apical BH groups of the boron cluster anions, additionally supported with a longer bifurcate B–H...H(N)...H–B dihydrogen bonding between a bridging NH-group and equatorial atoms of boron cages.
A similar reaction with the closo-dodecaborate anion and Phen resulted in isolation of salt 2·2H2O, which contained two monoprotonated HPhen+ molecules per one closo-dodecaborate anion. Asymmetric unit of this salt contains one cation, two water molecules and half of anion (Figure 2). Water molecules form a dimer through a H-bond (r(O...O) = 2.742(5) Å, OHO = 173.2°). One of two water molecules is involved in H-bonding with two HPhen+ cations (r(N...O) = 2.802(3)–2.125(4) Å, OHN = 120.9–152.6°). The second forms dihydrogen bonds with two anions.
As it follows from crystal structures of 1·2H2O and 2·2H2O, water molecules both act as likely acceptors of hydrogen bonding towards H(N) atoms to form dihydrogen bonds, and as more likely donor groups towards boron cages as compared with protonated ligands. Thus, a series of salts was obtained in anhydrous reaction conditions. When the reaction under discussion was carried out in the acetonitrile/CF3COOH system, compounds (Et3NH)(HBPA)[B10H10]·2CH3CN (3·2CH3CN) and [PhenH)]2[B10H10] (4) were obtained for BPA and Phen, respectively. No diprotonated cations were found in the compounds synthesized.
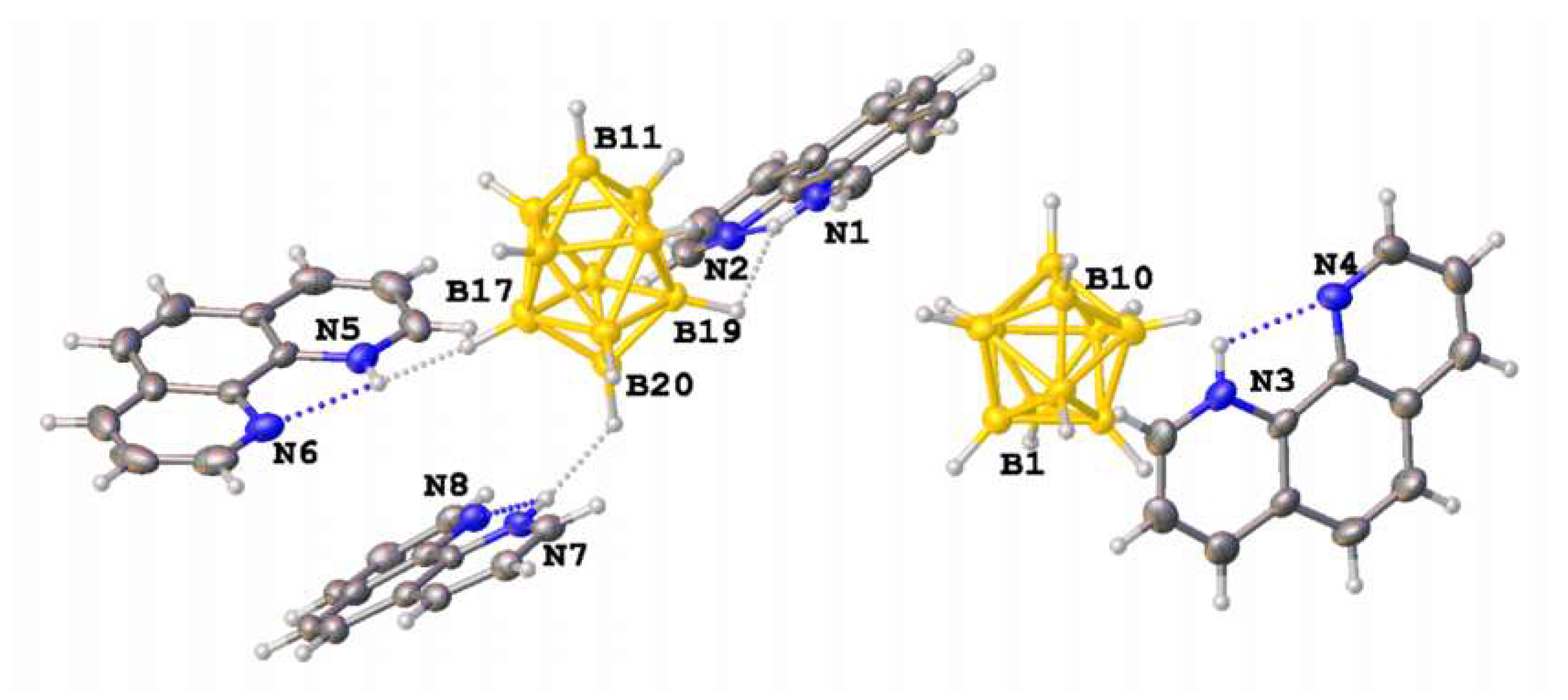
Asymmetric unit of 3·2CH3CN contains two organic cations, HBPA+ and Et3NH+, the closo-decaborate anion and two acetonitrile molecules (Figure 3). Both cations were found to be disordered, and the disorder could not be resolved in non-centrosymmetric groups of lower symmetry. The distorted geometry and high errors do not allow us to unambiguously determine positions of H(N) atoms in the amino-pyridilium cation, but planar conformation of HBPA+ allows us to propose intramolecular hydrogen bonding N–H...N, while the amino groups N(3)–H and N(1)–H form dihydrogen N–H...H–B bonds with equatorial atoms of the closo-decaborate anion. The Et3NH+ cation interacts with apical atoms of the anion. Acetonitrile molecules are involved in weak C–H...N bonding with methyl groups of Et3NH+.
