Theory of the Flower Micelle Formation of Amphiphilic Random and Periodic Copolymers in Solution


Department of Macromolecular Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
Polymers 2018, 10(1), 73; https://doi.org/10.3390/polym10010073
Submission received: 9 December 2017
/
Revised: 10 January 2018
/
Accepted: 11 January 2018
/
Published: 14 January 2018
(This article belongs to the Special Issue Polymer Micelles)
Abstract
:The mixing Gibbs energy Δgm for the flower-micelle phase of amphiphilic random and periodic (including alternating) copolymers was formulated on the basis of the lattice model. The formulated Δgm predicts (1) the inverse proportionality of the aggregation number to the degree of polymerization of the copolymer, (2) the increase of the critical micelle concentration with decreasing the hydrophobe content, and (3) the crossover from the micellization to the liquid–liquid phase separation as the hydrophobe content increases. The transition from the uni-core flower micelle to the multi-core flower necklace as the degree of polymerization increases was also implicitly indicated by the theory. These theoretical results were compared with experimental results for amphiphilic random and alternating copolymers reported so far.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Borisov and Halperin [1,2,3,4,5] proposed theoretical models of flower micelles, flower necklaces, and bouquets of polymer micelles formed by amphiphilic periodic copolymers composed of hydrophilic and hydrophobic monomer units in aqueous solutions. They assumed that the main chain of the periodic copolymer is perfectly flexible, and all hydrophobes in the copolymer chain are included in the hydrophobic core(s) of the micelle.
Afterward, experimental studies on amphiphilic random and periodic (including alternating) copolymers bearing hydrophobic side chains demonstrated the formation of flower micelles and flower necklaces in aqueous solutions [6,7,8,9,10,11]. However, experimental results indicated that not all hydrophobic side chains on the copolymer chain are included in the hydrophobic core(s) of the micelle, being different from the Borisov–Halperin model, and that the loop-chain size is determined by the main-chain stiffness rather than the content and sequence of the hydrophobic side chain on the copolymer chain. Thus, we need a new theory to discuss the micellization behavior of such flower micelles and flower necklaces.
The present paper proposes a lattice-model theory for dilute aqueous solutions of amphiphilic random and periodic copolymers bearing hydrophobic linear side chains, which can be regarded as graft copolymer chains bearing hydrophobic graft (side) chains to demonstrate the formation of the flower micelle. Recently, Sato and Takahashi [12] presented a similar lattice-model theory for amphiphilic block copolymer solutions to discuss the competition between micellization and liquid–liquid phase separation in the solutions. The present theory is the random and periodic copolymer version of this theory.
2. Theory
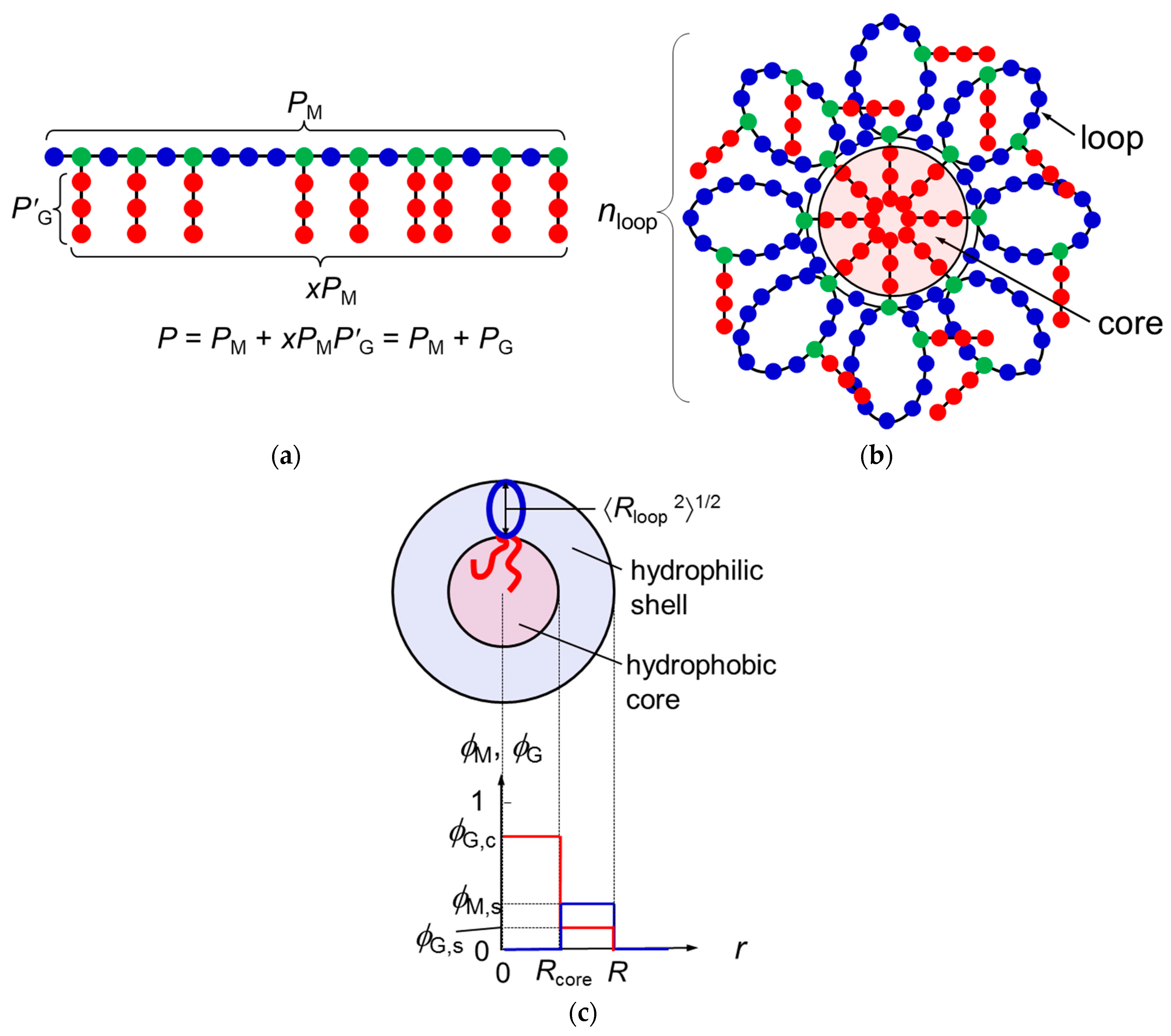
Let us consider the graft copolymer illustrated in Figure 1a. The main chain and graft chains consist of PM units and P′G units, respectively. The mole fraction of the branch units on the main chain is denoted as x, and the distribution of the branch units along the main chain is assumed to be random or periodic (not block-like). The total number of the graft-chain units per copolymer chain is PG = xPMP′G, and the total degree of polymerization of the graft copolymer chain is P = PM + PG = PM(1 + xP′G). It is assumed that the main-chain and graft-chain units as well as the solvent S molecule occupy lattice sites with a common size a.
If the graft-chain unit is sufficiently hydrophobic, graft chains of the copolymer tend to aggregate to form a hydrophobic core, and the main chain tends to form loop chains in aqueous medium. As a result, the m copolymer chains construct a flower micelle illustrated in Figure 1b; m is the copolymer-chain aggregation number of the micelle. Only graft chains attaching to roots of the loops can enter the hydrophobic core, and the remaining graft chains are outside the core.
According to the wormlike chain model [13,14,15], the ring closure probability of the chain rapidly diminishes to zero at the chain contour length reducing to ca. 1.6q, where q is the persistence length. This means that the main chain portion shorter than 1.6q cannot form the loop because of the chain stiffness. In what follows, we consider the flower micelle consisting of loop chains with this “minimum loop size” [6]. The number of main-chain units per the minimum loop chain Ploop, and the number of loop chains per chain nloop are calculated by
The numbers of graft chains included in the hydrophobic core and outside of the core, xcPM and xsPM, respectively, are calculated by
where λ is the number of side chains included in the core at each root of the loop. (Figure 1b illustrates the case of λ = 1). It has been assumed in Equation (2) that nloop is much larger than unity.
