Single-Molecule Dynamics and Discrimination between Hydrophilic and Hydrophobic Amino Acids in Peptides, through Controllable, Stepwise Translocation across Nanopores

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Electrophysiology
3. Results and Discussion
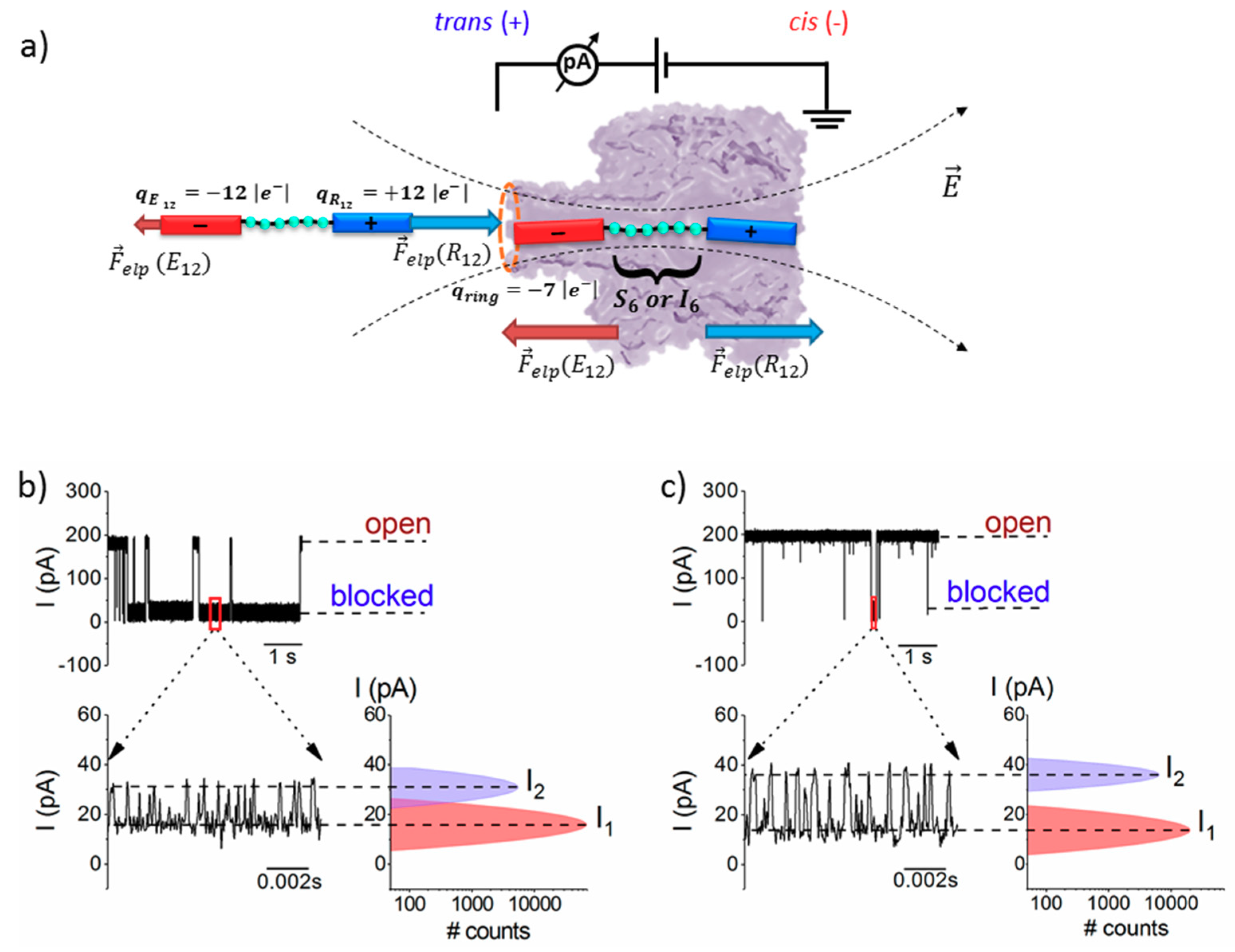
3.1. Steric- and Hydrophilic-Based Discrimination of Amino Acids at the Most Constricted Region of the Nanopore
