Insight into the Structure and Dynamics of Polymers by Neutron Scattering Combined with Atomistic Molecular Dynamics Simulations


1
Centro de Física de Materiales (CSIC, UPV/EHU) and Materials Physics Center MPC, Paseo Manuel de Lardizabal 5, E-20018 San Sebastián, Spain
2
Departamento de Polímeros y Materiales Avanzados, Física, Química y Tecnología (UPV/EHU), Apartado 1072, E-20080 San Sebastián, Spain
3
Donostia International Physics Center (DIPC), Paseo Manuel de Lardizabal 4, E-20018 San Sebastián, Spain
*
Author to whom correspondence should be addressed.
Polymers 2020, 12(12), 3067; https://doi.org/10.3390/polym12123067
Submission received: 20 November 2020
/
Revised: 4 December 2020
/
Accepted: 11 December 2020
/
Published: 21 December 2020
(This article belongs to the Special Issue State-of-the-Art Polymer Science and Technology in Spain (2020,2021))
Abstract
:Combining neutron scattering and fully atomistic molecular dynamics simulations allows unraveling structural and dynamical features of polymer melts at different length scales, mainly in the intermolecular and monomeric range. Here we present the methodology developed by us and the results of its application during the last years in a variety of polymers. This methodology is based on two pillars: (i) both techniques cover approximately the same length and time scales and (ii) the classical van Hove formalism allows easily calculating the magnitudes measured by neutron scattering from the simulated atomic trajectories. By direct comparison with experimental results, the simulated cell is validated. Thereafter, the information of the simulations can be exploited, calculating magnitudes that are experimentally inaccessible or extending the parameters range beyond the experimental capabilities. We show how detailed microscopic insight on structural features and dynamical processes of various kinds has been gained in polymeric systems with different degrees of complexity, and how intriguing questions as the collective behavior at intermediate length scales have been faced.

1. Introduction
The structural units of polymers are macromolecules. For this reason, their structural and dynamical properties strongly depend on a hierarchy of length and time scales. Considering structural aspects, nowadays it is well established the random coil conformation of linear polymer chains in the melt and the glassy state. This conformation was envisioned by Flory in the 50’s [1]. Reducing the length scale of observation, the local arrangements of the atoms leading to short-range order give rise to broad maxima in the diffraction patterns. Intermolecular correlations are revealed by the ‘amorphous halo’ in the range of scattering vector 1 (see Figure 1). Associated to the structural complexity, the dynamical processes in polymers also depend on the length scale considered. At large length scales polymer melts display unique properties driven by the chain-like character and the atomic decoration of the chain plays a secondary role. At such large scales, the relevant dynamic processes (chain diffusion, reptation and Rouse dynamics) exhibit characteristic times that depend on the macromolecular size and can be very long. At intermolecular length scales the so-called -relaxation is the main dynamical process [2]. The characteristic time of this relaxation—the structural relaxation time—raises enormously when the temperature decreases towards the glass transition temperature . The behavior of polymers at these length scales is dominated by the universal features of glass-forming systems. At even smaller length scales ≈ 1 Å the most relevant motions are usually localized and activated and persist in the glassy state. The structural disorder leads to the emergence of broad barrier distributions for such processes. This kind of dynamics comprises, among others, tunneling processes [3], secondary relaxations like the Johari–Goldstein or -relaxation [4] and vibrations including the Boson peak [5].
Unveiling the structural and dynamical properties of polymeric systems at the different levels and identifying their interplay is a great challenge. In addition, the consideration of different ingredients that increase the complexity of the system (e.g., chemically more complex monomeric units, different macromolecular architectures, polymer blends, nanocomposites, …) can lead to profound modifications of the features identified and characterized in chemically simple linear homopolymer melts. Even novel properties can emerge as consequence of complexity. In the following we present the strategy developed by us in order to tackle these questions [6]. It rests on the synergetic combination of two techniques: neutron scattering (NS) and fully atomistic molecular dynamics (MD) simulations. After setting the methodological aspects, we review the main achievements we have obtained by applying it to the investigation of structural and dynamical features of ‘simple’ linear homopolymers. This is followed by a section where more complex and intricate systems and situations are addressed. Finally, we present our conclusions and considerations about the extension of this kind of strategy to tackle problems in systems displaying other kinds of complexity.
2. Grounds of the Strategy
Neutrons interact with the atomic nuclei [7,8]. The involved interactions are important only at very short distances ( m) and can be approximately described by
with m the mass of the neutron and the scattering length. The scattering length depends on the isotope (: H, D, C, O, …) and also on the relative orientation of the neutron-nuclear spin pairs. The scattering length can be positive, negative or complex. Table 1 shows its mean value, , for the isotopes commonly present in soft materials.
The magnitude measured in NS experiments is the cross section (see Figure 2). The number of neutrons scattered into a solid angle comprised between and and which have changed their energy by an amount , with respect to the total number of incident neutrons [7], is the double differential scattering cross section . The difference between the wavevectors of the scattered () and incident () neutron determines the scattering vector (equivalently, the momentum transfer ). In an elastic experiment (), since , the modulus of this vector is given by . Here is the scattering angle. This expression can be considered as a good approximation for small energy transfers—as usually involved in the so-called quasielastic experiments, where small energy transfers are considered. The interpretation of is straightforward in terms of the so-called van Hove correlation functions, as shown in the scheme of Figure 2. In this scheme, indexes and run over all the possible kinds of isotopes (: H, D, C, O, N, …) in the sample. contains a coherent and an incoherent contribution. The latter has its origin in the random distribution of the deviations of the scattering lengths from their mean value, i.e., it is defined as . The - and -dependencies of coherent and incoherent contributions are determined by the corresponding scattering functions. They are respectively that involves nuclei of kinds and , and that involves nuclei of kind . The scattering functions are related, via Fourier transformation, with the intermediate scattering functions [ and ] and the van Hove correlation functions [ and its self-part ]. We note that the functions and are also denoted as and , respectively. The label ‘inc’ is sometimes substituted by ‘self’ or simply ‘s’ denoting ‘self’. In the classical limit, is expressed as:
Here [] is the position vector of the ith atom of kind [jth atom of kind ] at time = t [time = 0]. All the different atoms of kinds and [ (): total number of atoms of kind (); ] are included in the sum. Thus, is the probability that any particle of kind is in the volume at position at time t, if a particle of kind is at the origin at time t = 0. In the static case , where is the static pair distribution function. Thus, neutrons provide structural information and, through the time-dependent correlation functions, they also reveal dynamical collective properties. On the other hand, the self-part of the van Hove correlation function is obtained by restricting the correlations considered in Equation (2) to those relating the positions of a single particle of kind at different times:
is the Fourier transform of in space: Incoherent scattering relates to single particle motions, i.e., informs about the motions of an individual nucleus (or ‘self’ motions).
The main equation in Figure 2 shows that the weights of the coherent and incoherent contributions to the scattered intensity are determined by the scattering lengths of the isotopes involved. We also define the so-called scattering cross sections of the sample, and , in terms of the scattering cross sections of its individual nuclei : coherent (incoherent) scattering cross section of nucleus i (see Table 1). The definition of the sample cross section is , while the atomic cross sections are expressed as , [7,9]. From Table 1 it is follows that:
- Due to the large value of (and, thus, of ), in H-containing systems and the signal is dominated by the incoherent scattering from hydrogens. It thus reveals their self-motions
- If H is substituted by D, this incoherent contribution is drastically reduced. Then, we obtain differently weighted coherent contributions
- Since in a fully deuterated sample, the intensity scattered is mainly coherent. Given that , all pair correlations are almost equally weighted
Neutrons also provide an elegant—and unique—way to experimentally access another kind of correlation functions. Thanks to the large difference between and , they can address the dynamic structure factor of isotopically labeled molecular groups or macromolecules in experiments sensitive to energy changes. In diffraction experiments, its static counterpart, the corresponding form factor, is accessed. We recall that the dynamic structure factor of a labeled ’object’ containing z scattering points is defined as . Here the sums extend to all the scattering points in the ’object’, and is the vector joining two scattering points. The static and dynamic structure factors are revealed by the cross section measured on a mixture of protonated and deuterated ‘objects’ at low scattering angles (see Figure 1 for the example of a polymer melt). The scattered intensity is then weighted by the scattering contrast factor . This unique opportunity offered by neutrons made it possible to experimentally prove [10,11] the random coil conformation of macromolecules in the bulk. Those pioneering experiments were realized in the 70’s, as soon as the first small angle neutron scattering diffractometers were available, and are one of the greatest historical milestones in polymer physics. Only then was Flory awarded with the Nobel Prize. Later, dynamic measurements have provided microscopic evidence for the Rouse-like [12] dynamics in unentangled chains [13] and the reptation mechanism for entangled chain dynamics [14]. This was predicted by Doi, Edward and deGennes [15,16] in the 80’s. Thus, with NS we can also ‘see’ how ensembles of atoms (like those in a macromolecule) look like and move!
To perform neutron scattering experiments, large facilities including a neutron source and dedicated instrumentation (see, e.g., [17]) are needed. Currently, the effective () region covered by NS techniques is roughly 0.01 < Q < 2 and 0.1 ps < t < 100 ns. Neutrons have been used to investigate at microscopic level all the different dynamical processes taking place in linear homopolymer materials [13] over the past decades.
Neutron scattering experiments provide thus a big deal of information and the magnitudes measured have a clear physical meaning. However, their analysis and interpretation is sometimes very difficult, given the complexity of soft materials. In addition to the intrinsic problem of properly modeling the structural and dynamical complexity, several problems arise from the limitation of NS techniques, namely:
- NS accesses correlation functions in the reciprocal space (), never in real space
- NSE does not distinguish the signals of different atoms, if they are of the same isotopic species (e.g., main-chain hydrogens vs side-group hydrogens)
- Self-motions of C and O are not accessible ( = = 0)
- With exception of the neutron spin echo (NSE) technique (F. Mezei, 1972 [18]), that directly accesses the intermediate scattering functions, experiments are performed in the frequency domain and the results are affected by the instrumental resolution through convolution
- Spectrometers cover relatively narrow dynamic windows and usually several instruments have to be combined
- Though polarization analysis (PA) allows separation of coherent and incoherent contributions, in the practice this is currently available only for diffraction experiments (structural information)
Some of these limitations (4–6) might be overcome with the development of the neutron sources and instrumentation, in particular with the spallation sources of new generation. In fact, recent progresses in instrumentation have allowed PA also in quasielastic neutron scattering experiments, though not yet in a routinely way [19]. However, points (1–3) are inherent to the scattering processes.
