Preparation of a Water-Based Photoreactive Azosulphonate-Doped Poly(Vinyl Alcohol) and the Investigation of Its UV Response



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
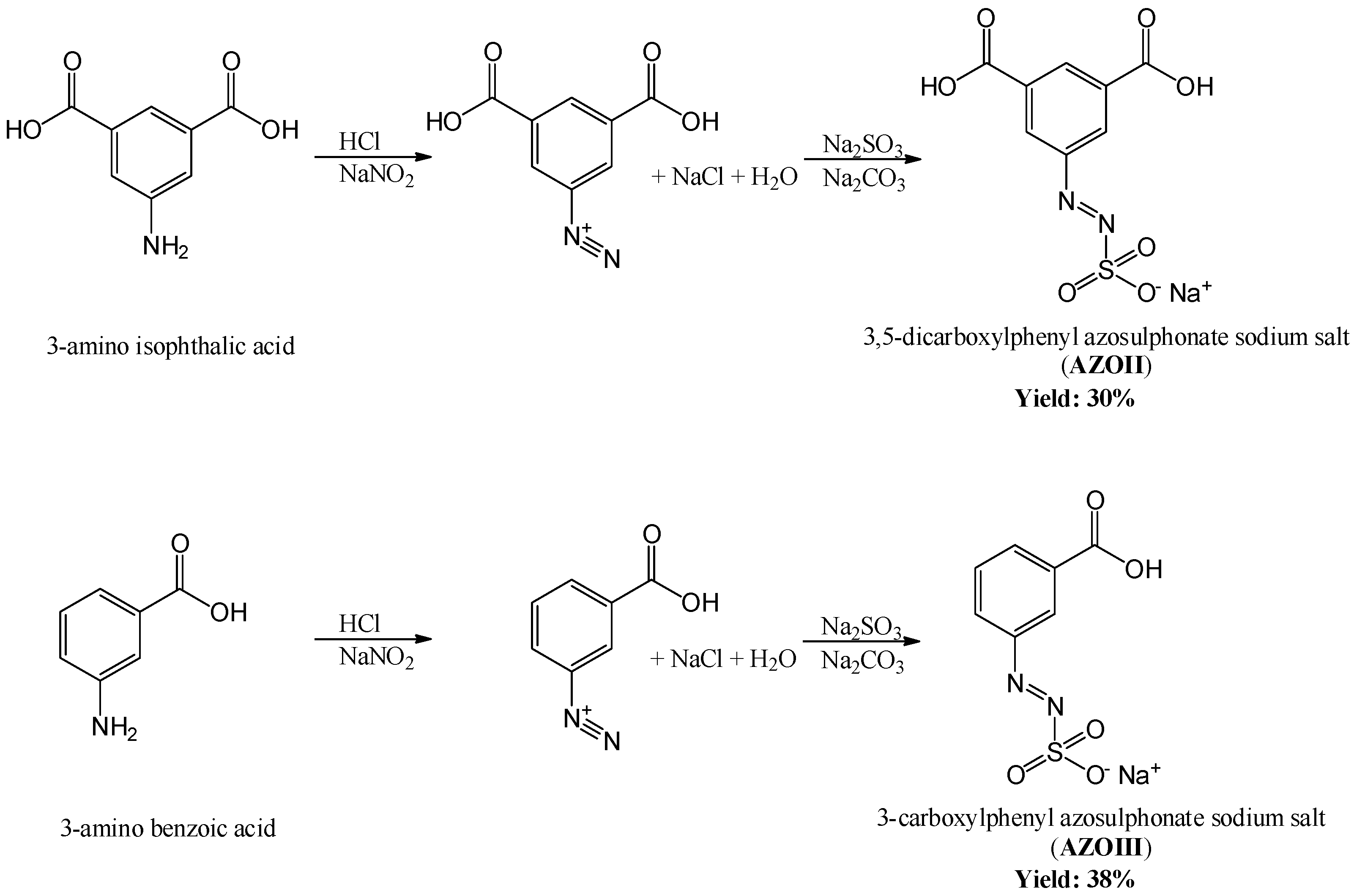
2.2.1. Synthesis of the Azosulphonate Sodium Salts
2.2.2. Photolysis
2.2.3. Thermolysis
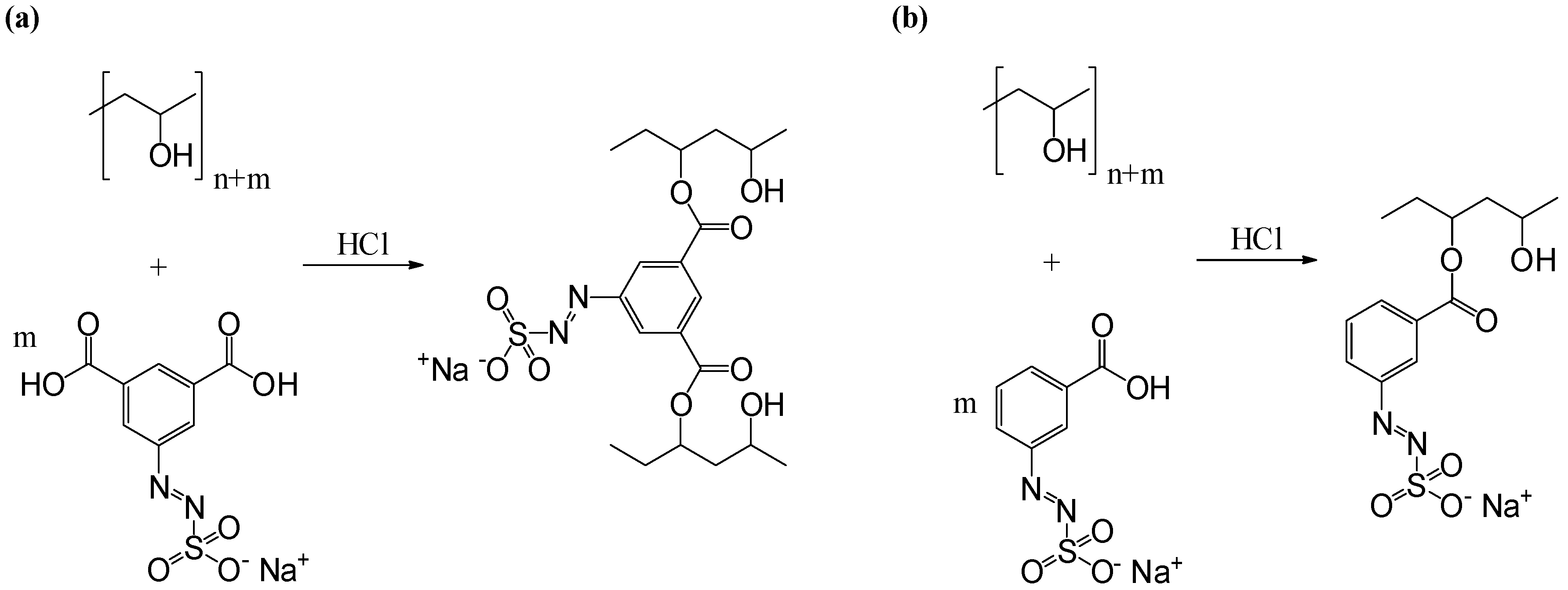
2.2.4. Preparation of Azosulphonate-Doped PVA
2.2.5. PVA-AZO Thin Film Preparation
2.2.6. Annealing
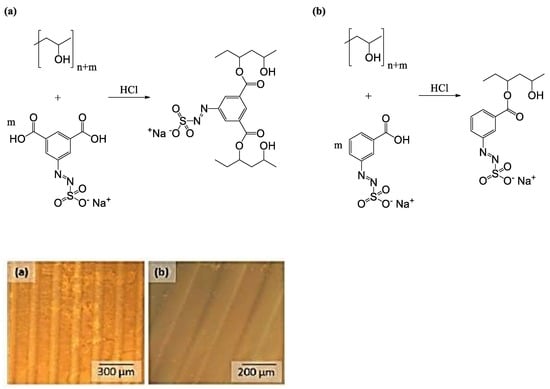
2.2.7. Photolithographic Patterning
3. Results and Discussion
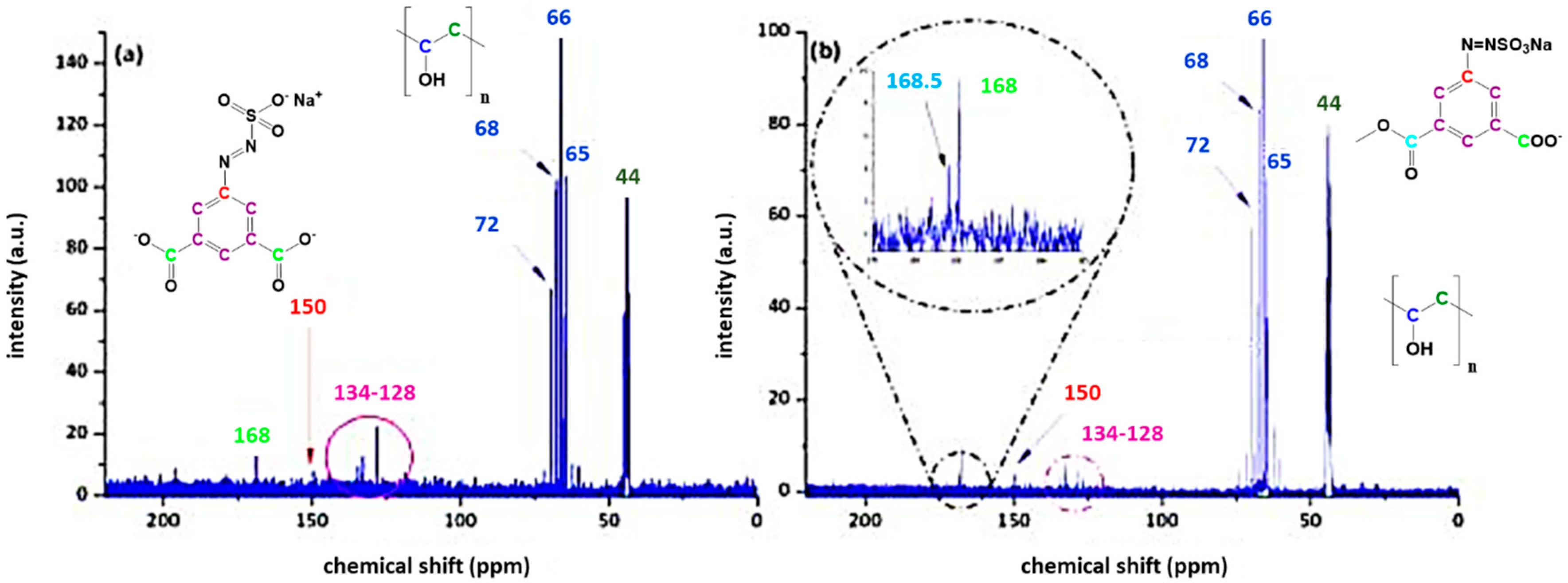
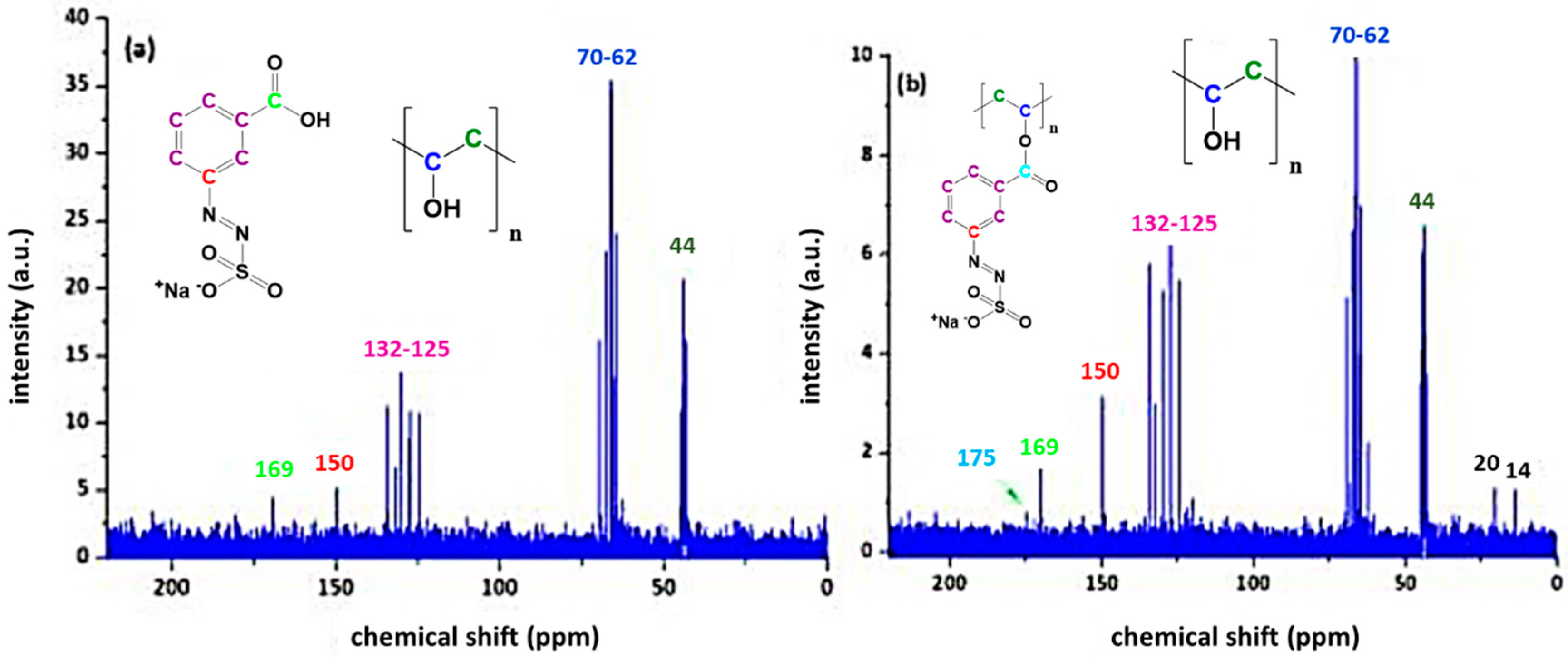
3.1. Characterisation of the Azosulphonate Sodium Salts
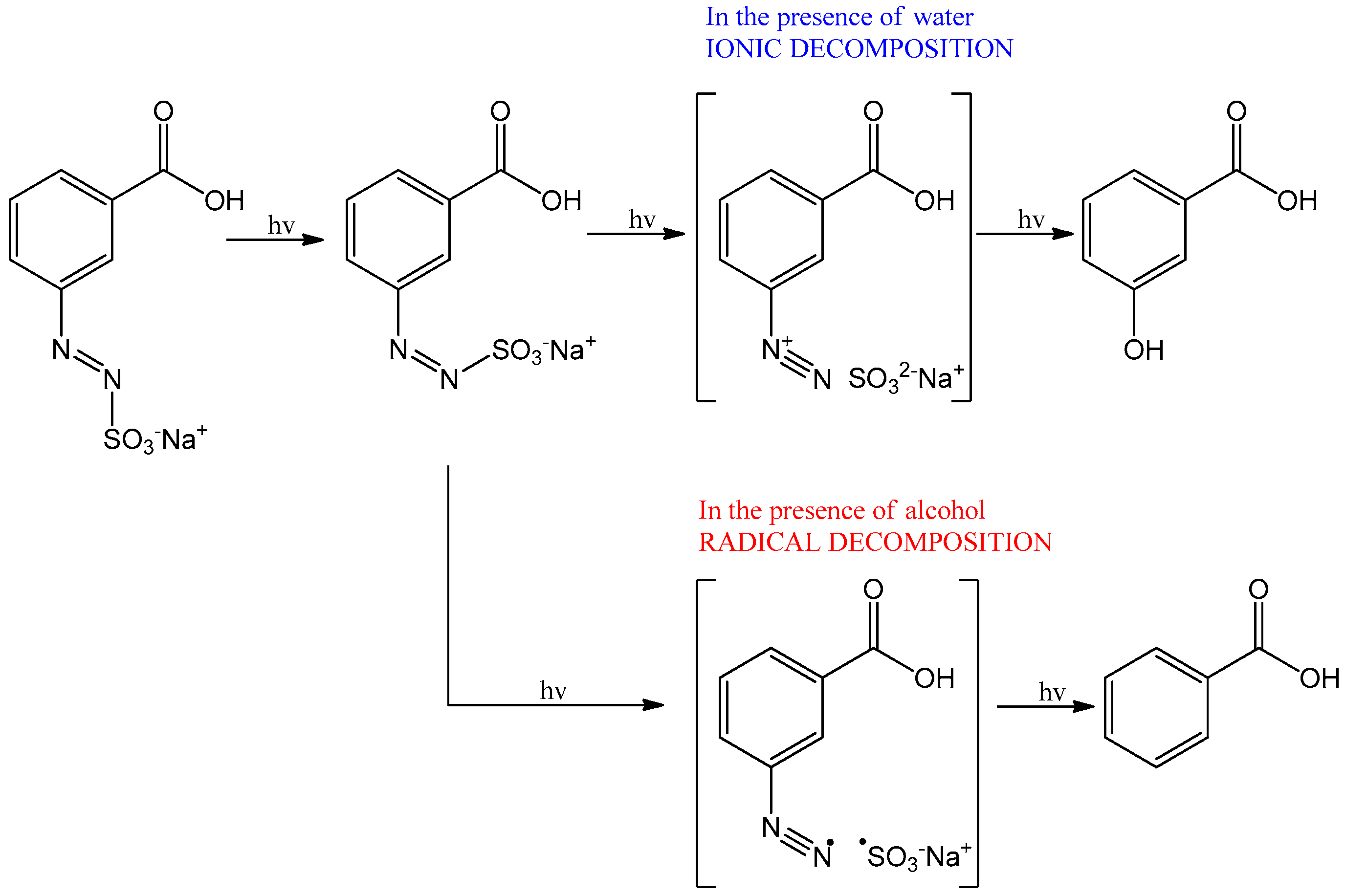
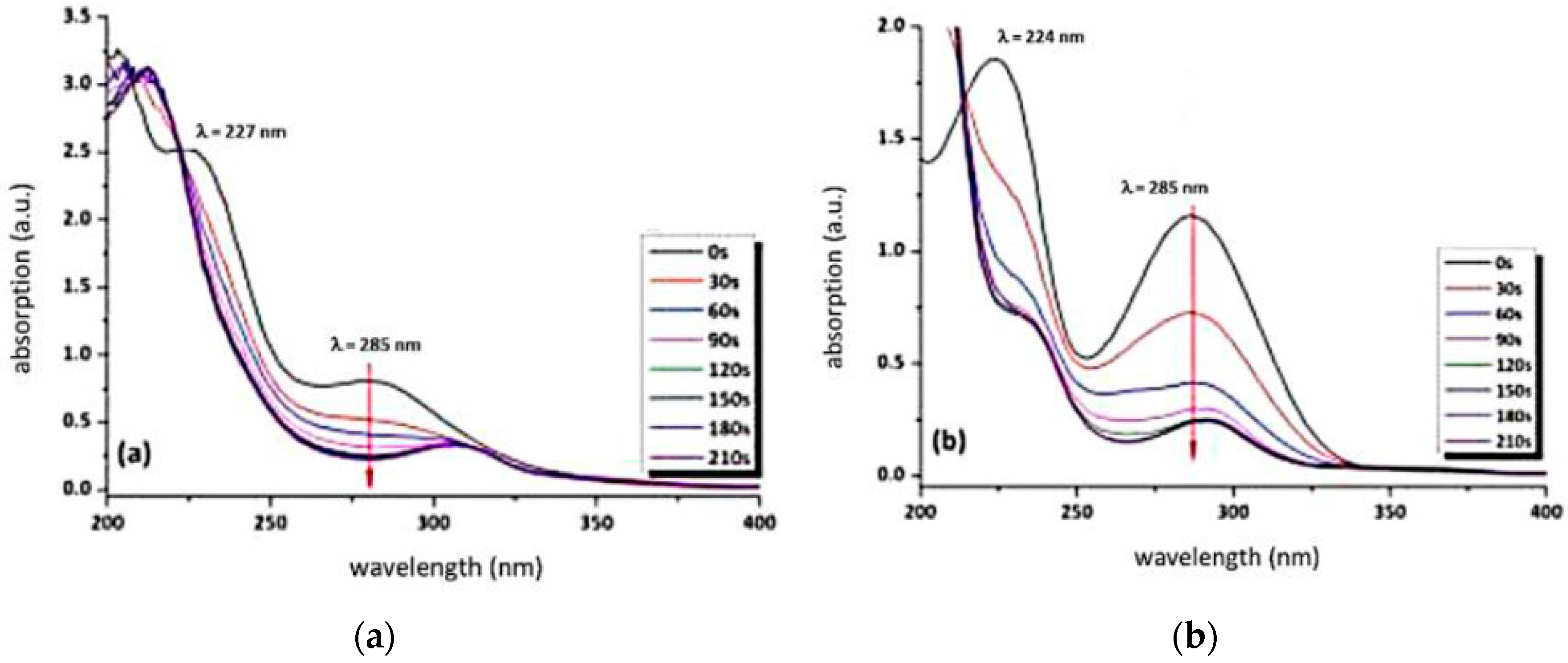
3.2. Behaviour of the Synthesised Azosulphonate Dyes under Thermal Load and UV Light
3.3. Characterisation of Azosulphonate-Doped PVA
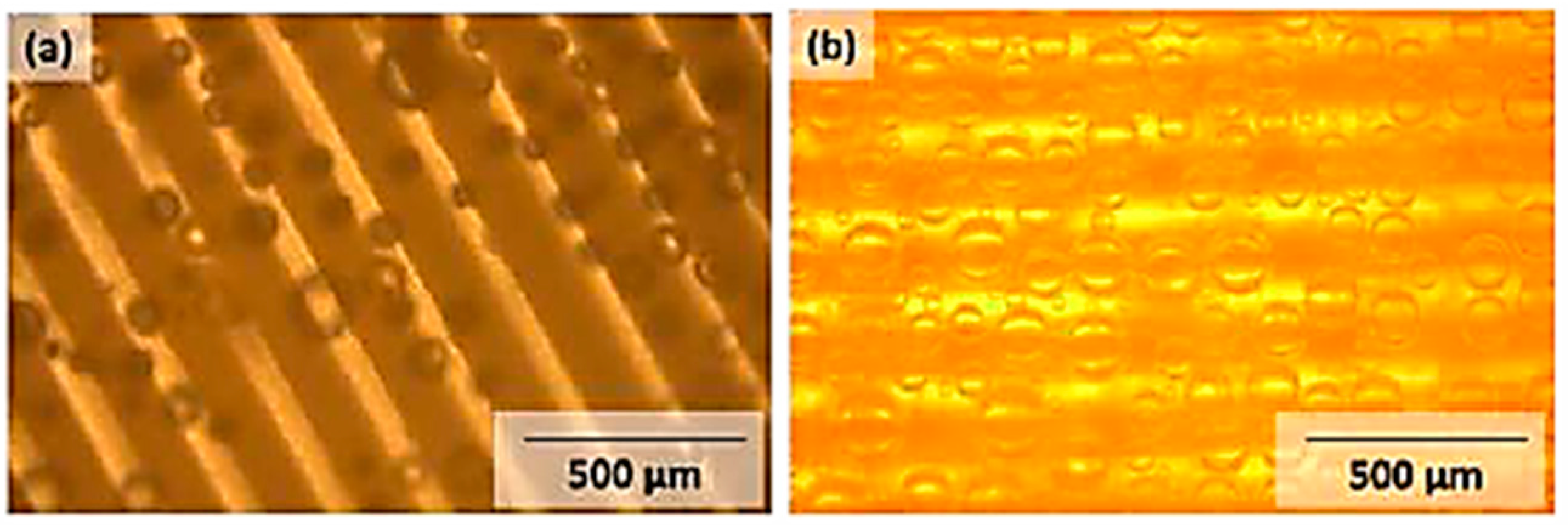
3.4. Photolithographic Patterning
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamadaa, S.; Medeiros, D.R.; Patterson, K.; Jen, W.L.K.; Rager, T.; Lin, Q.; Lenci, C.; Byers, J.D.; Havard, J.M.; Pasini, D.; et al. Positive and Negative Tone Water Processable Photoresists: A Progress Report. Proc. SPIE Int. Soc. Opt. Eng. 1998, 3333, 245–253. [Google Scholar]
