Morphology, Nucleation, and Isothermal Crystallization Kinetics of Poly(Butylene Succinate) Mixed with a Polycarbonate/MWCNT Masterbatch

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Sample Characterization
- (a)
- The sample was heated from 25 to 270 °C at 20 °C min−1 and maintained at that temperature for 3 min to erase thermal history.
- (b)
- It was then cooled from 270 to −20 °C at 20 °C min−1 to create the initial “standard” state, and held at that temperature for 3 min.
- (c)
- It was then heated from −20 °C to a selected self-nucleation temperature (Ts), located in the final melting temperature range of the sample, and held at that temperature for 5 min.
- (d)
- It was again cooled to −20 °C, where the effects of thermal treatment are reflected in the crystallization behaviour of the sample.
- (e)
- Finally, it was heated to 270 °C, where the effects of thermal treatment are reflected in the melting behaviour of the sample.
3. Results and Discussion
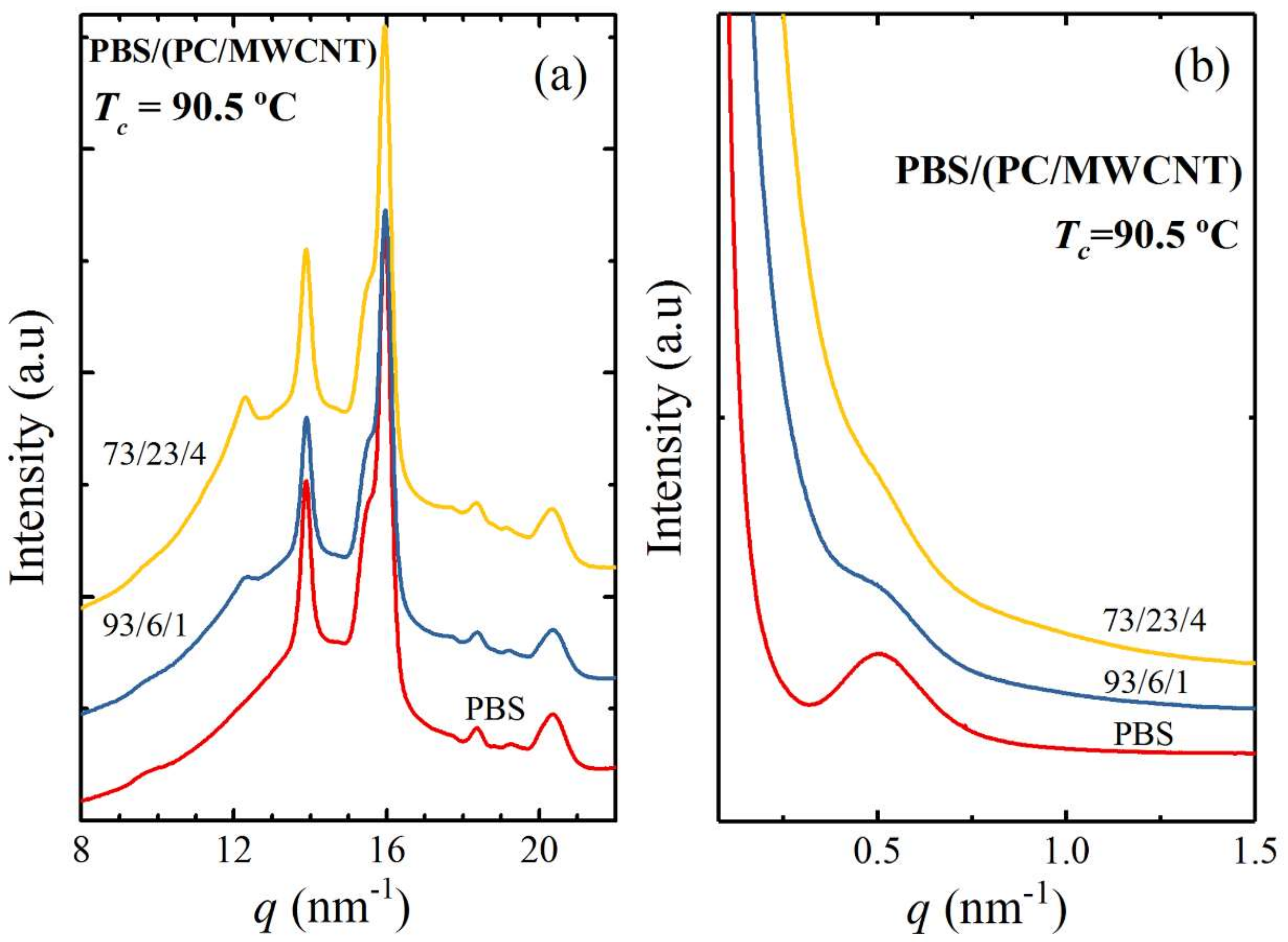
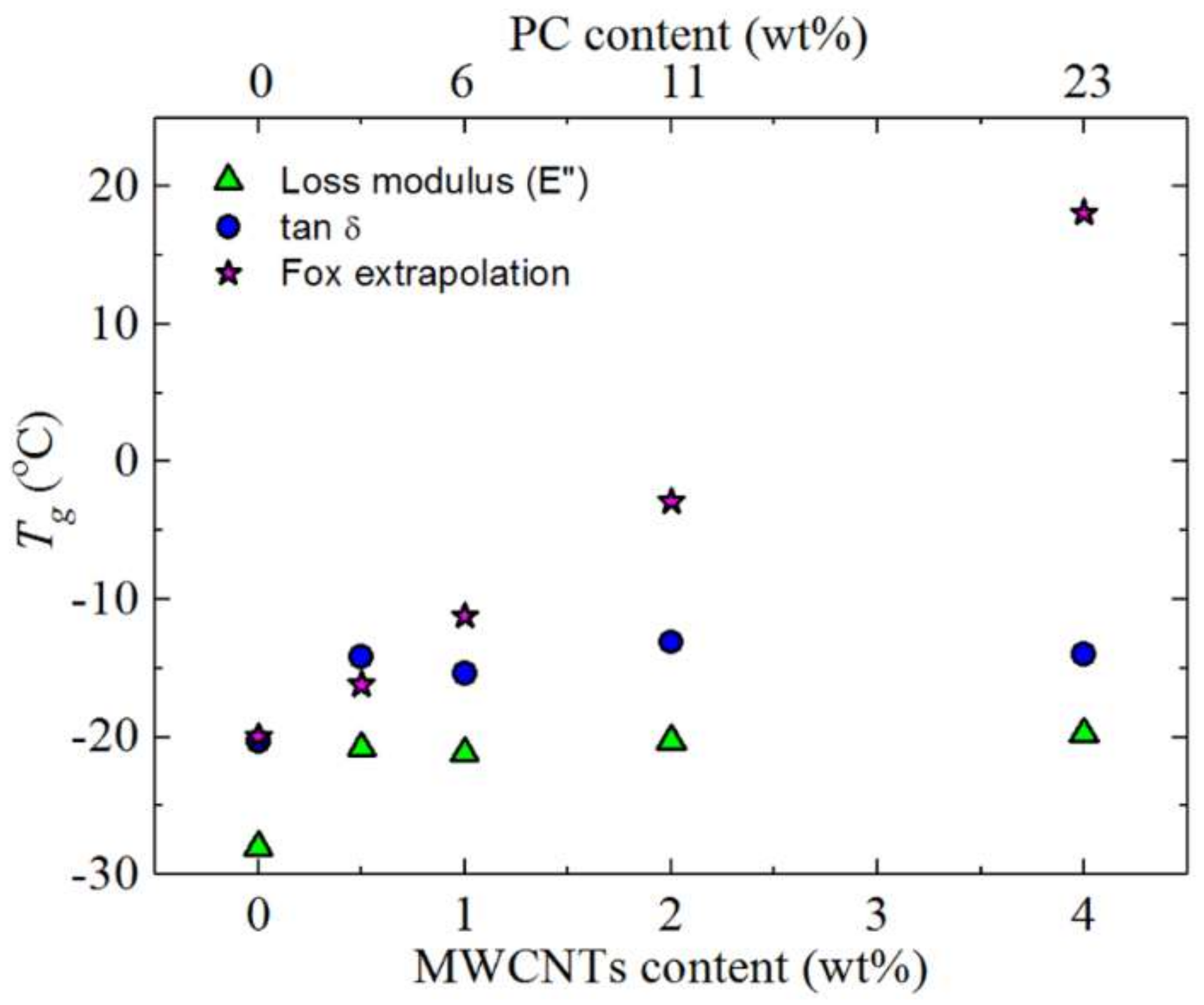
3.1. Miscibility Assessment
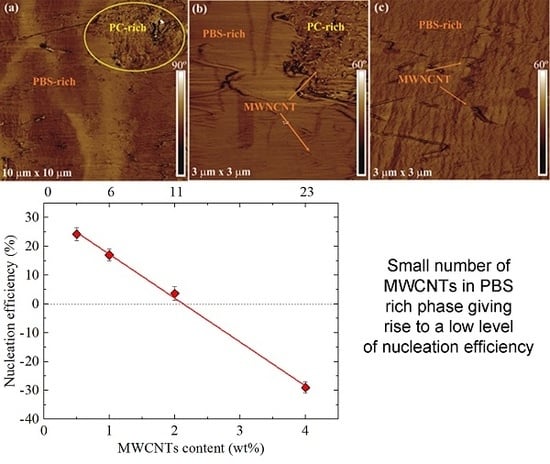
3.2. Morphology
3.3. Non-Isothermal DSC
3.4. Self-Nucleation (SN)
3.5. Overall Isothermal Crystallization Studied by DSC
3.5.1. Fitting DSC Isothermal Data to the Avrami Model
3.5.2. Overall Isothermal Crystallization Data Analysed by the Lauritzen–Hoffman Model
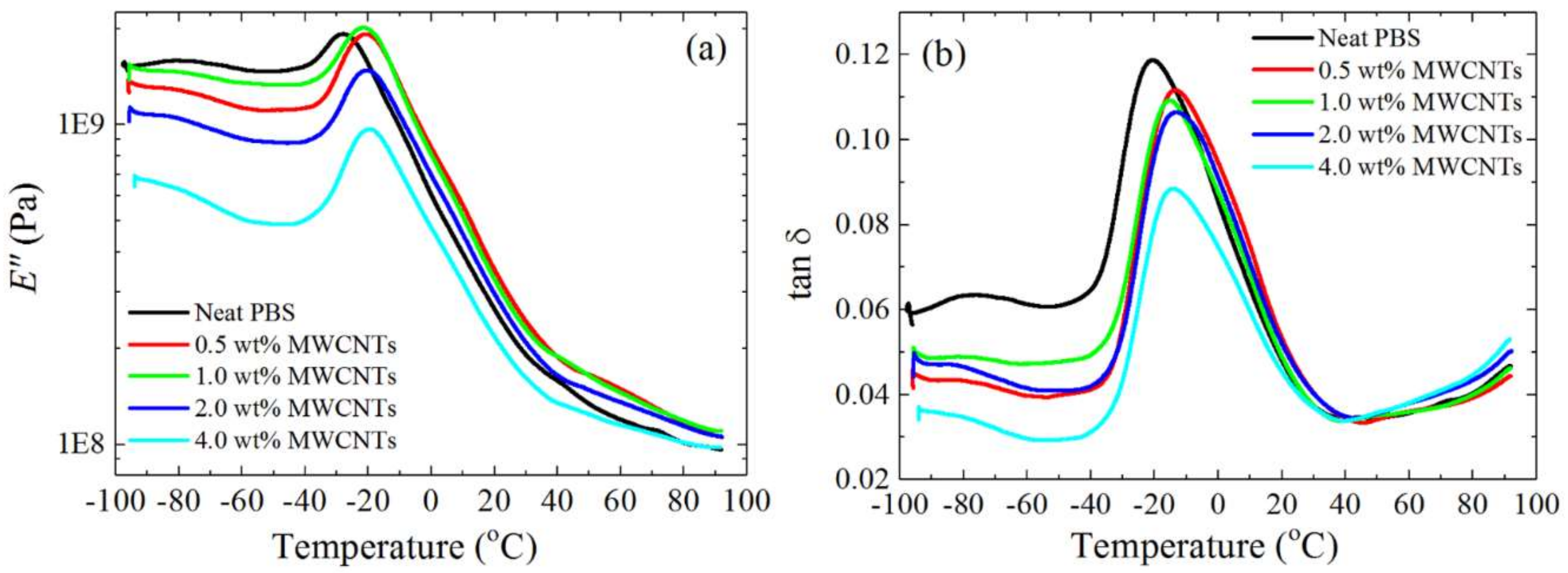
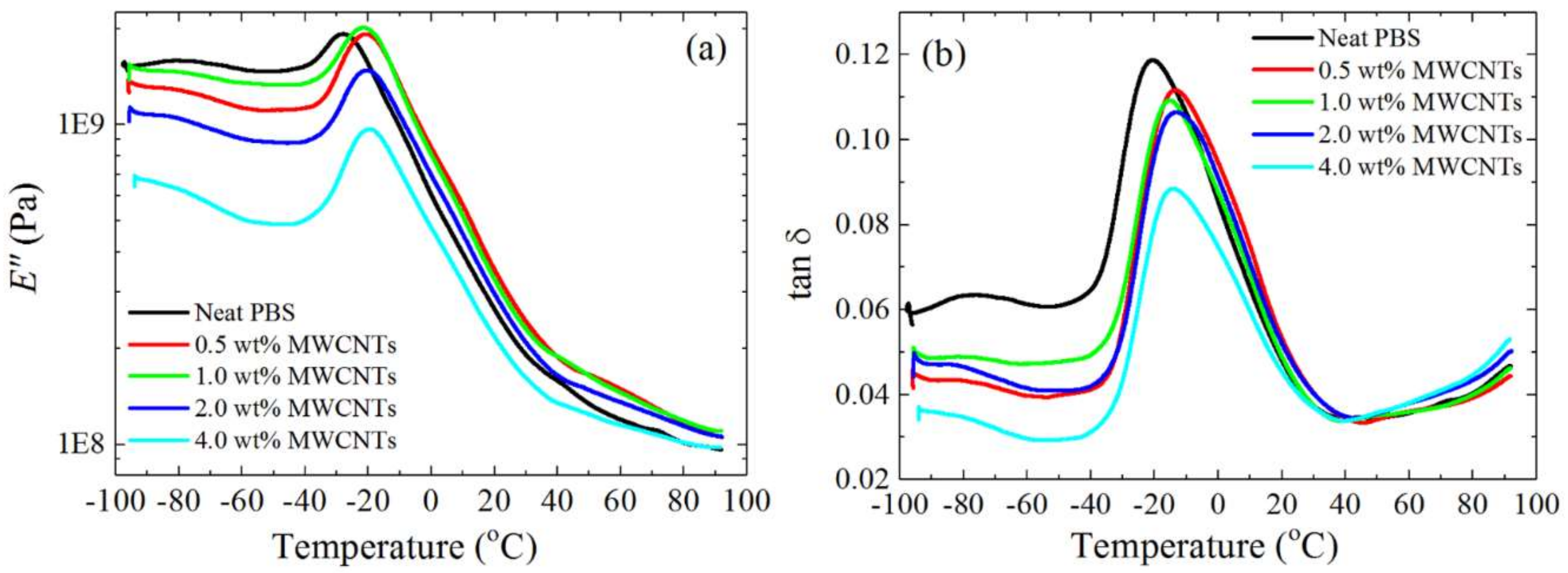
3.6. Thermal Conductivity
3.7. Tensile Properties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lin, C.S.; Shih, Y.F.; Jeng, R.J.; Dai, S.A.; Lin, J.J.; Lee, C.C. Nanocomposites with enhanced electrical properties based on biodegradable poly(butylene succinate) and polyetheramine modified carbon nanotube. J. Taiwan Inst. Chem. Eng. 2012, 43, 322–328. [Google Scholar] [CrossRef]
