Comparative Study on Kinetics of Ethylene and Propylene Polymerizations with Supported Ziegler–Natta Catalyst: Catalyst Fragmentation Promoted by Polymer Crystalline Lamellae


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Polymerization and Quenching Reaction
2.3. Characterization
3. Results and Discussion
3.1. Polymerization Kinetics
3.2. Morphology of the Polymer/Catalyst Particles
3.3. Polymer Aggregation State in Nascent Polymer Particle
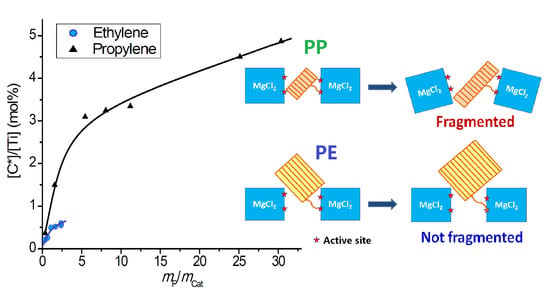
- Changes of active center concentration in the initial stage (0–10 min) clearly show that the lower rate of ethylene polymerization compared with that of propylene can be attributed to a much slower build-up of [C*] in the former system.
- Both polymerization systems experienced a similar degree of diffusion limitation in the first 0–3 min, as shown by the larger apparent propagation rate constant in ethylene polymerization and similar slopes of the kp versus mP/mCat curves. The porosity of PE/catalyst particles was larger than that of the PP/catalyst particles. The lower activity of ethylene polymerization cannot be attributed to its stronger diffusion limitation.