- Lin, Q.; Steinhäusler, T.; Simpson, L.; Wilder, M.; Medeiros, D.R.; Willson, C.G. A Water-Castable, Water-Developable Chemically Amplified Negative-Tone Resist. Chem. Mater. 1997, 9, 1725–1730. [Google Scholar] [CrossRef]
- Aoki, H.; Tokuda, T.; Nagasaki, Y.; Kato, M. Water-soluble silicon containing polymer resist. J. Polym. Sci. Part A Polym. Chem. 2000, 35, 2827–2833. [Google Scholar] [CrossRef]
- Shirai, M.; Katsuta, N.; Tsunooka, M.; Tanaka, M.; Nishijima, K. Photosensitive polymers bearing iminooxysulfonyl groups. A water soluble positive-type photoresist. Die Makromol. Chem. 1989, 190, 2099–2107. [Google Scholar] [CrossRef]
- Barker, I.C.; Allen, N.S.; Edge, M.; Sperry, J.A.; Batten, R.J. Synthesis, photochemistry and cross-linking of visibly sensitised photopolymers of PVA based on (E)-2-(4-Formylstyryl)-3,4-dimethylthiazolium Methylsulfate. J. Chem. Soc. Faraday Trans. 1994, 90, 3677–3683. [Google Scholar] [CrossRef]
- Dauth, J.; Nuyken, O.; Strohriegl, P.; Voit, B. Water-soluble photoresins based on azosulfonates. Die Makromol. Chem. 1992, 193, 723–734. [Google Scholar] [CrossRef]