- Gigli, M.; Negroni, A.; Zanaroli, G.; Lotti, N.; Fava, F.; Munari, A. Environmentally friendly PBS-copolyesters containing PEG-like subunit: Effect of block length on solid-state properties and enzymatic degradation. React. Funct. Polym. 2013, 73, 764–771. [Google Scholar] [CrossRef]
- Zeng, R.T.; Hu, W.; Wang, M.; Zhang, S.D.; Zeng, J.B. Morphology, rheological and crystallization behavior in non-covalently functionalized carbon nanotube reinforced poly(butylene succinate) nanocomposites with low percolation threshold. Polym. Test. 2016, 50, 182–190. [Google Scholar] [CrossRef]
- Shih, Y.F.; Chen, L.S.; Jeng, R.J. Preparation and properties of biodegradable PBS/multi-walled carbon nanotube nanocomposites. Polymer 2008, 49, 4602–4611. [Google Scholar] [CrossRef]
- Potapov, A.G.; Shundrina, I.K. The influence of different fillers on mechanical and physical properties of high-molecular-weight biodegradable aliphatic polyesters. Polym. Sci. Ser. A 2016, 58, 585–592. [Google Scholar] [CrossRef]
- Fujimaki, T. Processability and properties of aliphatic polyesters, ‘BIONOLLE’, synthesized by polycondensation reaction. Polym. Degrad. Stab. 1998, 59, 209–214. [Google Scholar] [CrossRef]
- Pramoda, K.P.; Linh, N.T.T.; Zhang, C.; Liu, T. Multiwalled carbon nanotube nucleated crystallization behavior of biodegradable poly(butylene succinate) nanocomposites. J. Appl. Polym. Sci. 2009, 111, 2938–3945. [Google Scholar] [CrossRef]
- Tan, L.; Chen, Y.; Zhou, W.; Ye, S.; Wei, J. Novel approach toward poly(butylene succinate)/single-walled carbon nanotubes nanocomposites with interfacial-induced crystallization behaviors and mechanical strength. Polymer 2011, 52, 3587–3596. [Google Scholar] [CrossRef]
- Yuan, L.; Wu, D.; Zhang, M.; Zhou, W.; Lin, D. Rheological percolation behavior and isothermal crystallization of poly(butyene succinate)/carbon nanotube composites. Ind. Eng. Chem. Res. 2011, 50, 14186–14192. [Google Scholar] [CrossRef]