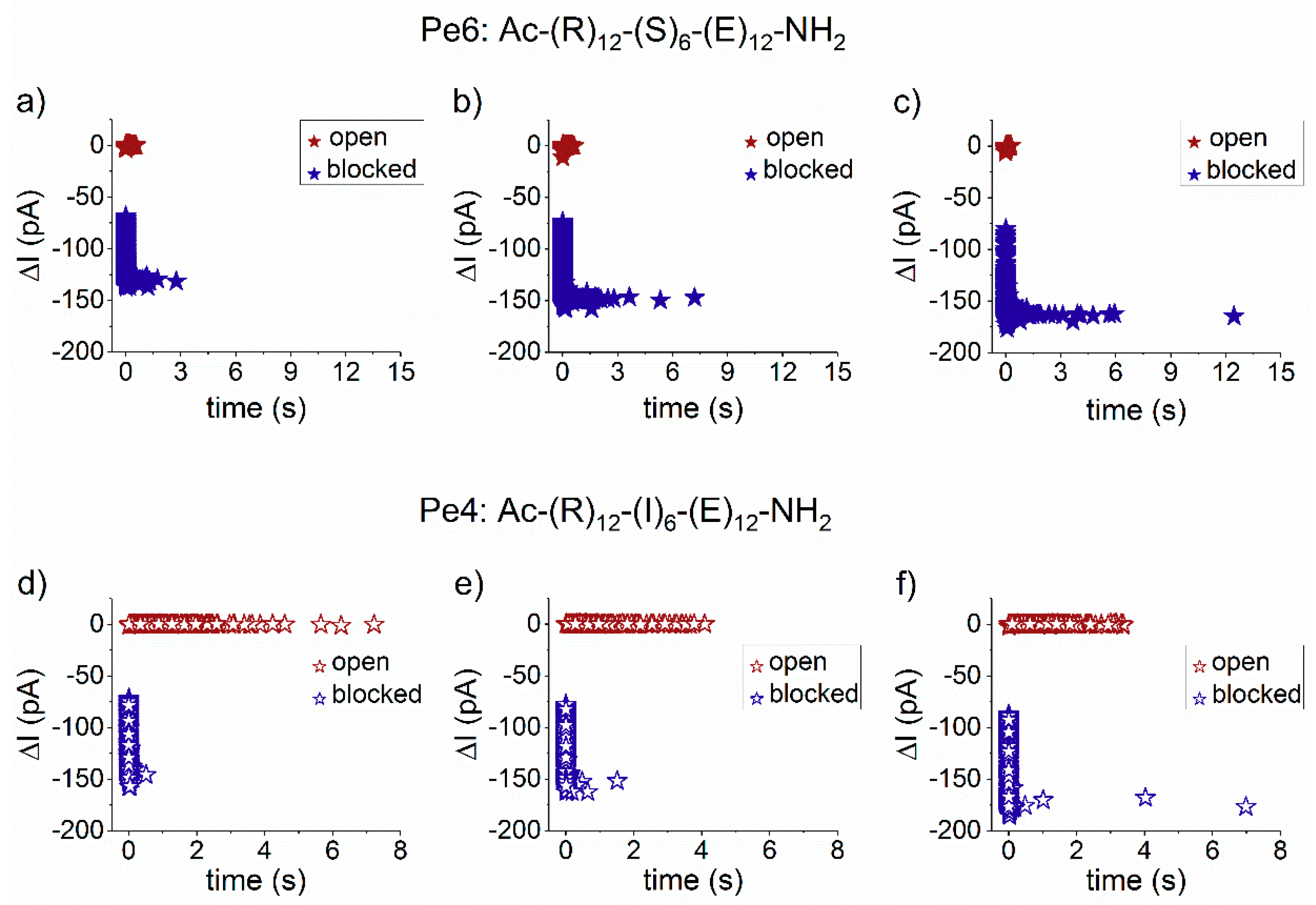
3.2. The Serine- and Isoleucine-Containing Peptides Interact Distinctly with the Nanopore, Despite Their Similar Net Charge
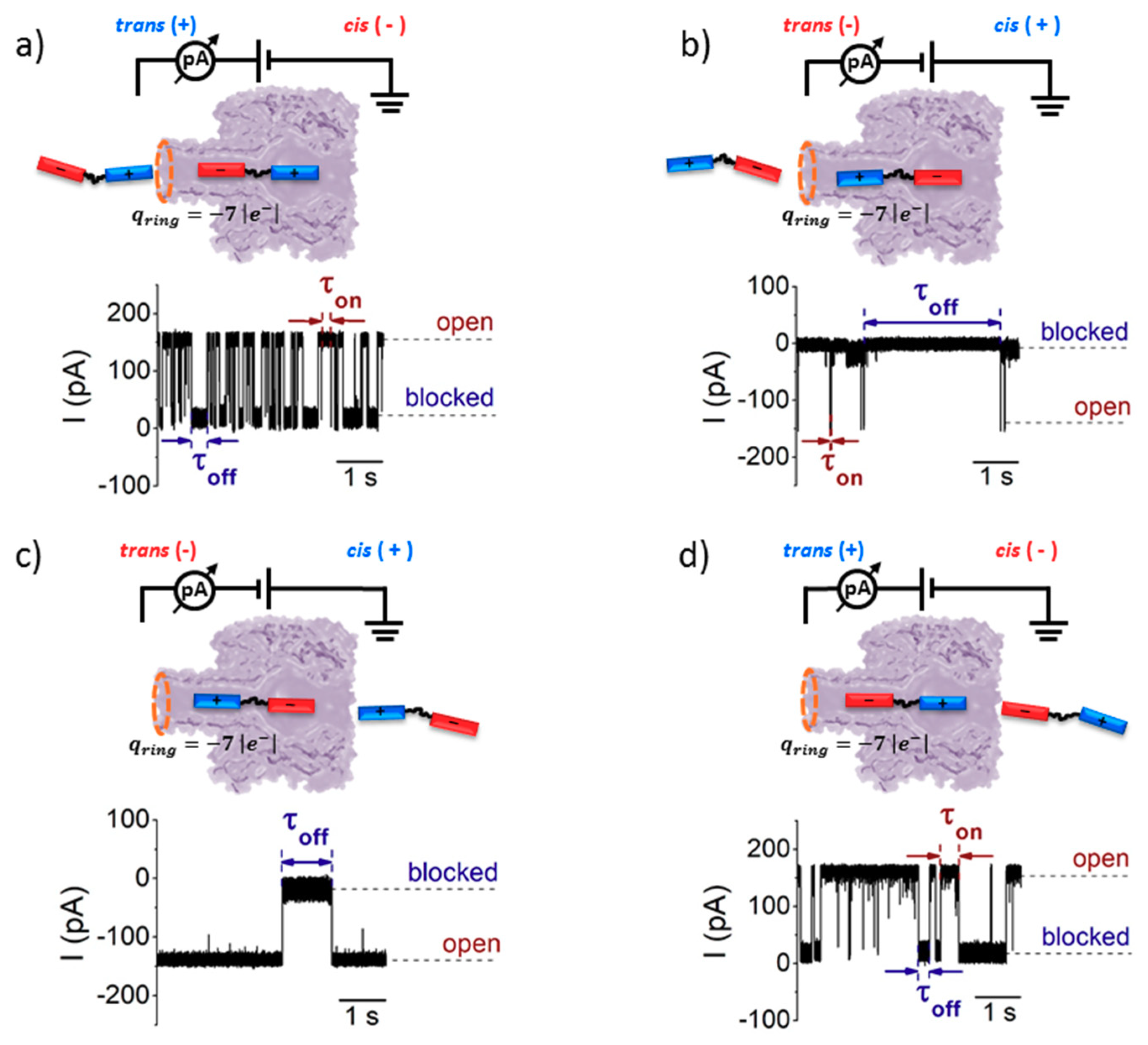
3.3. Sidedness-Dependence of Current Fluctuations Caused by Serine-Containing Peptides When Added from either Cis or Trans Side of the Nanopore
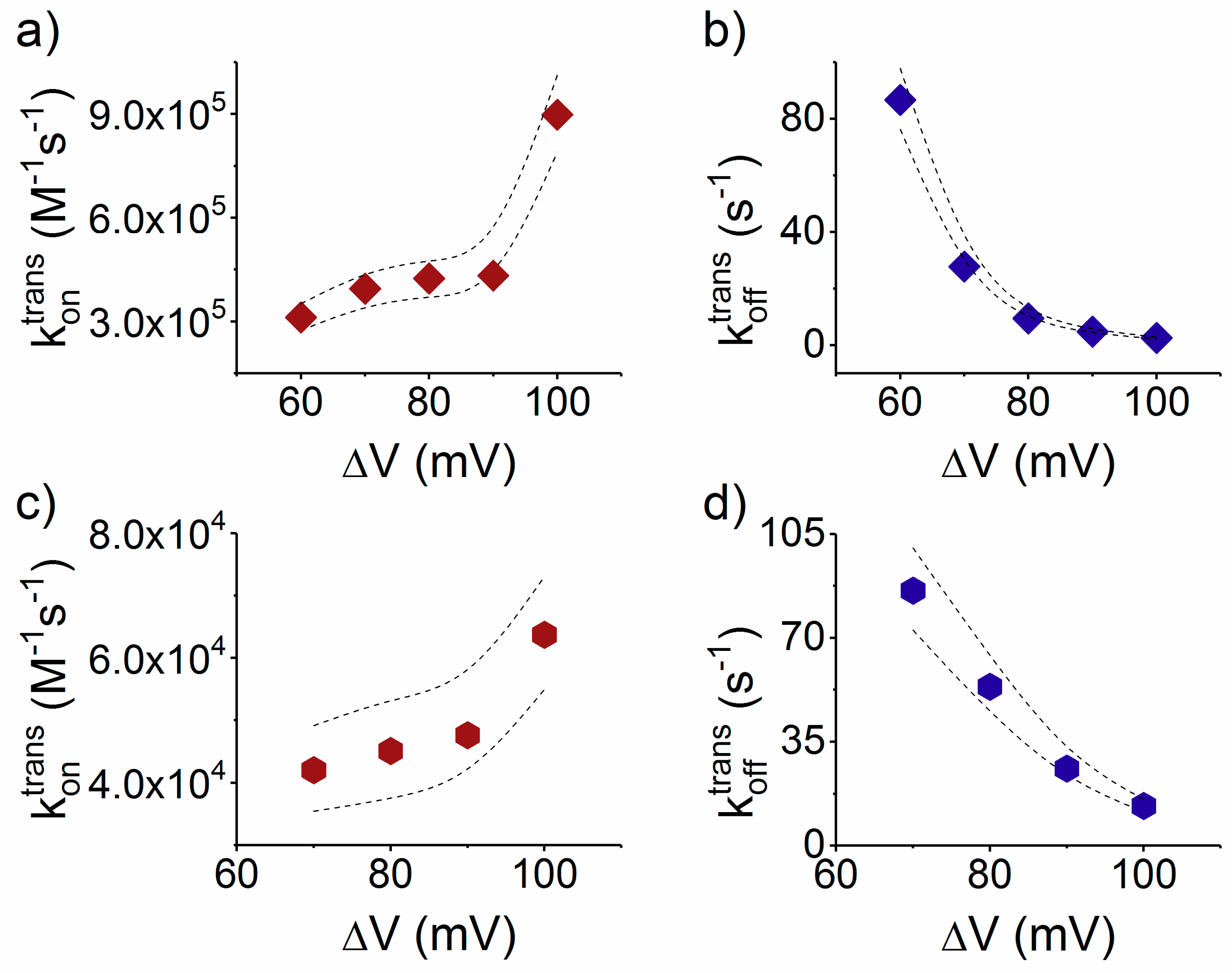
3.3.1. The Case of Peptide Association to the Nanopore