- Aslam, M.; Kalyar, M.A.; Zulfiqar Ali Raza, Z.A. Polyvinyl alcohol: A review of research status and use of polyvinyl alcohol based nanocomposites. Polym. Eng. Sci. 2018, 58, 2119–2132. [Google Scholar] [CrossRef]
- Suganthi, S.; Mohanapriya, S.; Raj, V.; Kanaga, S.; Dhandapani, R.; Vignesh, S.; Sundar, K.J. Tunable Physicochemical and Bactericidal Activity of Multicarboxylic-Acids-Crosslinked Polyvinyl Alcohol Membrane for Food Packaging Applications. Chem. Sel. 2018, 3, 11167–11176. [Google Scholar] [CrossRef]
- Desmaisons, J.; Rueff, M.; Brasa, J.; Dufresne, A. Impregnation of paper with cellulose nanocrystal reinforced polyvinyl alcohol: Synergistic effect of infrared drying and CNC content on crystallinity. Soft Matter 2018, 14, 9425–9435. [Google Scholar] [CrossRef]
- Finch, C.A. Polyvinyl Alcohol: Properties and Applications; Finch, C.A., Ed.; Wiley: New York, NY, USA, 1973. [Google Scholar]
- So, J.-H.; Oh, M.-H.; Lee, J.-D.; Yang, S.-M. Effects of Polyvinyl Alcohol on the Rheological Behavior and Phase Stability of Aqueous Silica Suspensions. J. Chem. Eng. 2001, 34, 262–268. [Google Scholar] [CrossRef]
- Mawad, D.; Martens, P.J.; Odell, R.A.; Poole-Warren, L.A. The effect of redox polymerisation on degradation and cell responses to poly (vinyl alcohol) hydrogels. Biomaterials 2007, 28, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Banthia, A.K.; Majumdar, D.K. Biomedical evaluation of polyvinyl alcohol–gelatin esterified hydrogel for wound dressing. J. Mater. Sci. Mater. Med. 2007, 18, 1889–1894. [Google Scholar] [CrossRef]
- Braun, D.; Walter, E. Intra- und intermolekulare Vernetzung von Polyvinylalkohol mit Dialdehyden. Colloid Polym. Sci. 1980, 258, 795–801. [Google Scholar] [CrossRef]
- Pal, K.; Banthia, A.K.; Majumdar, D.K. Preparation and characterization of polyvinyl alcohol-gelatin hydrogel membranes for biomedical applications. AAPS PharmSciTech 2007, 8, E142–E146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauberger, J.G.; Riess, G.; Kern, W. UV crosslinking of Fe3+-doped poly(vinyl alcohol)—Characterization of optical properties and swelling behavior. J. Appl. Polym. Sci. 2013, 129, 3623–3628. [Google Scholar] [CrossRef]
- Filoti, G.; Kuncser, V.; Franke, H.; Kardinahl, T.; Manivannan, G. Optical induced modifications in thin films of Fe:PVA. J. Radioanal. Nucl. Chem. 1995, 190, 315–320. [Google Scholar] [CrossRef]
- Guiot, O.; Tighzert, L.; Coqueret, X. Electron beam crosslinking of extrusion-blown LDPE films: 1. Mechanical properties. Eur. Polym. J. 1999, 35, 565–570. [Google Scholar] [CrossRef]