In the previous paper [12], we regarded the spherical micelle formed by di-block copolymer chains as a thermodynamic phase, assuming that the aggregation number of the micelle is sufficiently large. Similarly, the present study regards the flower micelle as a thermodynamic phase to demonstrate the micellization of the graft copolymer in a selective solvent. Furthermore, we use a simple model for the flower micellar phase, of which radial concentration profiles (volume fractions) of the main-chain and graft-chain units are given by
(cf. Figure 1c). Here, Rcore and R are the radii of the micelle core and the whole micelle, respectively, and the solvent volume fraction is given by S = 1 − M − G at each radial distance r. Furthermore, using the wormlike chain model, Rcore2 and the mean square distance from the end to the midpoint of the loop 〈Rloop2〉 (cf. Figure 1c) are expressed in terms of the persistence lengths of the graft chain (qG) and of the copolymer main chain (q), respectively, by [15]
(cf. Appendix A). The radius R of the whole micelle is calculated by
The average volume fraction ϕP of the copolymer in the flower micelle phase is given by
and the volume fractions M,s, G,s, and G,c are related to P by
From the last equation for G,c in Equation (8), it can be seen that P must be equal to or less than PRcore3/xcPMP′GR3, because G,c does not exceed unity. Furthermore, since m must be larger than unity, PM must be smaller than 4πR3P/3a3(1 + xP′G) from Equation (7).
For amphiphilic random or periodic copolymers, the ionizable group or hydrophilic side-chain group of each hydrophilic monomer unit is substituted by the hydrophobic graft chain. Thus, the branch unit in the main chain (green circles in Figure 1a,b) may be hydrophobic, having interaction parameters much different from those of the non-branch unit (i.e., the hydrophilic monomer unit) in the main chain but similar to those of the graft-chain unit. We refer to the non-branch unit in the main chain as the A unit and to the graft-chain unit as well as the branch unit in the main chain as the B unit, neglecting the difference in the interaction between the graft-chain unit and the branch unit in the main chain. The volume fractions of the A and B units in the shell and core phases are given by
and the mole fractions of the A and B units in the copolymer chain are written as
where xB,s and xB,c are the mole fractions of the B unit in the shell and core regions, respectively. The solvent volume fractions in the shell and core regions are given by S,s = 1 − A,s − B,s and S,c = 1 − B,c, respectively.
We apply the Flory–Huggins theory [16] to the flower micelle phase to formulate the mixing Gibbs energy per lattice site Δgm of the micelle phase, which consists of the mixing entropy ΔS, the mixing enthalpy ΔH, and the interfacial Gibbs energy 4πRcore2γ (γ: the interfacial tension between the core and shell regions of the micelle). The formulation method is described in Appendix B. The final result is written as
where χAS, χBS, and χAB are the interaction parameters between S and A, between S and B, and between A and B, respectively, κ is defined by Equation (B11), and (a2/kBT)γ is calculated by Equation (B13) with Equation (B14). The term ln κ includes the conformational entropy loss at the formation of the flower micelle.
3. Results and Discussion
Because we did not consider above the interaction among flower micellar phases in the solution, the following discussion is limited to dilute solutions of random and periodic copolymers. Ueda et al. [9] reported the molecular weight dependence of the micellization behavior for the amphiphilic alternating copolymer of sodium maleate and dodecyl vinyl ether, P(MAL/C12), in dilute aqueous solutions including 0.05 M NaCl. First, we examine theoretically the micellization behavior of an alternating copolymer mimicking P(MAL/C12).
In the lattice theory, the choice of the unit lattice site is rather arbitrary. Here, we assume the main-chain portion (the C2 unit) of maleate or dodecyl vinyl ether monomer unit is chosen as the unit lattice site. Then, the hydrophobic dodecyl side chain is assumed to occupy six lattice sites, i.e., P′G = 6. (The carboxy group and the ether oxygen atom in the maleate and dodecyl vinyl ether monomer units are not considered explicitly; they are assumed to be included in the main-chain portions). In aqueous solutions, a strong electrostatic repulsion acts among maleate units (the A unit), while a hydrophobic attraction acts among the C2 units of the dodecyl group (the B unit). The strong electrostatic repulsion and hydrophobic attraction are expressed using a negative χAS and positive χBS, respectively. (To account for the long range electrostatic interaction, the unit lattice site may have to be larger than the C2 unit, but the following results do not essentially change by the choice of the unit lattice site). Since we here focus on the amphiphilicity of the graft copolymer, we assume χAB to be zero, as in the previous study [12]. (The change of the χAB value may be compensated by adjusting values of χAS and χBS).
Figure 2 shows the copolymer concentration dependences of Δgm (red curve) and Δgh (black curve) calculated by Equations (11) and (12). We have chosen PM = 50, x = 0.5, χAS = −15, χBS = 3, and χAB = 0 ( = 0.75). All remaining parameters included in Equation (11) can be calculated from a = 0.25 nm (the contour length per the main-chain monomer (C2) units), and q = 3 nm, qG = 0.53 nm, and λ = 3 determined previously [9]. We can draw a common tangent (the thin line) to the dilute side of the black curve and red curve. (It is seen that the black curve has a downward convex shape around P = 0, if it is enlarged). The copolymer volume fractions at the two points of contact of the common tangent, denoted as P,d and P,m, are binodal concentrations of the coexisting dilute and micellar phases, respectively. The tangent line is below the common tangent line (the thin broken line) for the thick black curve for Δgh, indicating that the micellization is thermodynamically more stable than the phase separation into two homogeneous phases.
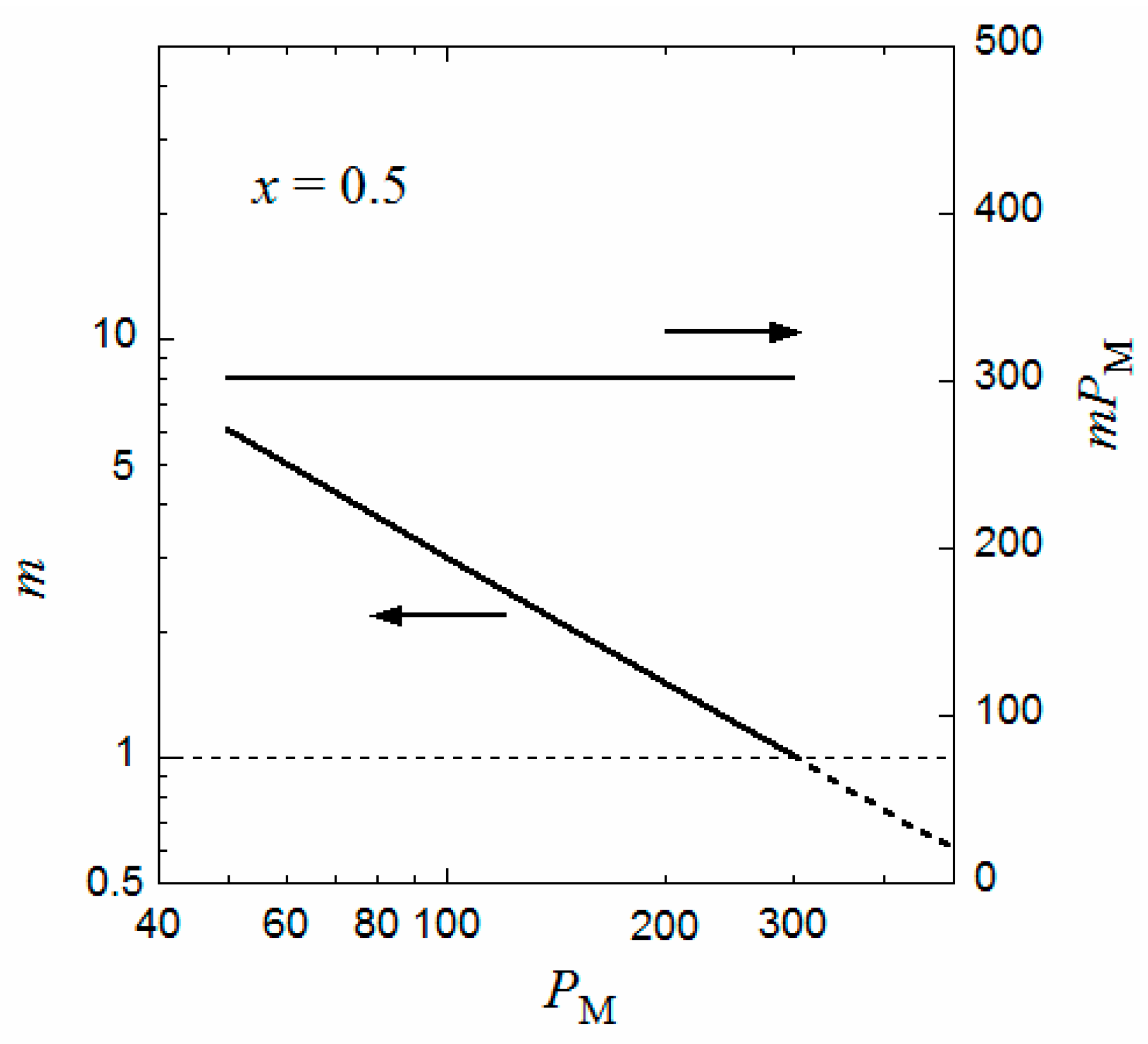
Similar curves for Δgm and Δgh were obtained for different PM, and the volume fraction P,m of the equilibrium micellar phase were determined by the above method. The aggregation number m of the copolymer chains per micelle can be calculated from Equation (7), i.e.,
Figure 3 shows the degree of polymerization PM dependence of m such obtained as well as the product mPM (the number of monomer units per micelle) at the interaction parameters identical to those in Figure 2. It is seen that m is inversely proportional to PM, and the product mPM is independent of PM. (Because P is proportional to PM and R is independent of PM, the inverse proportionality of m to PM comes from the PM independence of P,m calculated from the comparison between of the Δgm and Δgh curves). This relation was observed experimentally for P(MAL/C12) in 0.05 M aqueous NaCl solution [9] as well as for a random copolymer of poly(ethylene glycol) methyl ether methacrylate and dodecyl methacrylate, P(PEGMA/DMA), in water [18]; however, for P(PEGMA/DMA) with x = 0.5, the constant mPM is slightly larger than 300. The value of mPM changes by values of q, λ, and the interaction parameters. It is noted that the formulation of Δgm in the previous section can apply both to periodic and random copolymers.
