Improving Thermal Stability of Polyurethane through the Addition of Hyperbranched Polysiloxane

 ,
, 
Abstract
1. Introduction
2. Experiment
2.1. Materials
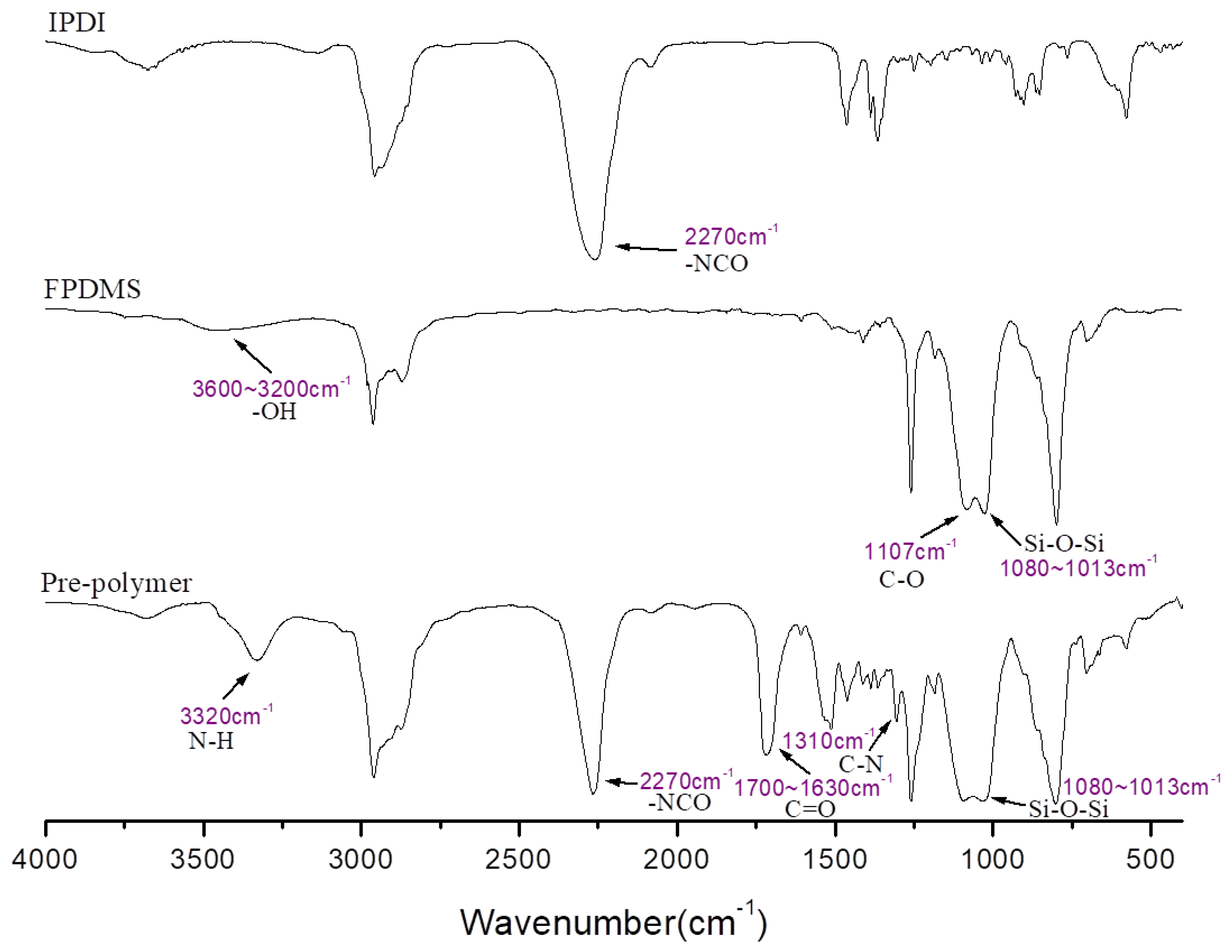
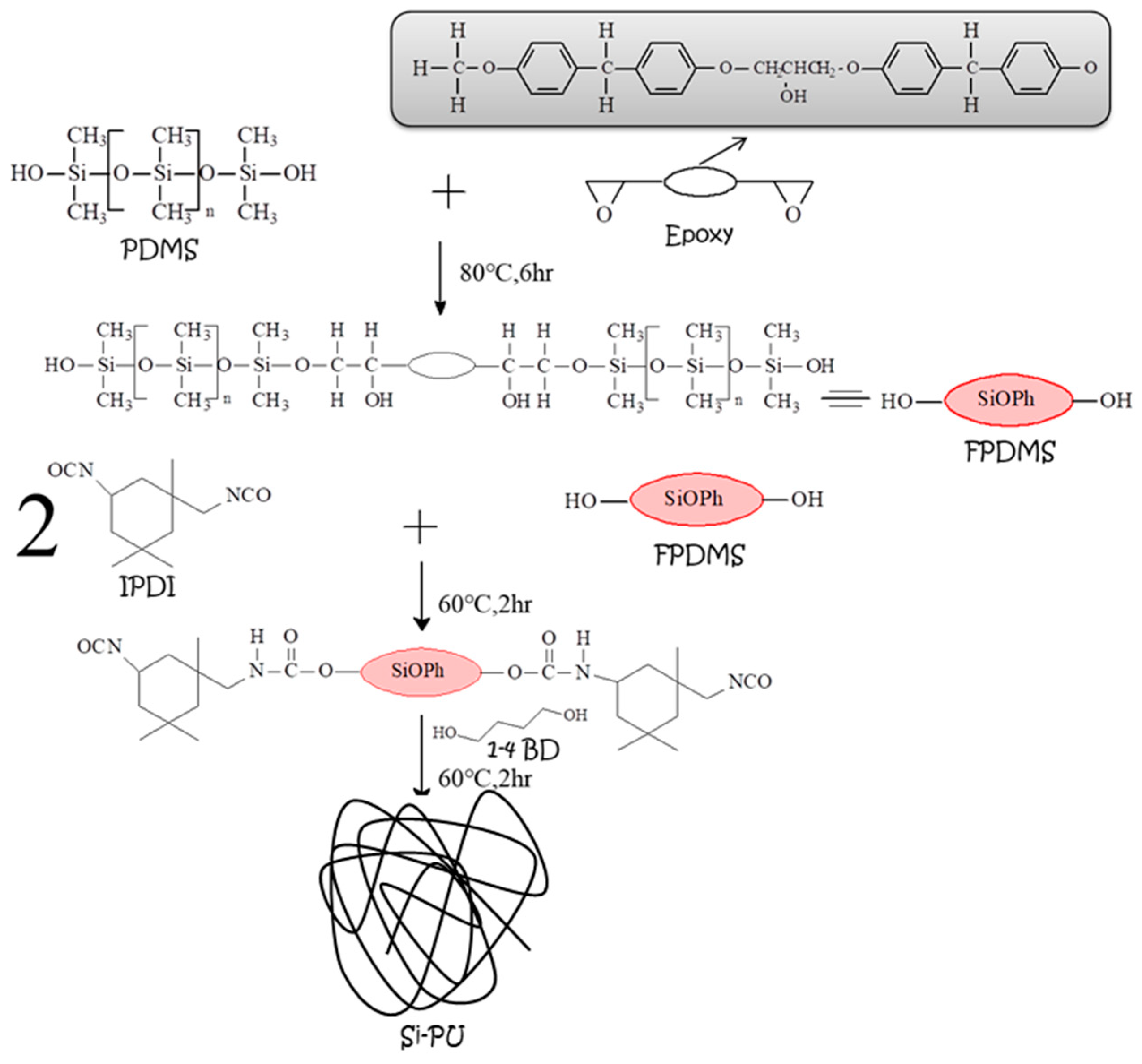
2.2. Preparation of Si-PU
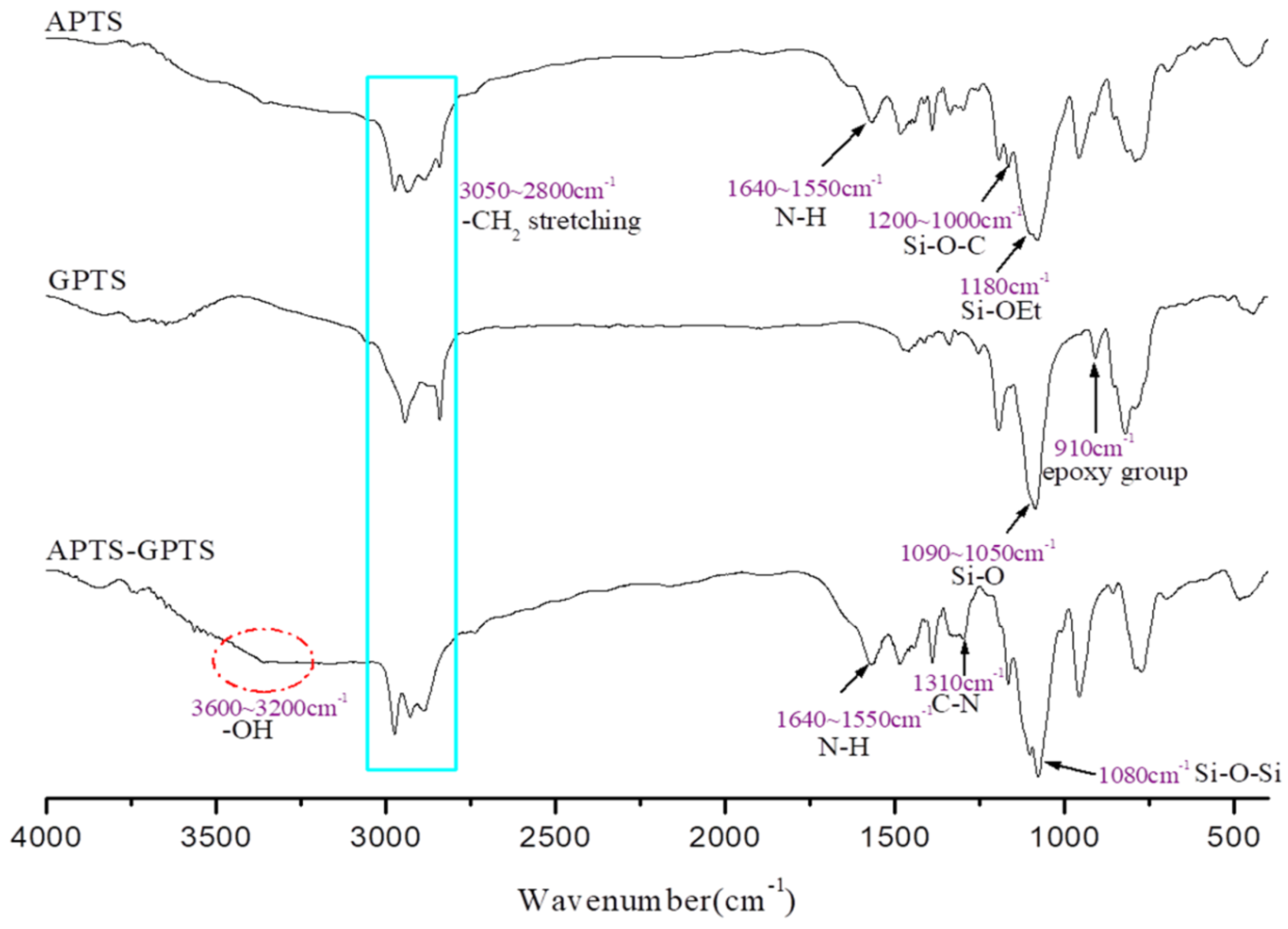
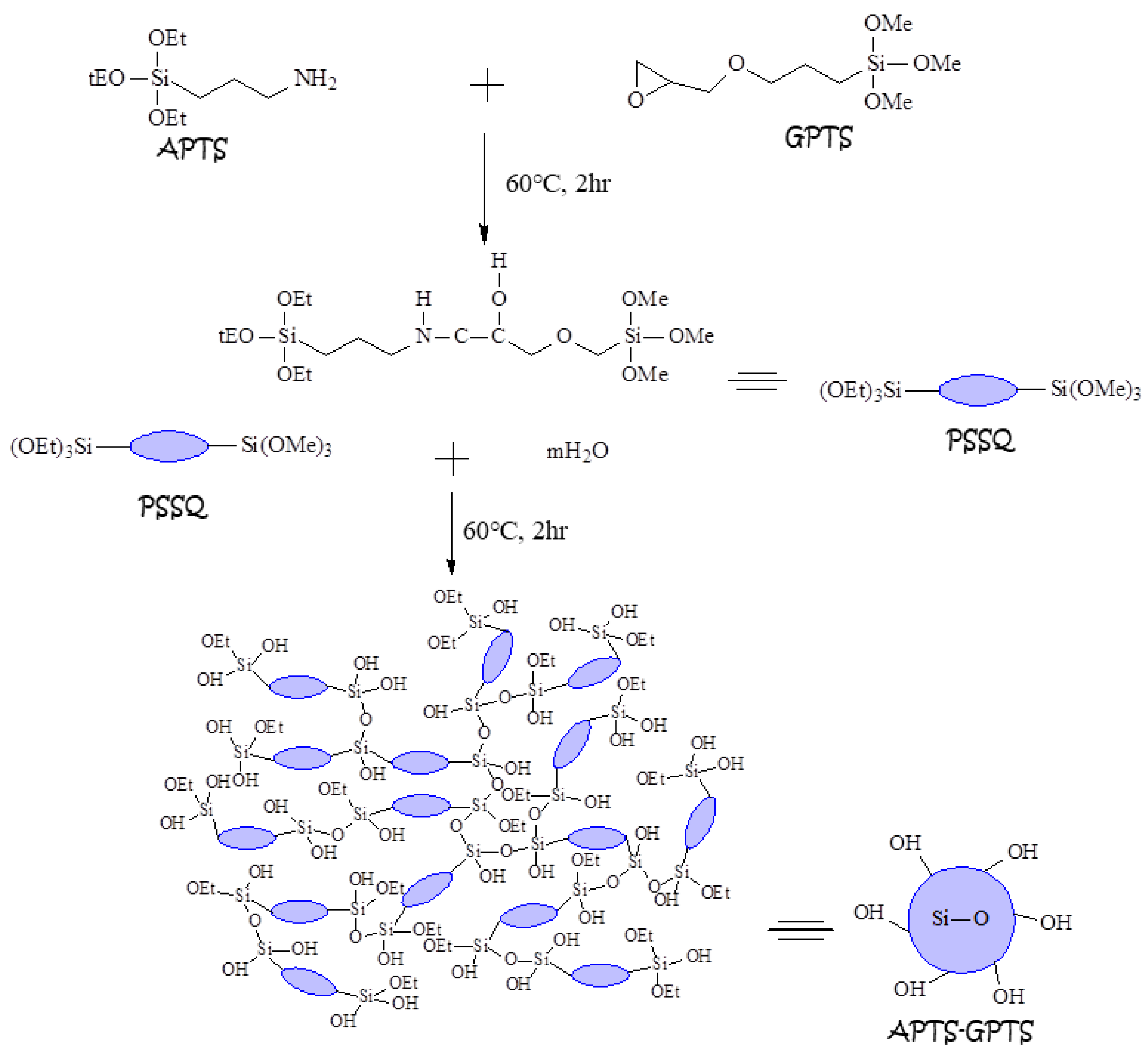
2.3. Preparation of APTS-GPTS
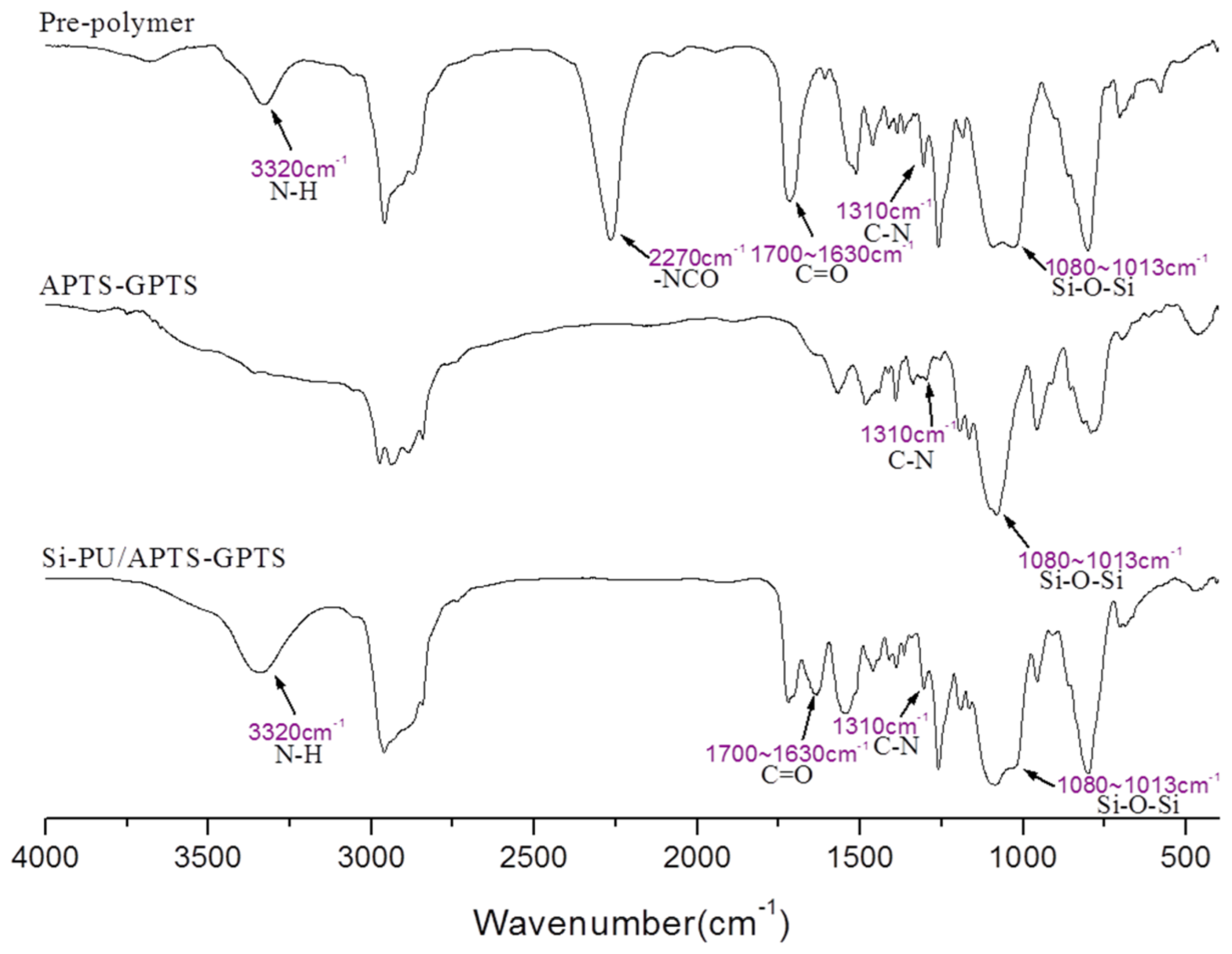
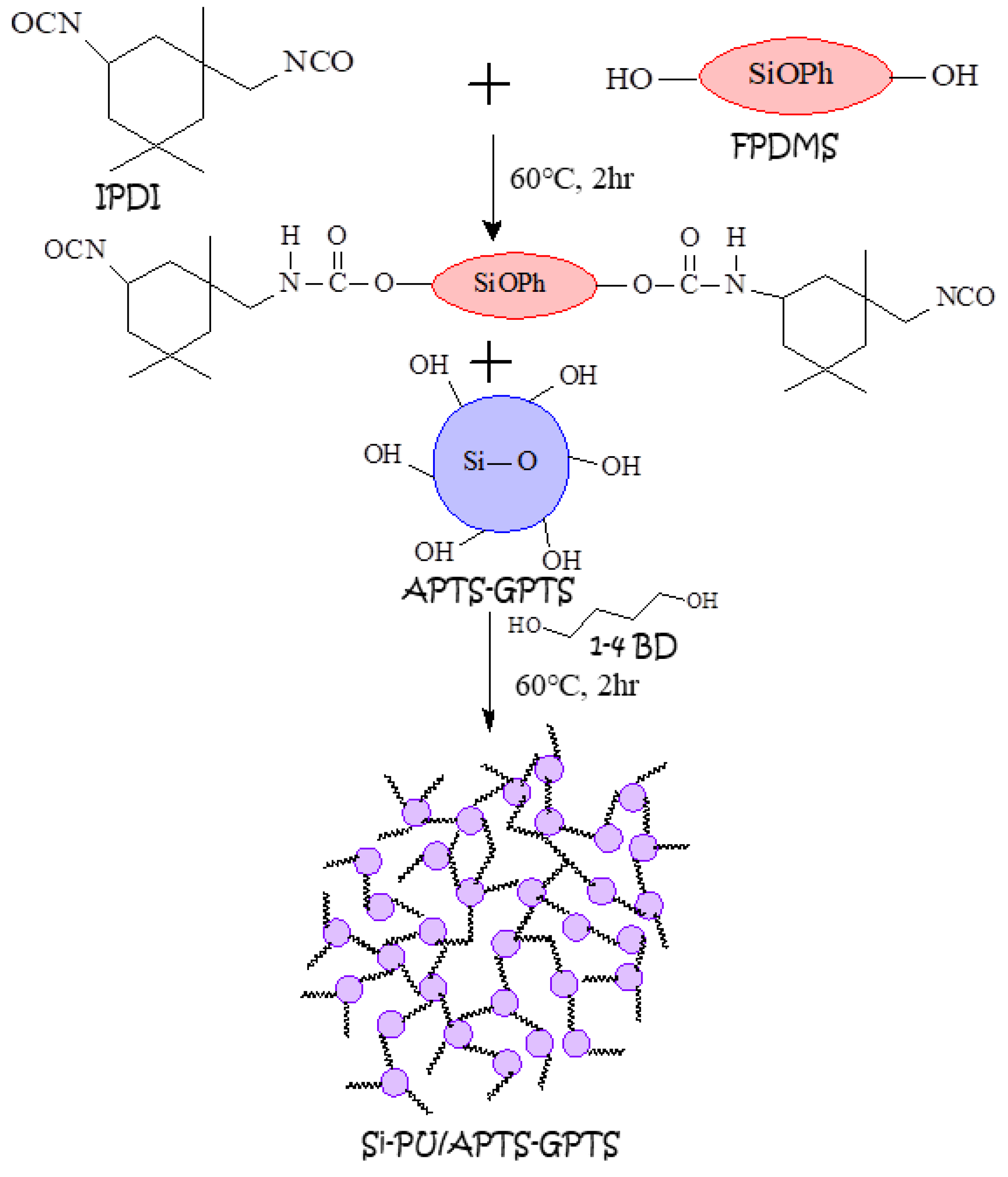
2.4. Preparation of Si-PU/APTS-GPTS
2.5. Measurements
3. Results and Discussion
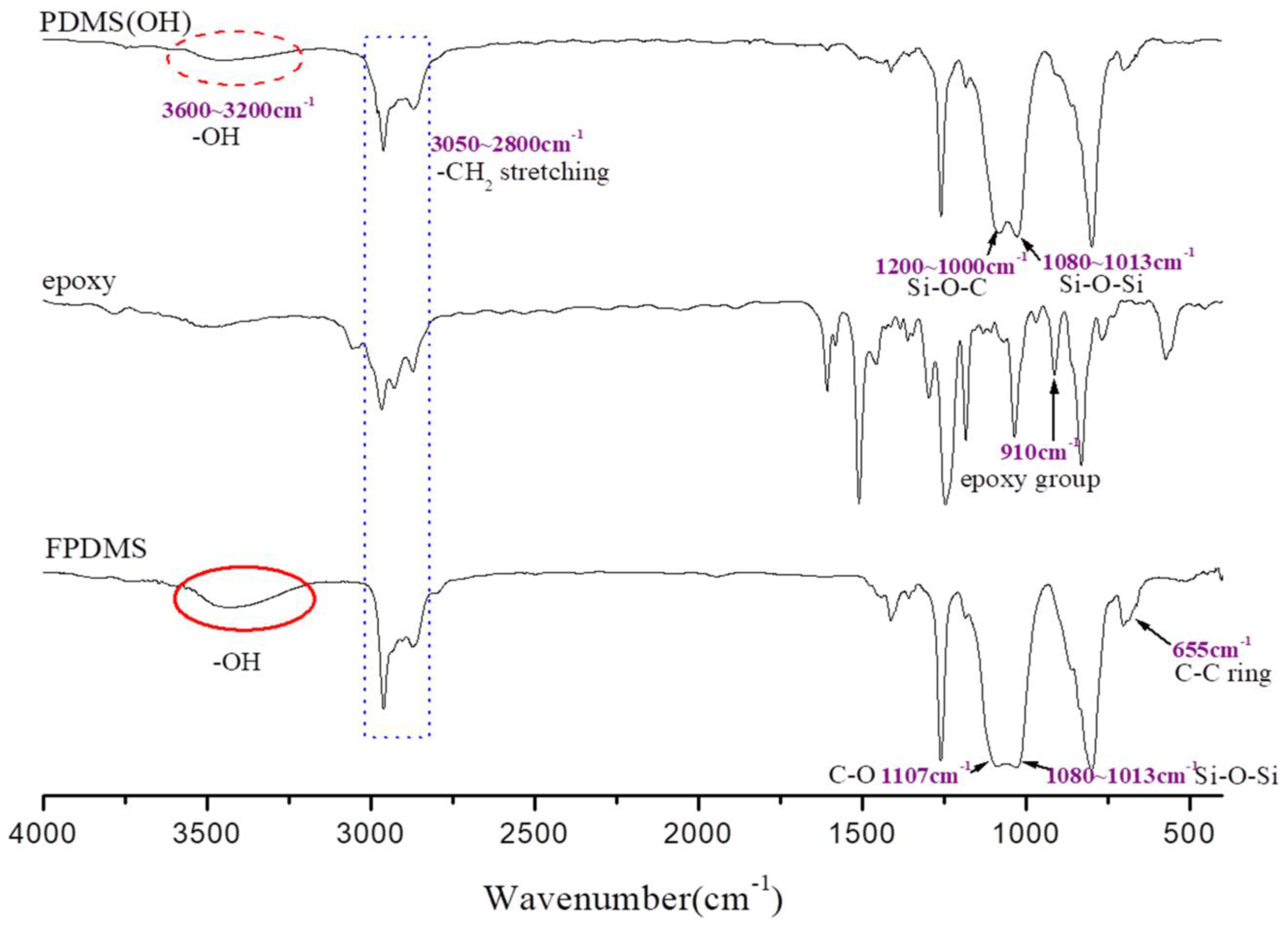
3.1. Characterization of Si-PU/APTS-GPTS Hybrid
3.2. Network Structure of Si-PU/APTS-GPTS Hybrid
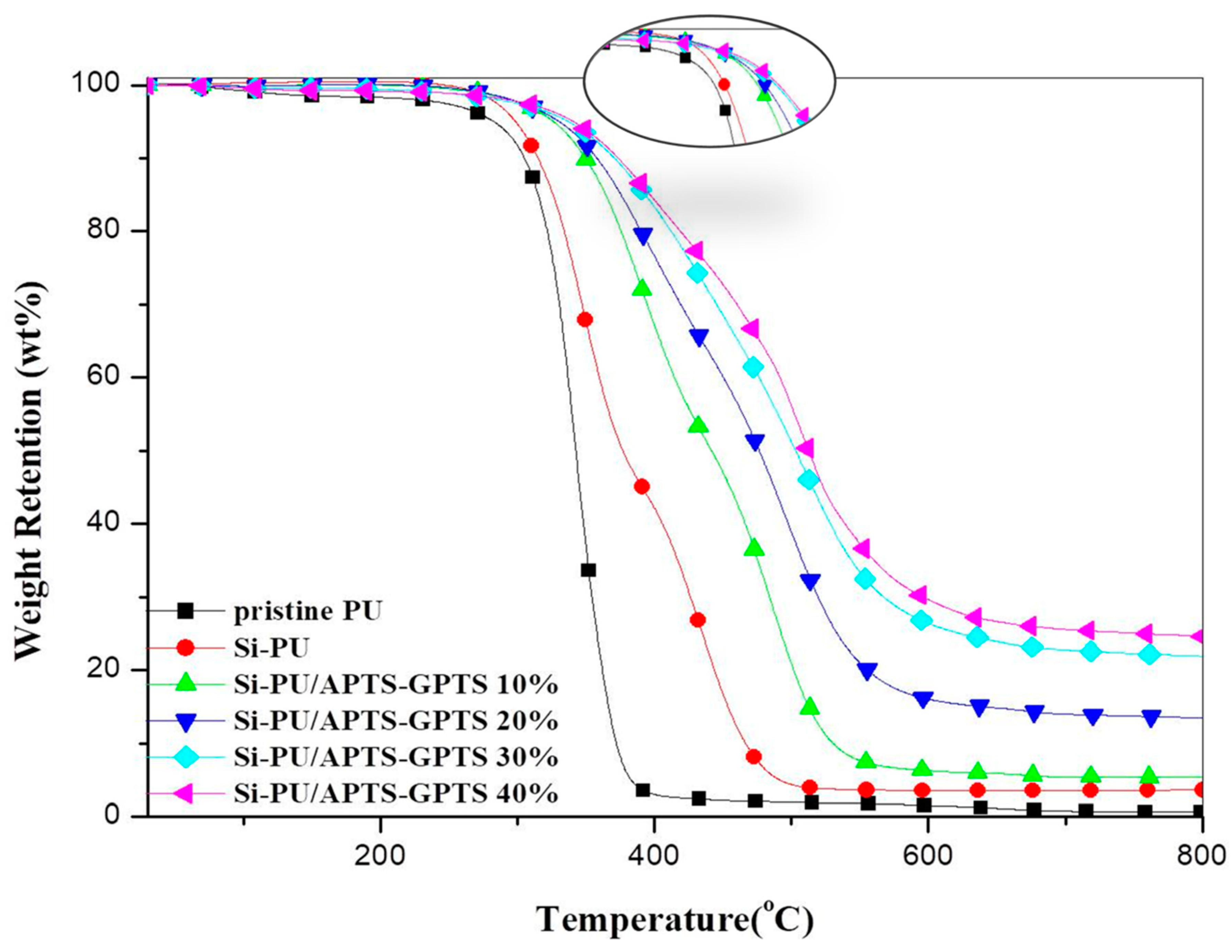
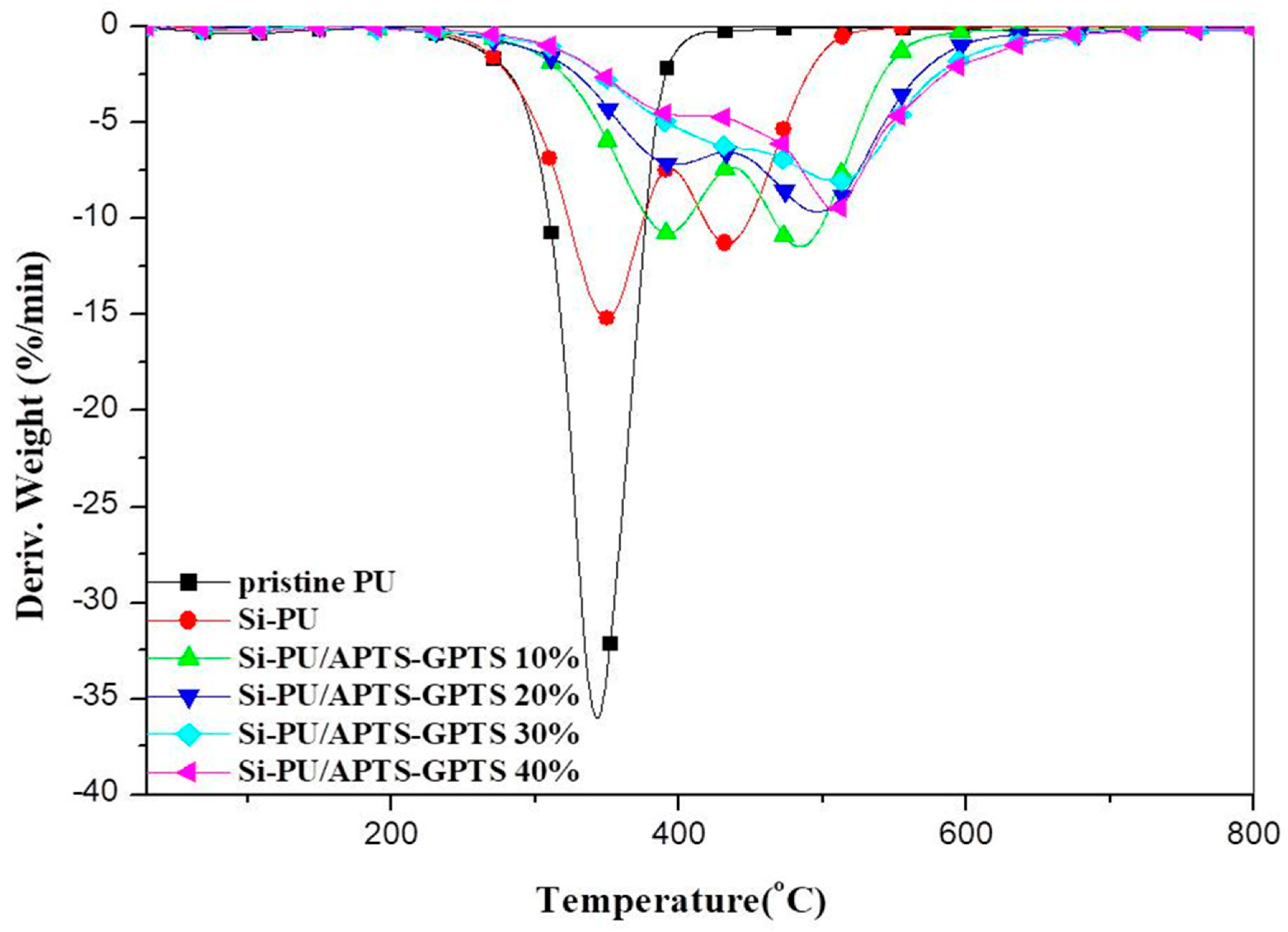
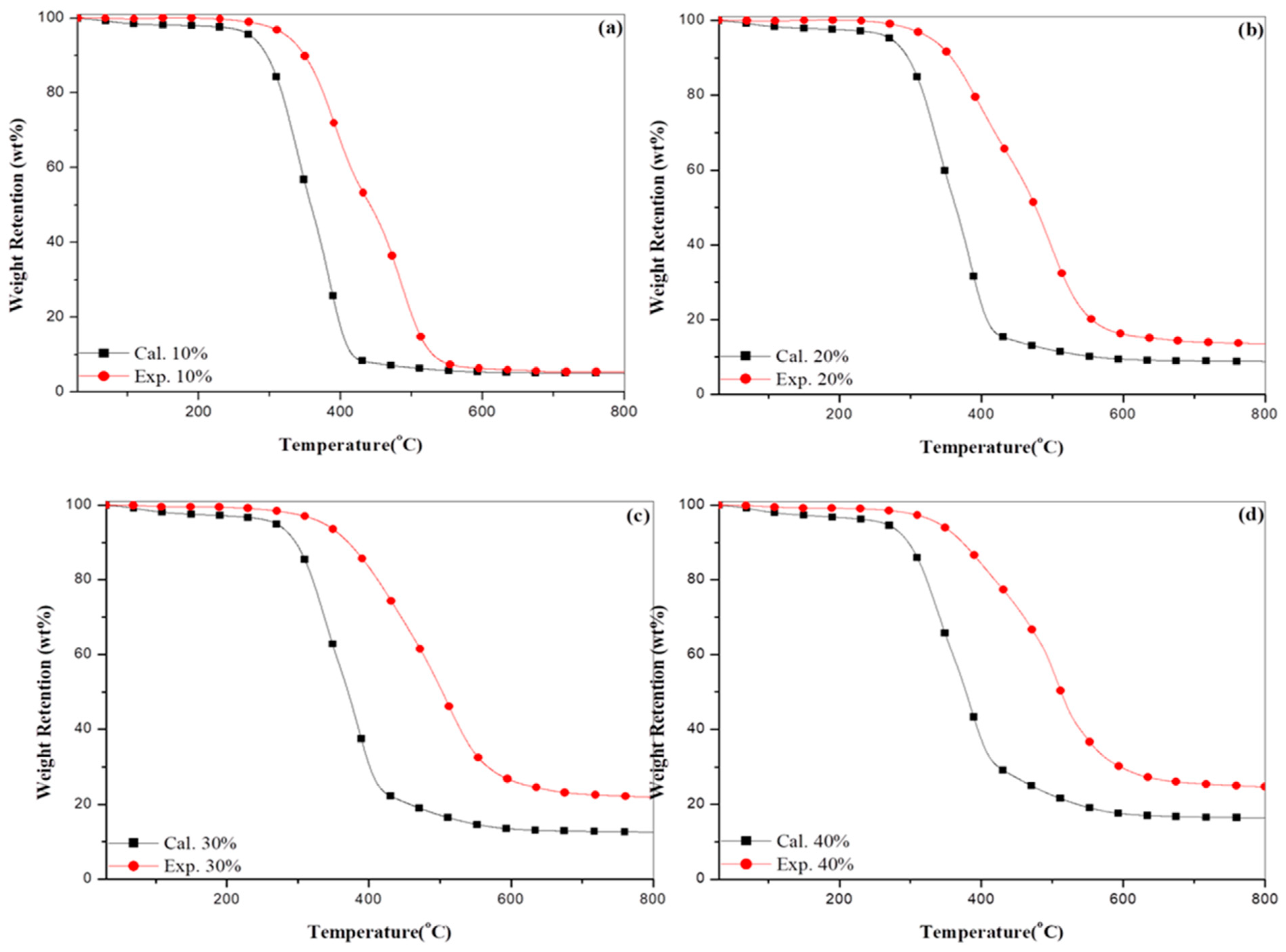
3.3. Thermogravimetric Analysis
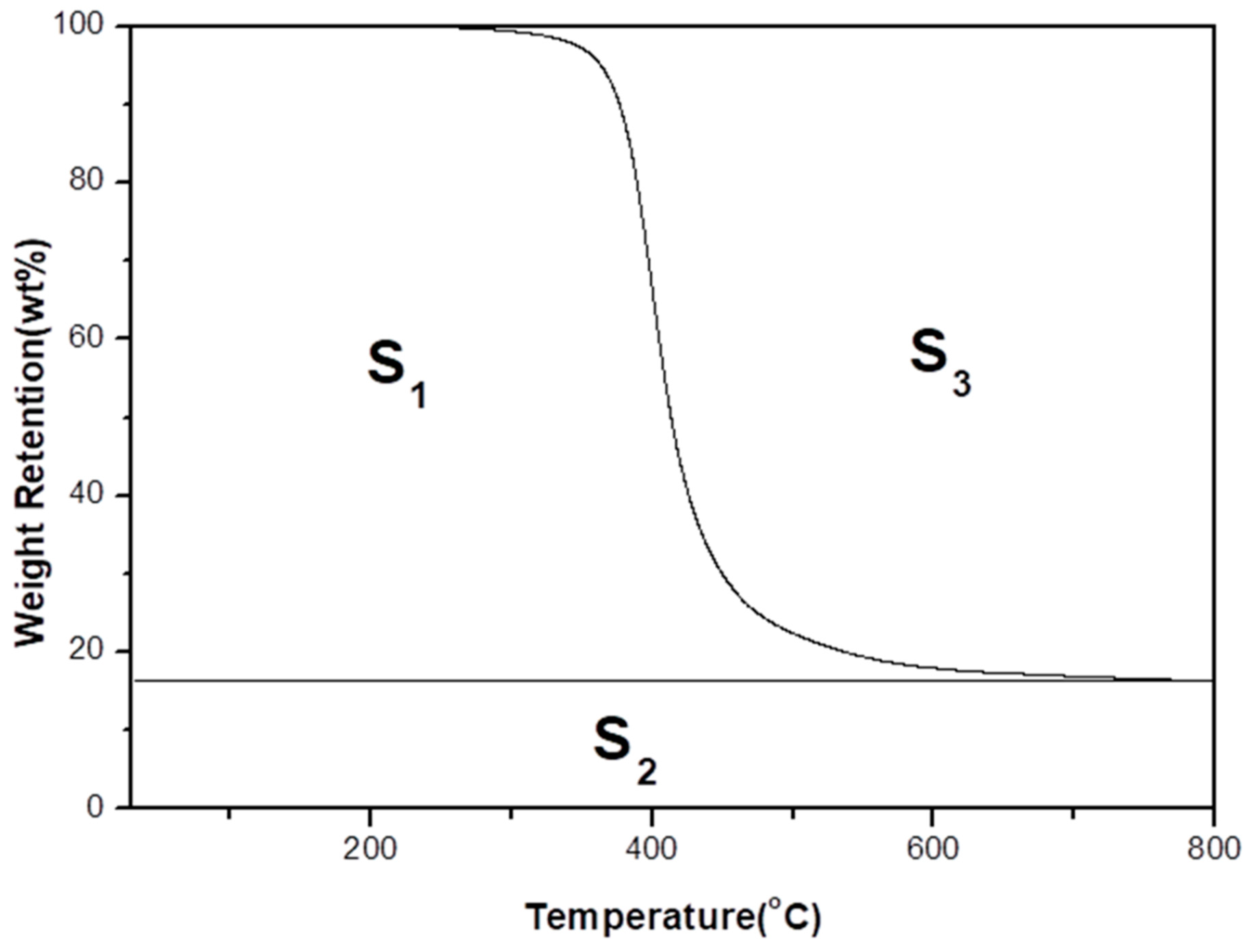
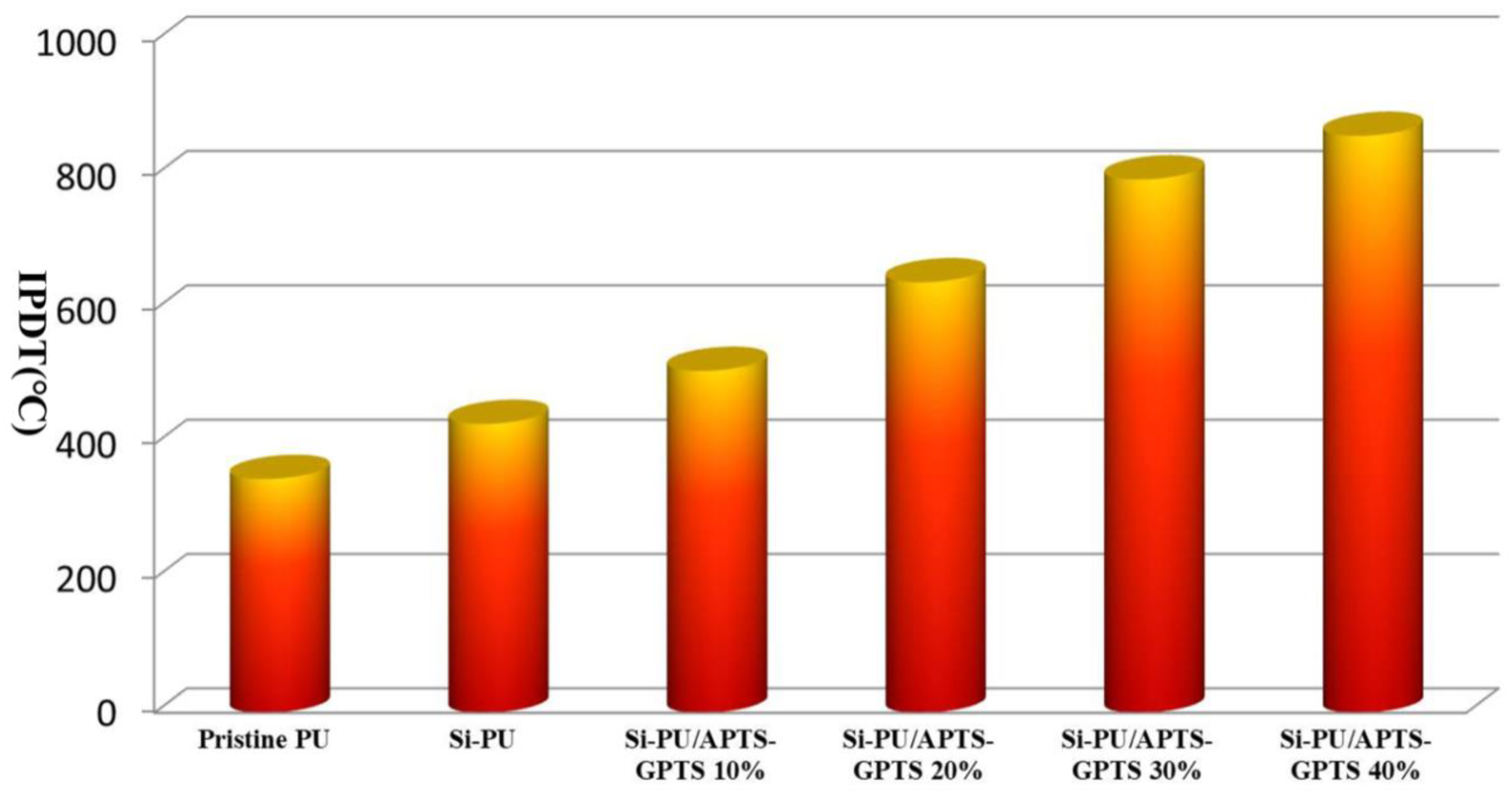
3.4. Integral Procedural Decomposition Temperature (IPDT)
3.5. Experimental and Calculated Data
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, R.; Yadav, R.; Kolhe, M.A.; Bhosale, R.S.; Narayan, R. 8-Hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS) based high fluorescent, pH stimuli waterborne polyurethane coatings. Polymer 2018, 136, 157–165. [Google Scholar] [CrossRef]