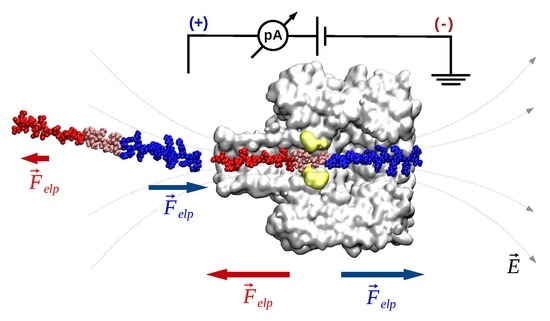
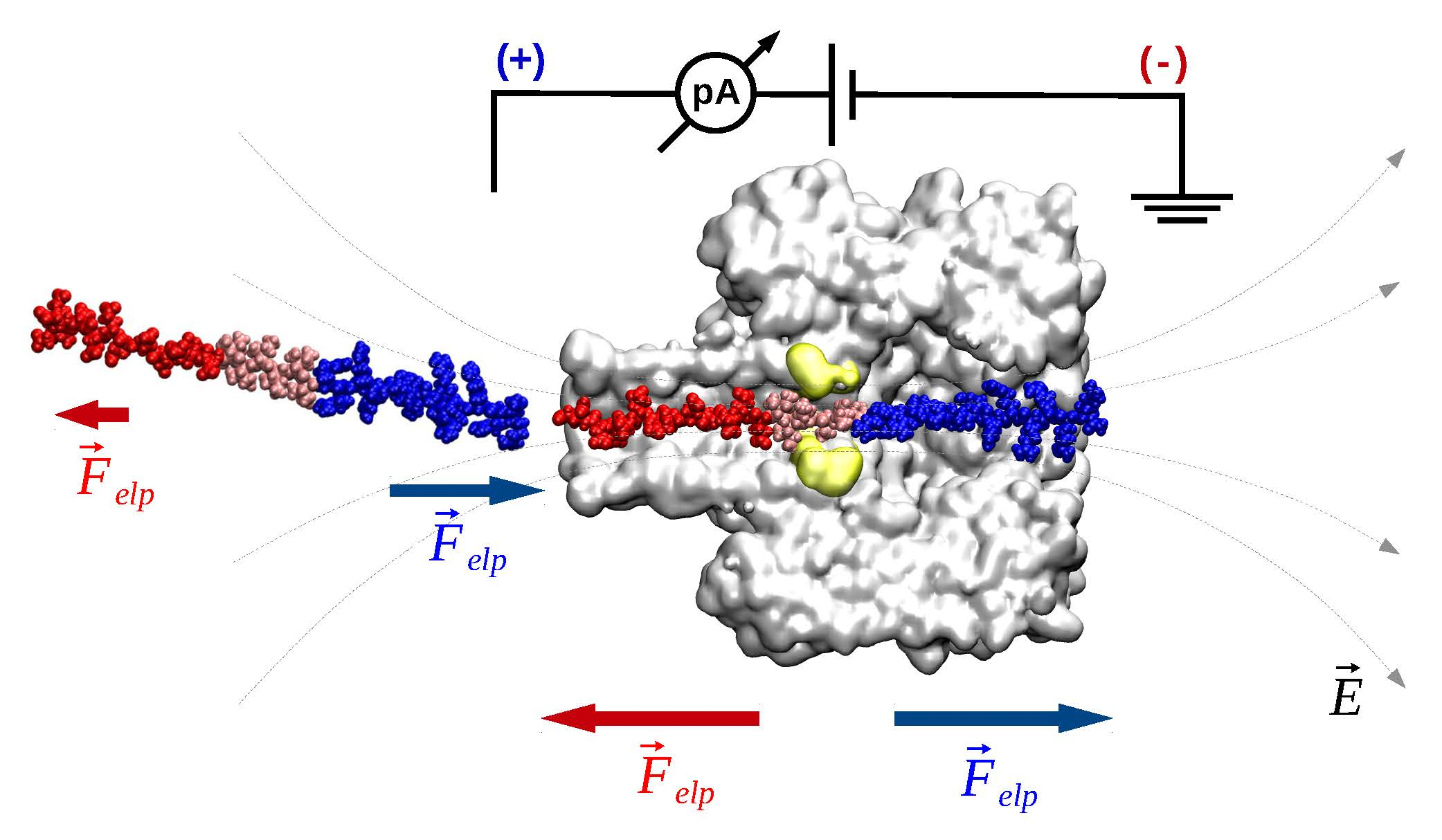
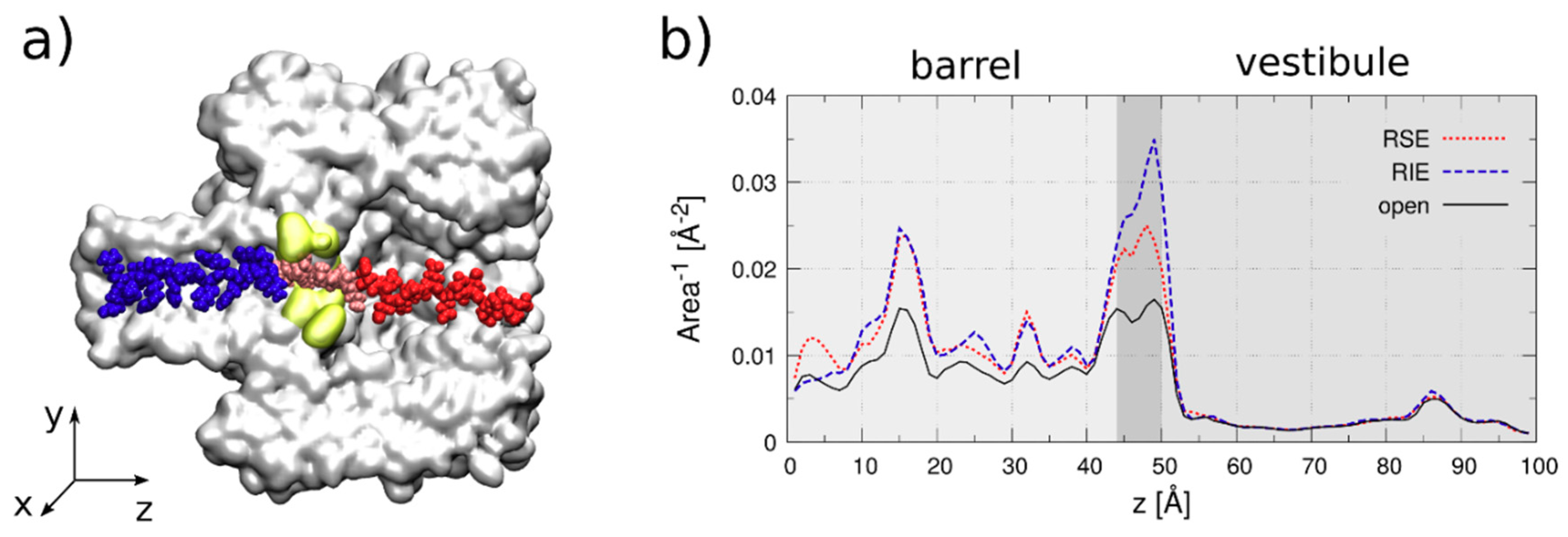
- Electrophoresis is more intense at the trans, β-barrel mouth than at the vestibule entry of the nanopore. Indeed, as a first approximation, considering the nanopore and the membrane as perfect isolators, in stationary state, the electrical field streamlines moves only in the electrolyte. In a quasi-1D approximation of the pore, the electrical field flux EzAz, with Ez the component of the electrical field parallel to the pore axis and Az the pore section, is constant along the pore. Hence, the electrical field is more intense in the narrower section of the pore. Consequently, the electrical field at the barrel mouth is larger than the one at the vestibule (see Supplementary Materials for physical details).