When PM approaches 300 in Figure 3, m tends to unity, and P,d corresponding to the critical micelle concentration (cmc) of the coexisting dilute phase becomes very low (not shown). That is, when PM approaches 300, the flower micelle is formed by one copolymer chain (the unimer micelle), and the cmc tends to zero. This situation resembles the liquid–liquid phase separation in a homopolymer polymer solution with an infinitely high-molecular-weight polymer, where the polymer volume fraction at the critical point is predicted to be zero by the conventional Flory–Huggins theory [16].
When the same calculation of m is performed where PM > 300, the inverse proportionality of m to PM still holds even if PM exceeds 300, as indicated by the dashed line in Figure 3. However, because the aggregation number is less than unity, some portion of the main chain is not included in the flower micelle at PM > 300. For example, at PM = 600 where m = 0.5, half of the main chain is not included in the flower micelle. This half main-chain portion may form another flower micelle. As a result, the whole copolymer chain forms a double-core flower necklace. (Strictly speaking, the double-core flower necklace needs a bridge chain connecting two unit flowers, so that PM must be slightly larger than 600 to form the double-core flower necklace). In fact, Ueda et al. [9] reported the transition from the flower micelle to the flower necklace at PM exceeding 300.
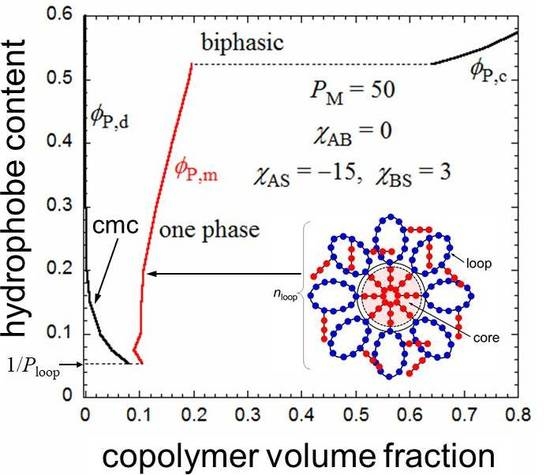
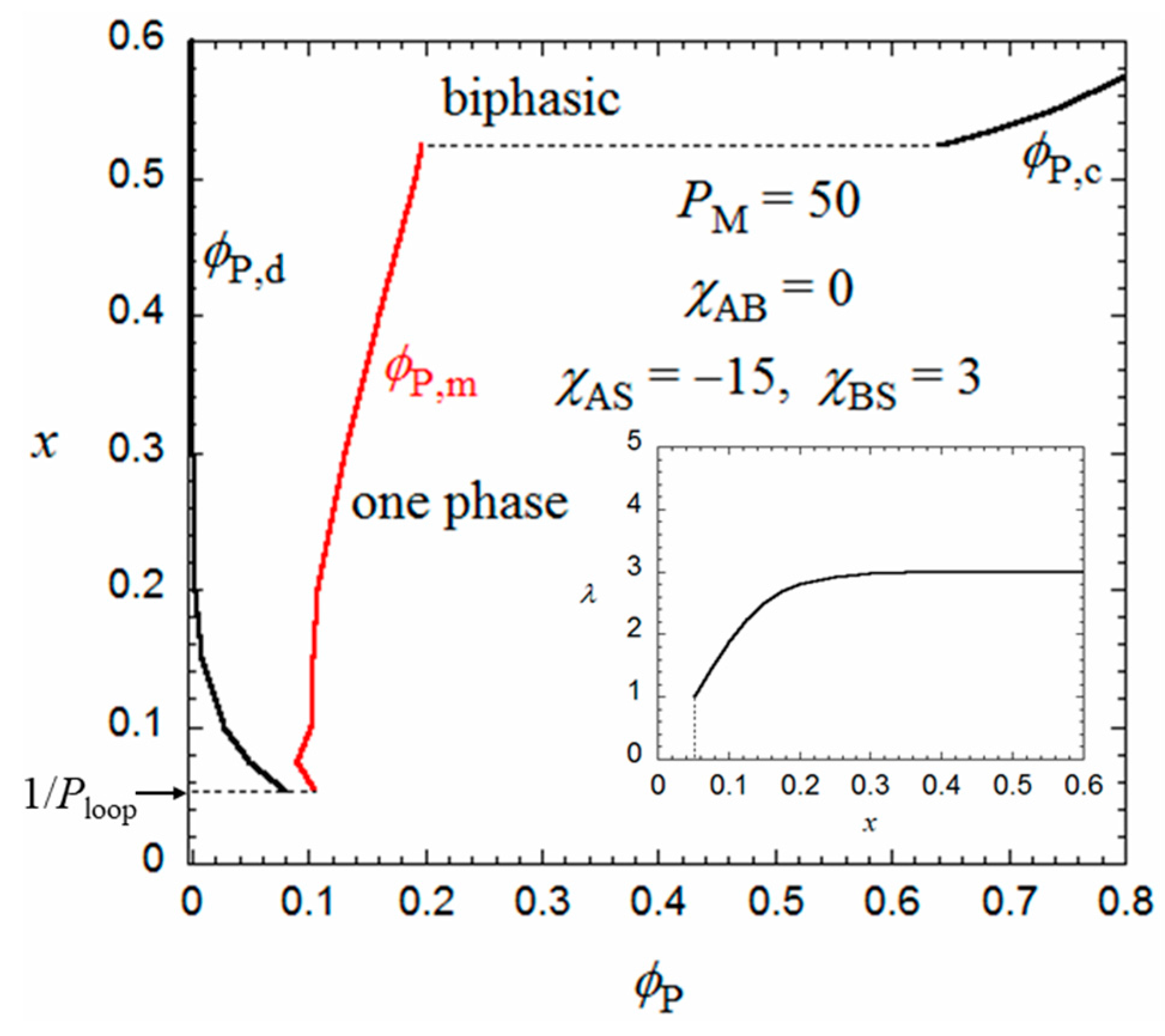
The flower micelle is formed also by amphiphilic random copolymers with hydrophobic dodecyl side chains of x < 0.5 in aqueous solutions. Next, we examine the hydrophobic monomer content dependence of the micellization for an aqueous solution of an amphiphilic random copolymer, calculated in the same way from the Δgm and Δgh curves as in Figure 2. The number λ of side chains included in the core at each root of the loop appearing in Equation (2) may be dependent on the monomer content x. In the limit of x = 1/Ploop, each loop chain has only one hydrophobic side chain on average. Thus, λ = 1 at x = 1/Ploop. When x increases, λ may first increase from unity and approach an asymptotic value. For a given value of λ, P,d and P,m of the coexisting dilute and micellar phases can be calculated as functions of x from the curves of Δgm and Δgh as mentioned above.