When Phen was allowed to react with the closo-decaborate anion in acetonitrile in the presence of trifluoroacetic acid, a solvent-free salt [PhenH)]2[B10H10] (4) was isolated. This compound contains two independent [B10H10]2− anions and four PhenH+ cations (Figure 4). The independent closo-decaborate anions have different environments. The B(1)–B(10) anion is situated in a hydrophobic 'cavity' formed by cations; and takes part in B–H…H–C and B–H…π interactions only. The B(11)–B(20) anion besides these hydrophobic interactions forms also contacts with the NH-groups via equatorial and apical boron atoms with three cations. The intramolecular N–H...N hydrogen bond is found in all cations (r(N...N) = 2.697(6)–2.723(6) Å, NHN = 104°–105°). In addition, the NH groups of the three cations form shortened contacts with the closo-borate anions (see above), and one cation does not participate in the contacts.
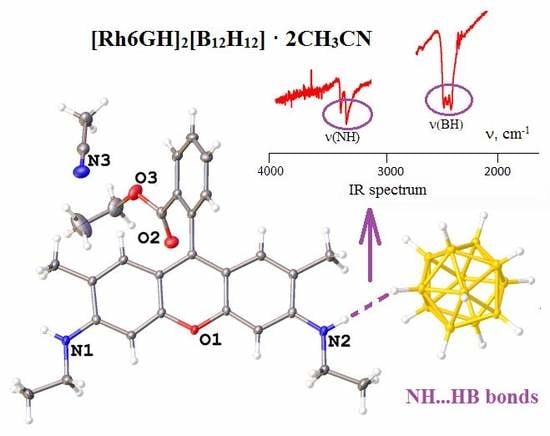
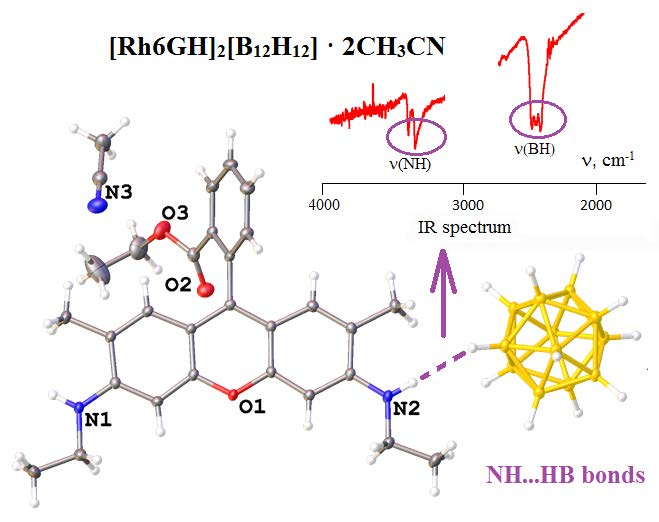
For rhodamine Rh6G, we obtained a salt with the [Rh6GH]2[B12H12]·2CH3CN composition (5·2CH3CN). This compound was synthesized in an acetonitrile/water solution when Rh6G·HCl was used and the precipitate obtained was recrystallized from acetonitrile. Asymmetric unit of this salt contains a protonated cation, half the closo-dodecaborate anion, and an acetonitrile molecule (Figure 5). The angle between mean planes of the phenyl ring and condensed rings of the Rh6GH+ is 116.9(1)°. Cations and anions from infinite chains connected through dihydrogen N−H...H−B bonds. There exist π...π stacking between parallel Rh6GH+ molecules situated 3.490(1) Å from each other, additionally supported with C=O...H−C interactions.
Note that acidic media can be created not only by the presence of an acid in reaction mixtures but also because of hydrolysis of metal cations in aqueous solutions. In particular, when we studied the cobalt(II) complexation with Phen in the presence of the closo-decaborate anion, two products were isolated, namely tris-chelate cobalt(II) complex [Co(phen)3][B10H10] (7) (main product) [36] and a salt of the closo-decaborate anion with Phen (6). The reaction proceeded in the water/acetonitrile system according to the scheme:
CoCl2 + 3 Phen + [B10H10]2− → [Co(Phen)3][B10H10] (7) + [H2(Phen)4.5][B10H10] (6)
Cobalt(II) complex 7 was characterized by elemental analysis, IR spectroscopy, and powder X-ray diffraction data. It was found that its powder X-ray diffraction pattern coincided with that calculated from the X-ray diffraction data reported [36].
After precipitation of complex 7, single crystals 6·0.5H2O were obtained from the reaction mixture in low yield. It was found that they contain salt of the closo-decaborate anion with non-protonated and monoprotonated Phen molecules [H2(Phen)4.5][B10H10]. Their preparation is explained by partial hydrolysis of cobalt(II) cation in water according to the following scheme:
[Co(H2O)6]2+ + H2O ↔ [Co(OH)(H2O)5]+ + H3O+
[B10H10]− + Phen + H3O+ → [H2(Phen)4.5][B10H10] (6)
As the concentration of protons in the resulting aqueous reaction solution was significantly lower than that in the case when we used trifluoroacetic acid, final salt 6 contained not only protonated ligand LH+ but neutral ligand L as well.
