Molecularly Imprinted Polymers for the Selective Extraction of Bisphenol A and Progesterone from Aqueous Media

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
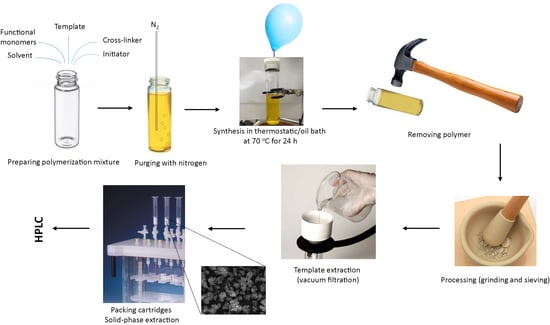
2.2.1. Synthesis of Molecularly Imprinted Polymers (MIPs) and Non-Imprinted Polymers (NIPs)
2.2.2. Removal of the Imprinting Molecules
2.2.3. Grinding and Sieving of MIPs and NIPs
2.2.4. Characterisation of MIPs and NIPs Using Infrared Spectroscopy (FT-IR) and Electron Scanning Microscope (SEM)
2.2.5. Detection of BPA and PG Using High Performance Liquid Chromatography (HPLC)
2.2.6. Retention Studies for MIPs and NIPs in Batch Method
2.2.7. Kinetics Studies
2.2.8. Adsorption Isotherms
2.2.9. Cross-Reactivity of the MIPs in Real Water Samples
2.2.10. Reusability Capacity of MIPs
3. Results
3.1. Synthesis and Characterisation of MIPs and NIPs
3.2. High Performance Liquid Chromatography (HPLC)
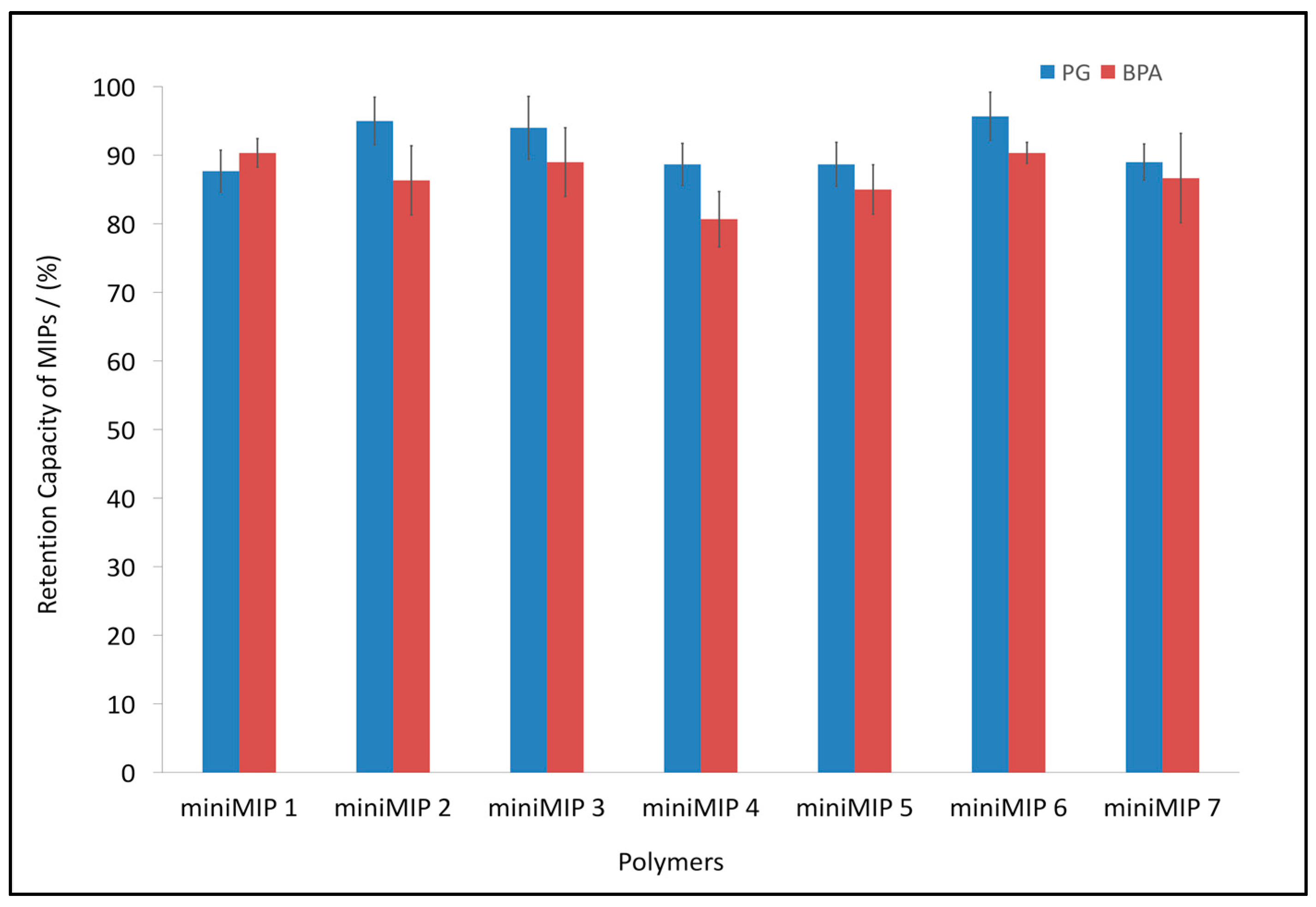
3.3. BPA and PG Retention Capacity of the Synthesised miniMIPs
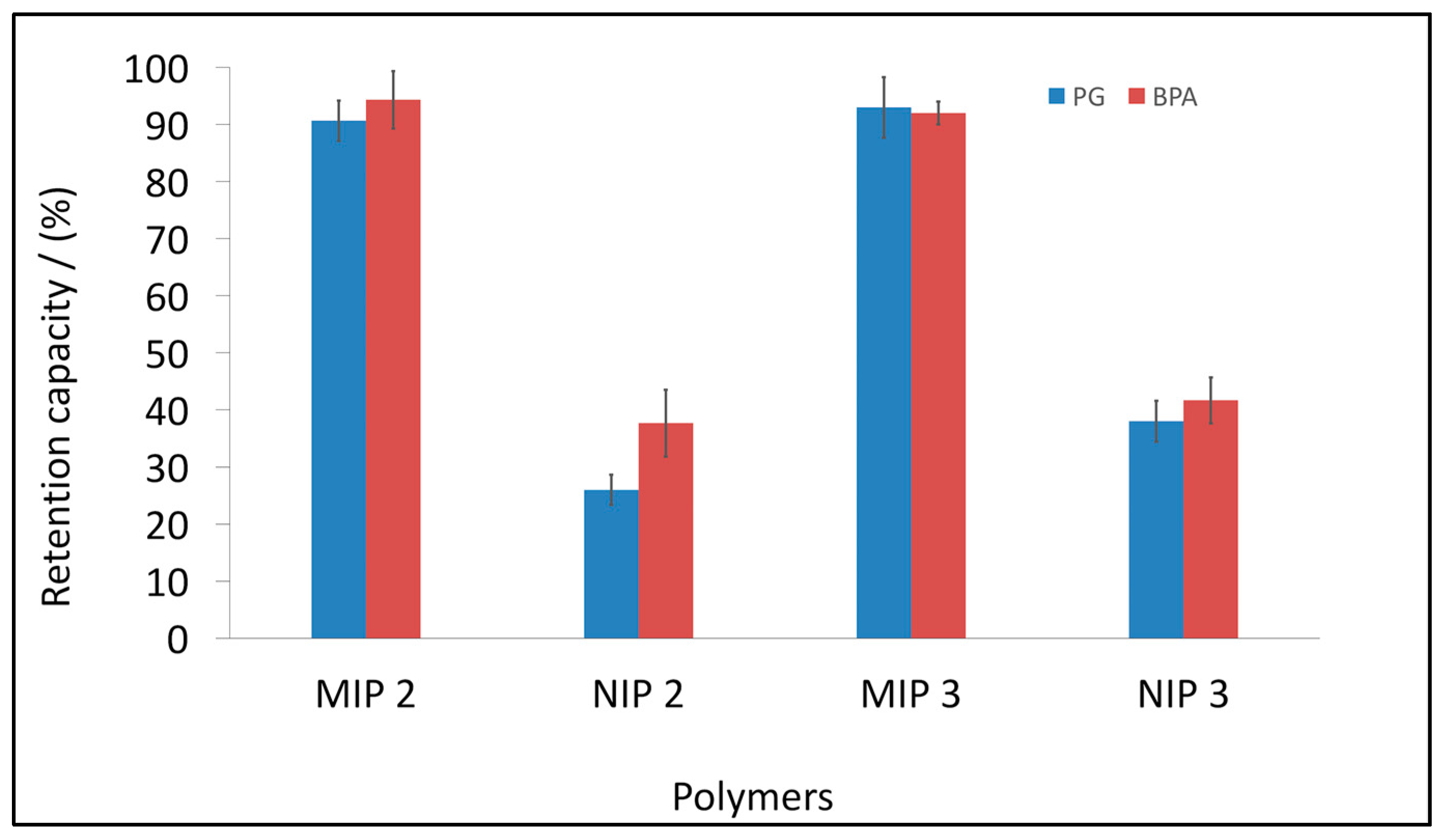
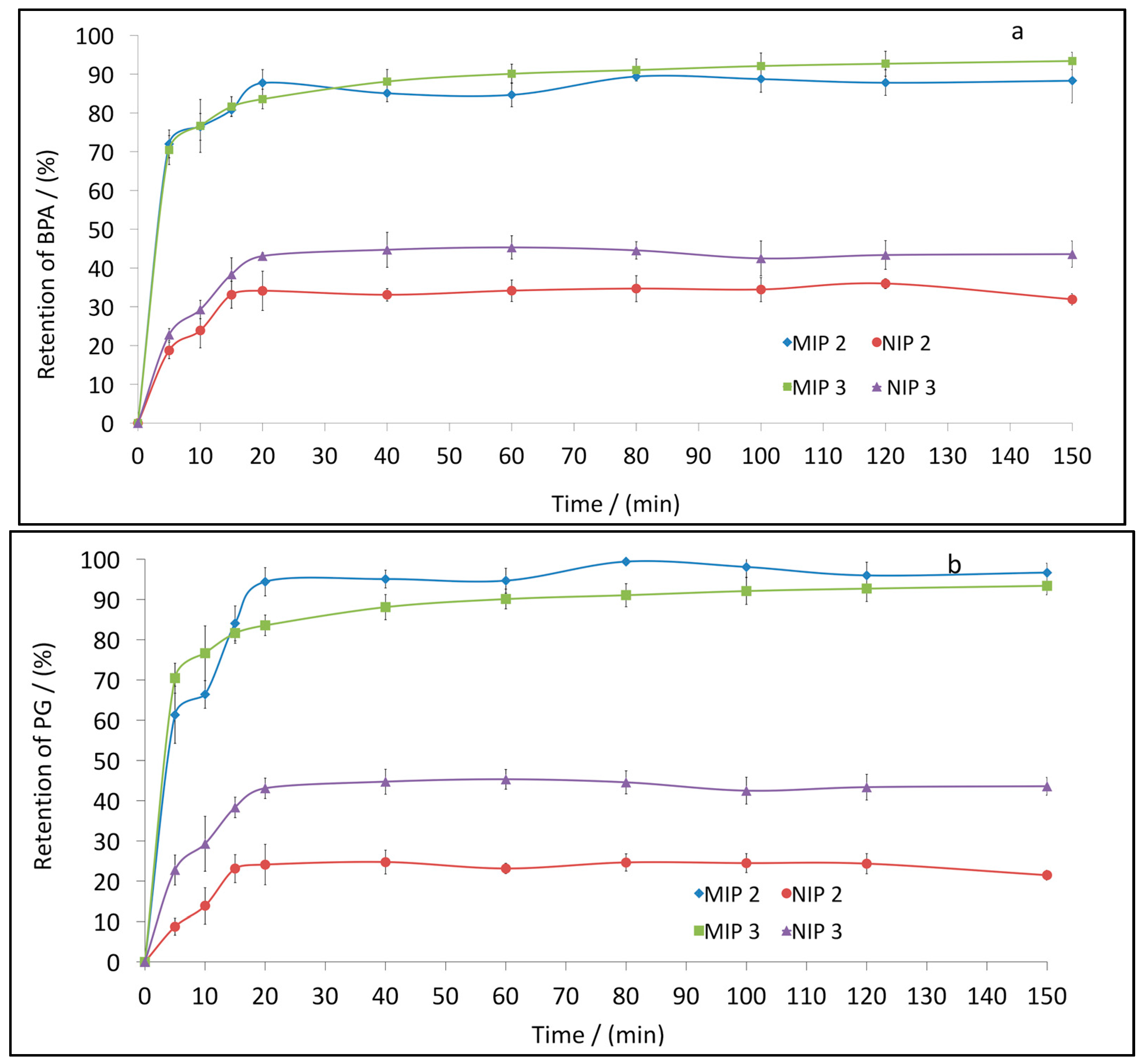
3.4. BPA and PG Retention Studies by Selected MIPs and NIPs
3.5. Adsorption Isotherms
3.6. Kinetic Study of the Retention of BPA and PG in MIPs
3.7. Cross-Reactivity of the MIPs in Water Samples
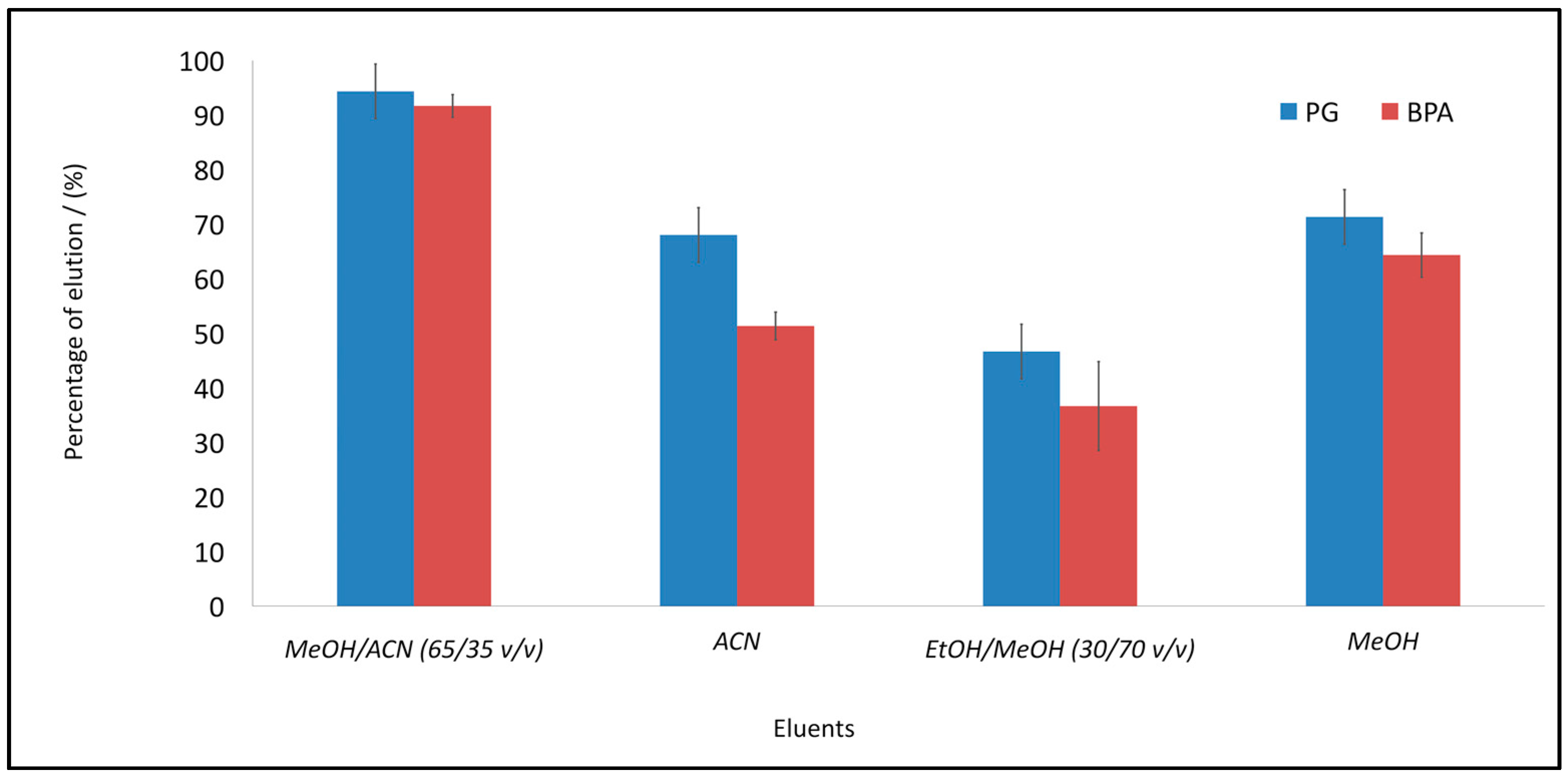
3.8. Evaluation of the Reusability Capacity of MIPs
3.9. Evaluation of the Adsorption Capacity in Real Water Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kavlock, R.J. Endocrine disrupting chemicals and human healthoverview of endocrine disruptor research activity in the united states. Chemosphere 1999, 39, 1227–1236. [Google Scholar] [CrossRef]
- Casey, F.X.M.; Larsen, G.L.; Hakk, H.; Šimůnek, J. Fate and transport of 17β-estradiol in soil−water systems. Environ. Sci. Technol. 2003, 37, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.R.; Williams, E.L.; Layton, A.C.; Burns, R.T.; Easter, J.P.; Daugherty, A.S.; Mullen, M.D.; Sayler, G.S. Estrogen content of dairy and swine wastes. Environ. Sci. Technol. 2004, 38, 3567–3573. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, T. Environmental endocrine disruptors. Nihon Rinsho 1998, 56, 2953–2962. [Google Scholar] [PubMed]