Figure 4 shows ϕP,d and ϕP,m obtained for the amphiphilic random copolymer with a PM of 50 and the same interaction parameters used in Figure 2 and Figure 3, in the x-P phase diagram. The x dependence of λ used is shown in the insert of Figure 4. When x is decreased from 0.5, P,d (cmc) increases, and the copolymer in a dilute solution (P < 0.08) transforms from the flower micelle to the random coil at passing the bimodal curve for P,d (cmc). At x < 1/Ploop, the loop size of the flower micelle should be larger than the minimum size given by Equation (1). We do not discuss such a loose flower micelle here.
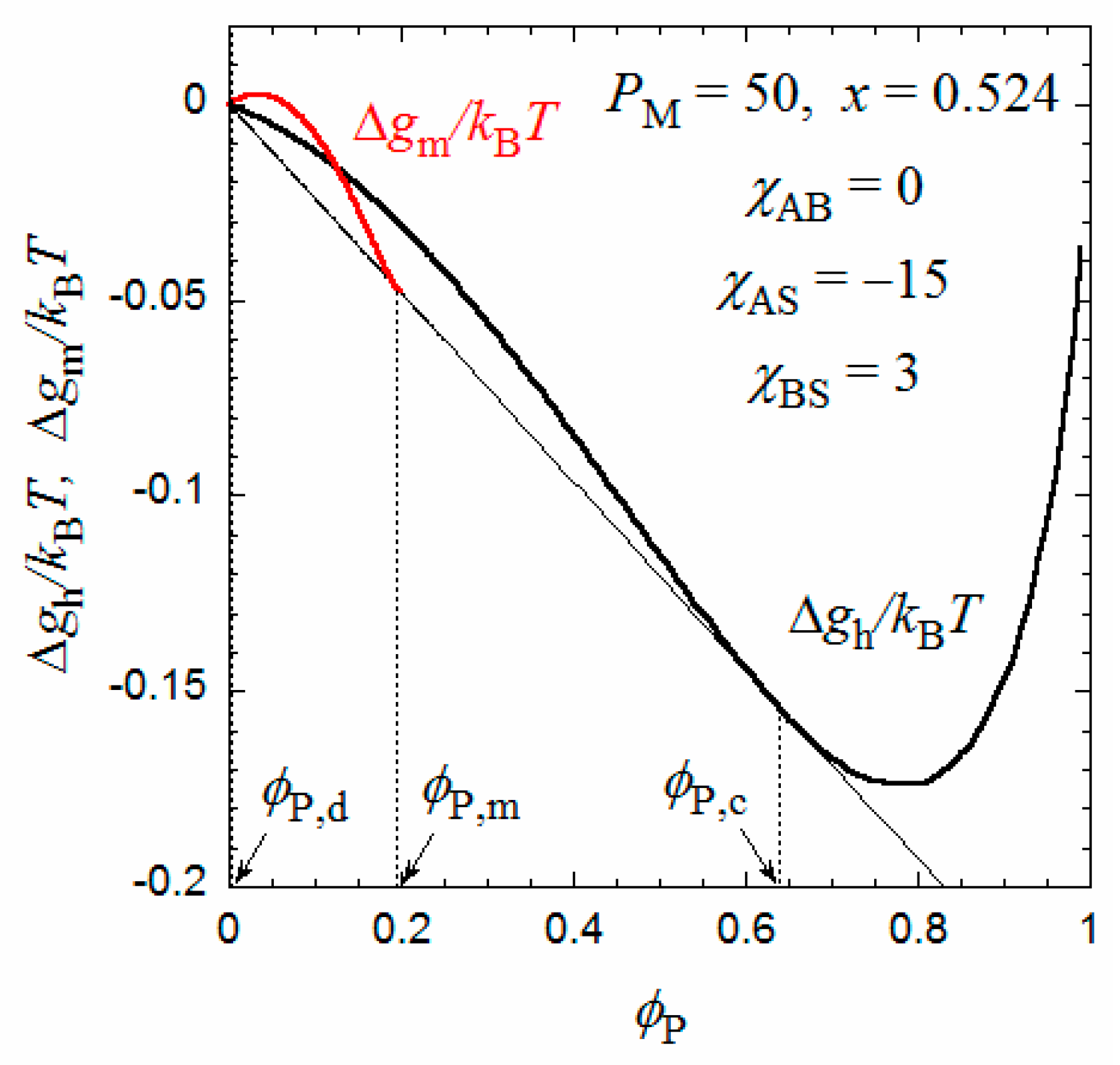
On the other hand, when x increases from 0.5, the Δgh − P curve goes down relative to the Δgm − P curve, and as shown in Figure 5, at x = 0.524, we can draw a common tangent (the thin line) to the dilute and concentrated sides of the black curve (Δgh) and the red curve (Δgm). When x > 0.524, the phase separation into dilute and concentrated homogeneous phases with concentrations P,d and P,c becomes thermodynamically more stable than the micellization. As a result, the phase gap in the x-P phase diagram is abruptly enlarged when x > 0.524, as shown in Figure 4. To the best of my knowledge, there have hitherto been no reports of the corresponding crossover from micellization to liquid–liquid phase separation as the hydrophobic content x increases.
Eisenberg et al. [19] investigated the random copolymer of styrene and methacrylic acid with x ~ 0.8, which was first dissolved in dioxane or tetrahydrofuran (THF), followed by the addition of water, and observed “large compound micelles” and “bowl-shaped aggregates” by transmission electronic microscopy (TEM). Here, the “large compound micelle” is the large homogeneous polymer-rich spheres, corresponding to the droplet of the concentrated homogeneous phase formed by the liquid–liquid phase separation, predicted in Figure 4, and a “bowl-shaped aggregate” may be formed from the concentrated-phase droplet in which solvent bubbles are trapped [19]. Wang et al. [20] reported the formation of uniform colloidal spheres by an amphiphilic random copolymer, poly{2-[4-(phenylazo)phenoxy]ethyl acrylate-co-acrylic acid}, where x = 0.5 in THF–water mixtures with high water concentrations. This may be another example of the liquid–liquid phase separation of the amphiphilic random copolymer in solution. Zhang et al. [21] studied the self-association of amphiphilic graft (periodic) copolymers in a hypothetical solution of the two-dimensional space by the self-consistent field theory. Although they assumed perfect flexibility and comparable chain lengths of the main and graft chains, being different conditions from the present study, they observed a “large compound micelle” at higher graft density (i.e., higher hydrophobic content x) under weaker amphiphilicity (cf. Figure 8a in [21], where the graft chain number = 5).
Yusa et al. [22] found a transition from the unimer micelle to the single random coil chain of a random copolymer of hydrophilic sodium 2-(acrylamido)-2-methylpropanesulfonate and hydrophobic 11-acrylamidoundecanoic acid (AmU) where x = 0.5 in 0.1 M aqueous NaCl solution by changing pH. At pH = 3, where the carboxy group is not ionized, AmU was strongly hydrophobic, and the copolymer formed a unimer micelle with m = 1. The degree of polymerization PM of the copolymer sample (= 475) was slightly larger than 300 (cf. Figure 3), maybe due to the difference in the parameters, e.g., P’G and λ, from those used in Figure 3. On the other hand, at pH = 9, where the carboxy group of AmU is ionized, the copolymer was transformed to a random coil. In Figure 2, the Δgm curve goes up and the Δgh has no inflection point when χBS is decreased from 3, i.e., AmU becomes more hydrophilic. As a result, the random coil conformation in the homogeneous phase becomes more stable than the flower micelle, which is consistent with Yusa et al.’s finding. The transition from the unimer micelle to the single random coil chain by decreasing x, predicted in Figure 4, was reported by Fujimoto and Sato [11].