- Wang, G.; Guo, B.; Xu, J.; Li, R. Rheology, crystallization behaviors, and thermal stabilities of poly(butylene succinate)/pristine multiwalled carbon nanotube composites obtained by melt compounding. J. Appl. Polym. Sci. 2011, 121, 59–67. [Google Scholar] [CrossRef]
- Bhattacharya, M. Review: Polymer nanocomposites—A comparison between carbon nanotubes, graphene, and clay as nanofillers. Materials 2016, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Jajam, K.C.; Rahman, M.M.; Hosur, M.V.; Tippur, H.V. Fracture behavior of epoxy nanocomposites modified with polyol diluent and amino-functionalized multi-walled carbon nanotubes: A loading rate study. Compos. A Appl. Sci. Manuf. 2014, 59, 57–69. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hosur, M.; Zainuddin, S.; Jajam, K.C.; Tippur, H.V.; Jeelani, S. Mechanical characterization of epoxy composites modified with reactivepolyol diluent and randomly-oriented amino-functionalized MWCNTs. Polym. Test. 2012, 31, 1083–1093. [Google Scholar] [CrossRef]
- Wu, T.; Chen, E.; Lin, Y.; Chiang, M.; Chang, G. Preparation and characterization of melt-processed polycarbonate/multiwalled carbon nanotube composites. Polym. Eng. Sci. 2008, 48, 1369–1375. [Google Scholar] [CrossRef]
- Pötschke, P.; Bhattacharyya, A.R.; Janke, A.; Goering, H. Melt mixing of polycarbonate/multi-wall carbon nanotube composites. Compos. Interfaces 2003, 10, 389–404. [Google Scholar] [CrossRef]
- Prashantha, K.; Soulestin, J.; Lacrampe, M.F.; Krawczak, P.; Dupin, G.; Claes, M. Masterbatch-based multi-walled carbon nanotube filled polypropylene nanocomposites: Assessment of rheological and mechanical properties. Compos. Sci. Technol. 2009, 69, 1756–1763. [Google Scholar] [CrossRef]
- Gumede, T.P.; Luyt, A.S.; Hassan, M.K.; Pérez-Camargo, R.A.; Tercjak, A.; Müller, A.J. Morphology, nucleation, and isothermal crystallization kinetics of poly(ε-caprolactone) mixed with a polycarbonate/MWCNTs masterbatch. Polymers 2017, 9, 709. [Google Scholar] [CrossRef]