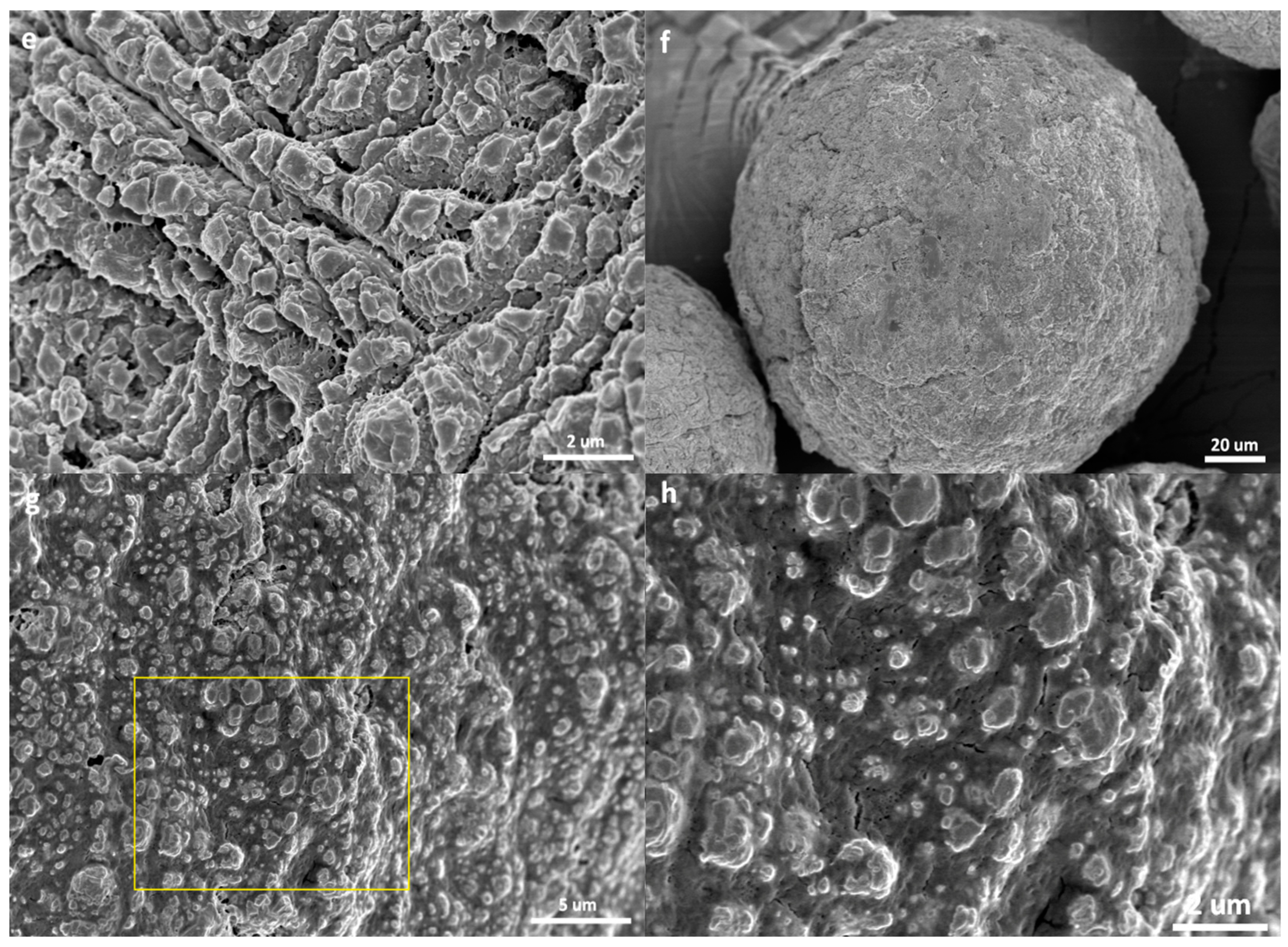
- The catalyst particles have rather compact solid structure, though they are composed of sub-particles with a size of about 200–500 nm, and there are cracks with widths ranging from 100 nm to 5 µm. Pore size distribution, determined by the nitrogen adsorption method, shows that the nanometer pores in the catalyst are concentrated in the range of 15–25 nm. There is a huge number of such small pores, which renders the catalyst a very large specific surface area (282 m2/g) and high porosity (0.32 cm3/g). These 20 nm pores should be uniformly scattered in the solid phase of the catalyst. Assuming that the sub-particles are dense cubes with edges of 200 nm and a density of 2.34 g/cm3 (density of MgCl2 crystal), their aggregate will have a specific surface area of 13 m2/g, which is far lower than the measured specific surface area. The measured value of 282 m2/g will correspond to MgCl2 crystallite size (length of cube edges) of about 9 nm. This size is quite close to that of MgCl2 crystallites (7 nm) in supported Z–N catalysts determined