Recently, several authors have reported that amphiphilic random copolymers form vesicles in dilute solutions [23,24,25,26], which was not considered in the present study. Zhu and Liu [24] investigated vinyl polymers bearing L-glutamic acid moieties and dodecyl groups in the random sequence to find the vesicle in water at a high hydrophobic content x (>0.75). Their random copolymer samples possess low degrees of polymerization (<36). For these samples to form the flower micelle, nloop should be less than 2 and one loop chain should bear many hydrophobes. The present theory may not be able to be applied to such random copolymers.
Tian et al. [25] observed vesicles as well as hollow tubes and wormlike rods formed by poly(hydroxyethyl methacrylate) (PHEMA) partially and randomly modified by the hydrophobic 2-diazo-1,2-naphthoquinone in solution. These copolymer samples were dissolved in dimethyl- formamide, followed by the addition of water, and finally dialyzed against water to form the vesicle. Because even PHEMA is insoluble in water, the vesicle formed must not be in the thermodynamically stable state, which cannot be treated in the present statistical thermodynamic theory.
Ghosh et al. [26] reported that an amphiphilic random copolymer of hydrophilic tri(oxyethylene) methacrylamide and hydrophobic n-octyl methacrylate exhibited a thermally induced vesicle to spherical micelle transition. However, it should be noted that the illustration of the spherical micelle by these authors (cf. Scheme 1 in [26]) was inconsistent with the experimental TEM observation of spherical aggregates (diameter in the range of 70–80 nm) at 60 °C. In the illustration, the hydrophilic and hydrophobic side chains were in the coronal and core regions of the micelle, respectively, and the whole copolymer main chain was confined to the corona-core interface. If this is the case, the diameter of the micelle must be equal to twice the sum of the hydrophilic and hydrophobic side chain lengths. Even if the side chains are fully extended, such an estimated diameter is as small as 6 nm, which is much smaller than the diameter of the spherical aggregate at 60 °C. Thus, the spherical aggregate at 60 °C may not be the spherical micelle indicated in their illustration, but the phase-separated concentrated phase droplet, because both kinds of side chains are hydrophobic at 60 °C above the lower critical solution temperature [26].
4. Conclusions
The flower micelle formed by amphiphilic random and periodic copolymers in solution was regarded as a thermodynamic phase to formulate the mixing Gibbs energy. The formulated mixing Gibbs energy of the micelle was compared with that of the homogeneous phase to calculate (1) the aggregation number m of the micelle as a function of the degree of polymerization PM of the copolymer chain, (2) the cmc as a function of the hydrophobic content x, and (3) the crossover x from micellization to liquid–liquid phase separation.
The above theoretical results were compared with experimental results for amphiphilic random and alternating copolymers reported previously. Prediction (1) was confirmed experimentally [9,18], and the experimentally observed transition from the uni-core flower micelle to the multi-core flower necklace [9] was also consistent with the present theory. The “large compound micelle” previously observed for amphiphilic random copolymers [19,20] may correspond to the concentrated-phase droplets produced by liquid–liquid phase separation, which was predicted to occur in this theory. The transition from the unimer micelle to the single random coil chain [11,22] was also predicted by this theory.
Acknowledgments
I thank Dr. Daichi Ida at Kyoto University for calculating Equation (A4) in Appendix A.
Conflicts of Interest
The author declares no conflict of interest.
Appendix A. Mean Square Distance from the End to the Midpoint of the Loop
The mean square distance from the end to the midpoint of the loop 〈Rloop〉 (cf. Figure 1c) near the rod and coil limits is calculated using the wormlike chain model. Yamakawa and Stockmayer [13] formulated 〈Rloop2〉 for the wormlike chain near the rod limit. Their result is written as
where q and NK is the persistence length and the Kuhn statistical segment number, respectively, and I2 and I3 are calculated by
with the angle θ formed by the tangent vectors at both chain ends and a constant C determined by the equation
Near the rod limit, the energetically most stable loop conformation gives us the results, θ = 1.7208, C = 0.6522, I2 = 3.29, and I3 = 2.58.
Using the first Daniels approximation, we can calculate 〈Rloop2〉 near the coil limit as [27]
Equation (5) in the text is the interpolation of 〈Rloop2〉 given by Equations (A1) and (A4) near rod and coil limits by use of the Padé approximation.
Appendix B. Mixing the Gibbs Energy of the Flower Micelle Phase
To calculate the mixing entropy of the flower micelle phase, we counted the number Ω of arrangements of m graft copolymer chains into the concentric spherical lattice with the inner and outer spherical radii Rcore and R, respectively, shown in Figure 1c in the text. Each main chain of the graft copolymer may form loops, trains, and tails on the core–shell interface, but we assume that both hydrophilicity of the main chain and hydrophobicity of the graft-chain are so strong that both train and tail chains are negligibly short.
The first unit of the main chain in the first copolymer chain must be located at one of the lattice sites on the core–shell interface. The number of such lattice sites is given by Nintf = 4π (Rcore/a)2. The number ω′i of lattice sites where the first unit of the i-th copolymer chain is given by
where f’i−1 is the probability of the vacancy for the lattice site on the core–shell interface when first units of i – 1 copolymer chains have been already arranged.
The flower micelle contains mnloop loop chains and mxsPM graft chains in the shell region. The first unit of the first copolymer chain is identical with the first unit of the first loop chain, and the last unit of the first loop chain must be located in the neighboring site of the first one of the same loop chain on the core–shell interface. Furthermore, the loop chain cannot be located in the core region, i.e., the core–shell interface acts as a reflecting barrier. The first loop chain possesses xsPloop graft chains with the degree of polymerization P’G. Thus, the number of arrangements ωs,1 of the first loop chain is given by [12,28]
where G(0,aPloop/2q) is the ring closure probability. Shimada and Yamakawa [14] proposed an expression of the probability for the wormlike chain as
Similarly, the number of arrangements ωs,i of the i-th loop chain is given by
Here, fs,j−1 is the probability of the vacancy for the lattice site in the shell region when j – 1 loop chains have been already arranged. The core region of the flower micelle consists of mxcPM graft chains. Numbers of arrangements ωc,1 and ωc,i of the first and i-th graft chains in the core region are written as
where fc,k−1 is the probability of the vacancy for the lattice site in the core region when k − 1 graft chains have been already arranged.
Using the above results, the number of arrangements Ω of the total m graft copolymer chains on the concentric spherical lattice is calculated by
or
The numbers of arrangements of the uniform bulk copolymer (ΩP) and the bulk solvent (ΩS) are given respectively by [16]
where
Therefore, the entropy of mixing ΔS in the micellar phase is given by
where S = 1 − M − G, kB is the Boltzmann constant, and κ is defined by
To obtain Equation (B10), we used Equation (5) in the text, approximated f’i−1 ≈ f’P,i−1, and replaced the summations with respect to j in Equations (B7) and (B8) by integrations on the assumption of mnloop >> 1. The parameter κ can be calculated from P’G, Ploop, and 2q/a using Equations (3), (4) and (B3).
At calculating the mixing enthalpy ΔH of the micelle phase and the interfacial tension γ between the core and shell regions of the flower micelle, we assume that the branch units in the main chain have the same interaction parameters as those of the graft-chain units. In what follows, the non-branch unit in the main chain is referred to as the A unit, and the graft-chain unit as well as the branch unit in the main chain are referred to as the B unit. Under the mean-field approximation [16], ΔH is given by
where χαβ (α, β = S, A, B) is the interaction parameter between species α and β (S stands for the solvent; the definition of χαβ is slightly different from that of [16]), and xA, xB,s, and xB,c are the mole fractions of the A and B units (existing in the shell and core regions) defined by Equation (8).
Noolandi and Hong [17] formulated the interfacial tension γ between the core and shell regions of the spherical micelle. We may extend their result to γ for the flower micelle, where the asymptotic volume fractions of the A and B units are A,s and B,s in the shell region and 0 and B,c in the core region, respectively. The result is given by
where the function f(x, y) is defined by
There is one more interface between the shell and solvent regions in Figure 1, but this interface is not so sharp that we did not consider its interfacial Gibbs energy.