- Pang, M.Z.; Qiao, J.J.; Jiao, J.; Wang, S.J.; Xiao, M.; Meng, Y.Z. Miscibility and properties of completely biodegradable blends of poly(propylene carbonate) and poly(butylene succinate). J. Appl. Polym. Sci. 2008, 107, 2854–2860. [Google Scholar] [CrossRef]
- Wang, T.; Wang, H.; Li, H.; Gan, Z.; Yan, S. Banded spherulitic structures of poly(ethlylene adipate), poly(butylene succinate) and in their blends. Phys. Chem. Chem. Phys. 2009, 11, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Kasaliwal, G.R.; Göldel, A.; Pötschke, P.; Heinrich, G. Influences of polymer matrix melt viscosity and molecular weight on MWCNT agglomerate dispersion. Polymer 2011, 52, 1027–1036. [Google Scholar] [CrossRef]
- Fillon, B.; Wittmann, J.C.; Lotz, B.; Thierry, A. Self-nucleation and recrystallization of isotactic polypropylene (α phase) investigated by differential scanning calorimetry. J. Polym. Sci. B Polym. Phys. 1993, 31, 1383–1393. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Arnal, M.A.; Sánchez, J.J.; Müller, A.J. Effect of annealing time on the self-nucleation behaviour of semicrystalline polymers. J. Polym. Sci. B Polym. Phys. 2006, 44, 1738–1750. [Google Scholar] [CrossRef]
- Müller, A.J.; Arnal, M.L. Thermal fractionation of polymers. Progr. Polym. Sci. 2005, 30, 559–603. [Google Scholar] [CrossRef]
- Michell, R.M.; Mugica, A.; Zubitur, M.; Müller, A.J. Self-nucleation of crystalline phases within homopolymers, polymer blends, copolymers, and nanocomposites. Adv. Polym. Sci. 2015, 276, 215–256. [Google Scholar] [CrossRef]