- Barichard, A.; Israëli, Y.; Rivaton, A. Photocrosslinking in dichromated poly(acrylic acid) during hologram recording and comparison with dichromated poly(vinyl alcohol). J. Polym. Sci. Part A Polym. Chem. 2007, 46, 636–642. [Google Scholar] [CrossRef]
- Bulinski, M.; Iova, I.; Belea, A.; Kuncser, V.; Filoti, G. Experimental investigation of the nonlinear optical response in Fe:PVA. J. Mater. Sci. Lett. 2000, 19, 27–28. [Google Scholar] [CrossRef]
- Schauberger, J.G.; Riess, G.; Kern, W. Preparation of UV reactive montmorillonite and characterization of its nanocomposites with poly(vinyl alcohol). J. Appl. Polym. Sci. 2013, 130, 665–672. [Google Scholar] [CrossRef]
- Tang, Z.; Wie, J.; Yung, L.; Ji, B.; Ma, H.; Qiu, C.; Yoon, K.; Wan, F.; Fang, D.; Hsiao, B.S.; et al. UV-cured poly(vinyl alcohol) ultrafiltration nanofibrous membrane based on electrospun nanofiber scaffolds. J. Memb. Sci. 2009, 328, 1–5. [Google Scholar] [CrossRef]
- Nuyken, O.; Voit, B. The photoactive diazosulfonate group and its role in polymer chemistry. Macromol. Chem. Phys. 2003, 198, 2337–2372. [Google Scholar] [CrossRef]
- Overberger, C.G.; O’Shaughnessy, M.T.; Shalit, H. The Preparation of Some Aliphatic Azo Nitriles and their Decomposition in Solution. J. Am. Chem. Soc. 1949, 71, 2661–2666. [Google Scholar] [CrossRef]