by E. Redzic et al. [53]. Therefore, the 200–500 nm sub-particles cannot be dense solid, but rather aggregates of smaller MgCl2 crystallites containing many nanopores. It is likely that there is a large number of pores and cracks of about 20 nm in the sub-particles.
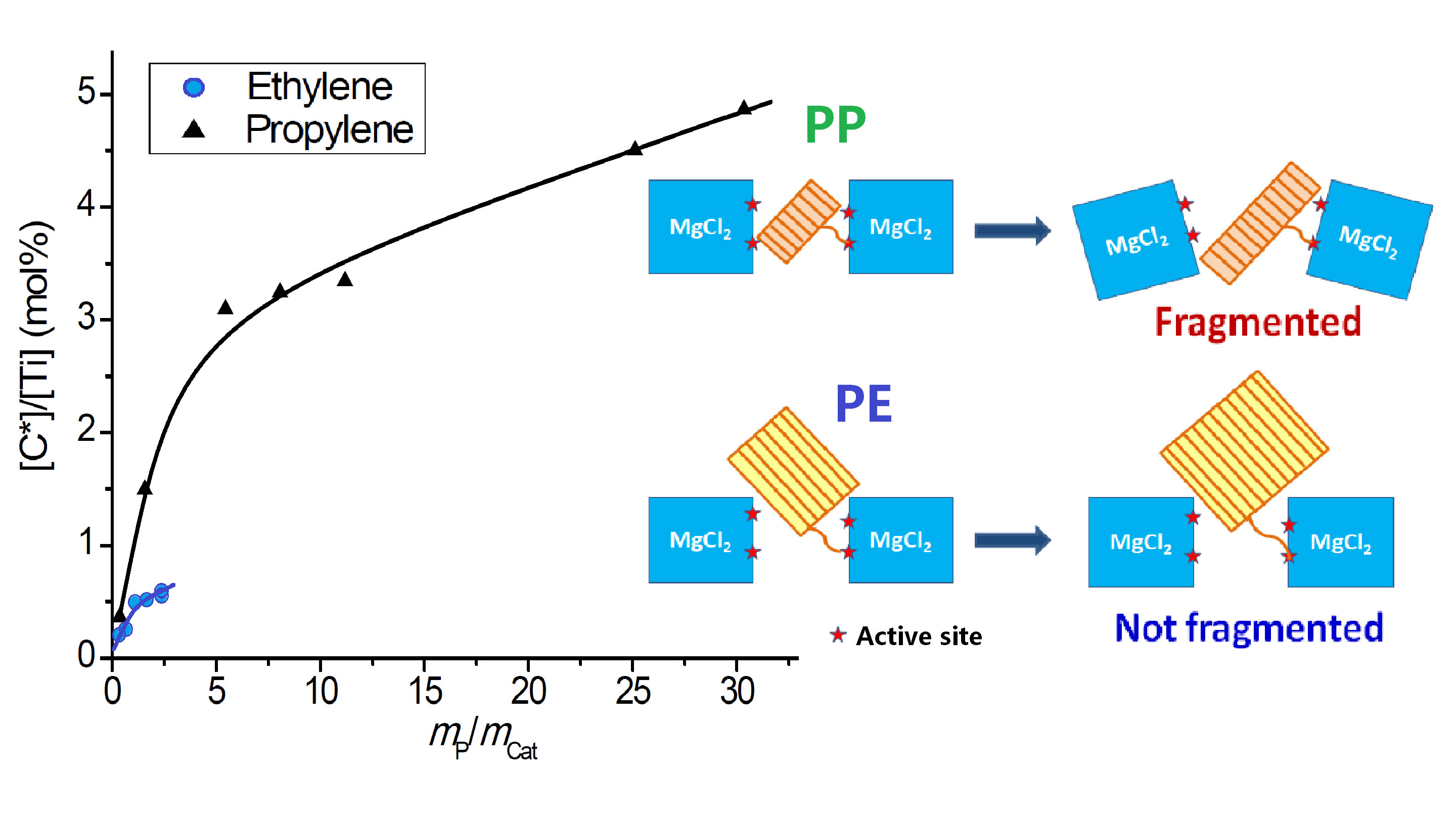
- Because the sizes of PE lamellae formed by the growing polymer chains are far larger than the size of nanopores in the catalyst’s sub-particles, these lamellae cannot grow inside the nanopores, leaving the porous sub-particles basically intact during ethylene polymerization. Only the active sites exposed on the outer surface of the sub-particles can be activated and work as catalytic centers, but a large proportion of active site precursors is buried in the sub-particles and thus becomes unavailable to the polymerization reaction, resulting in a low [C*]/[Ti] ratio of ethylene polymerization. With the proceeding of polymerization, the PE layer covering the sub-particle will form a diffusion barrier that grows quickly with the increase of the mP/mCat ratio, and finally leads to ceasing of the polymerization.