- Lorenzo, A.T.; Arnal, M.L.; Albuerne, J.; Müller, A.J. DSC isothermal polymer crystallization kinetics measurements and the use of the Avrami equation to fit the data: Guidelines to avoid common problems. Polym. Test. 2007, 26, 222–231. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Weeks, J.J. Melting process and the equilibrium melting temperature of polychlorotrifluoroethylene. J. Res. Nat. Bur. Stand. A Phys. Chem. 1962, 66, 13–28. [Google Scholar] [CrossRef]
- Menczel, J.D. Prime, R.B. Thermal Analysis of Polymers: Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Müller, A.J.; Paredes, E. Melting behavior, mechanical properties and fracture of crystallized polycarbonates. Lat. Am. J. Metall. Mater. 1985, 5, 130–141. [Google Scholar]
- Balsamo, V.; Calzadilla, N.; Mora, G.; Müller, A.J. Thermal characterization of polycarbonate/polycaprolactone blends. J. Polym. Sci. B Polym. Phys. 2001, 39, 771–785. [Google Scholar] [CrossRef]
- Guo, J.; Liu, Y.; Prada-Silvy, R.; Tan, Y.; Azad, S.; Krause, B.; Pötschke, P.; Grady, B.P. Aspect ratio effects of multi-walled carbon nanotubes on electrical, mechanical, and thermal properties of polycarbonate/MWCNT composites. J. Polym. Sci. B Polym. Phys. 2014, 52, 73–83. [Google Scholar] [CrossRef]
- Castillo, F.Y.; Socher, R.; Krause, B.; Headrick, R.; Grady, B.P.; Prada-Silvy, R.; Pötschke, P. Electrical, mechanical, and glass transition behavior of polycarbonate-based nanocomposites with different multi-walled carbon nanotubes. Polymer 2011, 52, 3835–3845. [Google Scholar] [CrossRef]
- Mukherjee, M.; Das, T.; Rajasekar, R.; Bose, S.; Kumar, S.; Das, C.K. Improvement of the properties of PC/LCP blends in the presence of carbon nanotubes. Compos. A Appl. Sci. Manuf. 2009, 40, 1291–1298. [Google Scholar] [CrossRef]
- Liu, G.; Zheng, L.; Zhang, X.; Li, C.; Jiang, S.; Wang, D. Reversible lamellar thickening induced by crystal transition in poly(butylene succinate). Macromolecules 2012, 45, 5487–5493. [Google Scholar] [CrossRef]