- Matusche, P.; Nuyken, O.; Voit, B.; Damme, M.V. Water Soluble and Photoactive Copolymers Containing Amidic Aryldiazosulfonate Groups. J. Macromol. Sci. Part A 1997, 34, 201–209. [Google Scholar] [CrossRef]
- Riess, G. Neue Arylazosulfonate und ihre Reaktionen. Master’s Thesis, University of Bayreuth, Bayreuth, Germany, 1990. [Google Scholar]
- Nuyken, O.; Presenz, U. Azo polymers—Syntheses and reactions. Polym. Bull. 1988, 20, 335–341. [Google Scholar] [CrossRef]
- Staško, A.; Szaboova, K.; Cholvad, V.; Nuyken, O.; Dauth, J. The photochemical decomposition of azo compounds (a spin trap study). J. Photochem. Photobiol. A Chem. 1993, 69, 295–304. [Google Scholar] [CrossRef]
- Franzke, D.; Scherer, C.; Nuyken, O.; Wokaun, A. Synthesis and photochemical properties of aromatic diazo-phosphonium salts. J. Photochcem. Photobiol. A Chem. 1997, 111, 47–50. [Google Scholar] [CrossRef]
- Voit, B.; Braun, F.; Gernert, M.; Sieczkowska, B.; Millaruelo, M.; Messerschmidt, M.; Mertig, M.; Opitz, J. Photolabile and thermally labile polymers as templates and for surface patterning. Polym. Adv. Technol. 2006, 17, 691–693. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie; Thieme Verlag: Stuttgart, Germany, 2005. [Google Scholar]