- In the propylene polymerization system, the PP lamellae with a size of 6–11 nm can enter the 20 nm pores in the sub-particles. Growth of these PP lamellae in the pores can exert hydraulic forces strong enough to break up the sub-particles and release their buried active site precursors. Subsequently, the 20 nm pores in the sub-particles will disappear, and the exposed surfaces carrying active sites will be covered by PP chains. After a short period of time, the whole polymer/catalyst particle will become rather compact, and the texture of the particle becomes rather smooth. Though the PP layer covering the MgCl2 crystallites (carrier of the active sites) can also cause serious diffusion barrier, for the much higher density of active sites in this system compared with the ethylene polymerization, the dynamically renewed carrier surface can allow for the presence of tiny pores in the PP layer. This will enable slow but stable diffusion of monomer stream in the PP layer, and a stable polymerization rate supported by a high [C*]/[Ti] ratio and low apparent rate constant.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Keii, T.; Terano, M.; Kimura, K.; Ishii, K. A kinetic argument for a quasi-living polymerization of propene with a MgCl2-supported catalyst. Macromol. Chem. Rapid Commun. 1987, 8, 583–587. [Google Scholar] [CrossRef]
- Marques, M.M.V.; Nunes, C.P.; Tait, P.J.T.; Dias, A.R. Polymerization of ethylene using a high-activity Ziegler-Natta catalyst. 1. Kinetic studies. J. Polym. Sci. Part A 1993, 31, 209–218. [Google Scholar] [CrossRef]
- Fan, Z.; Feng, L.; Yang, S. Distribution of active centers on TiCl4/MgCl2 catalyst for olefin polymerization. J. Polym. Sci. Part A 1996, 34, 3329–3335. [Google Scholar] [CrossRef]
- Han-Adebekun, G.C.; Hamba, M.; Ray, W.H. Kinetic study of gas phase olefin polymerization with a TiCl4/MgCl2 catalyst I. Effect of polymerization conditions. J. Polym. Sci. Part A 1997, 35, 2063–2074. [Google Scholar] [CrossRef]
- Wu, L.; Lynch, D.T.; Wanke, S.E. Kinetics of gas-phase ethylene polymerization with morphology-controlled MgCl2-supported TiCl4 catalyst. Macromolecules 1999, 32, 7990–7998. [Google Scholar] [CrossRef]
- Kissin, Y.V.; Mink, R.I.; Nowlin, T.E. Ethylene polymerization reactions with Ziegler–Natta catalysts. I. Ethylene polymerization kinetics and kinetic mechanism. J. Polym. Sci. Part A 1999, 37, 4255–4272. [Google Scholar] [CrossRef]
- Pimplapure, M.S.; Zheng, X.; Loos, J.; Weickert, G. Low-Rate Propylene Slurry Polymerization: Morphology and Kinetics. Macromol. Rapid. Commun. 2005, 26, 1155–1158. [Google Scholar] [CrossRef]
- Mirzaei, A.; Vakili, M.; Tafi, N. Prepolymerization of ethylene with a Ziegler–Natta catalyst. J. Appl. Polym. Sci. 2007, 105, 2703–2711. [Google Scholar] [CrossRef]
- Chen, K.; Liu, B.; Soares, J.B.P. Effect of prepolymerization on the kinetics of ethylene polymerization and ethylene/1-hexene copolymerization with a Ziegler-Natta Catalyst in slurry reactors. Macromol. React. Eng. 2016, 10, 463–478. [Google Scholar] [CrossRef]
- McKenna, T.F.L.; Tioni, E.; Ranieri, M.M.; Alizadeh, A.; Boisson, C.; Monteil, V. Catalytic olefin polymerisation at short times: Studies using specially adapted reactors. Can. J. Chem. Eng. 2013, 91, 669–686. [Google Scholar] [CrossRef]