Computer simulations can be of utmost help for the interpretation of NS results. In the following we introduce these techniques and the way their combination with NS allows a full exploitation of their capabilities.
It is considered that computer simulations in general—i.e., ab-initio methods, classical molecular dynamics simulations, Monte Carlo methods, etc—are in between theoretical approaches and experimental tools. We could distinguish two points of view when doing simulations: (i) that of a theoretician and (ii) that of an experimentalist. In the first case, the goal is to capture the essence of a given problem and use the simulated system to check theories and theoretical concepts. In this framework, no direct connection with real systems is usually invoked. In the case of the point of view of an experimentalist, the aim is to mimic a real system as faithfully as possible. Then, the best choice should be, in principle, fully atomistic simulations. The thorough validation of the simulated system by comparison with experimental results is crucial in such a case. Once the validation is confirmed—at least for the particular problem in the focus—the simulations can be exploited for e.g., calculating magnitudes that are not experimentally accessible (see Figure 3). Extending the parameters range beyond the experimental capabilities is another opportunity. This feedback involving experiments and simulations allows to understand a given problem and check theories as well.
A classical MD-simulation is just the solution of the classical equations of motion, i.e., Newton’s equations, for a set of N particles. The three main elements are (i) to define the system. This involves the construction of the simulated cell at the desired conditions of temperature, pressure, etc.; (ii) to define the interactions between the particles. This consists of selecting the potential energy or ’force-field’, from which forces acting on the particles can be calculated; and (iii) to integrate the equations of motion by a suitable computer algorithm. Figure 3 (blue boxes) shows a typical block-diagram for an atomistic simulation. In such a simulation the particles are atoms. In the first simulation done on a simple liquid ’Argon’ [20] (1954), the number of atoms was 864 in a cubic cell of size 35. The force-field was a simple Lennard-Jones potential. The simulation time was only a few ps and periodic boundary conditions were used. In that first work, noteworthy, some of the magnitudes measured by NS were computed, and some comparison (’validation’) with some available NS data was performed. In this sense, that pioneering work could be considered as the first example of the strategy here developed, applied to a very simple liquid. For a polymer melt (a dense ’liquid’ of macromolecules), some specific ingredients have to be additionally considered in order to perform a classical MD-simulation.
First, the big size of macromolecules has to be considered in order to define the construction of the simulated cell. The conformation of a macromolecule in the melt follows more or less the Gaussian statistics. As an example to estimate the required dimensions of a typical simulation cell, we may consider the simplest macromolecule, polyethylene (PE). For this polymer, the ratio between the average squared end-to-end distance and the molecular mass, M, is given by 1.25gmol [21]. For a short chain of 40 monomers (M = 1120 g mol) this gives . Thus, the side of the cubic cell must be at least . A number of atoms 6500 would be required, if we consider the density of PE (0.784 g/cm at 413 K). This implies that it is really difficult to construct simulation cells for polymer melts that contain the whole macromolecules, even for modest molecular weights. Usually, a ’folding’ procedure involving replicas of the chain and periodic boundary conditions is used. One of them is the so-called amorphous cell protocol proposed many years ago by Suter and Theodorou [22,23]. However, this type of methods limits the capabilities of atomistic MD-simulations for investigating large-scale dynamics even of not very large macromolecules.
Regarding the force-field, the potential energy of an ensemble of atoms belonging to macromolecules can be described as a sum of contributions due to all bond, valence bend and dihedral interactions, and non-bonded interactions. The latter are similar to those used for other organic molecular liquids and glasses. Therefore, the main difference is the diversity of intramolecular contributions associated to the complex macromolecular microstructure. Such a complexity usually makes difficult the actual equilibration of the simulated cell, in particular for large macromolecules, and specially close to glass-transition of the polymer. Different models are being developed to overcome these problems. In particular, those based on the end-bridging and double-bridging Monte Carlo algorithms (see, e.g., [24,25]). Despite these difficulties, different force fields are currently available for polymers, which yield quite reasonable results—within the limits of applicability commented above. We can cite e.g., the current versions of AMBER [26], COMPASS [27] CHARMM [28], GROMOS [29], OPLS-AA [30], GAFF [31] and DREIDING [32] force fields.
As for the computer algorithm, there are no qualitative differences with respect to MD-simulations of other non-polymeric liquids and glass-forming systems [33]. Different software packages are available—either commercial as for instance Materials Studio (Accelrys, now BIOVIA) [34] or of free access as e.g., Gromacs [35], DL POLY [36], LAMMPS [37], NAMD [38] or OCTA [39]. They allow performing fully atomistic MD-simulations on polymer systems by using different force-fields.
Last, it is worth considering how the simulation time compares with the characteristic time scale of the different dynamic processes in polymer melts. For example, at 100 K above its glass-transition temperature, the time scale of vibrations in a polymer melt would be about 0.1 ps, for methyl-group rotations of the order of 1 ps and for segmental (-) relaxation can be in the range 1–10 ns. Conversely, large-scale chain relaxation—as for instance the end-to-end relaxation–, involves characteristic times that depend on the molecular mass of the macromolecular structural units (i.e., on the number of monomers ). For non-entangled polymers, this characteristic time scales with , while for well entangled polymers, with [15]. Taking e.g., , the characteristic time would be about 10 s. The standard computing capabilities involve simulation times of the order of 100 ns for simulation cells of about 15,000 atoms. Thus, the characteristic times for large-scale chain relaxation are beyond these capabilities. These problems together with those above mentioned related to the equilibration of the simulation cells, imply that fully atomistic MD-simulations (even united atom simulations) of well-entangled polymer systems are really challenging (see, e.g., [40,41]). For this purpose, MD-simulations based on coarse-grained models are more suitable. There, a blob or bead represents several monomers—as for instance those forming a Kuhn segment, and, in principle, larger macromolecules can be simulated in this way. However, if the level of coarse-graining is increased, other problems can emerge, as e.g., unphysical bond crossing due to the softening of the coarse potentials. In the works described in this review, polymer systems well below the entanglement regime are always considered. Further information about MD-simulations in polymers can be found in the general references [42,43].
Now we address the combination of the two methods—neutron scattering and fully atomistic MD-simulations—in the strategy described here. it is clear that NS techniques are the right tools to validate fully atomistic MD-simulations of diverse systems like liquids and soft materials, and in particular in polymer melts. First of all, these methods cover the relevant length and time scales. In addition, the classical van Hove formalism allows easily calculating the magnitudes measured by NS from the simulated atomic trajectories. Different correlation functions can be experimentally addressed (in particular, thanks to deuterium labelling) and used for exhaustive check of the reliability of the simulated cell. We note that X-ray experiments can also be used as a reference for validation. However, the energy of X-rays is too high to resolve the small energy changes associated to the relaxational processes in polymers, and therefore this kind of studies are restricted to diffraction experiments ( = 0). Therefore, they only give structural information. In addition, since X-photons interact with the electron cloud, they are mostly sensitive to C and O atoms. Labelling techniques are no longer applicable and only one partial structure factor is accessed. As can be appreciated, neutrons provide much more demanding tests for the validation of the simulation.
Neutron scattering techniques on one hand and molecular dynamics simulations on the other hand have been extensively employed to study polymer structural and dynamical properties in the past. However, the synergetic combination we propose here has only been rarely applied. We can also mention some representative works by other authors [44,45,46,47,48,49,50,51,52,53,54,55]. We may emphasize here again that the exploitation of fully atomistic MD-simulations is trustworthy, of course, within the limits above discussed, and for the particular problem they can be validated by direct comparison with experimental results. In the following section we illustrate how this strategy has allowed us disentangling structural and dynamical properties of glass-forming polymers spanning over different length scales.
3. Results on ‘Simple’ Linear Homopolymers
3.1. Structural Properties
The local structure of many polymer materials is still poorly known. This structure emerges in a diffraction experiment in the Q-range . There, the results show two or more broad peaks, sometimes with shoulders. Taking into account their temperature dependence and from qualitative arguments, the peaks in the range between 1 and 1.5 approx. are assumed to have an intermolecular character. Conversely, those appearing at higher Q-values are intra-molecular in nature [56] (see e.g., Figure 1). However, in many cases, the particular atomic or molecular correlations behind these peaks, i.e., the actual short-range order in the real space, are not known.
X-ray diffraction delivering only one partial structure factor has been traditionally used for investigating the local structure of polymer systems. Neutron diffraction combined with selective deuteration allows to access different partial structure factors. They include the actual (total) structure factor . This is obtained in the case of fully deuterated samples. In all the atoms are equally weighted (see Figure 2 and Table 1). Experimentally, there is a problem for obtaining the coherent contribution to the diffraction patterns, in particular in protonated or partially protonated samples: the high level of incoherent scattering from the hydrogen atoms. This difficulty is overcome by neutron diffraction with PA, since it nicely allows separating coherent and incoherent contributions. We will present the outcome of the study presented in Ref. [57] on syndiotactic poly(methyl methacrylate) (PMMA, Plexiglass) as a representative example of the potential of the synergetic combination here proposed for structural investigations. Figure 4a,b show two different partial structure factors corresponding to different deuteration levels of PMMA. Some information about the particular atomic correlations behind these peaks can be deduced from a comparative analysis of these partial structure factors. However, MD-simulations on a realistic sample allow going beyond this qualitative analysis. A first step has to be given: to validate the cell ensuring that it reproduces well the experiments. Figure 4a,b show the direct comparison of both kinds of results without any adjustment. The agreement is highly satisfactory. Now the simulations can be exploited to address the short-range structural features in PMMA through the information provided by the partial structure factors. For example, we can easily unveil the different correlations contributing to the structure factors. To gain deep insight into the structure, three molecular substructures can be defined, in terms of which the simulation results may be grouped [57]: the main chain (MC), the -methyl group (MG), and the ester side group (SG). Figure 4c,d display the resulting decomposition of the partial structure factors shown in panels (a) and (b). The separate correlations can now be analyzed in depth. For example, a characteristic MC/MC distance of could be deduced in the Bragg approximation from the position of the MC/MC main peak . Also, a strong anticorrelation of MCs and SGs suggests a precursor effect of the nano-phase separation between the main chains and the side groups observed for methacrylates with larger alkyl side-groups [58].