- Soto, A.M.; Sonnenschein, C. Environmental causes of cancer: Endocrine disruptors as carcinogens. Nat. Rev. Endocrinol. 2010, 6, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, J.P.; Johnson, A.C. Lessons from endocrine disruption and their application to other issues concerning trace organics in the aquatic environment. Environ. Sci. Technol. 2005, 39, 4321–4332. [Google Scholar] [CrossRef] [PubMed]
- Newbold, R.R.; Padilla-Banks, E.; Snyder, R.J.; Phillips, T.M.; Jefferson, W.N. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod. Toxicol. 2007, 23, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anway, M.D.; Cupp, A.S.; Uzumcu, M.; Skinner, M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005, 308, 1466–1469. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.A.; Ho, S.-M.; Hunt, P.A.; Knudsen, K.E.; Soto, A.M.; Prins, G.S. An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod. Toxicol. 2007, 24, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commission Regulation (EU). Available online: http://data.europa.eu/eli/reg/2018/213/oj (accessed on 3 May 2018).
- Canadian Environmental Protection Act, 1999 Draft Federal Environmental Quality Guidelines, Bisphenol A. Available online: http://www.ec.gc.ca/ese-ees/default.asp?lang=En&n=CB1CEFFB-1 (accessed on 3 May 2018).
- Welshons, W.V.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006, 147, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Sheng, N.; Zhu, R.; Wei, F.; Cai, Z.; Zhai, M.; Du, S.; Hu, Q. Preparation of dummy template imprinted polymers at surface of silica microparticles for the selective extraction of trace bisphenol a from water samples. J. Hazard. Mater. 2010, 179, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Erickson, G.F.; Ryan, K.J. The effect of LH/FSH, dibutyryl cyclic amp, and prostaglandins on the production of estrogens by rabbit granulosa cells in vitro. Endocrinology 1975, 97, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.E. The pharmacological basis of therapeutics. Occup. Environ. Med. 2007, 64, e2. [Google Scholar] [CrossRef]