- Fisch, A.G.; Santos, J.H.Z.D.; Secchi, A.R.; Cardozo, N.S.M. Heterogeneous catalysts for olefin polymerization: Mathematical model for catalyst particle fragmentation. Ind. Eng. Chem. Res. 2015, 54, 11997–12010. [Google Scholar] [CrossRef]
- Skoumal, M.; Cejpek, I.; Cheng, C.P. Diffusion interface technique for determination of initial polymerization kinetics. Macromol. Rapid Commun. 2005, 26, 357–360. [Google Scholar] [CrossRef]
- Heuvelsland, A.; Wichmann, S.; Schellenberg, J. Investigations of the initial state polymerization of propylene with Ziegler–Natta catalysts in slurry. J. Appl. Polym. Sci. 2007, 106, 354–359. [Google Scholar] [CrossRef]
- Machado, F.; Lima, E.L.; Pinto, J.C.; McKenna, T.F. Evaluation of the initial stages of gas-phase ethylene polymerizations with a SiO2-supported Ziegler–Natta catalyst. Macromol. React. Eng. 2009, 3, 47–57. [Google Scholar] [CrossRef]
- Dwivedi, S.; Taniike, T.; Terano, M. Understanding the chemical and physical transformations of a Ziegler–Natta catalyst at the initial stage of polymerization kinetics: The key role of alkylaluminum in the catalyst activation process. Macromol. Chem. Phys. 2014, 215, 1698–1706. [Google Scholar] [CrossRef]
- Yu, Y.; Busico, V.; Budzelaar, P.H.M.; Vittoria, A.; Cipullo, R. Of poisons and antidotes in polypropylene catalysis. Angew. Chem. Intl. Ed. 2016, 55, 8590–8594. [Google Scholar] [CrossRef] [PubMed]
- Taniike, T.; Sano, S.; Ikeya, M.; Thang, V.Q.; Terano, M. Development of a large-scale stopped-flow system for heterogeneous olefin polymerization kinetics. Macromol. React. Eng. 2012, 6, 275–279. [Google Scholar] [CrossRef]
- Mori, H.; Yoshitome, M.; Terano, M. Investigation of a fine-grain MgC12-supported Ziegler catalyst by stopped-flow propene polymerization: model for the formation of active sites induced by catalyst fragmentation during polymerization. Macromol. Chem. Phys. 1997, 198, 3207–3214. [Google Scholar] [CrossRef]
- Kissin, Y.V. Multicenter nature of titanium-based Ziegler–Natta catalysts: Comparison of ethylene and propylene polymerization reactions. J. Polym. Sci. Part A 2003, 41, 1745–1758. [Google Scholar] [CrossRef]
- Kissin, Y.V. Active centers in Ziegler–Natta catalysts: Formation kinetics and structure. J. Catal. 2012, 292, 188–200. [Google Scholar] [CrossRef]
- Khan, A.; Guo, Y.; Fu, Z.; Fan, Z. Kinetics of short-duration ethylene polymerization with MgCl2-supported Ziegler–Natta catalyst: Two-stage initiation evidenced by changes in active center concentration. J. Appl. Polym. Sci. 2017, 134, 45187:1–45187:6. [Google Scholar] [CrossRef]
- Khan, A.; Guo, Y.; Zhang, Z.; Ali, A.; Fu, Z.; Fan, Z. Kinetics of short-duration ethylene–propylene copolymerization with MgCl2-supported Ziegler–Natta catalyst: Differentiation of active centers on the external and internal surfaces of the catalyst particles. J. Appl. Polym. Sci. 2018, 135, 46030:1–460301:8. [Google Scholar] [CrossRef]
- Jiang, B.; Weng, Y.; Zhang, S.; Zhang, Z.; Fu, Z.; Fan, Z. Kinetics and mechanism of ethylene polymerization with TiCl4/MgCl2 model catalysts: Effects of titanium content. J. Catal. 2018, 360, 57–65. [Google Scholar] [CrossRef]
- Nitta, T.; Liu, B.; Nakatani, H.; Terano, M. Formation, deactivation and transformation of stereospecific active sites on TiCl4/dibutylphthalate/Mg(OEt)2 catalyst induced by short time reaction with Al-alkyl cocatalyst. J. Mol. Catal. A 2002, 180, 25–34. [Google Scholar] [CrossRef]