Furthermore, directly accessing the real space (impossible with NS techniques), the radial probability distribution function corresponding to different key atoms could be inspected [57]. Such analysis pointed to a strong short-range order. In addition, the analysis of the sequence of intra-chain peaks indicated a persistent all-trans structure, in agreement with a locally stiff chain as indicated by the high values reported for the characteristic ratio [59]. We could also see that intra-chain correlations contribute as well at rather large distances. The analysis of the radial distribution of the ester methyl group carbon suggested some interdigitation of the side groups which again is in agreement with the nano-phase separation precursor effect previously mentioned. We will come back later to this interesting problem.
3.2. Dynamical Behavior
3.2.1. Localized Motions
Among the different dynamical processes that take place in polymers, methyl group rotation is perhaps the simplest one, since all the relevant interactions on the methyl group can be condensed in an effective mean-field one-dimensional potential. By means of neutron scattering it is possible to access experimentally the different manifestations of this process, namely quantum tunnelling, classical hopping as well as its fingerprint in the vibrational density of states. The results obtained in many chemically and structurally different polymers [66] can be consistently described in terms of the Rotation Rate Distribution Model (RRDM), which was first proposed in 1994 [67]. This model introduces a distribution of potential barriers for methyl group rotation, which is associated to the disorder present in any structural glass. Interestingly enough, this model predicts, instead of inelastic peaks, an apparent quasielastic broadening for the tunnelling spectrum. This was experimentally found in poly(vinyl acetate) (PVAc) in 1998 [3]. Later, MD-simulations provided further support to the RRDM and allowed extracting interesting information about this process, as we are detailing in the following. Molecular dynamics simulations on PI carried ot at very low temperature [68] were first validated through the comparison of the density of states for methyl group torsional librations, as calculated from the time evolution of the dihedral angles, with previous inelastic neutron scattering results. The librational peak showed a broad feature reflecting a distribution of potential barriers. This distribution was quantified in the framework of the threefold approximation—which was corroborated by the simulations. Then, the capabilities of the computational method were further exploited performing similar simulations under different conditions for the nonbond interactions considered in the used force field. This led to conclude that the width of this distribution is mainly controlled by the nonbond interactions. Moreover, these nonbond interactions also contribute significantly to the value of the average barrier for methyl group reorientation.
In a later work [69], the methyl group dynamics in PI was investigated well above the glass-transition temperature . Starting from the self part of the van Hove correlation function, the incoherent intermediate scattering function was calculated for the protons in the main chain and in the methyl groups. The dynamics of the latter ones could be well described assuming decoupled segmental dynamics and rotations in a threefold potential. The distribution of potential barriers for methyl group rotation at such high temperature was found to be very similar to that deduced from low temperature MD-simulation results and inelastic neutron scattering measurements above mentioned [68]. Other works combining NS and simulations on poly(propylene oxide) (PPO) [53] and PMMA [70] well above the glass transition have also shown that the assumption of statistically independence for methyl group rotations and -relaxation is a good approximation. The glass transition thus hardly modifies the energy landscape for this local process.
Beyond methyl group dynamics, the rich microstructure of polymers—even in systems as ‘simple’ as PB—leads to a variety of dynamic processes active below as well as above their glass transition. These processes are behind the so-called secondary relaxations detectable by relaxation techniques like e.g., dielectric spectroscopy. In fact, for PB, there was a controversial situation regarding the origin of the dynamics observed by different techniques, mainly dielectric spectroscopy and NSE measurements close to the glass transition [71,72,73,74]. To disentangle the local motions of PB in this range, MD-simulations [75,76] of a realistic sample with similar microstructure as that of the real chains were carried out. The simulations were run for a very long time (≈1 s!) to equilibrate the system as well as possible in the difficult temperature range under consideration (about 20 K above ). The results were carefully validated against NS data addressing structural as well as dynamical properties on both, protonated and deuterated samples [64,76]. Once the cell was validated, the radial probability distribution functions corresponding to the different kinds of hydrogens in PB were calculated. Figure 5a shows that a second peak emerges in the distribution function, which is a are clear evidence for localized motions. Moreover, different hydrogens in the different units move in a rather different way. Having a look on longer times, contributions of diffusive kind (broadening of the distribution and shift to longer distances) can also be envisaged at this low temperature (see Figure 5b). These contributions become more dominant as the temperature increases. The underlying diffusive motion is the -relaxation process we will address in the next section. Based on all these results, a simple model was constructed which combines hopping in an asymmetric double-well potential and an anomalous diffusion process representing the -relaxation (see below) [75,76]. The model nicely fits the results in the real space, as can be seen in Figure 5b. Note that the assumption in the model to combine the motions was statistical independence of local and diffusive processes—the same as found for the simultaneous occurrence of methyl group rotations and segmental relaxation above commented. From the fits of the model, the hopping times of cis- and trans units were obtained. They turn out to be rather different. The reported dielectric results on the secondary -relaxation are compatible with the timescales observed for the jumps involving the cis-units. This result is sensible, since the cis-unit is the moeity carrying the dipole moment of 1,4-PB. Conversely, the NSE values are rather close to those obtained for the local process of the trans-units. The short-time window accessed by NSE in the explored Q-range (below approx. 10 ns) is apparently more sensitive to the fastest motions occurring in the sample. Thus, MD-simulations allowed putting into a context the controversial puzzle. The intrinsic heterogeneous dynamics was the key question: cis- and trans-units behave in a rather different way and with different relaxation times.
3.2.2. The Structural Relaxation
At the beginning of this century, through extensive NS investigations on protonated polymers, a universal behavior for the self-atomic (H) motions in the -relaxation regime was established. Most experiments yielded in the frequency domain. Roughly speaking, the width of the spectra is inversely proportional to the characteristic time of the process involved. This width depends on Q, what is a signature of an underlying diffusive-like process. However, contrarily to the case of simple diffusion, the results are not compatible with a simple exponential decay for the intermediate scattering function. In general, they can be accounted for by the Fourier Transform of a stretched exponential function for :
This function is characterized by the stretching exponent and a Q- and T-dependent characteristic time (see, e.g., [77]). This function is also commonly known as Kohlrausch-Williams-Watts (KWW). Furthermore, results on polymers reported over more than ten years [77,78] had shown that in the Q-regime approx. 0.2 follows the law: (see e.g., Figure 6a). Considering this expression in Equation (4), one immediately arrives to the Gaussian Q-dependence of :
where the mean squared displacement of Hs in the -relaxation increases sublinearly with time as . This result qualifies the motions as an ’anomalous sublinear diffusion’. A perfect Gaussian behavior was in apparent contradiction to MD-simulations results on glass forming systems other than polymers (Lennard-Jones liquids [79,80], water [81], ortho-terphenyl [82] and Selenium [83,84]). Thus the question arose: Do glass forming polymers behave in a different way? Noteworthy, a close inspection to the high Q-data in Figure 6a already suggested deviations from the Gaussian law .
At this point, a combination of fully atomistic MD-simulations [87] and NS measurements on the same polymer, PI, was used to clarify the situation [85,86]. Several spectrometers were used to extend as much as possible the investigated Q-range. NSE was particularly important. It allowed direct access to the deconvoluted intermediate scattering function in the time domain, for an accurate determination of its functional form. The sample investigated had deuterated methyl groups (PId3), such that the self motions of the main chain protons were the main contribution to the experimental results. These motions were also followed from the atomic trajectories delivered by the MD-simulations. Figure 6b shows the obtained Q-dependence of from the two methods. The results univocally confirm the Gaussian-like behavior in the Q-range approx. . At higher Q-values, clear signatures of deviations become evident.
The question then was the origin of the deviations observed. It was noted the strong reminiscence of the way the characteristic time departs from the Gaussian expectation (Figure 6) with the manifestation of the discrete nature of diffusion in simple jump diffusion models [88]. Basing on this similarity, a model was proposed, where a distribution of elementary jump lengths was assumed behind the anomalous diffusion carried out by the atoms in the -process [85,86]. As shown in Figure 6b, such a simple approach provides a very good description of the Q-dependence of in the whole Q-range explored. The associated distribution of jump lengths has a maximum at about 0.4 Å for PI (inset of Figure 6b). This suggests that a distribution of discrete step lengths is the origin of the universal deviations from Gaussian behavior in the self-motions of atoms during the structural relaxation regime. This model has also been successfully applied on other polymers in latter works [PB [89], poly(vinyl ethylene) (PVE) [89,90], poly(vinyl methyl ether) (PVME) [91], PMMA [70], poly(ethyl methacrylate) (PEMA) [92] and poly(tetrahydrofurane) (PTHF) [93]].
The information about H-self motions in the -relaxation regime is of utmost interest in the field of glass-forming systems. However, in the framework of important theories for the glass transition as the Mode Coupling Theory (MCT) [2,94,95], the most relevant magnitude is the dynamic structure factor at the intermolecular correlations. This function is accessible by means of NS—in this case, on fully deuterated samples. The earliest experimental work by NSE on PB [96] was followed by others on PI [97], PIB [98,99], atactic polypropylene (a-PP) [100], polyurethene (PU) [101], poly(vinyl chloride) (PVC) [102], PVE [13], poly(ethylene propylene) (PEP) [103] PTHF [104]. In these works, the universality of the properties of the structural relaxation—stretching, scaling—was usually checked. The stretched character of the decay of the inter-molecular correlations (KWW-like functional form, Equation (4), see Figure 7) is nowadays a well established feature; however, the scaling property (time/temperature superposition principle) is not clearly always fulfilled (see, e.g., [103,104,105]).
Obviously the interpretation of collective motions is much more complicated than for the self-motions. Molecular Dynamics simulations have been used to complement experimental investigations on the dynamic structure factor in several cases [65,70,75,103]. Magnitudes that cannot be experimentally observed could be computed from the simulations, as they are the partial dynamic structure factors. An illustrative example is depicted for the case of deuterated PMMA in Figure 8 [70]. The observed temporal evolution of the total dynamic structure factor is the result of a complex interplay of the different partial contributions. At the first structure factor peak (), the timescales for collective motion strongly depend on the molecular groups considered, and are spread over more than one order of magnitude. At the second peak (, basically all the relaxation times are within a factor of 2. The simulations also allow easily comparing collective and self-correlation functions, addressing the coherency of the relaxation process. For PMMA it was found that, while coherency effects are observed for all correlations at the first structure factor peak, at the second peak coherency remains only for correlations involving the main chains.