The asymmetric unit of compound 6·0.5H2O contains four and a half independent Phen molecules, a disordered closo-decaborate anion, and partially occupied position of water molecule (Figure 6a). None of H(N) atoms could be located on difference Fourier maps due to low crystal quality, and at the same time their positions can be proposed from the most likely intermolecular hydrogen bonds. Three of Phen molecules are packed in stacks with an interplanar distance of ~3.5 Å, and these stacks are connected by means of N−H...N bonds (r(N...N) = 2.82(2) Å, NHN = 159.4°, only of two H-bonded stacks should be charged in this case, thus, positions of H(N) protons are only half occupied, Figure 6b). Besides, there exists a Phen molecule connected with a water molecule by means of H-bonds (r(N...O) = 2.55(2) Å, NHO = 168.9°), while hydrogen atoms of the water molecule should be directed towards boron atoms of two cages. Thus, the only Phen molecule surrounded by H(C) and H(B) atoms is neutral.
Therefore, when studying complexation of metals in water in the presence of ligands capable to be protonated, it should be taken into account that the corresponding salts can be obtained with anions present in reaction mixtures rather than metal complexes because metals can be reversible hydrolyzed in aqueous solutions.
3.2. Hirshfeld Surface Analysis
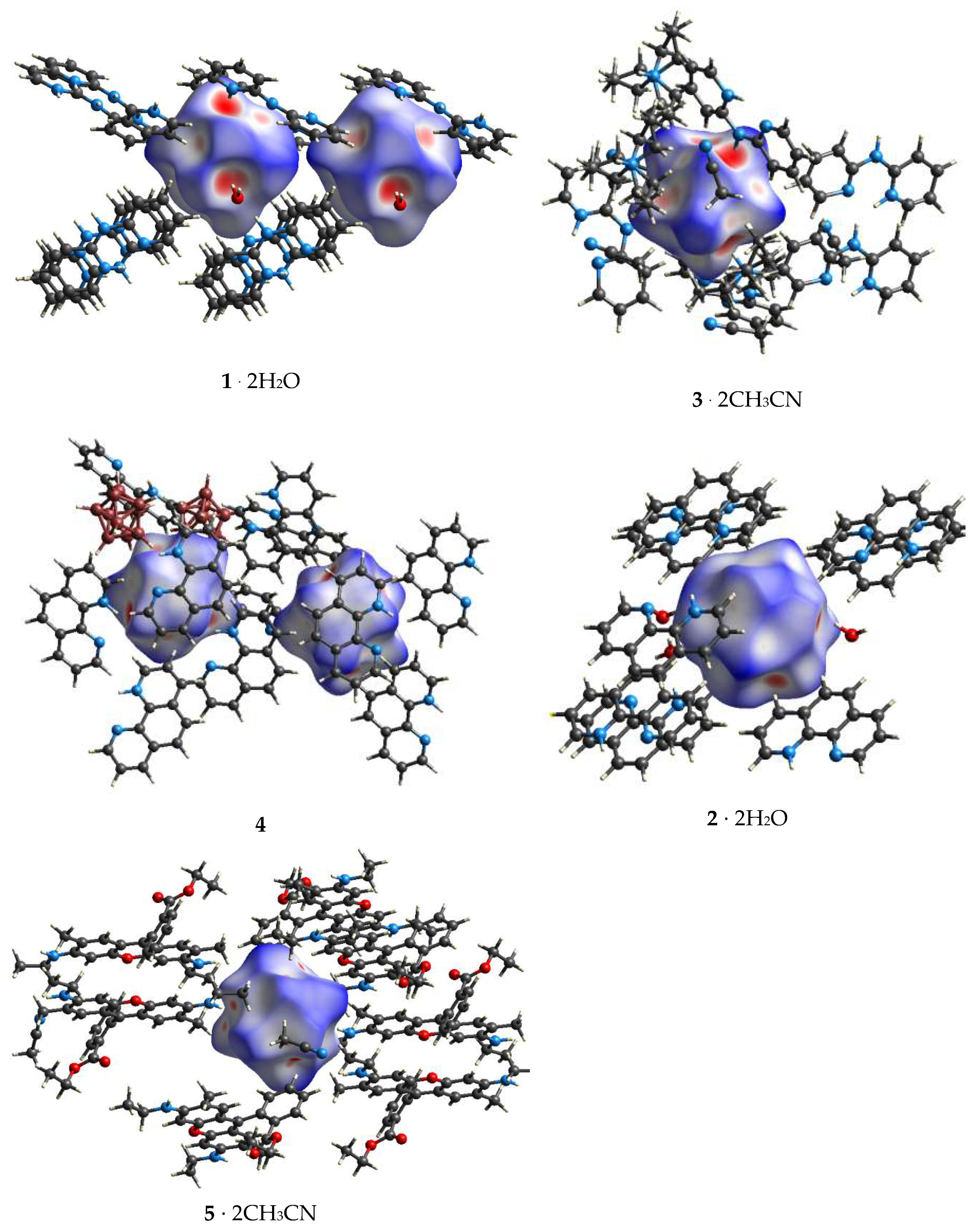
Molecular Hirshfeld surface analysis is a convenient tool to rank the week intermolecular interactions. The surfaces were previously used to investigate dihydrogen bonding in borane salts [40,41] and metallocarboranes [42,43]. For the compounds studied, dihydrogen bonds form 86–99% of molecular surface. At the same time, both the nature of donor groups, and that of a boron cage manifest themselves on Hirshfeld surfaces mapped with dnorm (Figure 7). The closest and strongest of intermolecular interactions are depicted in red. In closo-decaboranes such interactions appear between the anion and cations or water molecules, which also supports our conclusion that water molecules are likely donors of H-bonding. All B−H...H−O interactions also satisfy criteria of dihydrogen bonding given in Reference [24], particularly, r(H...H) < 2.2 Å and OHH > BHH. B−H...H−N interactions in 1·2H2O, 3·2CH3CN and 4 also manifest themselves as red regions on highly curved Hirshfeld surface mapped with dnorm. In closo-dodecaborates, the area of red regions seems to be smaller than that for the same types of interactions in closo-decaborates and is more similar to the Hirshfeld surface of metallocarboranes mapped with dnorm [43]. Besides B−H...H−N bonds, some B−H...H−C interactions are also among the closest in crystals of 2·2H2O and 5·2CH3CN which was characteristic of borane and bisborane salts [40,41].