- Torres, N.H.; Aguiar, M.M.; Ferreira, L.F.R.; Américo, J.H.P.; Machado, Â.M.; Cavalcanti, E.B.; Tornisielo, V.L. Detection of hormones in surface and drinking water in brazil by lc-esi-ms/ms and ecotoxicological assessment with daphnia magna. Environ. Monit. Assess. 2015, 187, 379. [Google Scholar] [CrossRef] [PubMed]
- Rubio, S.; Pérez-Bendito, D. Recent advances in environmental analysis. Anal. Chem. 2009, 81, 4601–4622. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.; Cáceres, C.; Rivera, F.; Rivas, B.; Sáez, P. Preparation of molecularly imprinted polymers for diphenylamine removal from organic gunshot residues. J. Chil. Chem. Soc. 2014, 59, 2731–2736. [Google Scholar] [CrossRef]
- Tamayo, F.G.; Turiel, E.; Martín-Esteban, A. Molecularly imprinted polymers for solid-phase extraction and solid-phase microextraction: Recent developments and future trends. J. Chromatogr. A 2007, 1152, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Benito-Peña, E.; Navarro-Villoslada, F.; Carrasco, S.; Jockusch, S.; Ottaviani, M.F.; Moreno-Bondi, M.C. Experimental mixture design as a tool for the synthesis of antimicrobial selective molecularly imprinted monodisperse microbeads. ACS Appl. Mater. Interfaces 2015, 7, 10966–10976. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, V.J.B.; Levisson, M.; Eppink, M.H.M.; Smidt, H.; Van Der Oost, J. Alternative affinity tools: More attractive than antibodies? Biochem. J. 2011, 436, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bakas, I.; Oujji, N.; Moczko, E.; Istamboulie, G.; Piletsky, S.; Piletska, E.; Ichou, I.; Addi, E.; Noguer, T. Molecular imprinting solid phase extraction for selective detection of methidathion in olive oil. Anal. Chim. Acta 2012, 739, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Chianella, I.; Guerreiro, A.; Moczko, E.; Caygill, J.S.; Piletska, E.V.; De Vargas Sansalvador, I.M.; Whitcombe, M.J.; Piletsky, S.A. Direct replacement of antibodies with molecularly imprinted polymer nanoparticles in elisa-development of a novel assay for vancomycin. Anal. Chem. 2013, 85, 8462–8472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guihen, E. Nanoparticles in modern separation science. TrAC Trends Anal. Chem. 2013, 46, 1–14. [Google Scholar] [CrossRef]
- Moczko, E.; Guerreiro, A.; Piletska, E.; Piletsky, S. Peg-stabilized core-shell surface-imprinted nanoparticles. Langmuir 2013, 29, 9891–9897. [Google Scholar] [CrossRef] [PubMed]
- Moczko, E.; Poma, A.; Guerreiro, A.; Perez de Vargas Sansalvador, I.; Caygill, S.; Canfarotta, F.; Whitcombe, M.J.; Piletsky, S. Surface-modified multifunctional MIP nanoparticles. Nanoscale 2013, 5, 3733–3741. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Fourty years of molecular imprinting in synthetic polymers: Origin, features and perspectives. Microchim. Acta 2013, 180, 1359–1370. [Google Scholar] [CrossRef]
- Poma, A.; Guerreiro, A.; Caygill, S.; Moczko, E.; Piletsky, S. Automatic reactor for solid-phase synthesis of molecularly imprinted polymeric nanoparticles (MIP NPs) in water. RSC Adv. 2014, 4, 4203–4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolinska-Kempisty, K.; Guerreiro, A.; Canfarotta, F.; Cáceres, C.; Whitcombe, M.J.; Piletsky, S. A comparison of the performance of molecularly imprinted polymer nanoparticles for small molecule targets and antibodies in the ELISA format. Sci. Rep. 2016, 6, 37638–37645. [Google Scholar] [CrossRef] [PubMed]
- Umpleby, R.J.; Baxter, S.C.; Chen, Y.; Shah, R.N.; Shimizu, K.D. Characterization of molecularly imprinted polymers with the Langmuir-Freundlich isotherm. Anal. Chem. 2001, 73, 4584–4591. [Google Scholar] [CrossRef] [PubMed]