The combined consideration of NS and MD-simulations has also been of utmost help in exploring the applicability of theoretical frameworks, like the MCT, in polymers [106,107,108]. Consistent results were found when applying MCT to these systems, though with rather high values of the exponent parameter (the key parameter in this theory). The -values reported from other works for real polymers like PB [109], poly(propylene glycol) (PPG) [110] or PS [111], and simple polymer models with intramolecular barriers [112] are similarly high. Such finding could be interpreted in the framework of a higher-order MCT transition for real polymers arising from the simultaneous occurrence of two mechanisms leading to dynamic arrest: (i) packing, of intermolecular character and present in all glass-forming systems, and (ii) barriers for conformational changes, of intramolecular origin, which combined with chain-connectivity are specific of macromolecular systems [106,112].
3.2.3. Chain Dynamics
At length scales much larger than the monomer size, polymer melts show unique dynamic processes that are controlled by chain connectivity and the size of the macromolecules. The rheological properties of polymer melts are ultimately determined by these processes. The so-called Rouse model [12] is the standard model to describe polymer chain dynamics in the melt. It considers the conformational entropy as the only source for restoring forces which stabilizes excursions from equilibrium. A coarse-grained model of N beads connected by entropic springs represents the chain, and a stochastic background gives account for the contribution of the surrounding chains. The created friction is characterized by the friction coefficient . The Langevin equation that results from this situation can be solved by transforming to the normal coordinates (Rouse modes)
with the position of the ith bead of the Gaussian chain and p the mode number (). In this model, the Rouse correlators relax independently and exponentially with p-dependent characteristic times. Conceptually, this model has two limitations. The first one arises at large distances. There, long chain melt topological constraints cause entanglements leading to the reptation mechanism. The other limitation is found at shorter distances. There, the assumptions allowing for a simplified coarse-grained chain cease to be valid. The local chain structure and dynamics become relevant, in particular approaching the intermolecular distances. As we have discussed above, there we approach the regime of the process. The way how the crossover from segmental to Rouse dynamics takes place is still an intriguing question in polymer physics. To answer it, one of the key ingredients is to determine when the Rouse model ceases to be valid. This was the subject of a number of NS investigations on different polymers [113,114,115] in the past.
However, a thorough analysis of the sources for Rouse deviations is very difficult due to two factors: the uncertainties involved in the experiments on the one hand, and the impossibility to directly access the Rouse correlators, on the other hand. These problems are overcome in the simulations. With the current computing capabilities, big enough cells can be built to address with atomistic detail the chain dynamics in unentangled polymer systems.
A combined NS/MD-simulations strategy to tackle this problem has been applied to PB [116], PE [117], poly(ethylene oxide) (PEO) [118] and PEP [119]. After validation, magnitudes that cannot be accessed experimentally, like the mean squared displacement (msd) of different atoms or groups of atoms, were computed from the simulations, leading to noteworthy observations. For instance, inspection of the msd of the center of mass of the chains in PEP revealed a sublinear increase with time below (the longest relaxation time of the chain), presumably due to the effect of intermolecular interactions neglected in the Rouse model.
In addition, by coarse graining the atomistic MD simulations a Rouse mode analysis of the simulated chains can be carried out. The results on the Rouse mode correlators are obtained defining a bead as the center of mass of a monomer. The such obtained correlators do not decay exponentially with time, but stretched exponential functions (like that in Equation (4)) have to be applied (Figure 9a). More pronounced stretching is observed when moving toward local length scales (small -values) (Figure 9b). Moreover, at large p-numbers the p-dependence of the characteristic times strongly deviates from the Rouse prediction. An ARS-like approach [120] can account for the deviations originating from chain stiffness. However, these corrections are not sufficient to reproduce the results. The additional deviations can be characterized invoking of a p-dependent effective friction coefficient. This parameter decreases with the length scale, reaching a constant value (corresponding to the pure Rouse limit) at . The influence of the local potentials in the chain dynamics is clearly evidenced by these results. In the functions accessed experimentally by neutron scattering, these effects are reflected at high Q values and mainly short times.
Taking advantage of these well-validated atomistic MD-simulations of PEP, a bottom-up coarse-grained model was built based on them. With this model, the simulations could be extended beyond the entanglement mass of PEP [121]. Establishing the limits of chemically simplified models for direct mapping with experimental results is important. It is worth mentioning that these limits were thoroughly explored by other authors by comparing different levels of modeling for MD-simulations. They included explicit atom, united atom and coarse-grained simulations for two polymers, PEO [54] and PMMA [55].
Finally, we note that the Rouse scattering functions are based on the Gaussian approximation. The influence of non-Gaussian effects on the Rouse behavior was also studied by NS combined with atomistic MD-simulations in PB [48] and later in PEO forming part of an asymmetric polymer blend with PMMA [122].
4. Beyond ‘Simple’ Polymers and ‘Standard’ Experiments
In the following we present studies tackling even more intricate problems than those addressed in the previous section. A first step in the complexity of the polymers investigated can be given by considering still homopolymers, but with long or bulky side groups. This, as we will show next, gives rise to an interesting phenomenon: the nano-segregation of the chain components, that can be addressed by combining neutron scattering and MD-simulations. Thereafter, we will focus on a general problem of high relevance for glass-forming systems and liquids in general: the collective dynamics at intermediate length scales. We will show recent progresses in this almost white field, emphasizing novel results obtained by state-of-the art instrumentation that allows for polarization analysis on quasielastic experiments with high energy resolution.
4.1. Nanosegregation in More Complex Polymers
The structural and dynamical features of the family on poly(n-alkyl methacrylates) (PnMAs) and other polymers containing alkyl side groups were the focus of great interest in the past [58,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147]. The main reason was the suggested formation of nano-domain structures by self-assembly of the side groups, that are segregated at nanometric scale from the main-chain subsytems. Such a morphology was deduced from the presence in the X-ray diffraction patterns of peaks at relatively low Q-values (’intermediate’-range), with side-group length dependent positions [134,138,146]. Neutron diffraction combined with isotopic labelling definitely proved such a scenario [58,143]. In addition, interesting peculiarities in the dynamics of the side groups were reported [58,138,143], in principle attributed to confinement effects induced by the surrounding main chains.
Later, the dynamics of the family of poly(alkylene oxides) (PAOs) were investigated by mechanical and dielectric spectroscopies [148] as well as by neutron scattering [149]. For these polymers, the side-groups motions did not show anomalies which could be attributed to confinement effects. If such effects emerged as a consequence of the nano-segregation of side groups and main chains, the question arose: Is the nano-segregation suppressed in PAOs by the large flexibility of the PEO-like main chain?
A combined approach including neutron diffraction and MD-simulations allowed deciphering the structure of PAOs at short and intermediate (from Å to few nanometers) length scales [150]. Clear experimental evidences were found for the presence of alkyl nano-domains within which the local structure is even more similar to bulk PE than in the case of PnMAs. In addition, the properly validated simulated samples (with = 2 and 4 alkyl carbons) provided direct proof of the nano-segregation and allowed calculating different magnitudes like the contributions of the main molecular groups (main chains and side groups) to the structure factor (see Figure 10 for the case of = 4). Thus, the lack of observation of confinement effects in the side-group dynamics of PAOs has to be attributed to other ingredients than the absence of structurally segregated phases. Large main chain flexibility leading to similar characteristic times for the dynamics of side groups and main chains could be invoked to rationalize the different dynamical behavior in PAOs with respect to PnMAs. Confinement effects in the latter would derive from the intrinsically different flexibility in main chains and side groups leading to a strong dynamic asymmetry. This effect is absent in PAOs. According to this scenario, the analysis of the dynamics from the simulations on the PAO with = 2 showed that for this polymer, full rotations of the pendant groups take place during the decaging of the main-chain segments [151].
It is worth noting that the universality of the emergence of nano-domains in polymers with long side groups was checked in a computational investigation of a simple bead-spring model for comb-like polymers [152]. Removing the particular chemical details, the systems constructed mimicked families of polymers like PAOs or PnMAs. That study showed that nanosegregation of main chains and side groups can arise as a purely entropic effect, provided that the density of branch points is large enough.
Another polymer with relatively complex composition, poly(vinylpyrrolidone) (PVP), was also investigated by combining NS, X-ray diffraction, and fully atomistic molecular dynamics simulations [153]. A “prepeak” in the X-ray diffraction pattern shows the typical features of a first amorphous halo. The experimentally accessed incoherent scattering function of hydrogens exhibited enhanced stretching and a Q-dependence of the characteristic time different from that usually reported for simpler polymers (main-chain polymers or polymers with small side groups). The comparison with both kinds of experimental results validated the simulations. Nanosegregation of side groups and main chains was suggested by the analysis of the simulated structure factor. Furthermore, the detailed insight provided by the simulations on the atomic trajectories revealed a partial and spatially localized decoupling of MC and SG dynamics at length scales between the average SG–SG distance and the characteristic length of the backbone interchain correlations. Correlators calculated for the SG subsystem showed anomalous behavior, like e.g., logarithmic-like decays of the density–density correlation function. They might be again a consequence of the existing large dynamic asymmetry between SG and MC subsystems. It was proposed that, as the SGs are spatially extended and chemically different from the backbone, they form transient nanosegregated domains. The dynamics of these domains showed similar behavior to that found in other systems displaying large dynamic asymmetry. We note that this ingredient has been found to be also crucial in e.g., polymer blends [154].