- Bedő, D.; Imre, B.; Domján, A.; Schöne, P.; Vancsoe, G.J.; Pukánszky, B. Coupling of poly(lactic acid) with a polyurethane elastomer by reactive processing. Eur. Polym. J. 2017, 97, 409–417. [Google Scholar] [CrossRef]
- Bing, H.; Lin, Y. Highly heat-resistant silicon-containing polyurethane-imide copolymers: Synthesis and thermal mechanical stability. Eur. Polym. J. 2017, 91, 337–353. [Google Scholar]
- Pagacz, J.; Hebdab, E.; Janowskic, B.; Sternikd, D.; Janciab, M.; Pielichowskib, K. Thermal decomposition studies on polyurethane elastomers reinforced with polyhedral silsesquioxanes by evolved gas analysis. Polym. Degrad. Stabil. 2018, 149, 129–142. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, J.; Zhu, T.; Hua, Z.; Chen, Q.; Yu, X. Blends of thermoplastic polyurethane and polyether–polyimide:preparation and properties. Polymer 2001, 42, 1493–1500. [Google Scholar] [CrossRef]
- Voronkov, A.V.; Kopylov, V.M.; Parsons, I.W. Furan-containing modified organosilicon polymers: Preparation, properties and some applications. Eur. Polym. J. 1997, 7, 979–990. [Google Scholar] [CrossRef]
- Macan, J. Thermal degradation of epoxy-silica organic-inorganic hybrid materials. Polym. Degrad. Stabil. 2006, 91, 122–127. [Google Scholar] [CrossRef]
- Heidsieck, S.U.H.; Dörrich, S.; Weidner, R.; Rieger, B. Branched siloxanes as possible new heat transfer fluids for application in parabolic through solar thermal power plants. Sol. Energy Mater. Sol. Cells 2017, 161, 278–284. [Google Scholar] [CrossRef]