- Sips, R. On the structure of a catalyst surface. J. Chem. Phys. 1948, 16, 490–495. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Chu, J.; Dong, C.; Qi, J.; Yuan, Y. Synthesis of core-shell magnetic molecular imprinted polymer by the surface raft polymerization for the fast and selective removal of endocrine disrupting chemicals from aqueous solutions. Environ. Poll. 2010, 158, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Mozaz, S.; Marco, M.-P.; Lopez de Alda, M.J.; Barceló, D. Biosensors for environmental monitoring of endocrine disruptors: A review article. Anal. Bioanal. Chem. 2004, 378, 588–598. [Google Scholar] [PubMed]
- Alenazi, N.A.; Manthorpe, J.M.; Lai, E.P.C. Selective extraction of BPA in milk analysis by capillary electrophoresis using a chemically modified molecularly imprinted polymer. Food Control 2015, 50, 778–783. [Google Scholar] [CrossRef]
- O’Mahony, J.; Moloney, M.; McCormack, M.; Nicholls, I.A.; Mizaikoff, B.; Danaher, M. Design and implementation of an imprinted material for the extraction of the endocrine disruptor bisphenol a from milk. J. Chromatogr. B 2013, 931, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Ričanyová, J.; Gadzała-Kopciuch, R.; Reiffova, K.; Bazel, Y.; Buszewski, B. Molecularly imprinted adsorbents for preconcentration and isolation of progesterone and testosterone by solid phase extraction combined with HPLC. Adsorption 2010, 16, 473–483. [Google Scholar] [CrossRef]
- Bianchi, V.D.N.; Silva, M.R.D.A.; Lamim, M.A.; Silva, C.L.D.; Lima, E.C.D. Solid phase extraction using molecular imprinting polymers (MISPE) for the determination of estrogens in surface water by HPLC. Rev. Ambient. Água 2017, 12, 380–389. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Dong, C.; Qi, J.; Han, X. A graphene oxide-based molecularly imprinted polymer platform for detecting endocrine disrupting chemicals. Carbon 2010, 48, 3427–3433. [Google Scholar] [CrossRef]
- Xu, Z.; Ding, L.; Long, Y.; Xu, L.; Wang, L.; Xu, C. Preparation and evaluation of superparamagnetic surface molecularly imprinted polymer nanoparticles for selective extraction of bisphenol a in packed food. Anal. Methods 2011, 3, 1737–1744. [Google Scholar] [CrossRef]