- Entropic penalty is larger on the trans side. In fact, the entropy cost of peptide squeezing inside the nanopore is larger for narrower pore sections.
- Due to the specific design of the studied peptides, which present opposite charges present at their ends, the enthalpy contribution to peptides capture depends on the sign of the applied voltage. At positive ΔVs, the trans-added peptide orients with the R12-containing moiety towards the negatively charged β-barrel α-HL’s opening. Consequently, the attractive electrostatic interactions manifested between the positively charged, R12-containing moiety of the peptide and the nanopore’s negatively charged β-barrel entry (at neutral pH, qring ~ −7|e−|) is expected to facilitate the peptide entry (Figure 5a). In contrast, at negative potentials, the trans-added peptides are driven with the negatively-charged, E12-containing moiety toward the β-barrel opening (Figure 5b), meaning that the peptide-nanopore electrostatic repulsions operate opposite to the electrophoretic force, and against peptide capture. On the other hand, the cis-added peptides are expected to associate to the nanopore with similar rates, regardless of the transmembrane potential polarity, as the vestibule entry of the nanopore is overall neutral at pH = 7 (Figure 5c,d), effectively nullifying the contribution of peptide–nanopore electrostatic interactions to the capture process.
- The electroosmotic flow (elo) favors peptide capture at negative potentials present on the peptide addition side. In such cases and judged from the peptide addition side perspective, the elo flow through the slightly anionic selective α-HL is directed toward the nanopore entry [33,43,52]. This implies that, for the trans-added peptides, electroosmosis favors the capture at negative ΔVs (i.e., the elo flow drives the peptide towards the nanopore) as compared to positive ΔVs (the elo flow drives the peptide away from the nanopore β-barrel and into the trans solution). Note however that, in the former situation (e.g., negative ΔVs), the electrophoretic force acting on the E12-containing moiety from peptides facilitate peptide migration toward the nanopore’s negatively charge β-barrel with the E12 tail head on, and this presents implications for the lumped force that determines peptides association to the nanopore (vide infra). The opposite occurs for cis added peptides, namely at negative ΔVs on the trans side, the elo flow drives the peptide away from the nanopore’s vestibule entry, while the elo flow elicited at positive ΔVs augment peptide association to the vestibule. As a side note, the elo flow is expected to be larger at the narrower, β-barrel section of the nanopore on the trans side (the mass flow rate in stationary state is constant, so that the smaller the cross-sectional area traversed by fluid, the higher the flow velocity).