4.2. The Collective Dynamics at Intermediate Length Scales: An Unknown Territory
A particularly interesting region, not only for polymers, but for glass-forming systems and liquids in general, is the so-called intermediate length scales (ILS) region—the range of length scales larger than the intermolecular distances but not yet in the hydrodynamic regime [65,155,156]. In polymers, this is also the region where the -relaxation crosses over toward the Rouse dynamics. This problem has been addressed above (Section 3.2.3) for the single chain dynamic structure factor. However, the general question how collective dynamics develop in this regime—in liquids and melts—is poorly understood. In fact, from a theoretical point of view, the connection between collective and self-motions is an unsolved problem in complex systems. The relationship proposed by deGennes [157], that the collective time is just the product of the self-correlation time and the structure factor, , would be strictly applicable only to monoatomic liquids in the range of the first maximum of . In the case of anomalous diffusion, where stretched exponentials are found instead of single exponentials, the ansatz proposed by Sköld [158] relating the intermediate scattering functions might be applied. This translates in a modified version of the deGennes narrowing as . From an experimental point of view, for polymers, this question has been addressed first only by NS [99,159], and, later, by the combination of NS and MD-Simulations [65,103,160]. Though the Sköld-like ansatz seems to work reasonably well in some cases close to the first structure factor peak, deviations are found away from this inter-molecular correlations region. For instance, in the case of PIB, a marked Q-dependence of the apparent activation energy was found for coherent scattering (see inset in Figure 7b) [65,99] rendering any ‘simple’ connection with incoherent scattering as inviable, even more at ILS.
Combining NS and MD-simulations it was possible to describe in a rather satisfactory way the collective behavior of PIB not only close to the first structure factor peak but also approaching the ILS. The starting point was the ansatz proposed in Ref. [111], where the collective time is given by
The non-diffusive (Q-independent) time should reflect the viscoelastic coupling of stress and density fluctuations on scales long enough compared to atomic dimensions, but not yet in the hydrodynamic limit [161]. Its contribution to the total collective time is affected by a Gaussian cutoff factor to ensure that it is present only on length scales beyond a characteristic length , which is assumed to be . The diffusive time was generalized by us to the case of anomalous diffusion as above described by the Sköld approximation. To apply the model to PIB, we used the information on the total self-motions available from MD-simulations properly validated by direct comparison with experimental results. The collective time at was extracted from the fits of the coherent NSE data. This time agrees very well with compiled results from different experimental techniques directly accessing such relaxation time. Figure 11 shows the different components involved in the model. This generalized model also gives account for the modulation of the apparent activation energy of the collective times with the static structure factor (inset of Figure 7b). It would mainly result from changes of the short-range order at inter-molecular length scales [65,156]. This model also allows a semiquantitative description of recent experimental results on PTHF [93,104].
Despite these advances, the ILS region is still hardly explored in glass-forming systems and liquids. A severe complication for the experiments derives from the difficulties in separating coherent and incoherent contributions from the measured signal. Though the coherent scattering cross section is much higher than the incoherent one in fully deuterated systems, the small amplitude of the density fluctuations away from the first amorphous halo, in particular at low-Q values, leads to very weak signals, comparable there to the incoherent contributions. The parasitic incoherent contribution is largely (but not completely) suppressed by the NSE technique [99]. As mentioned above, currently, polarization analysis (PA) allowing for a full separation is not routinely implemented in neutron spectrometers due to a series of technical difficulties [162,163]. For this reason, extending the experimental investigations toward the ILS is a real challenge.
4.3. A Breakthrough Experiment on Water: A ‘Simple’ and Yet Enormously Complex System
Recently, a few multi-detector spectrometers have demonstrated the capability of performing neutron spectroscopy with PA and sub-meV resolution [164,165,166]. We took this unique opportunity to scrutinize the coherent structural relaxation in a wide Q-range from meso- to inter-molecular scales in the most relevant liquid: water [19]. We note the relevance of water also in different polymer and bio-polymer systems. The experiments were carried out on the LET direct geometry time-of-flight spectrometer at the ISIS Neutron and Muon Source, Oxfordshire, UK. Figure 12a shows the coherent and incoherent contributions to the total spectrum, for a Q-value in the ILS region (see structure factor of water in the inset of Figure 12b). The quality of the results is absolutely amazing, considering the extremely low coherent signal in this regime. As can be appreciated just at first sight, coherent and incoherent scattering are extremely different. The coherent contribution is very broad in comparison with the incoherent one. As this broadening is a measure of the inverse of the corresponding relaxation time (FWHM ∼ 1/), this tells us—without any fitting—that the coherent characteristic time is of the order of 10 times smaller than the incoherent (diffusive) relaxation time. The latter is close to that deduced from the total (incoherent plus coherent) spectra that is measured if PA is not used. Since the relaxation time at 0.5 Å reported from measurements on DO at 298 K without PA is ≈20 ps [167], then should be ≈2 ps.
In the low-Q meso-scale, the observed decorrelation of the collective fluctuations hardly depended on Q. These results were also corroborated by MD-simulations on a big cell (see Figure 12b). In the crossover range towards inter-molecular scales—where the structural relaxation is dominated by diffusion–, the experimental results were nicely described by the convolution of a Q-independent mode and diffusion. The model applied is very general, and had been used previously to describe the merging of the and processes in glass-forming polymers [71,72,168] and more recently to analyze the incoherent NS and NMR results of water [167,169]. We address the interested reader to Ref. [19] for more details about this work.
5. Conclusions
We have presented a general and robust methodology consisting of the synergetic combination of neutron scattering experiments on real samples and fully atomistic MD-simulations, to face challenges in diverse areas, including in particular the field of polymer melts and glass-forming polymers. The success of the methodology here presented has been illustrated with examples covering a wide range of systems of diverse complexity and problems of different nature. We have shown the added value of the combination of neutron scattering and fully atomistic MD-simulations in deciphering the structural properties of chemically simple polymers as well as of other systems with long or bulky side-groups. The potential of this strategy has also been highlighted in the case of the dynamical studies; we have shown a substantial increase in the understanding of different processes characteristic for polymers, ranging from the simplest motions (methyl group rotations) to the Rouse dynamics, passing through the local processes involved in the secondary relaxations and the structural relaxation. The power and versatility of the tandem composed of neutron scattering and fully atomistic MD-simulations has thus largely been demonstrated along these years of application by our group.
Many interesting questions in the field of polymers, however, remain of course open and are still challenging. A large number of them relate to multi-component systems and/or to systems containing macromolecules with complex architectures or displaying relevant features at mesoscopic scales. These problems are in general not affordable by the combination here presented. Still in these cases, neutron scattering on properly H/D labeled systems can provide the right experimental tool to scrutinize such systems. However, the design of the adequate samples is usually very delicate and in general demands for advanced chemical synthesis capabilities involving deuteration. The experiments involve then small angle neutron scattering (SANS) and NSE techniques at low Q-values, addressing the static and dynamical features at the mesoscale, and the interpretation of the results can also be very complicated. Support of simulations becomes then also extremely useful. But in such cases, coarse-grained simulations are the good choice. Forgetting about the chemical details, they can be used to explore generic features and/or very big systems. In this review, when presenting the PAOs studies, we have already mentioned the interesting result of coarse-grained investigations on comb-like polymers addressing the generality of the nano-segregation effect [152,170]. Thus, the methodology here proposed can be extended to this kind of investigations but using coarse-grained models instead of fully atomistic simulations. Another case where this synergy has recently been exploited is in the field of single-chain nano-particles (SCNPs) [171,172,173,174,175]. These unimolecular nano-objects obtained by intramolecular cross-linking of individual macromolecular chains are highly promising in the field of nanotechnology. Combining these two tools—SANS and/or NSE on H/D labelled samples and coarse-grained simulations–, deep insight into the fractal dimension and the internal friction characterizing these fascinating macromolecules has been gained.
Special mention deserve, concerning the use of coarse-grained models, the studies where the microstructure of the polymer plays a relevant role, even though the focus is on relatively large length scales as compared with the intermolecular distances. This could be for example the case of random copolymers. In such works, fully atomistic simulations can be of utmost help providing characteristic parameters of polymer chains as, for instance, the statistical segment length. We have already mentioned the case of PEP, where atomistic MD-simulations validated with experiments were used to build a bottom-up coarse-grained model to extend the simulations beyond the entanglement mass [121].
Finally, we have also shown that to tackle some intriguing problems, new experimental developments are crucial, as, for instance, the implementation of polarization analysis in quasielastic neutron scattering instruments. In fact, our recent work on DO [19] has opened a new way of approaching the unknown territory of coherent scattering—from meso- to inter-molecular scales—not only in water under different conditions but also in H-bonded liquids, glass-forming liquids and biological systems where water plays an important role. It also convincingly proved the power of these capabilities, that can hugely impact the progress of microscopic dynamics investigations in fields like soft matter or biology.
Author Contributions
Conceptualization, J.C.; methodology, J.C.; software, F.A.; experiments, A.A. and J.C.; validation, J.C., F.A. and A.A.; writing–original draft preparation, A.A.; writing–review and editing, J.C. and A.A.; funding acquisition, J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Basque Government, code: IT-1175-19 and the Ministerio de Economía y Competitividad code: PGC2018-094548-B-I00 (MCIU/AEI/FEDER, UE).