- Socrates, G. Infrared Characteristic Group Frequencies, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- McKnight, D.M.; Kimball, B.A.; Runkel, R.L. pH dependence of iron photoreduction in a rocky mountain stream affected by acid mine drainage. Hydrol. Process. 2001, 15, 1979–1992. [Google Scholar] [CrossRef]
- Kocaokutgen, H.; Gümrükçüoèlu, I.E. Thermal Characterization of Some Azo Dyes Containing Intramolecular Hydrogen Bonds and Non-Bonds. J. Therm. Anal. Calorim. 2003, 71, 675–679. [Google Scholar] [CrossRef]
- Holt, B.D.; Engelkemeir, A.G. Thermal decomposition of barium sulfate to sulfur dioxide for mass spectrometric analysis. Anal. Chem. 1970, 42, 1451–1453. [Google Scholar] [CrossRef]
- Schauberger, J.G. Polymere als Barrierematerialien für Kartonverpackungen. Master’s Thesis, Montanuniversitaet Leoben, Leoben, Austria, 2009. [Google Scholar]
- Knopp, D.; Tang, D.; Niessner, R. Review: Bioanalytical applications of biomolecule-functionalized nanometer-sized doped silica particles. Anal. Chim. Acta 2009, 647, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, H.; Epperlein, J. Untersuchungen zum UV/Vis-Spektralverhalten von Azofarbstoffen. V Quantitative Beschreibung der Absorptionsmaxima mehrfach substituierter Azobenzene mit einem Inkrementsystem. J. Prakt. Chem. 1981, 323, 755–775. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | PVA (wt %) | AZOII (wt %) | AZOIII (wt %) |
|---|---|---|---|
| PAII.1 | 97.5 | 2.5 | |
| PAII.2 | 95 | 5 | |
| PAII.3 | 92.5 | 7.5 | |
| PAII.4 | 90 | 10 | |
| PAII.5 | 80 | 20 | |
| PAIII.1 | 97.5 | 2.5 | |
| PAIII.2 | 95 | 5 | |
| PAIII.3 | 92.5 | 7.5 | |
| PAIII.4 | 90 | 10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nothdurft, P.; Schauberger, J.G.; Riess, G.; Kern, W. Preparation of a Water-Based Photoreactive Azosulphonate-Doped Poly(Vinyl Alcohol) and the Investigation of Its UV Response. Polymers 2019, 11, 169. https://doi.org/10.3390/polym11010169
Nothdurft P, Schauberger JG, Riess G, Kern W. Preparation of a Water-Based Photoreactive Azosulphonate-Doped Poly(Vinyl Alcohol) and the Investigation of Its UV Response. Polymers. 2019; 11(1):169. https://doi.org/10.3390/polym11010169
Chicago/Turabian StyleNothdurft, Philipp, Jörg Guido Schauberger, Gisbert Riess, and Wolfgang Kern. 2019. "Preparation of a Water-Based Photoreactive Azosulphonate-Doped Poly(Vinyl Alcohol) and the Investigation of Its UV Response" Polymers 11, no. 1: 169. https://doi.org/10.3390/polym11010169