- Zhao, Z.; Jin, Q.; Zhang, N.; Guo, X.; Yan, H. Preparation of a novel polysiloxane and its synergistic effect with ammonium polyphosphate on the flame retardancy of polypropylene. Polym. Degrad. Stabil. 2018, 150, 73–85. [Google Scholar] [CrossRef]
- Chiang, C.L.; Chang, R.C. Synthesis, characterization, and thermal properties of bridged polysilsesquioxanes-molecular nanocomposites. In Proceedings of the 13th European Conference on Composite Materials (ECCM 2008), Stockholm, Sweden, 2–5 June 2008. [Google Scholar]
- Kusumoto, T.; Mori, Y.; Kanasaki, M.; Ueno, T.; Kameda, Y.; Oda, K.; Kodaira, S.; Kitamura, H.; Barillon, R.; Yamauchi, T. Yields on the formation of OH groups and the loss of CH groups along nuclear tracks in PADC films. Radiat. Meas. 2015, 83, 59–62. [Google Scholar] [CrossRef]
- Sritham, E.; Gunasekaran, S. FTIR spectroscopic evaluation of sucrose-maltodextrin-sodium citrate bioglass. Food Hydrocoll. 2017, 70, 371–382. [Google Scholar] [CrossRef]
- Kim, J.; Jung, D.; Park, Y.; Kim, Y.; Moon, D.W.; Lee, T.G. Quantitative analysis of surface amine groups on plasma-polymerized ethylenediamine films using UV–visible spectroscopy compared to chemical derivatization with FT-IR spectroscopy, XPS and TOF-SIMS. Appl. Surf. Sci. 2007, 253, 4112–4118. [Google Scholar] [CrossRef]