- Ray, S.S.; Vaudreuil, S.; Maazouz, A.; Bousmina, M. Dispersion of multi-walled carbon nanotubes in biodegradable poly(butylene succinate) matrix. J. Nanosci. Nanotechnol. 2006, 6, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.B.; Mohan, R. Thermal, mechanical, and rheological properties of biodegradable polybutylene succinate/carbon nanotubes nanocomposites. Polym. Compos. 2010, 31, 1309–1314. [Google Scholar] [CrossRef]
- Arandia, I.; Mugica, A.; Zubitur, M.; Arbe, A.; Liu, G.; Wang, D.; Mincheva, R.; Dubois, P.; Müller, A.J. How composition determines the properties of isodimorphic poly(butylene succinate-ran-butylene azelate) random biobased copolymers: From single tp double crystalline random copolymers. Macromolecules 2015, 48, 43–57. [Google Scholar] [CrossRef]
- Fillon, B.; Lotz, B.; Thierry, A.; Wittmann, J.C. Self-nucleation and enhanced nucleation of polymers. Definition of a convenient calorimetric “efficiency scale” and evaluation of nucleating additives in isotactic polypropylene (α phase). J. Polym. Sci. B Polym. Phys. 1993, 31, 1395–1405. [Google Scholar] [CrossRef]
- Pérez, R.A.; López, J.V.; Hoskins, J.N.; Zhang, B.; Grayson, S.M.; Casas, M.T.; Puiggalí, J.; Müller, A.J. Nucleation and antinucleation effects of functionalized carbon nanotubes on cyclic and linear poly(ε-caprolactones). Macromolecules 2014, 47, 3553–3566. [Google Scholar] [CrossRef]
- Chen, J.; Wu, D.; Tam, K.C.; Pan, K.; Zheng, Z. Effect of surface modification of cellulose nanocrystal on nonisothermal crystallization of poly(β-hydroxybutyrate) composites. Carbohydr. Polym. 2017, 157, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Camargo, R.A.; Saenz, G.; Laurichesse, S.; Casas, M.T.; Puiggalí, J.; Avérous, L.; Müller, A.J. Nucleation, crystallization, and thermal fractionation of poly(ε-caprolactone)-grafted-lignin: Effects of grafted chains length and lignin content. J. Polym. Sci. B Polym. Phys. 2015, 53, 1736–1750. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure kinetics of phase change III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Reiter, G.; Strobl, G.R. Progress in Understanding Polymer Crystallization; Lecture Notes Physics 714; Springer: Heidelberg/Berlin, Germany, 2007. [Google Scholar]