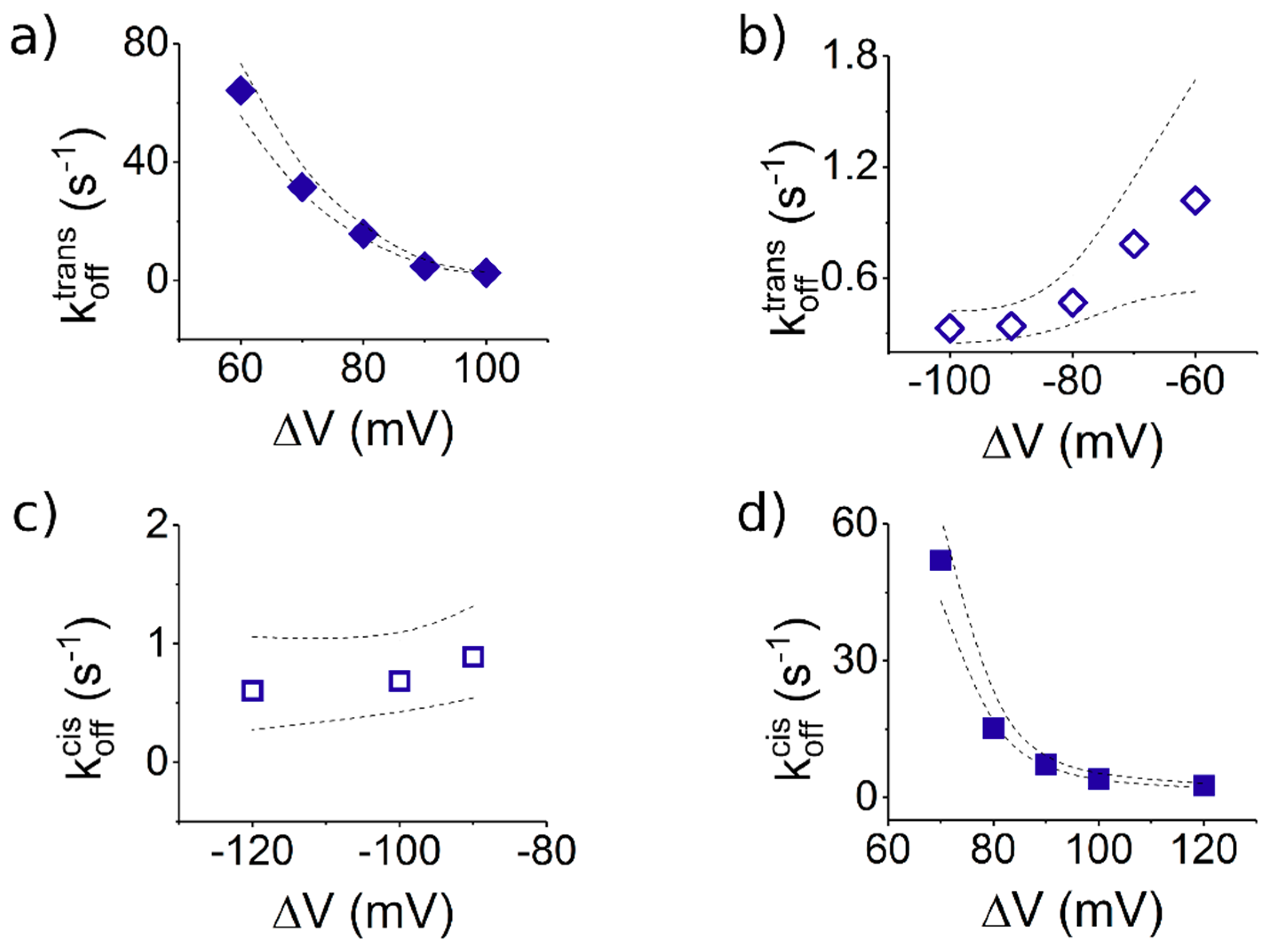
3.3.2. The Case of Peptide Dissociation from the Nanopore
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bayley, H.; Cremer, P.S. Stochastic Sensors Inspired by Biology. Nature 2001, 413, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Balijepalli, A.K.; Ettedgui, J.; Forstater, J.H.; Wang, H.; Zhang, H.; Robertson, J.W.F. Analytical Applications for Pore-Forming Proteins. Biochim. Biophys. Acta 2016, 1858, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Shim, J.W. Single Molecule Sensing by Nanopores and Nanopore Devices. Analyst 2010, 135, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S.; Siwy, Z. Nanopore Analytics: Sensing of Single Molecules. Chem. Soc. Rev. 2009, 38, 2360–2384. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Long, Y.T. Biological Nanopores: Confined Spaces for Electrochemical Single-Molecule Analysis. Acc. Chem. Res. 2018, 51, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Dekker, C. Solid-State Nanopores. Nat. Nanotechnol. 2007, 2, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of Individual Polynucleotide Molecules Using a Membrane Channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The Potential and Challenges of Nanopore Sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meller, A.; Nivon, L.; Brandin, E.; Golovchenko, J.; Branton, D. Rapid Nanopore Discrimination between Single Polynucleotide Molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Ying, Y.L.; Hu, Z.L.; Liao, D.F.; Tian, H.; Long, Y.T. Discrimination of Oligonucleotides of Different Lengths with a Wild-Type Aerolysin Nanopore. Nat. Nanotechnol. 2016, 11, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Nivala, J.; Marks, D.B.; Akeson, M. Unfoldase-Mediated Protein Translocation through an α-Hemolysin Nanopore. Nat. Biotechnol. 2013, 31, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Pastoriza-Gallego, M.; Rabah, L.; Gibrat, G.; Thiebot, B.; van der Goot, F.G.; Auvray, L.; Betton, J.M.; Pelta, J. Dynamics of Unfolded Protein Transport through an Aerolysin Pore. J. Am. Chem. Soc. 2011, 133, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Mereuta, L.; Asandei, A.; Seo, C.H.; Park, Y.; Luchian, T. Quantitative Understanding of pH- and Salt-Mediated Conformational Folding of Histidine-Containing, β-Hairpin-Like Peptides, through Single-Molecule Probing with Protein Nanopores. ACS Appl. Mater. Interfaces 2014, 6, 13242–13256. [Google Scholar] [CrossRef] [PubMed]
- Talaga, D.S.; Li, J. Single-Molecule Protein Unfolding in Solid State Nanopores. J. Am. Chem. Soc. 2009, 131, 9287–9297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piguet, F.; Ouldali, H.; Pastoriza-Gallego, M.; Manivet, P.; Pelta, J.; Oukhaled, A. Identification of Single Amino Acid Differences in Uniformly Charged Homopolymeric Peptides with Aerolysin Nanopore. Nat. Commun. 2018, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.D.; Bricker, L.; Nebel, L.; Miller, J.; Menges, S.; D’Arcangelo, G.; Kramer, A.; Drahushuk, L.; Benck, J.; Shimizu, S.; et al. Transport of Amino Acid Cations through a 2.25-nm-Diameter Carbon Nanotube Nanopore: Electrokinetic Motion and Trapping/Desorption. J. Phys. Chem. C 2017, 121, 27709–27720. [Google Scholar] [CrossRef]
- Kennedy, E.; Dong, Z.; Tennant, C.; Timp, G. Reading the Primary Structure of a Protein with 0.07 nm3 Resolution Using a Subnanometre-Diameter Pore. Nat. Nanotechnol. 2016, 11, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ashcroft, B.; Zhang, P.; Liu, H.; Sen, S.; Song, W.; Im, J.; Gyarfas, B.; Manna, S.; Biswas, S.; et al. Single-Molecule Spectroscopy of Amino Acids and Peptides by Recognition Tunneling. Nat. Nanotechnol. 2014, 9, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Chinappi, M.; Cecconi, F. Protein Sequencing via Nanopore Based Devices: A Nanofluidic Perspective. J. Phys. Condens. Matter 2018, 30, 30. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Sloman, L.; He, Z.; Askimentiev, A. Grafene Nanopores for Protein Sequencing. Adv. Funct. Mater. 2016, 26, 4830–4838. [Google Scholar] [CrossRef] [PubMed]
- Farimani, A.B.; Heiranian, M.; Aluru, N.R. Identification of Amino Acids with Sensitive Nanoporous MoS2: Towards Machine Learning-Based Predition. NPJ 2D Mater. Appl. 2018, 14. [Google Scholar] [CrossRef]