Note that other types of dihydrogen bound could be found in boron-containing compounds. In particular, the M∙∙∙H–B interactions (M = Li, K) and homopolar CH∙∙∙HC and BH∙∙∙HB dihydrogen bonds were found to exist in hydrogen storage materials LiN(CH3)2BH3 and KN(CH3)2BH3 [44]; they were described by the charge and energy decomposition method and the Interacting Quantum Atoms approach. The BH∙∙∙HB bonds were discussed by the authors as destabilizing, whereas the CH∙∙∙HC bonds should be considered stabilizing.
3.3. IR Spectroscopy Data of Compounds Synthesized
IR spectroscopy is a perfect tool to determine the presence of dihydrogen bonds in compounds with the boron cluster anions. For compounds containing neutral organic ligands, IR spectroscopy allows one to determine whether an organic ligand is present in a free form, is coordinated by metal atoms or is protonated. IR spectra of salts containing protonated N-containing organic ligands have pronounced changes in regions of characteristic bands of the closo-borate anions and ligands L, particularly in both B–H and N–H stretching vibrations, as compared to compounds with free ligands and alkaline closo-borates. IR spectra of all compounds discussed are present in SI; selected IR data are shown in Table 1.
In salts with cations [PhenH]+, a new band is observed in the ν(NH) region (3350–3100 cm–1), which is an unambiguous evidence for the protonation of Phen. This band is observed for compound 6 with protons not localized by X-ray diffraction, indicating that NH groups are present in the structure.
Unlike Phen, BPA ligand in the free, non-protonated state contains NH groups which are manifested as three ν(NH) bands. Salts with monoprotonated and diprotonated BPA contain four (for 1) or five (3·2CH3CN) bands in the IR spectra, respectively, which reflects DHB between the BH groups of the boron cluster anions and NH groups of the ligand. In addition, a ν(NH) band with a maximum at 3467 cm−1 is observed in the spectrum of compound 3·2CH3CN which is assigned to the triethylammonium cation from the starting salt.
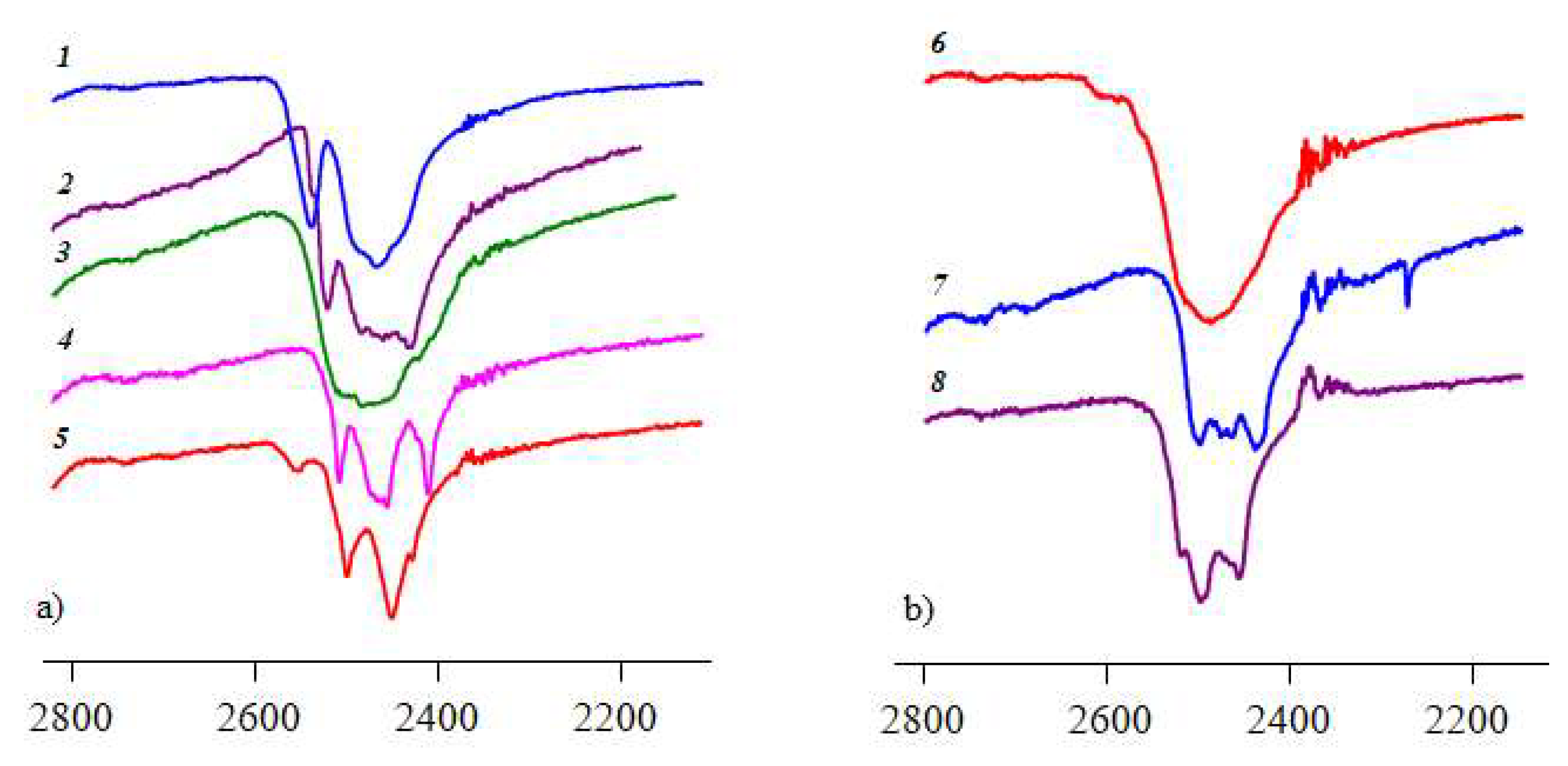
Pronounced changes are observed in IR spectra of the final compounds in the ν(BH) region (2550–2400 cm−1). Boron cluster anions are known to form numerous X–H…H–B interactions in complexes and salts with cations and neutral molecules containing NH, CH, OH groups [28]. This leads to splitting of the ν(BH) band to several components indicating the violation of the initial state of the BH bonds in “free” (non-coordinated) boron clusters (such as alkaline closo-borates).