Flory [29] extended the Flory–Huggins theory [16] to solutions of a semiflxible polymer with energetically unfavorable “bend” conformations and demonstrated that the equilibrium degree of the bending of the chain is independent of the polymer concentration. This result indicates that the chain stiffness does not contribute to the mixing Gibbs energy ΔG because of the cancelation of the bending energy in the solution and bulk states. Therefore, the above formulations of ΔS and ΔH can be applied to semiflexible polymer solutions.
References
- Borisov, O.V.; Halperin, A. Micelles of polysoaps. Langmuir 1995, 11, 2911–2919. [Google Scholar] [CrossRef]
- Borisov, O.V.; Halperin, A. Micelles of polysoaps: The role of bridging interactions. Macromolecules 1996, 29, 2612–2617. [Google Scholar] [CrossRef]
- Borisov, O.V.; Halperin, A. Polysoaps: Extension and compression. Macromolecules 1997, 30, 4432–4444. [Google Scholar] [CrossRef]
- Borisov, O.V.; Halperin, A. Self-assembly of polysoaps. Curr. Opin. Colloid Interface Sci. 1998, 3, 415–421. [Google Scholar] [CrossRef]
- Halperin, A. Supramolecular Polymers; Ciferri, A., Ed.; Marcel Dekker Ltd.: New York, NY, USA; Basel, Switzerland, 2000; ISBN 9780824723316-CAT# DK3116. [Google Scholar]
- Kawata, T.; Hashidzume, A.; Sato, T. Micellar Structure of Amphiphilic Statistical Copolymers Bearing Dodecyl Hydrophobes in Aqueous Media. Macromolecules 2007, 40, 1174–1180. [Google Scholar] [CrossRef]
- Sato, T.; Kimura, T.; Hashidzume, A. Hierarchical Structures in Amphiphilic Random Copolymer Solutions. Prog. Theor. Phys. Suppl. 2008, 175, 54–63. [Google Scholar] [CrossRef]
- Tominaga, Y.; Mizuse, M.; Hashidzume, A.; Morishima, Y.; Sato, T. Flower Micelle of Amphiphilic Random Copolymers in Aqueous Media. J. Phys. Chem. B 2010, 114, 11403–11408. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Hashidzume, A.; Sato, T. Unicore-Multicore Transition of the Micelle Formed by an Amphiphilic Alternating Copolymer in Aqueous Media by Changing Molecular Weight. Macromolecules 2011, 44, 2970–2977. [Google Scholar] [CrossRef]
- Uramoto, K.; Takahashi, R.; Terao, K.; Sato, T. Local and Global Conformations of Flower Micelles and Flower necklaces Formed by an Amphiphilic Alternating Copolymer in Aqueous Solution. Polym. J. 2016, 48, 863–867. [Google Scholar] [CrossRef]
- Fujimoto, M.; Sato, T. Aggregation and Micellization Behavior of Amphiphilic Random Copolymers Bearing Various Hydrophobic Groups in Aqueous Solution. Kobunshi Ronbunshu 2016, 73, 547–555. [Google Scholar] [CrossRef]
- Sato, T.; Takahashi, R. Competition between the micellization and the liquid–liquid phase separation in amphiphilic block copolymer solutions. Polym. J. 2017, 49, 273–277. [Google Scholar] [CrossRef]
- Yamakawa, H.; Stackmayer, W.H. Statistical Mechanics of Wormlike Chains. II. Excluded Volume Effects. J. Chem. Phys. 1972, 57, 2843–2854. [Google Scholar] [CrossRef]
- Shimada, J.; Yamakawa, H. Statistical mechanics of helical worm-like chains. XV. Excluded-volume effects. J. Chem. Phys. 1986, 85, 591–600. [Google Scholar] [CrossRef]
- Yamakawa, H.; Yoshizaki, T. Helical Wormlike Chains in Polymer Solutions, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 978-3-662–48714–3. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: New York, NY, USA, 1953; ISBN 978-0-8014-0134-3. [Google Scholar]
- Noolandi, J.; Hong, K.M. Theory of Block Copolymer Micelles in Solution. Macromolecules 1983, 16, 1443–1448. [Google Scholar] [CrossRef]
- Hirai, Y.; Terashima, T.; Takenaka, M.; Sawamoto, M. Precision Self-Assembly of Amphiphilic Random Copolymers into Uniform and Self-Sorting Nanocompartments in Water. Macromolecules 2016, 49, 5084–5091. [Google Scholar] [CrossRef]
- Liu, X.; Kim, J.-S.; Wu, J.; Eisenberg, A. Bowl-Shaped Aggregates from the Self-Assembly of an Amphiphilic Random Copolymer of Poly(styrene-co-methacrylic acid). Macromolecules 2005, 38, 6749–6751. [Google Scholar] [CrossRef]
- Li, Y.; Deng, Y.; Tong, X.; Wang, X. Formation of Photoresponsive Uniform Colloidal Spheres from an Amphiphilic Azobenzene-Containing Random Copolymer. Macromolecules 2006, 39, 1108–1115. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, J.; Lin, S. Aggregate Morphologies of Amphiphilic Graft Copolymers in Dilute Solution Studied by Self-Consistent Field Theory. J. Phys. Chem. B 2007, 111, 9209–9217. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Sakakibara, A.; Yamamoto, T.; Morishima, Y. Reversible pH-Induced Formation and Disruption of Unimolecular Micelles of an Amphiphilic Polyelectrolyte. Macromolecules 2002, 35, 5243–5249. [Google Scholar] [CrossRef]
- Li, L.; Raghupathi, K.; Song, C.; Prasad, P.; Thayumanavan, S. Self-Assembly of Random Copolymers. Chem. Commun. 2014, 50, 13417–13432. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, M. Self-Assembly and Morphology Control of New l-Glutamic Acid-Based Amphiphilic Random Copolymers: Giant Vesicles, Vesicles, Spheres, and Honeycomb Film. Langmuir 2011, 27, 12844–12850. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Yu, Y.; Wang, C.; Yang, S. Consecutive Morphological Transitions in Nanoaggregates Assembled from Amphiphilic Random Copolymer via Water-Driven Micellization and Light-Triggered Dissociation. Macromolecules 2008, 41, 3385–3388. [Google Scholar] [CrossRef]
- Dan, K.; Bose, N.; Ghosh, S. Vesicular Assembly and Thermo-Responsive Vesicle-to-Micelle Transition from an Amphiphilic Random Copolymer. Chem. Commun. 2011, 47, 12491–12493. [Google Scholar] [CrossRef] [PubMed]
- Ida, D. Private communication, 2017.
- Hoeve, C.A.J. Density Distribution of Polymer Segments in the Vicinity of an Adsorbing Interface. J. Chem. Phys. 1965, 43, 3007–3008. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Thermodynamics of Semi-Flexible Chain Molecules. Proc. Royal Soc. London A 1956, 234, 60–89. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagrams of the graft copolymer chain (a), the flower micelle formed by the copolymer chain (b), and the radial concentration profiles of the main-chain and graft-chain units in the flower micelle (c). In Panel b, the flower micelle is constructed by m copolymer chains. In Panel a, blue and green circles are referred to as the main chain, and red circles as the graft chain. In Panels a and b, blue circles are called the A unit, and red and green circles as the B unit to discuss the mixing enthalpy (cf. Equations (9)–(11)).
Figure 1.
Schematic diagrams of the graft copolymer chain (a), the flower micelle formed by the copolymer chain (b), and the radial concentration profiles of the main-chain and graft-chain units in the flower micelle (c). In Panel b, the flower micelle is constructed by m copolymer chains. In Panel a, blue and green circles are referred to as the main chain, and red circles as the graft chain. In Panels a and b, blue circles are called the A unit, and red and green circles as the B unit to discuss the mixing enthalpy (cf. Equations (9)–(11)).