Acknowledgments
We thank all our collaborators in the neutron scattering facilities all over the world who, along the past decades, have participated in the experiments. We also would like to thank the rest of the members of our research group in San Sebastián as well as Dieter Richter and his group in Jülich for the long-standing and fruitful collaboration.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| NS | Neutron scattering |
| MD | Molecular dynamics |
| NSE | Neutron spin echo |
| PA | Polarization analysis |
| PE | Polyethylene |
| AMBER | Assisted Model Building with Energy Refinement |
| COMPASS | Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies |
| CHARMM | Chemistry at Harvard Macromolecular Mechanics |
| GROMOS | Groningen Molecular Simulation |
| OPLS | Optimized Potentials for Liquid Simulations |
| NAMD | Nanoscale Molecular Dynamics |
| GAFF | Generalized AMBER Force Field |
| GROMACS | Groningen Machine for Chemical Simulations |
| LAMMPS | Large-scale Atomic/Molecular Massively Parallel Simulator |
| OCTA | Open Computational Tool for Advanced material technology |
| PMMA | Poly(methyl metacrylate) |
| MC | Main chain |
| MG | Methyl group |
| SG | Side group |
| PI | Polyisoprene |
| PS | Polystyrene |
| PB | 1,4-Polybutadiene |
| PIB | Polyisobutylene |
| RRDM | Rotational rate distribution model |
| PVAc | Poly(vinyl acetate) |
| PPO | poly(propylene oxide) |
| KWW | Kohlrausch-Williams-Watts |
| PId3 | Polyisoprene with deuterated methyl groups |
| PVE | Poly(vinyl ethylene) |
| PVME | poly(vinyl methyl ether) |
| PEMA | Poly(ethyl methacrylate) |
| PTHF | Poly(tetrahydrofurane) |
| a-PP | atactic Polypropylene |
| PU | Polyurethene |
| PVC | Poly(vinyl chloride) |
| PEP | Poly(ethylene propylene) |
| MCT | Mode Coupling Theory |
| PPG | Poly(propylene glycol) |
| ARS | All rotational state model |
| PEO | Poly(ethylene oxide) |
| msd | mean squared displacement |
| PnMAs | Poly(n-alkyl methacrylates) |
| PAOs | Poly(alkylene oxides) |
| PVP | Poly(vinylpyrrolidone) |
| ILS | Intermediate length scales |
| NMR | Nuclear magnetic resonance |
| SANS | Small angle neutron scattering |
| SCNPs | Single-chain nano-particles |
References
- Flory, P. Principles of Polymer Chemistry; Cornell University Press: London, UK, 1953. [Google Scholar]
- Götze, W. Liquids, Freezing, Glass Transition; Elsevier: Amsterdam, The Netherlands, 1991; p. 287. [Google Scholar]
- Colmenero, J.; Mukhopadhyay, R.; Alegría, A.; Frick, B. Quantum rotational tunneling of methyl groups in polymers. Phys. Rev. Lett. 1998, 80, 2350–2353. [Google Scholar]
- Johari, G.; Goldstein, M. Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules. J. Chem. Phys. 1970, 53, 2372. [Google Scholar]
- Götze, M.; Mayr, M.R. Evolution of vibrational excitations in glassy systems. Phys. Rev. E 2000, 61, 587. [Google Scholar]
- Arbe, A.; Alvarez, F.; Colmenero, J. Neutron scattering and molecular dynamics simulations: Synergetic tools to unravel structure and dynamics in polymers. Soft Matter 2012, 8, 8257–8270. [Google Scholar]
- Lovesey, S.W. Theory of Neutron Scattering from Condensed Matter; Clarendon Press: Oxford, UK, 1984. [Google Scholar]
- Squires, G.L. Introduction to the Theory of Thermal Neutron Scattering; Dover Publication Inc.: New York, NY, USA, 1996. [Google Scholar]
- Squires, G.L. Introduction to Thermal Neutron Scattering; Cambridge University Press: Cambridge, UK, 1988. [Google Scholar]
- Kirste, R.G.; Kruse, W.A.; Schelten, J. Die Bestimmung des Trägheitsradius von Polymethylmethacrylat in Glaszustand durch Neutronenbeugung. Die Makromolekulare Chemie 1973, 162, 299–303. [Google Scholar]
- Benoit, H.; Decker, D.; Higgins, J.S.; Picot, C.; Cotton, J.P.; Farnoux, B.; Jannink, G.; Ober, R. Dimensions of a flexible polymer chain in the bulk and in solution. Nat. Phys. Sci. 1973, 245, 13. [Google Scholar]
- Rouse, P.E. A theory of the linear viscoelastic properties of dilute solutions of coiling polymers. J. Chem. Phys. 1953, 21, 1272. [Google Scholar]
- Richter, D.; Monkenbusch, M.; Arbe, A.; Colmenero, J. Neutron Spin Echo Investigations on Polymer Dynamics. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2005; Volume 174. [Google Scholar]
- Schleger, P.; Farago, B.; Lartigue, C.; Kollmar, A.; Richter, D. Clear evidence of reptation in polyethylene from neutron spin-echo spectroscopy. Phys. Rev. Lett. 1998, 81, 124. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572. [Google Scholar]
- Available online: http://neutronsources.org (accessed on 17 December 2020).
- Mezei, F. Neutron Spin Echo, Lecture Notes in Physics; Springer: Heidelberg, Germany, 1980; Volume 28. [Google Scholar]
- Arbe, A.; Nilsen, G.J.; Stewart, J.R.; Alvarez, F.; Sakai, V.G.; Colmenero, J. Coherent structural relaxation of water from meso- to intermolecular scales measured using neutron spectroscopy with polarization analysis. Phys. Rev. Res. 2020, 2, 022015. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A. Correlations in the motion of atoms in liquid argon. Phys. Rev. 1964, 136, A405. [Google Scholar]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639. [Google Scholar]
- Theodorou, D.; Suter, U. Atomistic modeling of mechanical properties of polymeric glasses. Macromolecules 1986, 19, 139. [Google Scholar]
- Theodorou, D.; Suter, U. Local structure and the mechanism of response to elastic deformation in a glassy polymer. Macromolecules 1986, 19, 379. [Google Scholar]
- Karayiannis, N.; Mavrantzas, V.; Theodorou, D. A novel Monte Carlo scheme for the rapid equilibration of atomistic model polymer systems of precisely defined molecular architecture. Phys. Rev. Lett. 2002, 88, 105503. [Google Scholar] [PubMed]
- Karayiannis, N.C.; Mavrantzas, V.G. Hierarchical modeling of the dynamics of polymers with a nonlinear molecular architecture: Calculation of branch point friction and chain reptation time of H-shaped polyethylene melts from long molecular dynamics simulations. Macromolecules 2005, 38, 8583–8596. [Google Scholar]
- Cornell, W.; Cieplak, P.; Bayly, C.; Gould, I.; Merz, K.J.; Ferguson, D.; Spellmeyer, D.; Fox, T.; Caldwell, J.; Kollman, P. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179. [Google Scholar]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586. [Google Scholar]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596. [Google Scholar]
- Jorgensen, W.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.; Olafson, B.; Goddard, W. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897. [Google Scholar] [CrossRef]
- Allen, M.; Tildesley, D. Computer Simulation of Liquids; Oxford Science Publications: Oxford, UK, 1987. [Google Scholar]
- Available online: www.accelrys.com (accessed on 17 December 2020).
- Available online: www.gromacs.org (accessed on 17 December 2020).
- Available online: www.ccp5.ac.uk (accessed on 17 December 2020).
- Large-Scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) is a Molecular Dynamics Program from Sandia National Laboratories.
- Available online: www.ks.uiuc.edu/Research/namd (accessed on 17 December 2020).
- Available online: octa.jp (accessed on 17 December 2020).
- Tsolou, G.; Mavrantzas, V.; Theodorou, D. Detailed atomistic molecular dynamics simulation of cis-1,4-poly(butadiene). Macromolecules 2005, 38, 1478. [Google Scholar] [CrossRef]
- Stephanou, P.; Baig, C.; Tsolou, G.; Mavrantzas, V.; Kröger. Quantifying chain reptation in entangled polymer melts: Topological and dynamical mapping of atomistic simulation results onto the tube model. J. Chem. Phys. 2010, 132, 124904. [Google Scholar] [CrossRef] [Green Version]
- Binder, K. (Ed.) Monte Carlo and Molecular Dynamics Simulations in Polymer Science; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Kotelyanski, M.; Theodorou, D. (Eds.) Simulation Methods for Polymers; Marcel Dekker: New York, NY, USA, 2004. [Google Scholar]
- Smith, G.D.; Paul, W.; Yoon, D.Y.; Zirkel, A.; Hendricks, J.; Richter, D.; Schober, H. Local dynamics in a long-chain alkane melt from molecular dynamics simulations and neutron scattering experiments. J. Chem. Phys. 1997, 107, 4751–4755. [Google Scholar] [CrossRef]
- Paul, W.; Smith, G.; Yoon, D.; Farago, B.; Rathgeber, S.; Zirkel, A.; Willner, L.; Richter, D. Chain motion in an unentangled polyethylene melt: A critical test of the Rouse model by molecular dynamics simulations and neutron spin echo spectroscopy. Phys. Rev. Lett. 1998, 80, 2346. [Google Scholar] [CrossRef]
- Eilhard, J.; Zirkel, A.; Tschop, W.; Hahn, O.; Kremer, K.; Scharpf, O.; Richter, D.; Buchenau, U. Spatial correlations in polycarbonates: Neutron scattering and simulation. J. Chem. Phys. 1999, 110, 1819. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.; Paul, W.; Monkenbusch, M.; Richter, D. A comparison of neutron scattering studies and computer simulations of polymer melts. Chem. Phys. 2000, 261, 61. [Google Scholar] [CrossRef]
- Smith, G.; Paul, W.; Monkenbusch, M.; Richter, D. On the non-Gaussianity of chain motion in unentangled polymer melts. J. Chem. Phys. 2001, 114, 4285. [Google Scholar] [CrossRef] [Green Version]
- Triolo, A.; Arrighi, V.; Triolo, R.; Passerini, S.; Mastragostino, M.; Lechner, R.; Ferguson, R.; Borodin, O.; Smith, G. Dynamic heterogenity in polymer electrolytes. Comparison between QENS data and MD simulations. Physica B 2001, 301, 163. [Google Scholar] [CrossRef]