- Sambe, H.; Hoshina, K.; Hosoya, K.; Haginaka, J. Direct injection analysis of bisphenol a in serum by combination of isotope imprinting with liquid chromatography-mass spectrometry. Analyst 2005, 130, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Casis, N.; Busatto, C.; Fidalgo de Cortalezzi, M.M.; Ravaine, S.; Estenoz, D.A. Molecularly imprinted hydrogels from colloidal crystals for the detection of progesterone. Polym. Int. 2015, 64, 773–779. [Google Scholar] [CrossRef]
- Lee, M.-H.; O’Hare, D.; Guo, H.-Z.; Yang, C.-H.; Lin, H.-Y. Electrochemical sensing of urinary progesterone with molecularly imprinted poly(aniline-co-metanilic acid)s. J. Mater. Chem. B 2016, 4, 3782–3787. [Google Scholar] [CrossRef]
- Luo, S.-C.; Thomas, J.L.; Guo, H.-Z.; Liao, W.-T.; Lee, M.-H.; Lin, H.-Y. Electrosynthesis of nanostructured, imprinted poly(hydroxymethyl 3,4-ethylenedioxythiophene) for the ultrasensitive electrochemical detection of urinary progesterone. ChemistrySelect 2017, 2, 7935–7939. [Google Scholar] [CrossRef]
- Rodriguez-Mozaz, S.; Lopez de Alda, M.J.; Barceló, D. Biosensors as useful tools for environmental analysis and monitoring. Anal. Bioanal. Chem. 2006, 386, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Székely, G.; Henriques, B.; Gil, M.; Alvarez, C. Experimental design for the optimization and robustness testing of a liquid chromatography tandem mass spectrometry method for the trace analysis of the potentially genotoxic 1,3-diisopropylurea. Drug Test. Anal. 2014, 6, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Analytical Methods, C. Recommendations for the definition, estimation and use of the detection limit. Analyst 1987, 112, 199–204. [Google Scholar]