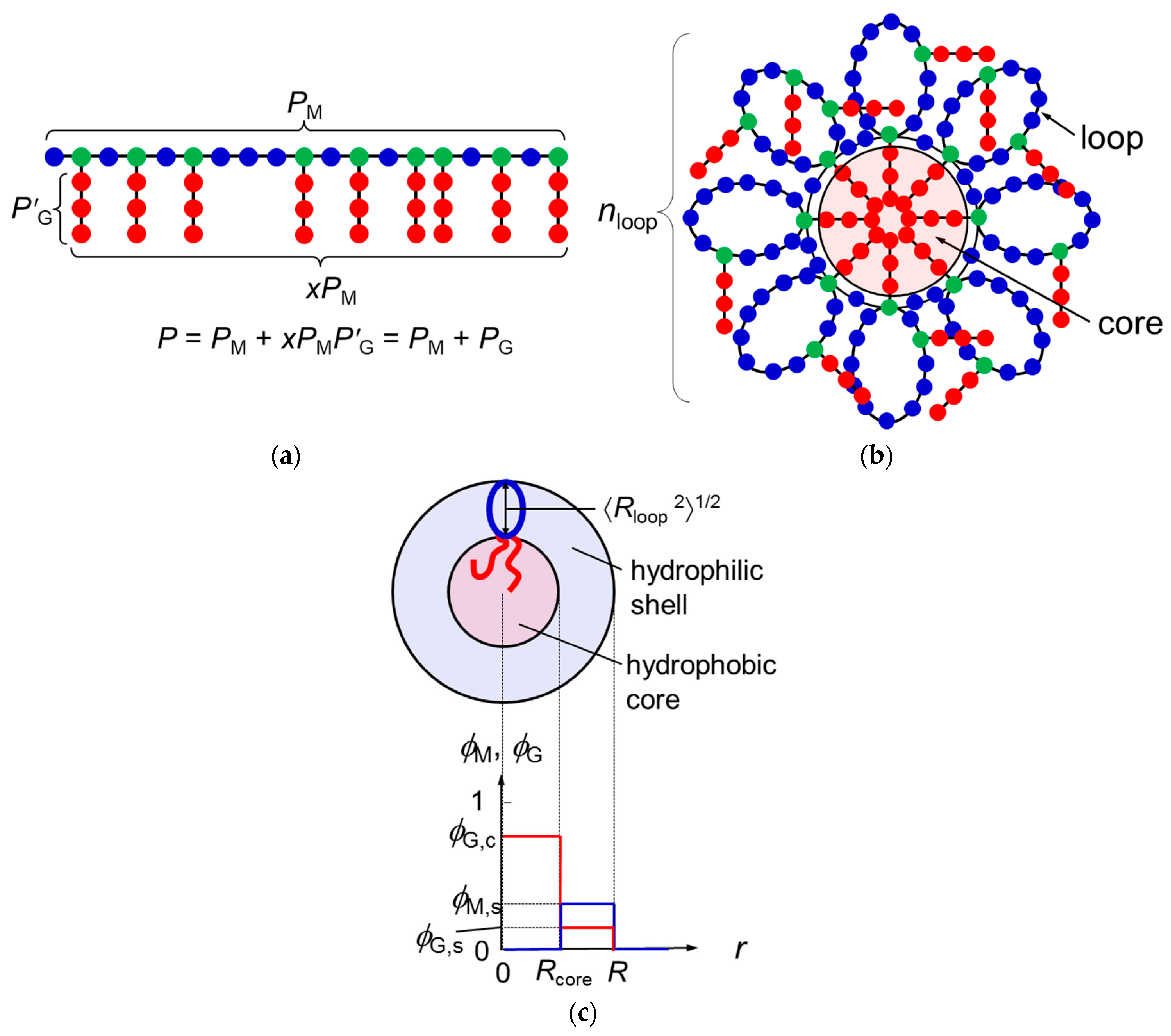
Figure 2.
Concentration dependences of Δgm and Δgh at x = 0.5 calculated by Equations (9) and (10).
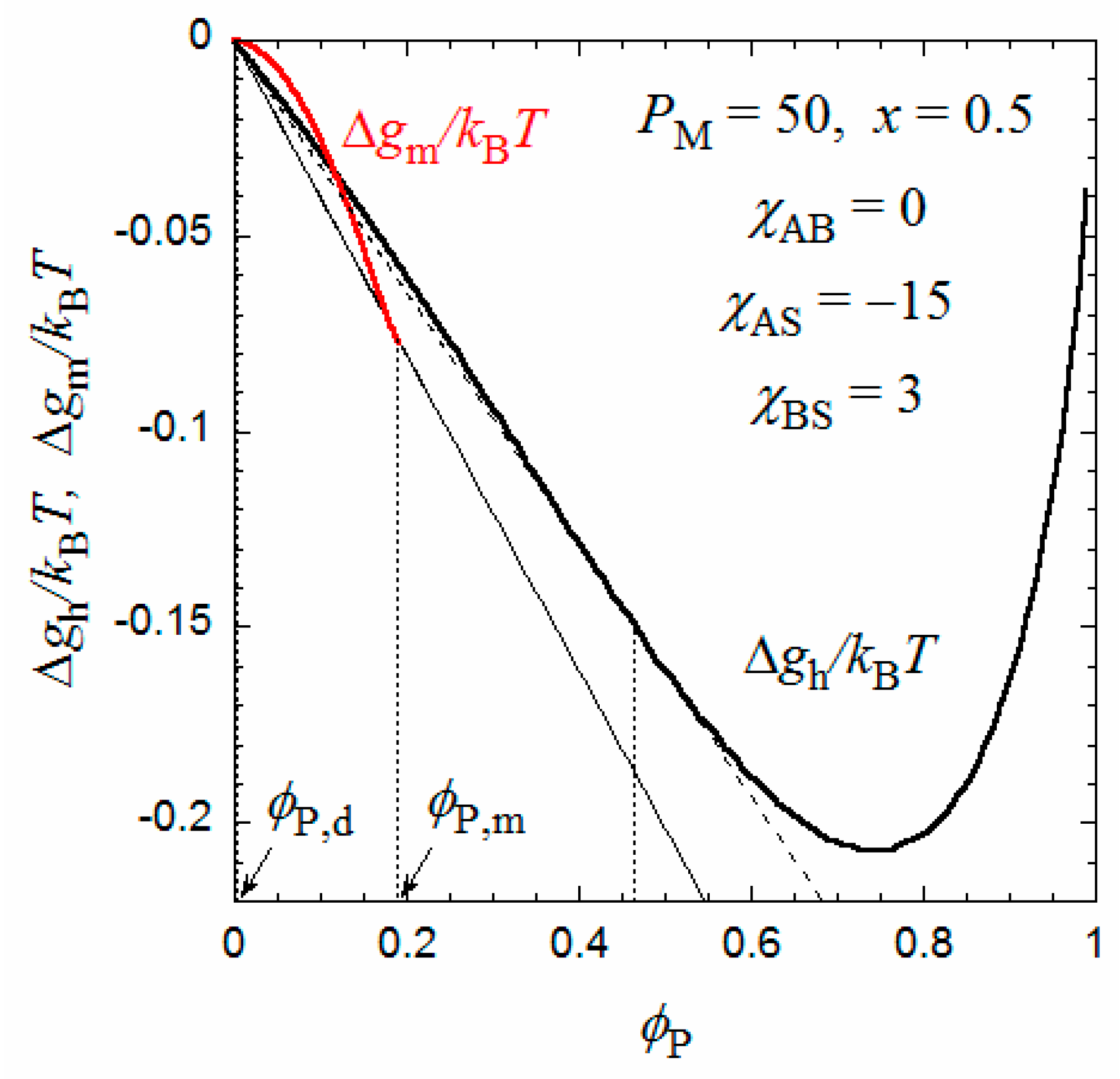
Figure 3.
Degree of polymerization dependences of the aggregation number m and mPM at the interaction parameters identical to those in Figure 2.
Figure 3.
Degree of polymerization dependences of the aggregation number m and mPM at the interaction parameters identical to those in Figure 2.