- Lorenzo, A.T.; Müller, A.J. Estimation of the nucleation and crystal growth contributions to the overall crystallization energy. J. Polym. Sci. B Polym. Phys. 2008, 46, 1478–1487. [Google Scholar] [CrossRef]
- Wurm, A.; Lellinger, D.; Minakov, A.A.; Skipa, T.; Pötschke, P.; Nicula, R.; Alig, I.; Schick, C. Crystallization of poly(ε-caprolactone)/MWCNT composites: A combined SAXS/WAXS, electrical and thermal conductivity study. Polymer 2014, 55, 2220–2232. [Google Scholar] [CrossRef]
- Chin, S.J.; Vempati, S.; Dawson, P.; Knite, M.; Linarts, A.; Ozols, K.; McNally, T. Electrical conduction and rheological behaviour of composites of poly(ε-caprolactone) and MWCNTs. Polymer 2015, 58, 209–221. [Google Scholar] [CrossRef]
- Pötschke, P.; Villmow, T.; Krause, B. Melt mixed PCL/MWCNT composites prepared at different rotation speeds: Characterization of rheological, thermal, and electrical properties, molecular weight, MWCNT macrodispersion, and MWCNT length distribution. Polymer 2013, 54, 3071–3078. [Google Scholar] [CrossRef]
- Ma, X.; Zare, Y.; Rhee, K.Y. A two-step methodology to study the influence of aggregation/agglomeration of nanoparticles on Young’s modulus of polymer nanocomposites. Nanoscale Res. Lett. 2017, 12, 621. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBS (wt%) | PC (wt%) | MWCNTs (wt%) |
|---|---|---|
| 100 | 0 | 0 |
| 97 | 2.55 | 0.45 |
| 93 | 5.95 | 1.05 |
| 87 | 11.05 | 1.95 |
| 73 | 22.95 | 4.05 |
| Sample | q (nm−1)/d-spacing (nm)/(Plane) | d* (nm) |
|---|---|---|
| Neat PBS | 13.88/0.426/(002) 15.54/0.381/(012) 15.96/0.371/(110) 18.38/0.322/(121) 20.36/0.291/(111) | 12.3 |