- Sideridou, I.D.; Vouvoudi, E.C.; Papadopoulos, G.D. Epoxy polymer Hxtal NYL-1TM used in restoration and conservation: Irradiation with short and long wavelengths and study of photo-oxidation by FT–IR spectroscopy. J. Cult. Herit. 2016, 18, 279–289. [Google Scholar] [CrossRef]
- Bagherzadeh, M.R.; Daneshvar, A.; Shariatpanahi, H. Novel water-based nanosiloxane epoxy coating for corrosion protection of carbon steel. Surf. Coat. Technol. 2012, 206, 2057–2063. [Google Scholar] [CrossRef]
- Colombani, J.; Chauvet, E.; Amat, S.; Dupuy, N.; Gigmes, D. A FTIR/chemometrics approach to characterize the gamma radiation effects on iodine/epoxy-paint interactions in Nuclear Power Plants. Anal. Chim. Acta 2017, 960, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Gallego, R.; Arteaga, J.F.; Valencia, C.; Franco, J.M. Thickening properties of several NCO-functionalized cellulose derivatives in castor oil. Chem. Eng. Sci. 2015, 134, 260–268. [Google Scholar] [CrossRef]
- Mishra, A.K.; Narayan, R.; Raju, K.V.S.N.; Aminabhavi, T.M. Hyperbranched polyurethane (HBPU)-urea and HBPU-imide coatings: Effect of chain extender and NCO/OH ratio on their properties. Prog. Org. Coat. 2012, 74, 134–141. [Google Scholar] [CrossRef]
- Sheikh, Z.; Khan, A.S.; Roohpour, N.; Glogauer, M.; Rehman, I.u. Protein adsorption capability on polyurethane and modified-polyurethane membrane for periodontal guided tissue regeneration applications. Mater. Sci. Eng. C 2016, 68, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, H.; Tian, X.; Zheng, K.; Cheng, Q. Effect of 3-Aminopropyltriethoxysilane on polycarbonate based waterborne polyurethane transparent coatings. Prog. Org. Coat. 2014, 77, 1073–1078. [Google Scholar] [CrossRef]
- Mahdavi, M.; Ahmad, M.B.; Haron, M.J.; Gharayebi, Y.; Shameli, K.; Nadi, B. Fabrication and characterization of SiO2/(3-aminopropyl) triethoxysilane coated magnetite nanoparticles for lead (II) removal from aqueous solution. J. Inorg. Organomet. Polym. 2013, 23, 599–607. [Google Scholar] [CrossRef]
- Pontón, P.I.; d’Almeida, J.R.M.; Marinkovic, B.A.; Savić, S.M.; Mancic, L.; Rey, N.A.; Morgado, E., Jr.; Rizzo, F.C. The effects of the chemical composition of titanate nanotubes and solvent type on 3-aminopropyltriethoxysilane grafting efficiency. Appl. Surf. Sci. 2014, 301, 315–322. [Google Scholar] [CrossRef]
- Ramírez, C.; Rico, M.; Torres, A.; Barral, L.; López, J.; Montero, B. Epoxy/POSS organic–inorganic hybrids: ATR-FTIR and DSC studies. Eur. Polym. J. 2008, 44, 3035–3045. [Google Scholar] [CrossRef]
- Jain, S.; Goossens, H.; Picchioni, F.; Magusin, P.; Mezari, B.; van Duin, M. Synthetic aspects and characterization of polypropylene–silica nanocomposites prepared via solid-state modification and sol–gel reactions. Polymer 2005, 46, 6666–6681. [Google Scholar] [CrossRef]