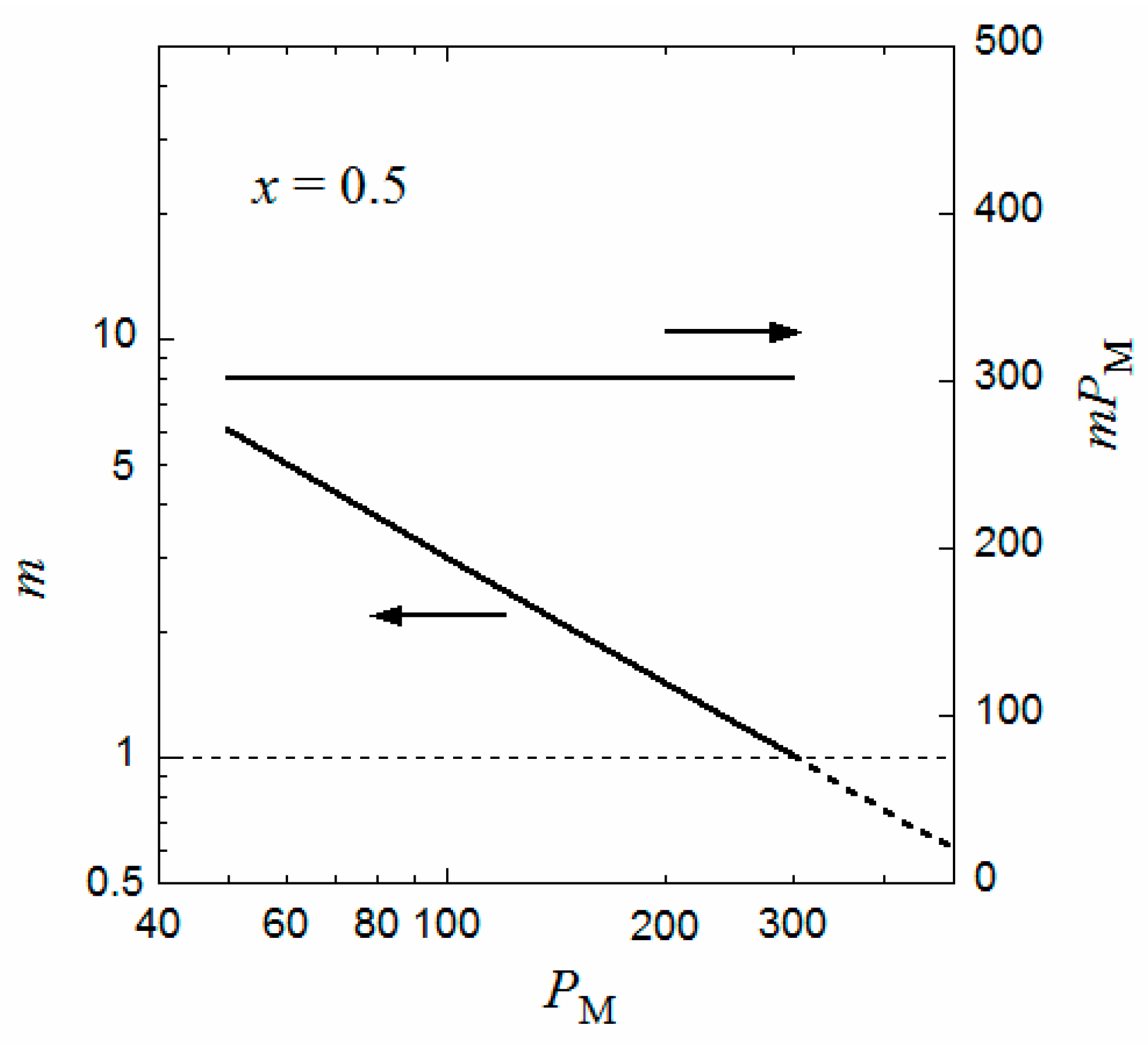
Figure 4.
Monomer content-concentration phase diagram for an aqueous solution of a random copolymer with PM = 50 and the same interaction parameters as those used in Figure 2 and Figure 3.
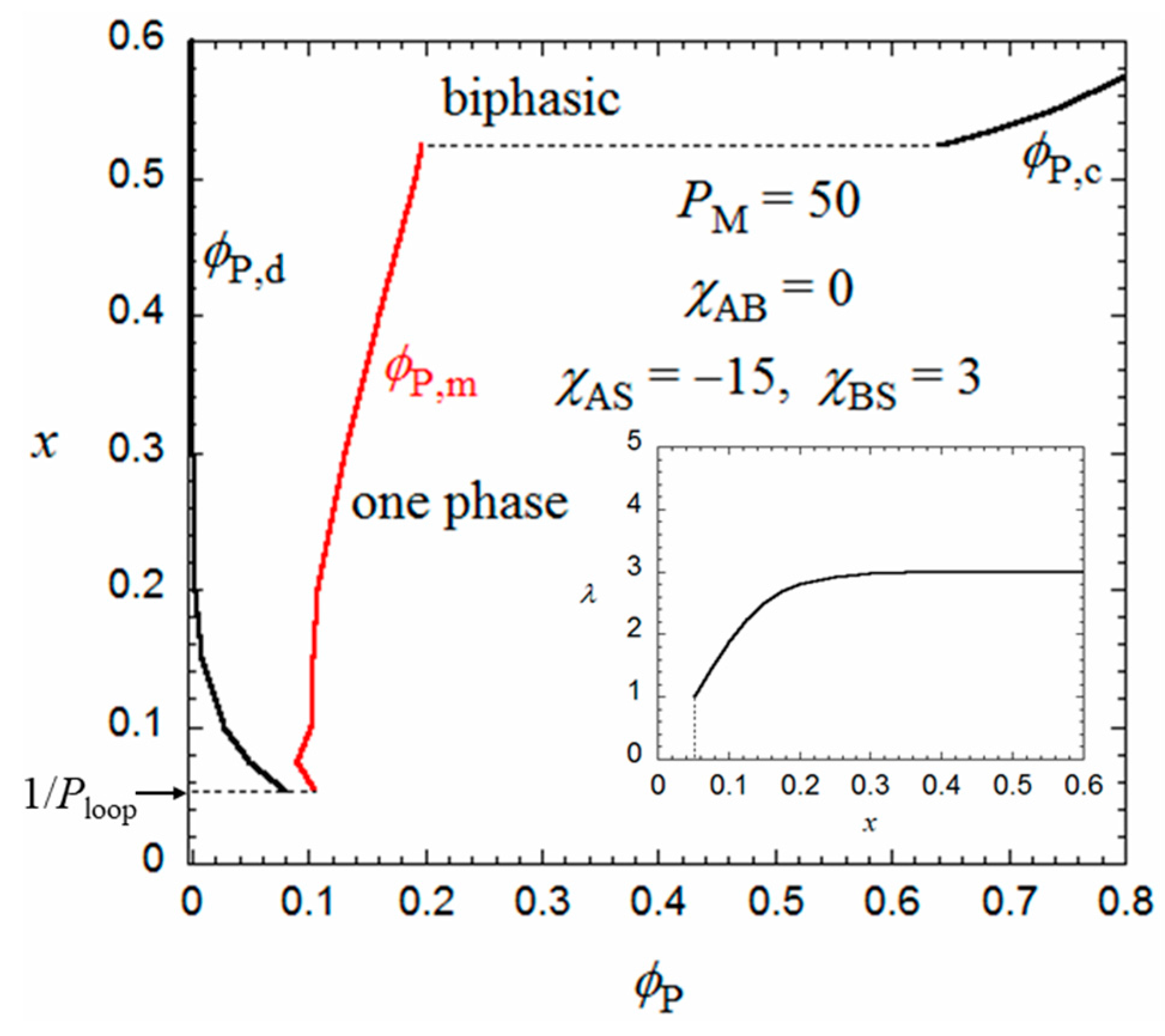
Figure 5.
Concentration dependences of Δgm and Δgh at x = 0.524 calculated by Equations (9) and (10).
Figure 5.
Concentration dependences of Δgm and Δgh at x = 0.524 calculated by Equations (9) and (10).
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sato, T. Theory of the Flower Micelle Formation of Amphiphilic Random and Periodic Copolymers in Solution. Polymers 2018, 10, 73. https://doi.org/10.3390/polym10010073
AMA Style
Sato T. Theory of the Flower Micelle Formation of Amphiphilic Random and Periodic Copolymers in Solution. Polymers. 2018; 10(1):73. https://doi.org/10.3390/polym10010073
Chicago/Turabian StyleSato, Takahiro. 2018. "Theory of the Flower Micelle Formation of Amphiphilic Random and Periodic Copolymers in Solution" Polymers 10, no. 1: 73. https://doi.org/10.3390/polym10010073
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.