| 93/(6/1) w/w PBS/(PC/MWCNTs) | 13.90/0.426/(002) 15.52/0.381/(012) 15.97/0.371/(110) 18.35/0.323/(12-1) 20.42/0.291/(111) 12.34/0.479 * | 11.9 ** |
| 73/(23/4) w/w PBS/(PC/MWCNTs) | 13.88/0.426/(002) 15.55/0.381/(012) 15.95/0.371/(110) 18.34/0.323/(121) 20.36/0.291/(111) 12.30/0.481 * | 8.3 ** |
| Sample | (K) | × 104 (K2) | σ (erg/cm2) | σe (erg/cm2) | q × 10−13 (erg) | R2 |
|---|---|---|---|---|---|---|
| 100/0 | 394.0 | 8.16 | 8.08 | 81.13 | 2.99 | 0.995 |
| 97/(2.5/0.5) | 393.0 | 8.01 | 8.08 | 79.83 | 2.95 | 0.998 |
| 93/(6/1) | 392.1 | 7.91 | 8.08 | 79.04 | 2.92 | 0.999 |
| 87/(11/2) | 391.1 | 6.99 | 8.08 | 70.01 | 2.58 | 0.987 |
| 73/(23/4) | 389.1 | 6.79 | 8.08 | 68.32 | 2.52 | 0.997 |
| w/w PBS/(PC/MWCNTs) | (MPa) | (%) | E (MPa) |
|---|---|---|---|
| 100/0 | 38.1 ± 2.1 | 210 ± 110 | 579 ± 139 |
| 97/(2.5/0.5) | 28.3 ± 6.1 | 7.0 ± 2.9 | 641 ± 50 |
| 93/(6/1) | 29.4 ± 0.5 | 5.0 ± 0.1 | 672 ± 44 |
| 87/(11/2) | 26.0 ± 6.6 | 5.0 ± 1.0 | 715 ± 84 |
| 73/(23/4) | 10.9 ± 0.6 | 4.0 ± 0.8 | 638 ± 79 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumede, T.P.; Luyt, A.S.; Pérez-Camargo, R.A.; Tercjak, A.; Müller, A.J. Morphology, Nucleation, and Isothermal Crystallization Kinetics of Poly(Butylene Succinate) Mixed with a Polycarbonate/MWCNT Masterbatch. Polymers 2018, 10, 424. https://doi.org/10.3390/polym10040424
Gumede TP, Luyt AS, Pérez-Camargo RA, Tercjak A, Müller AJ. Morphology, Nucleation, and Isothermal Crystallization Kinetics of Poly(Butylene Succinate) Mixed with a Polycarbonate/MWCNT Masterbatch. Polymers. 2018; 10(4):424. https://doi.org/10.3390/polym10040424
Chicago/Turabian StyleGumede, Thandi P., Adriaan S. Luyt, Ricardo A. Pérez-Camargo, Agnieszka Tercjak, and Alejandro J. Müller. 2018. "Morphology, Nucleation, and Isothermal Crystallization Kinetics of Poly(Butylene Succinate) Mixed with a Polycarbonate/MWCNT Masterbatch" Polymers 10, no. 4: 424. https://doi.org/10.3390/polym10040424