- Zakharov, V.A.; Bukatov, G.D.; Barabanov, A.A. Recent data on the number of active centers and propagation rate constants in olefin polymerization with supported ZN catalysts. Macromol. Symp. 2004, 213, 19–28. [Google Scholar] [CrossRef]
- Qi, M.; Zhang, B.; Fu, Z.; Xu, J.; Fan, Z. Millimeter-size polyethylene hollow spheres synthesized with MgCl2-supported Ziegler-Natta catalyst. J. Appl. Polym. Sci. 2016. [Google Scholar] [CrossRef]
- Fu, Z.; Xu, J.; Zhang, Y.; Fan, Z. Chain Structure and mechanical properties of polyethylene/polypropylene/poly(ethylene-co-propylene) in-reactor alloys synthesized with spherical Ziegler-Natta catalyst by gas-phase polymerization. J. Appl. Polym. Sci. 2005, 97, 640–647. [Google Scholar] [CrossRef]
- Fu, Z.; Fan, Z.; Zhang, Y.; Xu, J. Chain structure of polyethylene/polypropylene in-reactor alloy synthesized in gas phase with spherical Ziegler–Natta catalyst. Polym. Int. 2004, 53, 1169–1175. [Google Scholar] [CrossRef]
- Shen, X.; Hu, J.; Fu, Z.; Lou, J.; Fan, Z. Counting the number of active centers in MgCl2-supported Ziegler–Natta catalysts by quenching with 2-thiophenecarbonyl chloride and study on the initial kinetics of propylene polymerization. Catal. Commun. 2013, 30, 66–69. [Google Scholar] [CrossRef]
- Shen, X.; Fu, Z.; Hu, J.; Wang, Q.; Fan, Z. Mechanism of propylene polymerization with MgCl2-supported Ziegler–Natta catalysts based on counting of active centers: The role of external electron donor. J. Phys. Chem. C 2013, 117, 15174–15182. [Google Scholar] [CrossRef]
- Xu, T.; Yang, H.; Fu, Z.; Fan, Z. Effects of comonomer on active center distribution of TCl4/MgCl2–AlEt3 catalyst in ethylene/1-hexene copolymerization. J. Organomet. Chem. 2015, 798, 328–334. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Z.; Guo, W.; Khan, A.; Fu, Z.; Xu, J.; Fan, Z. Kinetics and mechanism of metallocene catalyzed olefin polymerization: Comparison of ethylene, propylene homopolymerizations and their copolymerization. J. Polym. Sci. Part A 2017, 55, 867–875. [Google Scholar] [CrossRef]
- Yang, P.; Fu, Z.; Fan, Z. 1-Hexene polymerization with supported Ziegler-Natta catalyst: Correlation between catalyst particle disintegration and active center distribution. Mol. Catal. 2018, 447C, 13–20. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, P.; Zhang, S.; Jiang, B.; Khan, A.; Zhu, L.; Fu, Z.; Fan, Z. Study on 2-thiophenecarbonyl chloride-quenched olefin polymerization with α-diimine nickel catalysts. Iran. Polym. J. 2018, 27, 153–159. [Google Scholar] [CrossRef]
- Floyd, S.; Choi, K.Y.; Taylor, T.W.; Ray, W.H. Polymerization of olefins through heterogeneous catalysis. III. Polymer particle modelling with an analysis of intraparticle heat and mass transfer effects. J. Appl. Polym. Sci. 1986, 32, 2935–2960. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.; Mann, G.E.; Ray, W.H. Heat and Mass Transfer Limitations and Catalyst Deactivation Effects in Olefin Polymerization for Gas Phase and Slurry Reactors. In Studies in Surface Science and Catalysis; Keii, T., Soga, K., Eds.; Elsevier: Amsterdam, The Netherlands, 1986; Volume 25, pp. 339–367. [Google Scholar]
- Soga, K.; Ohgizawa, M.; Shiono, T.; Lee, D.H. Possibility of mass-transfer resistance in ethylene polymerization with magnesium chloride-supported catalysts. Macromolecules 1991, 24, 1699–1700. [Google Scholar] [CrossRef]