- Kasianowicz, J.J.; Robertson, J.W.F.; Chan, E.R.; Reiner, J.E.; Stanford, V.M. Nanoscopic Porous Sensors. Annu. Rev. Anal. Chem. 2008, 1, 737–766. [Google Scholar] [CrossRef] [PubMed]
- Reiner, J.E.; Robertson, J.W.; Burden, D.L.; Burden, L.K.; Balijepalli, A.; Kasianowicz, J.J. Temperature Sculpting in Yoctoliter Volumes. J. Am. Chem. Soc. 2013, 135, 3087–3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fologea, D.; Uplinger, J.; Thomas, B.; McNabb, D.S.; Li, J. Slowing DNA Translocation in a Solid-State Nanopore. Nano Lett. 2005, 5, 1734–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanunu, M.; Meller, A. Chemically Modified Solid-State Nanopores. Nano Lett. 2007, 7, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Schiel, M.; Siwy, Z.S. Diffusion and Trapping of Single Particles in Pores with Combined Pressure and Dynamic Voltage. J. Phys. Chem. C 2014, 118, 19214–19223. [Google Scholar] [CrossRef]
- Di Fiori, N.; Squires, A.; Bar, D.; Gilboa, T.; Moustakas, T.D.; Meller, A. Optoelectronic Control of Surface Charge and Translocation Dynamics in Solid-State Nanopores. Nat. Nanotechnol. 2013, 8, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, S.W.; Wells, D.B.; Aksimentiev, A.; Dekker, C. Slowing Down DNA Translocation through a Nanopore in Lithium Chloride. Nano Lett. 2012, 12, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Yusko, E.C.; Johnson, J.M.; Majd, S.; Prangkio, P.; Rollings, R.C.; Li, J.; Yang, J.; Mayer, M. Controlling Protein Translocation through Nanopores with Bio-Inspired Fluid Walls. Nat. Nanotechnol. 2011, 6, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Keyser, U.F. Controlling Molecular Transport through Nanopores. J. R. Soc. Interface 2011, 8, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Mereuta, L.; Roy, M.; Asandei, A.; Lee, J.K.; Park, Y.; Andricioaei, I.; Luchian, T. Slowing Down Single-Molecule Trafficking through a Protein Nanopore Reveals Intermediates for Peptide Translocation. Sci. Rep. 2014, 4, 3885. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Schiopu, I.; Chinappi, M.; Seo, C.H.; Park, Y.; Luchian, T. Electroosmotic Trap Against the Electrophoretic Force Near a Protein Nanopore Reveals Peptide Dynamics During Capture and Translocation. ACS Appl. Mater. Interfaces 2016, 8, 13166–13179. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Chinappi, M.; Lee, J.K.; Chang, H.S.; Mereuta, L.; Park, Y.; Luchian, T. Placement of Oppositely Charged Aminoacids at a Polypeptide Termini Determines the Voltage-Controlled Braking of Polymer Transport through Nanometer-Scale Pores. Sci. Rep. 2015, 5, 10419. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Chinappi, M.; Kang, H.K.; Seo, C.H.; Mereuta, L.; Park, Y.; Luchian, T. Acidity-Mediated, Electrostatic Tuning of Asymmetrically Charged Peptides Interactions with Protein Nanopores. ACS Appl. Mater. Interfaces 2015, 7, 16706–16714. [Google Scholar] [CrossRef] [PubMed]
- Chinappi, M.; Luchian, T.; Cecconi, F. Nanopore Tweezers: Voltage Controlled Trapping and Releasing of Analytes. Phys. Rev. E 2015, 92, 032714. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Rossini, A.E.; Chinappi, M.; Park, Y.; Luchian, T. Protein Nanopore-Based Discrimination between Selected Neutral Amino Acids from Polypeptides. Langmuir 2017, 33, 14451–14459. [Google Scholar] [CrossRef] [PubMed]
- Ferro, E.S.; Rioli, V.; Castro, L.M.; Fricker, L.D. Intracellular peptides: From Discovery to Function. EuPA Open Proteom. 2014, 3, 143–151. [Google Scholar] [CrossRef]
- Montal, M.; Mueller, P. Formation of Bimolecular Membranes from Lipid Monolayers and a Study of their Electrical Properties. Proc. Natl. Acad. Sci. USA 1972, 69, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Apetrei, A.; Park, Y.; Hahm, K.S.; Luchian, T. Investigation of Single-Molecule Kinetics Mediated by Weak Hydrogen-Bonds Within a Biological Nanopore. Langmuir 2011, 27, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.Y.; Rabin, Y.J. DNA Capture into a Nanopore: Interplay of Diffusion and Electrohydrodynamics. Chem. Phys. 2010, 133, 165102. [Google Scholar] [CrossRef] [PubMed]
- Counterman, A.E.; Clemmer, D.E. Volumes of Individual Amino Acid Residues in Gas-Phase Peptide Ions. J. Am. Chem. Soc. 1999, 121, 4031–4039. [Google Scholar] [CrossRef]