In the IR spectrum of K2[B10H10], two bands are observed which correspond to ν(BH) stretching vibrations of apical and equatorial BH groups (Figure 8, curve 1). In the IR spectra of salts with the [B10H10]2− anion and protonated ligands (curves 2–5), there is a pronounced splitting of this band, therefore no bands corresponding to stretching vibrations of apical and equatorial BH groups can be defined. In the spectrum of salts with the [B12H12]2− anion (curves 7 and 8), the ν(BH) band is also split to several components because of DHB, while in the spectrum of Cs2[B12H12] (curve 6), one band is observed indicating equivalence of all BH bonds in the B12 icosahedron.
In the IR spectra of salts under discussion there are changes in the intensities and redistribution of bands in regions corresponding to vibrations of the phenyl rings of the heterocycles (1700–600 cm−1), namely in the ν(CC) and ν(CN) regions (1650–1500 cm−1) as well as out-of-plane π(CH) vibrations (850–700 cm−1) (see Table S2).
4. Conclusions
Six novel salts of the closo-decaborate and closo-dodecaborate anions with cations of 2,2’-dipyridylamine, 1,10-phenanthroline, and rhodamine 6G were synthesized and studied by IR spectroscopy and X-ray diffraction. Compounds contain monoprotonated and/or diprotonated organic cations depending on the synthetic method. Numerous dihydrogen B−H...H−O and B−H…H−N interactions between the boron clusters, water molecules and N-containing cations were detected. Characteristic changes were found in the IR spectra of the compounds indicating unambiguously the formation of dihydrogen bonds.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/9/7/330/s1, Table S1: Crystallographic data and refinement parameters for crystals 1–6, Table S2: Maxima of selected absorption bands in IR spectra of compounds 1–6 as compared to alkali metal closo-borates, Figure S1: IR spectrum of K2[B10H10]·nH2O, Figure S2: IR spectrum of bpa, Figure S3: IR spectrum of (Hbpa)2(H2bpa)[B10H10]2 (1), Figure S4: IR spectrum of (NHEt3)(Hbpa)[B10H10] (3), Figure S5: IR spectrum of phen, Figure S6: IR spectrum of [Hphen)]2[B10H10] (4), Figure S7: IR spectrum of [H2(phen)4.5][B10H10] (6), Figure S8: IR spectrum of Cs2[B12H12], Figure S9: IR spectrum of [PhenH]2[B12H12] (2), Figure S10: IR spectrum of [Rh6GH]2[B12H12]·CH3CN (5), Figure S11: Experimental and calculated X-ray diffraction patterns of [PhenH)]2[B10H10] (4); Figure S12: Experimental and calculated X-ray diffraction patterns of [Rh6GH]2[B12H12]·CH3CN (5); Figure S13: Experimental and calculated X-ray diffraction patterns of complex 7.
Author Contributions
Synthesis, writing—original draft preparation, review and editing – V.V.A and E.A.M; X-ray diffraction studies and Hirshfeld surface analysis – A.V.V.; conceptualization, supervision – N.T.K.
Funding
“This research received no external funding”.