- McKenna, T.F.; Soares, J.B.P. Single particle modelling for olefin polymerization on supported catalysts: A review and proposals for future developments. Chem. Eng. Sci. 2001, 56, 3931–3949. [Google Scholar] [CrossRef]
- Di Martino, A.; Weickert, G.; McKenna, T.F.L. Contributions to the experimental investigation of the nascent polymerisation of ethylene on supported catalysts. 1. Macromol. React. Eng. 2007, 1, 165–184. [Google Scholar] [CrossRef]
- Taniike, T.; Thang, V.Q.; Binh, N.T.; Hiraoka, Y.; Uozumi, T.; Terano, M. Initial particle morphology development in Ziegler-Natta propylene polymerization tracked with stopped-flow technique. Macromol. Chem. Phys. 2011, 212, 723–729. [Google Scholar] [CrossRef]
- Thang, V.Q.; Taniike, T.; Umemori, M.; Ikeya, M.; Hiraoka, Y.; Nghia, N.D.; Terano, M. New quenching procedure for preservation of initial polymer/catalyst particle morphology in Ziegler-Natta olefin polymerization. Macromol. React. Eng. 2009, 3, 467–472. [Google Scholar] [CrossRef]
- Galli, P.; Haylock, J.C. Advances in Ziegler-Natta polymerization – Unique polyolefin copolymers, alloys and blends made directly in the reactor. Makromol. Chem. Macromol. Symp. 1992, 63, 19–54. [Google Scholar] [CrossRef]
- Cecchin, G.; Marchetti, E.; Baruzzi, G. On the mechanism of polypropene growth over MgCl2/TiCl4 catalyst systems. Macromol. Chem. Phys. 2001, 202, 1987–1994. [Google Scholar] [CrossRef]
- Fan, Z.; Zhang, Y.; Xu, J.; Wang, H.; Feng, L. Structure and properties of polypropylene/poly(ethylene-co-propylene) in-situ blends synthesized by spherical Ziegler-Natta catalyst. Polymer 2001, 42, 5559–5566. [Google Scholar] [CrossRef]
- Mehtarani, R.; Fu, Z.; Tu, S.; Fan, Z.; Tian, Z.; Feng, L. Synthesis of polypropylene/poly(ethylene-co-propylene) in-reactor alloys by periodic switching polymerization process — Dynamic change of gas phase monomer composition and its influences on polymer structure and properties. Ind. Eng. Chem. Res. 2013, 52, 9775–9782. [Google Scholar] [CrossRef]
- Mehtarani, R.; Fu, Z.; Fan, Z.; Tu, S.; Feng, L. Synthesis of polypropylene/poly(ethylene-co-propylene) in-reactor alloys by periodic switching polymerization process — Effects of gas-phase polymerization time on polymer properties. Ind. Eng. Chem. Res. 2013, 52, 13556–13563. [Google Scholar] [CrossRef]
- Flory, P.J.; Vrij, A. Melting points of linear-chain homologs. The normal paraffin hydrocarbons. J. Am. Chem. Soc. 1963, 85, 3548–3553. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited: Theory and experiment. Polymer 1997, 38, 3151–3212. [Google Scholar] [CrossRef]
- Mirabella, F.M.; Bafna, A. Determination of the crystallinity of polyethylene/α-olefin copolymers by thermal analysis: Relationship of the heat of fusion of 100% polyethylene crystal and the density. J. Polym. Sci. B 2002, 40, 1637–1643. [Google Scholar] [CrossRef]
- Fatou, J.G. Melting temperature and enthalpy of isotactic polypropylene. Eur. Polym. J. 1971, 7, 1057–1064. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Janimak, J.J.; Zhang, A.; Cheng, H.N. Regime transitions in fractions of isotactic polypropylene. Macromolecules 1990, 23, 298–303. [Google Scholar] [CrossRef]
- Grebowicz, J.; Lau, S.F.; Wunderlich, B. The thermal properties of polypropylene. J. Polym. Sci. Polym. Symp. 1984, 71, 19–37. [Google Scholar] [CrossRef]