- Ahumada, O.; Theodorou, D.; Triolo, A.; Arrighi, V.; Karatasos, C.; Ryckaert, J. Segmental dynamics of atactic polypropylene as revealed by molecular simulations and quasielastic neutron scattering. Macromolecules 2002, 35, 7110. [Google Scholar] [CrossRef] [Green Version]
- Arialdi, G.; Karatasos, C.; Ryckaert, J.; Arrighi, V.; Saggio, F.; Triolo, A.; Desmedt, A.; Pieper, J.; Lechner, R. Local dynamics of polyethylene and its oligomers: A molecular dynamics interpretation of the incoherent dynamic structure factor. Macromolecules 2003, 36, 8864. [Google Scholar] [CrossRef]
- Paul, W.; Smith, G. Structure and dynamics of amorphous polymers: Computer simulations compared to experiment and theory. Rep. Prog. Phys. 2004, 67, 1117. [Google Scholar] [CrossRef]
- Tengroth, C.; Engberg, D.; Wahnström, G.; Börjesson, L.; Carlsson, P.; Ahlström, P.; Howells, W.S. The segmental and rotational dynamics of PPO, above the glass-transition, investigated by neutron scattering and molecular dynamics simulations. Soft Mater. 2005, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Depa, P.; García-Sakai, V.; Maranas, J.; Lynn, J.; Peral, I.; Copley, J. A comparison of united atom, explicit atom, and coarse-grained simulation models for poly(ethylene oxide). J. Chem. Phys. 2006, 124, 234901. [Google Scholar] [CrossRef]
- Chen, C.; Depa, P.; Maranas, J.; García-Sakai, V. Comparison of explicit atom, united atom, and coarse-grained simulations of poly(methyl methacrylate). J. Chem. Phys. 2008, 128, 124906. [Google Scholar] [CrossRef]
- Frick, B.; Richter, D.; Ritter, C. Structural changes near the glass transition – neutron diffraction on a simple polymer. Europhys. Lett. 1989, 9, 557. [Google Scholar] [CrossRef]
- Genix, A.C.; Arbe, A.; Alvarez, F.; Colmenero, J.; Schweika, W.; Richter, D. Local structure of syndiotactic poly(methyl methacrylate). A combined study by neutron diffraction with polarization analysis and atomistic molecular dynamics simulations. Macromolecules 2006, 39, 3947–3958. [Google Scholar] [CrossRef] [Green Version]
- Arbe, A.; Genix, A.C.; Colmenero, J.; Richter, D.; Fouquet, P. Anomalous relaxation of self-assembled alkyl nanodomains in high-order poly(n-alkyl methacrylates). Soft Matter 2008, 4, 1792–1795. [Google Scholar] [CrossRef]
- Sundarajan, P.; Flory, P. Configurational characteristics of poly (methyl methacrylate). J. Am. Chem. Soc. 1974, 96, 5025. [Google Scholar] [CrossRef]
- Alvarez, F.; Colmenero, J.; Zorn, R.; Willner, L.; Richter, D. Partial structure factors of polyisoprene: Neutron scattering and molecular dynamics simulation. Macromolecules 2003, 36, 238. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, F.; Arbe, A.; Colmenero, J.; Zorn, R.; Richter, D. Partial structure factors of a simulated polymer melt. Comput. Mater. Sci. 2002, 25, 596–606. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A.; Alvarez, F. Short-range order of glassy polymers by neutron diffraction with polarization analysis and MD-simulations. Comm. Powder Diffr. Int. Union Crystallogr. Newsl. 2004, 31, 35. [Google Scholar]
- Iradi, I.; Alvarez, F.; Colmenero, J.; Arbe, A. Structure factors in polystyrene: A neutron scattering and MD-simulation study. Physica B 2004, 350, 881. [Google Scholar] [CrossRef]
- Narros, A.; Arbe, A.; Alvarez, F.; Colmenero, J.; Zorn, R.; Schweika, W.; Richter, D. Partial Structure Factors in 1,4-Polybutadiene. A Combined Neutron Scattering and Molecular Dynamics Simulations Study. Macromolecules 2005, 38, 9847–9853. [Google Scholar] [CrossRef] [Green Version]
- Khairy, Y.; Alvarez, F.; Arbe, A.; Colmenero, J. Collective features in polyisobutylene. A study of the static and dynamic structure factor by molecular dynamics simulations. Macromolecules 2014, 47, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Colmenero, J.; Moreno, A.J.; Alegría, A. Neutron scattering investigations on methyl group dynamics in polymers. Progr. Polym. Sci. 2005, 30, 1147–1184. [Google Scholar] [CrossRef] [Green Version]
- Chahid, A.; Alegría, A.; Colmenero, J. Methyl group dynamics in poly(vinyl methyl ether). A rotation rate distribution model. Macromolecules 1994, 27, 3282–3288. [Google Scholar] [CrossRef]
- Alvarez, F.; Alegría, A.; Colmenero, J.; Nicholson, T.M.; Davis, G. Origin of the distribution of potential barriers for methyl group dynamics in glassy polymers: A molecular dynamics simulation in polyisoprene. Macromolecules 2000, 33, 8077–8084. [Google Scholar] [CrossRef]
- Alvarez, F.; Arbe, A.; Colmenero, J. Methyl group dynamics above the glass transition temperature: A molecular dynamics simulation in polyisoprene. Chem. Phys. 2000, 261, 47–59. [Google Scholar] [CrossRef]
- Genix, A.C.; Arbe, A.; Alvarez, F.; Colmenero, J.; Farago, B.; Wischnewski, A.; Richter, D. Self- and collective dynamics of syndiotactic poly(methyl methacrylate). A combined study by quasielastic neutron scattering and atomistic molecular dynamics simulations. Macromolecules 2006, 39, 6260–6272. [Google Scholar] [CrossRef] [Green Version]
- Arbe, A.; Buchenau, U.; Willner, L.; Richter, D.; Farago, B.; Colmenero, J. Study of the dynamic structure factor in the β relaxation regime of polybutadiene. Phys. Rev. Lett. 1996, 76, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Arbe, A.; Richter, D.; Colmenero, J.; Farago, B. Merging of the α and β relaxations in polybutadiene: A neutron spin echo and dielectric study. Phys. Rev. E 1996, 54, 3853–3869. [Google Scholar] [CrossRef]
- Aouadi, A.; Lebon, M.J.; Dreyfus, C.; Strube, B.; Steffen, W.; Patkowski, A.; Pick, R.M. A light-scattering stdy of 1,4 cis-trans polybutadiene. J. Phys. Cond. Mat. 1997, 9, 3803. [Google Scholar] [CrossRef]
- Ding, Y.; Novikov, V.; Sokolov, A. Unusual relaxation process in polybutadiene: Resolving the controversy. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 994. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A.; Alvarez, F.; Narros, A.; Monkenbusch, M.; Richter, D. The decisive influence of local chain dynamics on the overall dynamic structure factor close to the glass transition. Europhys. Lett. 2005, 71, 262–268. [Google Scholar] [CrossRef]
- Narros, A.; Arbe, A.; Alvarez, F.; Colmenero, J.; Richter, D. Atomic motions in the αβ-merging region of 1,4-polybutadiene: A molecular dynamics simulation study. J. Chem. Phys. 2008, 128, 224905. [Google Scholar] [CrossRef]
- Colmenero, J.; Alegría, A.; Arbe, A.; Frick, B. Correlation between non-Debye behavior and Q-behavior of the α relaxation in glass-forming polymeric systems. Phys. Rev. Lett. 1992, 69, 478–481. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J.; Monkenbusch, M.; Richter, D. Dynamics of glass-forming polymers: “homogeneous” versus “heterogeneous” scenario. Phys. Rev. Lett. 1998, 81, 590–593. [Google Scholar] [CrossRef]
- Kob, W.; Andersen, H.C. Testing mode-coupling theory for a supercooled binary Lennard-Jones mixture I: The van Hove correlation function. Phys. Rev. E 1995, 51, 4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kob, W.; Donati, C.; Plimpton, S.J.; Poole, P.H.; Glotzer, S.C. Dynamical heterogeneities in a supercooled Lennard-Jones liquid. Phys. Rev. Lett. 1997, 79, 2827. [Google Scholar] [CrossRef] [Green Version]
- Sciortino, F.; Gallo, P.; Tartaglia, P.; Chen, S.H. Supercooled water and the kinetic glass transition. Phys. Rev. E 1996, 54, 6331. [Google Scholar] [CrossRef] [PubMed]
- Mossa, S.; Leonardo, R.D.; Ruocco, G.; Sampoli, M. Molecular dynamics simulation of the fragile glass-former orthoterphenyl: A flexible molecule model. Phys. Rev. E 2000, 62, 612. [Google Scholar] [CrossRef] [Green Version]
- Caprion, D.; Matsui, J.; Schober, H.R. Dynamic heterogeneity of relaxations in glasses and liquids. Phys. Rev. Lett. 2000, 85, 4293. [Google Scholar] [CrossRef]
- Caprion, D.; Matsui, J.; Schober, H.R. Structure and relaxation in liquid and amorphous selenium. Phys. Rev. B 2000, 62, 3709. [Google Scholar] [CrossRef] [Green Version]
- Arbe, A.; Colmenero, J.; Alvarez, F.; Monkenbusch, M.; Richter, D.; Farago, B.; Frick, B. Non-Gaussian nature of the α relaxation of glass-forming polyisoprene. Phys. Rev. Lett. 2002, 89, 245701. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J.; Alvarez, F.; Monkenbusch, M.; Richter, D.; Farago, B.; Frick, B. Experimental evidence by neutron scattering of a crossover from Gaussian to non-Gaussian behavior in the α relaxation of polyisoprene. Phys. Rev. E 2003, 67, 051802. [Google Scholar] [CrossRef] [Green Version]
- Colmenero, J.; Alvarez, F.; Arbe, A. Self-motion and the α relaxation in a simulated glass-forming polymer: Crossover from Gaussian to non-Gaussian dynamic behavior. Phys. Rev. E 2002, 65, 041804. [Google Scholar] [CrossRef]
- Singwi, K.S.; Sjölander, A. Diffusive motions in water and cold neutron scattering. Phys. Rev. 1960, 119, 863. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A.; Alvarez, F.; Narros, A.; Richter, D.; Monkenbusch, M.; Farago, B. Hydrogen motions and the α-relaxation in glass-forming polymers: Molecular dynamics simulation and quasi-elastic neutron scattering results. PRAMANA J. Phys. 2004, 63, 25. [Google Scholar] [CrossRef]
- Narros, A.; Alvarez, F.; Arbe, A.; Colmenero, J.; Richter, D.; Farago, B. Hydrogen motions in the α-relaxation regime of poly(vinyl ethylene): A molecular dynamics simulation and neutron scattering study. J. Chem. Phys. 2004, 121, 3282–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capponi, S.; Arbe, A.; Cerveny, S.; Busselez, R.; Frick, B.; Embs, J.P.; Colmenero, J. Quasielastic neutron scattering study of hydrogen motions in an aqueous poly(vinyl methyl ether) solution. J. Chem. Phys. 2011, 134, 204906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genix, A.C.; Arbe, A.; Colmenero, J.; Wuttke, J.; Richter, D. Neutron scattering and X-ray Investigation of the structure and dynamics of poly(ethyl methacrylate). Macromolecules 2012, 45, 2522–2536. [Google Scholar] [CrossRef]