Acknowledgments
This work was performed within the framework of the State Assignment of the Kurnakov Institute (IGIC RAS) in the field of fundamental scientific research. The X-ray diffraction studies were performed within the framework of the State Assignment of the Nesmeyanov Institute (INEOS RAS). X-ray diffraction studies were performed at the Centre for Molecular Studies of the Nesmeyanov Institute; X-ray powder diffraction studies were performed at the Shared Facility Centre of the Kurnakov Institute.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alcock, N.W. Secondary Bonding to Nonmetallic Elements. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus, H.J., Sharpe, A.G., Eds.; Elsevier: Amsterdam, Netherlands, 1972; Volume 15, pp. 1–444. [Google Scholar]
- Glidewell, C. Some chemical and structural consequences of non-bonded interactions. Inorg. Chim. Acta 1975, 12, 219–227. [Google Scholar] [CrossRef]
- Kuz'mina, L.G.; Struchkov, Y.T. Structural chemistry of organomercury compounds – role of secondary interactions. Croat. Chim. Acta 1984, 57, 701–724. [Google Scholar]
- Kuz'mina, L.G. Secondary bonds and their role in chemistry. Russ. J. Coord. Chem. 1999, 25, 599–617. [Google Scholar]
- Virovets, A.V.; Podberezskaya, N.V. Specific nonbonded interactions in the structures of M3X74+ and M3X44+ (M = Mo, W; X = O, S, Se) clusters. J. Struct. Chem. 1993, 34, 306–322. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- George, A.J. An Introduction to Hydrogen Bonding (Topics in Physical Chemistry); Oxford University Press: New York, NY, USA, 1997; pp. 4–320. [Google Scholar]
- Muetterties, E.L.; Knoth, W.H. Polyhedral Boranes; Marcel Dekker: New York, NY, USA, 1968; pp. 104–168. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth–Heinemann: Oxford, UK, 1997; pp. 4–1600. [Google Scholar]
- Malinina, E.A.; Avdeeva, V.V.; Goeva, L.V.; Kuznetsov, N.T. Coordination compounds of electron-deficient boron cluster anions BnHn2− (n = 6, 10, 12). Russ. J. Inorg. Chem. 2010, 55, 2148–2202. [Google Scholar] [CrossRef]
- Avdeeva, V.V.; Malinina, E.A.; Sivaev, I.B.; Bregadze, V.I.; Kuznetsov, N.T. Silver and Copper Complexes with closo-Polyhedral Borane, Carborane and Metallacarborane Anions: Synthesis and X-ray Structure. Crystals 2016, 6, 60. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Prikaznov, A.V.; Naoufal, D. Fifty years of the closo-decaborate anion chemistry. Collect. Czech. Chem. Commun. 2010, 75, 1149–1199. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I.; Sjoberg, S. Chemistry of closo-Dodecaborate Anion [B12H12]2-: A Review. Collect. Czech. Chem. Commun. 2002, 67, 679–727. [Google Scholar] [CrossRef]
- Zhizhin, K.Y.; Zhdanov, A.P.; Kuznetsov, N.T. Derivatives of closo-decaborate anion [B10H10]2− with exo-polyhedral substituents. Russ. J. Inorg. Chem. 2010, 55, 2089–2127. [Google Scholar] [CrossRef]
- Grimes, R. Carboranes, 2nd ed.; Academic Press: Cambridge, MA, USA, 2011; 1139p. [Google Scholar]
- Klanberg, F.; Muetterties, E.L.; Guggenberger, L.J. Metalloboranes. I. Metal complexes of B3, B9, B9S, B10, and B11 borane anions. Inorg. Chem. 1968, 7, 2272–2278. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Fehlner, T.P. Metalloboranes: Their Relationships to Metal-Hydrocarbon Complexes and Clusters. Adv. Organomet. Chem. 1982, 21, 57–112. [Google Scholar]
- Shubina, E.S.; Bakhmutova, E.V.; Filin, A.M.; Sivaev, I.B.; Teplitskaya, L.N.; Chistyakov, A.L.; Stankevich, I.V.; Bakhmutov, V.I.; Bregadze, V.I.; Epstein, L.M. Dihydrogen bonding of decahydro-closo-decaborate(2-) and dodecahydro-closo-dodecaborate(2-) anions with proton donors: Experimental and theoretical investigation. J. Organomet. Chem. 2002, 657, 155–162. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, J.-C.; Shore, S.G. The Roles of Dihydrogen Bonds in Amine Borane Chemistry. Acc. Chem. Res. 2013, 46, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Mebs, S.; Kalinowski, R.; Grabowsky, S.; Förster, D.; Kickbusch, R.; Justus, E.; Morgenroth, W.; Paulmann, C.; Luger, P.; Gabel, D.; et al. Charge transfer via the dative N-B bond and dihydrogen contacts. Experimental and theoretical electron density studies of four deltahedral boranes. J. Phys. Chem. A. 2011, 115, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H.; Siegbahn, P.E.M.; Eisenstein, O.; Rheingold, A.L.; Koetzle, T.F. A new intermolecular interaction: unconventional hydrogen bonds with element-hydride bonds as proton acceptor. Acc. Chem. Res. 1996, 29, 348–354. [Google Scholar] [CrossRef]