- Han, Y.H.; Taylor, A.; Mantle, M.D.; Knowles, K.M. Sol–gel-derived organic–inorganic hybrid materials. J. Non-Cryst. Solids 2007, 353, 313–320. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, G.; Huang, X.; Bu, J.; Sun, X.; Jiang, P. Molecular structures of (3-aminopropyl)trialkoxysilane on hydroxylated barium titanate nanoparticle surfaces induced by different solvents and their effect on electrical properties of barium titanate based polymer nanocomposites. Appl. Surf. Sci. 2016, 364, 798–807. [Google Scholar]
- Jin, Q.F.; Liao, G.X.; Jian, X.G. Synthesis and characterization of trimethoxysilyl-functionalized poly(phthalazinone ether ketone). Chin. Chem. Lett. 2007, 8, 1137–1140. [Google Scholar] [CrossRef]
- Paquet, O.; Brochier Salon, M.C.; Zeno, E.; Belgacem, M.N. Hydrolysis-condensation kinetics of 3-(2-amino-ethylamino)propyl-trimethoxysilane. Mater. Sci. Eng. C 2012, 32, 487–493. [Google Scholar] [CrossRef]
- Shea, K.J.; Loy, D.A.; Webster, O. Arylsilsesquioxane Gels and Related Materials. New Hybrids of Organic and Inorganic Networks. J. Am. Chem. Soc. 1992, 114, 6700–6710. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G. The novel silicon-containing epoxy/PEPA phosphate flame retardantfor transparent intumescent fire resistant coating. Appl. Surf. Sci. 2016, 385, 453–463. [Google Scholar] [CrossRef]
- Wu, C.S.; Liu, Y.L.; Chiu, Y.S. Epoxy resins possessing flame retardant elements from silicon incorporated epoxy compounds cured with phosphorus or nitrogen containing curing agents. Polymer 2002, 43, 4277–4284. [Google Scholar] [CrossRef]