- Lu, L.; Alamo, R.G.; Mandelkern, L. Lamellar thickness distributions in linear polyethylene and ethylene copolymers. Macromolecules 1994, 27, 6571–6576. [Google Scholar] [CrossRef]
- Rashedi, R.; Sharif, F. Variation of comonomer content in LLDPE particles with different sizes from an industrial fluidized bed reactor. Ind. Eng. Chem. Res. 2015, 54, 9870–9876. [Google Scholar] [CrossRef]
- Xue, B.; Hui, L.; Yang, H.; Zhao, Y.; Hou, L.; Li, W. Immobilization of Ziegler−Natta catalyst for ethylene polymerization on macropores SiO2 with an open-framework structure. Ind. Eng. Chem. Res. 2017, 56, 135–142. [Google Scholar] [CrossRef]
- Zheng, X.; Pimplapure, M.S.; Weickert, G.; Loos, J. Influence of copolymerization on fragmentation behavior using Ziegler-Natta catalysts. Macromol. Rapid Commun. 2006, 27, 15–20. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | tpb (s) | mP/mCatc (g/g) | Activity (kg/g Ti·h) | Mwd (105) | Đ d | Rp (10−3 mol/L·s) | [C*]/[Ti] (%) | kp (L/mol·s) |
|---|---|---|---|---|---|---|---|---|
| E1 | 30 | 0.17 | 0.76 | 3.34 | 10.6 | - e | - e | - e |
| E2 | 60 | 0.35 | 0.78 | 3.71 | 8.1 | 0.32 | 0.20 | 2035 |
| E3 | 120 | 0.65 | 0.72 | 4.26 | 16.7 | 0.48 | 0.25 | 2410 |
| E4 | 180 | 1.12 | 0.83 | 5.79 | 12.6 | 0.54 | 0.49 | 1380 |
| E5 | 240 | 1.67 | 0.93 | 5.51 | 9.5 | 0.45 | 0.51 | 1103 |
| E6 | 480 | 2.38 | 0.66 | 5.67 | 14.0 | 0.04 | 0.55 | 85 |
| E7 | 600 | 2.39 | 0.53 | 6.21 | 10.8 | 0.01 | 0.59 | 17 |
| P1 | 30 | 0.36 | 1.61 | 1.51 | 5.3 | 2.27 | 0.37 | 1433 |
| P2 | 60 | 1.56 | 3.48 | 1.57 | 5.7 | 2.27 | 1.50 | 356 |
| P3 | 120 | 5.44 | 6.05 | 1.59 | 6.2 | 2.27 | 3.10 | 172 |
| P4 | 180 | 8.05 | 5.97 | 1.49 | 5.3 | 2.27 | 3.25 | 164 |
| P5 | 240 | 11.17 | 6.20 | 1.24 | 5.6 | 2.27 | 3.35 | 160 |
| P6 | 480 | 25.12 | 6.98 | 1.25 | 5.3 | 2.27 | 4.51 | 118 |
| P7 | 600 | 30.34 | 6.74 | 1.27 | 5.5 | 2.27 | 4.87 | 110 |
| Sample | Specific Surface Area (m2/g) | Total Pore Volume (cm3/g) | Average Pore Size (nm) |
|---|---|---|---|
| Cat. | 281.55 | 0.320 | 22.37 |
| E3 | 34.84 | 0.065 | 37.95 |
| E4 | 51.85 | 0.090 | 33.36 |
| P1 | 2.79 | 0.013 | 95.80 |
| P2 | 1.55 | 0.009 | 117.44 |
| Run | Polymer | Tma (°C) | ∆Hf b (J/g) | Xcc (%) |
|---|---|---|---|---|
| E1 | PE | 139.5 | 228.8 | 79.4 |
| E2 | 140.1 | 208.5 | 72.4 | |
| E3 | 139.9 | 201.1 | 69.8 | |
| E4 | 141.8 | 200.7 | 69.7 | |
| P1 | PP | 161.0 | 86.7 | 56.3 |
| P2 | 161.6 | 83.4 | 54.2 | |
| P3 | 161.0 | 79.8 | 51.8 | |
| P4 | 161.1 | 69.2 | 44.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Jiang, B.; He, F.; Fu, Z.; Xu, J.; Fan, Z. Comparative Study on Kinetics of Ethylene and Propylene Polymerizations with Supported Ziegler–Natta Catalyst: Catalyst Fragmentation Promoted by Polymer Crystalline Lamellae. Polymers 2019, 11, 358. https://doi.org/10.3390/polym11020358
Zhang Z, Jiang B, He F, Fu Z, Xu J, Fan Z. Comparative Study on Kinetics of Ethylene and Propylene Polymerizations with Supported Ziegler–Natta Catalyst: Catalyst Fragmentation Promoted by Polymer Crystalline Lamellae. Polymers. 2019; 11(2):358. https://doi.org/10.3390/polym11020358
Chicago/Turabian StyleZhang, Zhen, Baiyu Jiang, Feng He, Zhisheng Fu, Junting Xu, and Zhiqiang Fan. 2019. "Comparative Study on Kinetics of Ethylene and Propylene Polymerizations with Supported Ziegler–Natta Catalyst: Catalyst Fragmentation Promoted by Polymer Crystalline Lamellae" Polymers 11, no. 2: 358. https://doi.org/10.3390/polym11020358