- Richardson, T.B.; de Gala, S.; Siegbahn, P.E.M.; Crabtree, R.H. Unconventional Hydrogen Bonds: Intermolecular B-H...H-N Interactions. J. Am. Chem. Soc. 1995, 117, 12875–12876. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Beno, C.L.; Harwell, D.E.; Jalisatgi, S.S.; Knobler, C.B. Intra- and inter-molecular hydrogen bonding in some cobaltacarboranes. J. Mol. Struct. (Theochem) 2003, 656, 239–247. [Google Scholar] [CrossRef]
- Virovets, A.V.; Vakulenko, N.N.; Volkov, V.V.; Podberezskaya, N.V. Crystal structure of di(1-n-dodecylpyridine) decahydro-closo-decaborate(2-) (C5H5N-C12H25)2[B10H10]. J. Struct. Chem 1994, 35, 339–344. [Google Scholar] [CrossRef]
- Custelcean, R.; Jackson, J.E. Dihydrogen Bonding: Structures, Energetics, and Dynamics. Chem. Rev. 2001, 101, 1963–1980. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Shubina, E.S. Hydrogen and dihydrogen bonds in the reactions of metal hydrides. Chem. Rev. 2016, 116, 8545–8587. [Google Scholar] [CrossRef] [PubMed]
- Bakhmutov, V.I. Dihydrogen Bonds: Principles, Experiments and Applications; Wiley-Interscience: Hoboken, NJ, USA, 2008. [Google Scholar]
- Malinina, E.A.; Avdeeva, V.V.; Goeva, L.V.; Polyakova, I.N.; Kuznetsov, N.T. Specific interactions in metal salts and complexes with cluster boron anions BnHn2− (n = 6, 10, 12). Russ. J. Inorg. Chem. 2011, 56, 687–697. [Google Scholar] [CrossRef]
- Avdeeva, V.V.; Kravchenko, E.A.; Gippius, A.A.; Vologzhanina, A.V.; Ugolkova, E.A.; Minin, V.V.; Malinina, E.A.; Kuznetsov, N.T. Synthesis, structure, and physicochemical properties of triply-bridged binuclear copper(II) complex [Cu2Phen2(μ-CH3CO2)2(μ-OH)]2[B10Cl10]. Inorg. Chim. Acta 2019, 487, 208–213. [Google Scholar] [CrossRef]
- Kravchenko, E.A.; Gippius, A.A.; Vologzhanina, A.V.; Avdeeva, V.V.; Malinina, E.A.; Ulitin, E.O.; Kuznetsov, N.T. Secondary interactions in decachloro-closo-decaborates of alkali metals M2[B10Cl10] (M = K+ and Cs+): 35Cl NQR and X-ray studies. Polyhedron 2016, 117, 561–568. [Google Scholar] [CrossRef]
- Kravchenko, E.A.; Gippius, A.A.; Vologzhanina, A.V.; Avdeeva, V.V.; Malinina, E.A.; Demikhov, E.I.; Kuznetsov, N.T. Secondary interactions as defined by 35Cl NQR spectra in cesium decachloro-closo-decaborates prepared in non-aqueous solutions. Polyhedron 2017, 138, 140–144. [Google Scholar] [CrossRef]
- Chantler, C.T.; Maslen, E.N. Charge transfer and three-centre bonding in monoprotonated and diprotonated 2,2'-bipyridylium decahydro-closo-decaborate(2-). Acta Crystallogr., Sect. B 1989, 45, 290–297. [Google Scholar] [CrossRef]
- Fuller, D.J.; Kepert, D.L.; Skelton, B.W.; White, A.H. Structure, Stereochemistry and Novel 'Hydrogen Bonding' in Two Bipyridinium Salts of the B10H102−Anion. Aust. J. Chem. 1987, 40, 2097–2105. [Google Scholar] [CrossRef]
- Miller, H.C.; Miller, N.E.; Muetterties, E.L. Synthesis of polyhedral boranes. J. Am. Chem. Soc. 1963, 85, 3885–3886. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Morris, J.H. Novel Synthesis of the B12H122− Anion. Proc. Chem. Soc. 1963, 11, 338. [Google Scholar]
- Avdeeva, V.V.; Vologzhanina, A.V.; Goeva, L.V.; Malinina, E.A.; Kuznetsov, N.T. Reactivity of boron cluster anions [B10H10]2−, [B10Cl10]2– and [B12H12]2– in cobalt(II)/cobalt(III) complexation with 1,10-phenanthroline. Inorg. Chim. Acta 2015, 428, 154–162. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17 (2017). University of Western Australia. Available online: http://hirshfeldsurface.net (accessed on 27 June 2019).
- Qi, G.; Wang, K.; Yang, K.; Zou, B. Pressure-Induced Phase Transition of Hydrogen Storage Material Hydrazine Bisborane: Evolution of Dihydrogen Bonds. J. Phys. Chem. C. 2016, 120, 21293–21298. [Google Scholar] [CrossRef]
- Qi, G.; Wang, K.; Xiao, G.; Zou, B. High pressure, a protocol to identify the weak dihydrogen bonds: experimental evidence of C–H···H–B interaction. Sci. Chin. Chem. 2018, 61, 276–280. [Google Scholar] [CrossRef]
- Smol'yakov, A.F.; Korlyukov, A.A.; Dolgushin, F.M.; Balagurova, E.V.; Chizhevsky, I.T.; Vologzhanina, A.V. Studies of Multicenter and Intermolecular Dihydrogen B–H···H–C Bonding in [4,8,8′-exo-{PPh3Cu}-4,8,8′-(μ-H)3-commo-3,3′-Co(1,2-C2B9H9)(1′,2′-C2B9H10)]. Eur. J. Inorg. Chem. 2015, 5847–5855. [Google Scholar] [CrossRef]
- Bennour, I.; Haukka, M.; Teixidor, F.; Vinas, C.; Kabadou, A. Crystal structure and Hirshfeld surface analysis of [N(CH3)4][2,2′-Fe(1,7-closo-C2B9H11)2]. J. Organomet. Chem. 2017, 846, 74–80. [Google Scholar] [CrossRef]
- Sagan, F.; Filas, R.; Mitoraj, M.P. Non-Covalent Interactions in Hydrogen Storage Materials LiN(CH3)2BH3 and KN(CH3)2BH3. Crystals 2016, 6. [Google Scholar] [CrossRef]
Figure 1.