- Qian, Y.; Wei, P.; Jiang, P.; Zhao, X.; Yu, H. Synthesis of a novel hybrid synergisticflame retardantand its application in PP/IFR. Polym. Degrad. Stabil. P 2011, 96, 1134–1140. [Google Scholar] [CrossRef]
- Gao, F.; Tong, L.; Fang, Z. Effect of a novel phosphorous–nitrogen containing intumescent flame retardant on the fire retardancy and the thermal behaviour of poly(butylene terephthalate). Polym. Degrad. Stabil. 2006, 91, 1295–1299. [Google Scholar] [CrossRef]
- Jakic, M.; Vrandecic, N.S.; Klari, I. Thermal degradation of poly(vinyl chloride)/poly(ethylene oxide) blends: Thermogravimetric analysis. Polym. Degrad. Stabil. 2013, 98, 1738–1743. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample NO. | Area (%) | Dc (%) | ||
|---|---|---|---|---|
| T1 | T2 | T3 | ||
| Si-PU/APTS-GPTS | 20.1 | 34.1 | 46.0 | 75.4 |
| Sample NO. | a Td5 (°C) | b Tmax (°C) | c Rmax (wt%/min) | IPDT (°C) | C.Y (wt %) | ||
|---|---|---|---|---|---|---|---|
| 1st | 2nd | 1st | 2nd | ||||
| Pristine PU | 273 | 343 | - | −36.0 | - | 348 | 0.7 |
| Si-PU | 294 | 349 | 434 | −15.2 | −11.3 | 430 | 3.7 |
| Si-PU/APTS-GPTS 10% | 318 | 392 | 484 | −10.7 | −11.5 | 509 | 5.3 |
| Si-PU/APTS-GPTS 20% | 321 | 397 | 496 | −7.2 | −9.6 | 641 | 13.5 |
| Si-PU/APTS-GPTS 30% | 326 | 510 | - | −8.0 | - | 794 | 21.9 |