- Székely, G.; Henriques, B.; Gil, M.; Ramos, A.; Alvarez, C. Design of experiments as a tool for LC–MS/MS method development for the trace analysis of the potentially genotoxic 4-dimethylaminopyridine impurity in glucocorticoids. J. Pharm. Biomed. Anal. 2012, 70, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kupai, J.; Razali, M.; Buyuktiryaki, S.; Kecili, R.; Szekely, G. Long-term stability and reusability of molecularly imprinted polymers. Polym. Chem. 2017, 8, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Cross-Linker (DVB)/mol % of Monomer | Solvent (Acetonitrile) mL | Template (BPA or PG)/mol % of Monomer |
|---|---|---|---|
| miniMIP1 | 50 | 10 | 10 |
| miniMIP2 | 50 | 2 | 30 |
| miniMIP3 | 300 | 2 | 10 |
| miniMIP4 | 300 | 10 | 30 |
| miniMIP5 | 175 | 6 | 20 |
| miniMIP6 | 175 | 6 | 20 |
| miniMIP7 | 175 | 6 | 20 |
| Parameters | Analysis of BPA | Analysis of PG |
|---|---|---|
| Mobile phase | H2O:Acetonitrile (55:45) | Methanol:H2O (70:30) |
| Column | Lichrospher RP-C8 Symmetry® column, 5 μm, 4.6 × 250 mm | Lichrospher RP-C8 Symmetry® column, 5 μm, 4.6 × 250 mm |
| Mobile phase flow (mL/min) | 1 | 1 |
| Column temperature (°C) | 25 | 25 |
| Time of analysis (min) | 5 | 15 |
| Injection volume (µL) | 10 | 50 |
| Detector | Fluorescence | UV-Vis |
| Wavelength excitation/emission (nm) | 230/315 | 248 |
| 298 K | 303 K | 308 K | 313 K | |||||
|---|---|---|---|---|---|---|---|---|
| Polymer | n | K (L/mg) | n | K (L/mg) | n | K (L/mg) | n | K (L/mg) |
| MIP 2 | 0.851 | 3.336 | 0.589 | 0.867 | 1.369 | 11.32 | 1.850 | 5.235 |
| NIP 2 | 0.780 | 2.940 | 0.661 | 0.579 | 0.876 | 2.677 | 0.851 | 0.620 |
| MIP 3 | 1.340 | 50.15 | 0.607 | 35.03 | 1.620 | 80.33 | 1.254 | 76.51 |
| NIP 3 | 0.490 | 29.02 | 0.540 | 28.67 | 0.473 | 29.85 | 0.320 | 26.42 |
| 298 K | 303 K | 308 K | 313 K | |||||
|---|---|---|---|---|---|---|---|---|
| Polymer | Qmax (mg/g) | b (L/mg) | Qmax (mg/g) | b (L/mg) | Qmax (mg/g) | b (L/mg) | Qmax (mg/g) | b (L/mg) |
| MIP 2 | 14.06 | 0.230 | 13.61 | 0.056 | 19.53 | 1.321 | 1.850 | 5.235 |
| NIP 2 | 2.49 | 0.076 | 4.02 | 0.096 | 1.876 | 2.677 | 0.851 | 0.620 |
| MIP 3 | 12.85 | 1.36 | 19.46 | 0.481 | 17.62 | 80.33 | 1.254 | 76.51 |
| NIP 3 | 2.501 | 0.020 | 2.584 | 0.018 | 2.473 | 29.85 | 0.320 | 24.42 |
| 298 K | 303 K | 308 K | 313 K | |||||