Asymmetric unit of 1·2H2O in representation of atoms with thermal ellipsoids (p = 50%). H-bonds are depicted with dotted lines.
Figure 1.
Asymmetric unit of 1·2H2O in representation of atoms with thermal ellipsoids (p = 50%). H-bonds are depicted with dotted lines.

Figure 2.
Fragment of the structure of 2·2H2O in representation of atoms with thermal ellipsoids (p = 50%). Only symmetrically independent boron atoms are labeled. H-bonds are depicted with dotted lines.
Figure 2.
Fragment of the structure of 2·2H2O in representation of atoms with thermal ellipsoids (p = 50%). Only symmetrically independent boron atoms are labeled. H-bonds are depicted with dotted lines.

Figure 3.
Asymmetric unit of 3·2CH3CN in representation of atoms with thermal ellipsoids (p = 50%). H-bonds are depicted with dotted lines.
Figure 3.
Asymmetric unit of 3·2CH3CN in representation of atoms with thermal ellipsoids (p = 50%). H-bonds are depicted with dotted lines.

Figure 4.
Asymmetric unit of 4 in representation of atoms with thermal ellipsoids (p = 50%). H-bonds are depicted with dotted lines.
Figure 4.
Asymmetric unit of 4 in representation of atoms with thermal ellipsoids (p = 50%). H-bonds are depicted with dotted lines.

Figure 5.
Fragment of the structure of 5·2CH3CN in representation of atoms with thermal ellipsoids (p = 50%). Only symmetrically independent atoms are labelled. H-bonds are depicted with dotted lines.
Figure 5.
Fragment of the structure of 5·2CH3CN in representation of atoms with thermal ellipsoids (p = 50%). Only symmetrically independent atoms are labelled. H-bonds are depicted with dotted lines.

Figure 6.
(a) Asymmetric unit of 6 ∙ 0.5H2O in representation of atoms with thermal ellipsoids (p = 50%). (b) Fragment of unit cell of 6 ∙ 0.5H2O along crystallographic axis c. H(C) atoms are omitted, N-H...N bonds are dashed.
Figure 6.
(a) Asymmetric unit of 6 ∙ 0.5H2O in representation of atoms with thermal ellipsoids (p = 50%). (b) Fragment of unit cell of 6 ∙ 0.5H2O along crystallographic axis c. H(C) atoms are omitted, N-H...N bonds are dashed.

Figure 7.
Molecular Hirshfeld surfaces of 1–5 mapped with dnorm.

Figure 8.
IR spectra (in the ν(BH) region) of compounds with (a) [B10H10]2− and (b) [B12H12]2− anions in comparison with alkali closo-borates: (1) K2[B10H10]; (2) [PhenH]2[B10H10] (4); (3) [H2(Phen)4.5][B10H10] (6); (4) (Et3NH)(BPAH)[B10H10] (3); (5) (BPAH)2(BPAH2)[B10H10]2 (1); (6) Cs2[B12H12]; (7) [Rh6GH]2[B12H12] (5); (8) [PhenH]2[B12H12] (2).
Figure 8.
IR spectra (in the ν(BH) region) of compounds with (a) [B10H10]2− and (b) [B12H12]2− anions in comparison with alkali closo-borates: (1) K2[B10H10]; (2) [PhenH]2[B10H10] (4); (3) [H2(Phen)4.5][B10H10] (6); (4) (Et3NH)(BPAH)[B10H10] (3); (5) (BPAH)2(BPAH2)[B10H10]2 (1); (6) Cs2[B12H12]; (7) [Rh6GH]2[B12H12] (5); (8) [PhenH]2[B12H12] (2).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Maxima of ν(NH) and ν(BH) absorption bands in IR spectra of compounds 1–6 as compared to alkali metal closo-borates (cm–1).
Table 1.
Maxima of ν(NH) and ν(BH) absorption bands in IR spectra of compounds 1–6 as compared to alkali metal closo-borates (cm–1).
| Compound | ν(NH) | ν(BH) |
|---|---|---|
| K2[B10H10] | — | 2529, 2461 |
| Phen | — | — |
| 4 | 3095 | 2510, 2477, 2451, 2421 |
| 6·0.5H2O | 3164, 3152 | 2495, 2476, 2414 |
| BPA | 3255, 3181, 3102 | — |
| 1·2H2O | 3286, 3236, 3204, 3136 | 2543, 2492, 2444, 2420 |
| 3·2CH3CN | 3467, 3316, 3255, 3212, 3145, 3111 | 2499, 2466, 2448, 2404 |
| Cs2[B12H12] | — | 2465 |
| 5·CH3CN | 3410, 3361 | 2484, 2461, 2447, 2422 |
| 2·2H2O | 3098, 3063 | 2487, 2443 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Avdeeva, V.V.; Vologzhanina, A.V.; Malinina, E.A.; Kuznetsov, N.T. Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases. Crystals 2019, 9, 330. https://doi.org/10.3390/cryst9070330
AMA Style
Avdeeva VV, Vologzhanina AV, Malinina EA, Kuznetsov NT. Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases. Crystals. 2019; 9(7):330. https://doi.org/10.3390/cryst9070330
Chicago/Turabian StyleAvdeeva, Varvara V., Anna V. Vologzhanina, Elena A. Malinina, and Nikolai T. Kuznetsov. 2019. "Dihydrogen Bonds in Salts of Boron Cluster Anions [BnHn]2− with Protonated Heterocyclic Organic Bases" Crystals 9, no. 7: 330. https://doi.org/10.3390/cryst9070330
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.