| Si-PU/APTS-GPTS 40% | 330 | 404 | 508 | −4.6 | −9.6 | 859 | 24.7 |
| Sample NO. | Cal. | Exp. | ||
|---|---|---|---|---|
| Td5 | C.Y. (wt%) at 800 °C | Td5 | C.Y. (wt%) at 800 °C | |
| Si-PU/APTS-GPTS 10% | 268 | 5.0 | 318 | 5.3 |
| Si-PU/APTS-GPTS 20% | 263 | 8.8 | 321 | 13.5 |
| Si-PU/APTS-GPTS 30% | 256 | 12.6 | 326 | 21.9 |
| Si-PU/APTS-GPTS 40% | 242 | 16.4 | 330 | 24.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.-H.; Shen, M.-Y.; Kuan, C.-F.; Kuan, H.-C.; Ke, C.-Y.; Chiang, C.-L. Improving Thermal Stability of Polyurethane through the Addition of Hyperbranched Polysiloxane. Polymers 2019, 11, 697. https://doi.org/10.3390/polym11040697
Liu S-H, Shen M-Y, Kuan C-F, Kuan H-C, Ke C-Y, Chiang C-L. Improving Thermal Stability of Polyurethane through the Addition of Hyperbranched Polysiloxane. Polymers. 2019; 11(4):697. https://doi.org/10.3390/polym11040697
Chicago/Turabian StyleLiu, Shang-Hao, Ming-Yuan Shen, Chen-Feng Kuan, Hsu-Chiang Kuan, Cing-Yu Ke, and Chin-Lung Chiang. 2019. "Improving Thermal Stability of Polyurethane through the Addition of Hyperbranched Polysiloxane" Polymers 11, no. 4: 697. https://doi.org/10.3390/polym11040697
APA StyleLiu, S.-H., Shen, M.-Y., Kuan, C.-F., Kuan, H.-C., Ke, C.-Y., & Chiang, C.-L. (2019). Improving Thermal Stability of Polyurethane through the Addition of Hyperbranched Polysiloxane. Polymers, 11(4), 697. https://doi.org/10.3390/polym11040697