|---|---|---|---|---|---|---|---|---|
| Polymer | n | K (L/mg) | n | K (L/mg) | n | K (L/mg) | n | K (L/mg) |
| MIP 2 | 0.636 | 2.167 | 0.462 | 1.046 | 1.618 | 10.72 | 2.010 | 4.351 |
| NIP 2 | 0.408 | 1.245 | 0.322 | 0.351 | 0.762 | 3.485 | 0.895 | 1.358 |
| MIP 3 | 1.070 | 20.24 | 0.701 | 3.423 | 1.981 | 28.13 | 1.387 | 21.69 |
| NIP 3 | 0.355 | 17.43 | 0.431 | 2.589 | 0.553 | 3.618 | 0.391 | 4.071 |
| 298 K | 303 K | 308 K | 313 K | |||||
|---|---|---|---|---|---|---|---|---|
| Polymer | Qmax (mg/g) | b (L/mg) | Qmax (mg/g) | b (L/mg) | Qmax (mg/g) | b (L/mg) | Qmax (mg/g) | b (L/mg) |
| MIP 2 | 12.18 | 0.184 | 16.17 | 0.174 | 21.49 | 1.047 | 6.850 | 17.35 |
| NIP 2 | 1.75 | 0.103 | 3.29 | 0.052 | 2.593 | 0.869 | 1.101 | 1.940 |
| MIP 3 | 10.19 | 0.272 | 18.62 | 0.218 | 17.63 | 15.84 | 2.061 | 31.25 |
| NIP 3 | 2.428 | 0.015 | 3.105 | 0.085 | 1.951 | 1.89 | 0.269 | 9.71 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cáceres, C.; Bravo, C.; Rivas, B.; Moczko, E.; Sáez, P.; García, Y.; Pereira, E. Molecularly Imprinted Polymers for the Selective Extraction of Bisphenol A and Progesterone from Aqueous Media. Polymers 2018, 10, 679. https://doi.org/10.3390/polym10060679
Cáceres C, Bravo C, Rivas B, Moczko E, Sáez P, García Y, Pereira E. Molecularly Imprinted Polymers for the Selective Extraction of Bisphenol A and Progesterone from Aqueous Media. Polymers. 2018; 10(6):679. https://doi.org/10.3390/polym10060679
Chicago/Turabian StyleCáceres, César, Catalina Bravo, Bernabé Rivas, Ewa Moczko, Pedro Sáez, Yadiris García, and Eduardo Pereira. 2018. "Molecularly Imprinted Polymers for the Selective Extraction of Bisphenol A and Progesterone from Aqueous Media" Polymers 10, no. 6: 679. https://doi.org/10.3390/polym10060679
APA StyleCáceres, C., Bravo, C., Rivas, B., Moczko, E., Sáez, P., García, Y., & Pereira, E. (2018). Molecularly Imprinted Polymers for the Selective Extraction of Bisphenol A and Progesterone from Aqueous Media. Polymers, 10(6), 679. https://doi.org/10.3390/polym10060679