- Aksimentiev, A.; Schulten, K. Imaging α-Hemolysin with Molecular Dynamics: Ionic Conductance, Osmotic Permeability, and the Electrostatic Potential Map. Biophys. J. 2005, 88, 3745–3761. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.J.; Mohammad, M.M.; Cheley, S.; Bayley, H.; Movileanu, L. Catalyzing the Translocation of Polypeptides through Attractive Interactions. J. Am. Chem. Soc. 2007, 129, 14034–14044. [Google Scholar] [CrossRef] [PubMed]
- Asandei, A.; Ciuca, A.; Apetrei, A.; Schiopu, I.; Mereuta, L.; Seo, C.H.; Park, Y.; Luchian, T. Nanoscale Investigation of Generation 1 PAMAM Dendrimers Interaction with a Protein Nanopore. Sci. Rep. 2017, 7, 6167. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.N.; Muthukumar, M.; Meller, A. pH Tuning of DNA Translocation Time through Organically Functionalized Nanopores. ACS Nano 2013, 7, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Maglia, G.; Restrepo, M.R.; Mikhailova, E.; Bayley, H. Enhanced Translocation of Single DNA Molecules through α-Hemolysin Nanopores by Manipulation of Internal Charge. Proc. Natl. Acad. Sci. USA 2008, 105, 19720–19725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnyk, R.A.; Collier, R.J. A Loop Network within the Anthrax Toxin Pore Positions the Phenylalanine Clamp in an Active Conformation. Proc. Natl. Acad. Sci. USA 2006, 103, 9802–9807. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. Microbiology. Translocation of Anthrax Toxin: Lord of the Rings. Science 2005, 309, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Matouschek, A.; Glick, B.S. Barreling through the Outer Membrane. Nat. Struct. Biol. 2001, 8, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.; Bannwarth, M.; Meisinger, C.; Hill, K.; Model, K.; Krimmer, T.; Casadio, R.; Truscott, K.N.; Schulz, G.E.; Pfanner, N.; et al. Preprotein Translocase of the Outer Mitochondrial Membrane: Reconstituted Tom40 Forms a Characteristic TOM Pore. Mol. Biol. 2005, 353, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Bonome, E.L.; Cecconi, F.; Chinappi, M. Electroosmotic Flow through an α-Hemolysin Nanopore. Microfluid. Nanofluid. 2017, 21, 96. [Google Scholar] [CrossRef]
- Muthukumar, M. Theory of Capture Rate in Polymer Translocation. J. Chem. Phys. 2010, 132, 195101. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, M.; Plesa, C.; Dekker, C. Single-Molecule Sensing with Nanopores. Phys. Today 2015, 68, 40. [Google Scholar] [CrossRef]
- Farahpour, F.; Maleknejad, A.; Varnik, F.; Ejtehadi, M.R. Chain Deformation in Translocation Phenomena. Soft Matter 2013, 9, 2750–2759. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Misakian, M.; Robertson, B.; Kasianowicz, J.J. Driven DNA Transport into an Asymmetric Nanometer-Scale Pore. Phys. Rev. Lett. 2000, 85, 3057–3060. [Google Scholar] [CrossRef] [PubMed]
- Pastoriza-Gallego, M.; Gibrat, G.; Thiebot, B.; Betton, J.M.; Pelta, J. Polyelectrolyte and Unfolded Protein Pore Entrance Depends on the Pore Geometry. Biochim. Biophys. Acta 2009, 1788, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Howorka, S.; Bayley, H. Probing Distance and Electrical Potential within a Protein Pore with Tethered DNA. Biophys. J. 2002, 83, 3202–3210. [Google Scholar] [CrossRef] [Green Version]
- Cressiot, B.; Braselmann, E.; Oukhaled, A.; Elcock, A.H.; Pelta, J.; Clark, P.L. Dynamics and Energy Contributions for Transport of Unfolded Pertactin through a Protein Nanopore. ACS Nano 2015, 9, 9050–9061. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asandei, A.; Dragomir, I.S.; Di Muccio, G.; Chinappi, M.; Park, Y.; Luchian, T. Single-Molecule Dynamics and Discrimination between Hydrophilic and Hydrophobic Amino Acids in Peptides, through Controllable, Stepwise Translocation across Nanopores. Polymers 2018, 10, 885. https://doi.org/10.3390/polym10080885
Asandei A, Dragomir IS, Di Muccio G, Chinappi M, Park Y, Luchian T. Single-Molecule Dynamics and Discrimination between Hydrophilic and Hydrophobic Amino Acids in Peptides, through Controllable, Stepwise Translocation across Nanopores. Polymers. 2018; 10(8):885. https://doi.org/10.3390/polym10080885
Chicago/Turabian StyleAsandei, Alina, Isabela S. Dragomir, Giovanni Di Muccio, Mauro Chinappi, Yoonkyung Park, and Tudor Luchian. 2018. "Single-Molecule Dynamics and Discrimination between Hydrophilic and Hydrophobic Amino Acids in Peptides, through Controllable, Stepwise Translocation across Nanopores" Polymers 10, no. 8: 885. https://doi.org/10.3390/polym10080885