- Arbe, A.; Rubio, J.; Malo de Molina, P.; Maiz, J.; Pomposo, J.A.; Fouquet, P.; Prevost, S.; Juranyi, F.; Khaneft, M.; Colmenero, J. Melts of single-chain nanoparticles: A neutron scattering investigation. J. Appl. Phys. 2020, 127, 044305. [Google Scholar] [CrossRef]
- Götze, W. Recent tests of the mode-coupling theory for glassy dynamics. J. Phys. Cond. Matter 1999, 11, A1–A45. [Google Scholar] [CrossRef]
- Götze, W. Complex Dynamics of Glass-Forming Liquids. A Mode Coupling Theory; Oxford University Press: New York, NY, USA, 2009. [Google Scholar]
- Richter, D.; Frick, B.; Farago, B. Neutron-spin-echo investigation on the dynamics of polybutadiene near the glass transition. Phys. Rev. Lett. 1988, 61, 2465–2468. [Google Scholar] [CrossRef]
- Zorn, R.; Richter, D.; Farago, B.; Frick, B.; Kremer, F.; Kirst, U.; Fetters, L. Comparative study of the segmental relaxation in polyisoprene by quasi-elastic neutron scattering and dielectric spectroscopy. Physica B 1992, 180, 534–536. [Google Scholar] [CrossRef]
- Richter, D.; Arbe, A.; Colmenero, J.; Monkenbusch, M.; Farago, B.; Faust, R. Molecular motions in polyisobutylene: A neutron spin-echo and dielectric investigation. Macromolecules 1998, 31, 1133–1143. [Google Scholar] [CrossRef]
- Farago, B.; Arbe, A.; Colmenero, J.; Faust, R.; Buchenau, U.; Richter, D. Intermediate length scale dynamics of polyisobutylene. Phys. Rev. E 2002, 65, 051803. [Google Scholar] [CrossRef] [Green Version]
- Arrighi, V.; Pappas, C.; Triolo, A.; Pouget, S. Temperature dependence of local chain dynamics in atactic polypropylene: A neutron spin-echo study. Physica B 2001, 301, 157–162. [Google Scholar] [CrossRef]
- Faivre, A.; Levelut, C.; Durand, D.; Longeville, S.; Ehlers, G. Influence of the microstructure of PPO-based glass-formers on their dynamics as investigated by neutron spin echo. J. Non Cryst. Solids 2002, 307–310, 712–718. [Google Scholar] [CrossRef]
- Arbe, A.; Moral, A.; Alegría, A.; Colmenero, J.; Pyckhout-Hintzen, W.; Richter, D.; Farago, B.; Frick, B. Heterogeneous structure of poly(vinyl chloride) as the origin of anomalous dynamical behavior. J. Chem. Phys. 2002, 117, 1336–1350. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Aparicio, R.; Arbe, A.; Alvarez, F.; Colmenero, J.; Willner, L. Quasielastic neutron scattering and molecular dynamics simulation study on the structure factor of poly(ethylene-alt-propylene). Macromolecules 2009, 42, 8271–8285. [Google Scholar] [CrossRef]
- Arbe, A.; Rubio-Cervilla, J.; Alegría, A.; Moreno, A.J.; Pomposo, J.A.; Robles-Hernández, B.; Malo de Molina, P.; Fouquet, P.; Juranyi, F.; Colmenero, J. Mesoscale dynamics in melts of single-chain polymeric nanoparticles. Macromolecules 2019, 52, 6935–6942. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A.; Richter, D.; Farago, B.; Monkenbusch, M. Neutron spin echo spectroscopy. Basics, trends and applications; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2003; Volume 601. [Google Scholar]
- Colmenero, J.; Narros, A.; Alvarez, F.; Arbe, A.; Moreno, A.J. Atomic motions in the αβ-region of glass-forming polymers: Molecular versus mode coupling theory approach. J. Phys. Cond. Matter 2006, 19, 205127. [Google Scholar] [CrossRef] [Green Version]
- Capponi, S.; Arbe, A.; Alvarez, F.; Colmenero, J.; Frick, B.; Embs, J.P. Atomic motions in poly(vinyl methyl ether): A combined study by quasielastic neutron scattering and molecular dynamics simulations in the light of the mode coupling theory. J. Chem. Phys. 2009, 131, 204901. [Google Scholar] [CrossRef] [Green Version]
- Khairy, Y.; Alvarez, F.; Arbe, A.; Colmenero, J. Applicability of mode-coupling theory to polyisobutylene: A molecular dynamics simulation study. Phys. Rev. E 2013, 88, 042302. [Google Scholar] [CrossRef] [Green Version]
- Zorn, R.; Richter, D.; Frick, B.; Farago, B. Neutron scattering experiments on the glass transition of polymers. Physica A 1993, 201, 52–66. [Google Scholar] [CrossRef]
- Bergman, R.; Borjesson, L.; Torell, L.M.; Fontana, A. Dynamics around the liquid-glass transition in poly(propylene-glycol) investigated by wide-frequency-range light-scattering techniques. Phys. Rev. B 1997, 56, 11619. [Google Scholar] [CrossRef]
- Kisliuk, A.; Mathers, R.T.; Sokolov, A.P. Crossover in dynamics of polymeric liquids: Back to Tll? J. Polym. Sci. Part B Polym. Phys. 2000, 38, 2785–2790. [Google Scholar] [CrossRef]
- Bernabei, M.; Moreno, A.J.; Colmenero, J. Dynamic arrest in polymer melts: Competition between packing and intramolecular barriers. Phys. Rev. Lett. 2008, 101, 255701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, D.; Monkenbusch, M.; Allgaier, J.; Arbe, A.; Colmenero, J.; Farago, B.; Cheol Bae, Y.; Faust, R. From Rouse dynamics to local relaxation: A neutron spin echo study on polyisobutylene melts. J. Chem. Phys. 1999, 111, 6107. [Google Scholar] [CrossRef]
- Arbe, A.; Monkenbusch, M.; Stellbrink, J.; Richter, D.; Farago, B.; Almdal, K.; Faust, R. Origin of internal viscosity effects in flexible polymers: A comparative neutron spin-echo and light scattering study on poly(dimethylsiloxane) and polyisobutylene. Macromolecules 2001, 34, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Richter, D.; Monkenbusch, M.; Willner, L.; Arbe, A.; Colmenero, J.; Farago, B. Direct observation of the crossover from α-relaxation to Rouse dynamics in a polymer melt. Europhys. Lett. 2004, 66, 239. [Google Scholar] [CrossRef]
- Smith, G.D.; Paul, W.; Monkenbusch, M.; Willner, L.; Richter, D.; Qiu, X.H.; Ediger, M.D. Molecular dynamics of a 1,4-polybutadiene melt. Comparison of experiment and simulation. Macromolecules 1999, 32, 8857. [Google Scholar] [CrossRef]
- Zamponi, M.; Wischnewski, A.; Monkenbusch, M.; Willner, L.; Richter, D.; Falus, P.; Farago, B.; Guenza, M.G. Cooperative dynamics in homopolymer melts: A comparison of theoretical predictions with neutron spin echo experiments. J. Phys. Chem. B 2008, 112, 16220. [Google Scholar] [CrossRef]
- Brodeck, M.; Alvarez, F.; Arbe, A.; Juranyi, F.; Unruh, T.; Holderer, O.; Colmenero, J.; Richter, D. Study of the dynamics of poly(ethylene oxide) by combining molecular dynamic simulations and neutron scattering experiments. J. Chem. Phys. 2009, 130, 094908. [Google Scholar] [CrossRef]
- Pérez-Aparicio, R.; Alvarez, F.; Arbe, A.; Willner, L.; Richter, D.; Falus, P.; Colmenero, J. Chain dynamics of unentangled poly(ethylene-alt-propylene) melts by means of neutron scattering and fully atomistic molecular dynamics simulations. Macromolecules 2011, 44, 3129–3139. [Google Scholar] [CrossRef] [Green Version]
- Brueckner, S. Configurational statistics of polymer chains accounting for all rotational states. General extension to finite chains and copolymers. Macromolecules 1981, 14, 449. [Google Scholar] [CrossRef]
- Pérez Aparicio, R.; Colmenero, J.; Alvarez, F.; Padding, J.T.; Briels, W.J. Chain dynamics of poly(ethylene-alt-propylene) melts by means of coarse-grained simulations based on atomistic molecular dynamics. J. Chem. Phys. 2010, 132, 024904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodeck, M.; Alvarez, F.; Colmenero, J.; Richter, D. Single chain dynamic structure factor of poly(ethylene oxide) in dynamically asymmetric blends with poly(methyl methacrylate). Neutron scattering and molecular dynamics simulations. Macromolecules 2011, 45, 536. [Google Scholar] [CrossRef]
- Heijboer, J. Physics of Non-Crystalline Solids; North-Holland: Amsterdam, The Netherlands, 1965; p. 231. [Google Scholar]
- McCrum, N.; Read, B.; Williams, G. Anelastic, Dielectric Effects in Polymeric Solids; Wiley: London, UK, 1967. [Google Scholar]
- Cowie, J. Relaxation processes in the glassy state: Molecular aspects. J. Macromol. Sci. Phys. B 1980, 18, 569. [Google Scholar] [CrossRef]
- Meier, G.; Fytas, G.; Dorfmüller, T. Photon correlation study of the relaxation processes in bulk poly(n-butyl methacrylate). Macromolecules 1984, 17, 957. [Google Scholar] [CrossRef]
- Giebel, L.; Meier, G.; Fytas, G.; Fischer, E.W. Dynamic light scattering from bulk poly(n-hexylmethacrylate) near and above the glass transition. J. Polym. Sci. B Polym. Phys. 1992, 30, 1291. [Google Scholar] [CrossRef]
- Garwe, F.; Schönhals, A.; Lockwenz, H.; Beiner, M.; Schröter, K.; Donth, E. Influence of cooperative α dynamics on local β relaxation during the development of the dynamic glass transition in poly(n-alkyl methacrylate)s. Macromolecules 1996, 29, 247. [Google Scholar] [CrossRef]
- Schröter, K.; Unger, R.; Reissig, S.; Garwe, F.; Kahle, S.; Beiner, M.; Donth, E. Dielectric spectroscopy in the αβ splitting region of glass transition in poly(ethyl methacrylate) and poly(n-butyl methacrylate): Different evaluation methods and experimental conditions. Macromolecules 1998, 31, 8966. [Google Scholar] [CrossRef]
- Floudas, G.; Stepanek, P. Structure and dynamics of poly(n-decyl methacrylate) below and above the glass transition. Macromolecules 1998, 31, 6951. [Google Scholar] [CrossRef]
- Beiner, M.; Schröter, K.; Hempel, E.; Reissig, S.; Donth, E. Multiple glass transition and nanophase separation in poly(n-alkyl methacrylate) homopolymers. Macromolecules 1999, 32, 6278. [Google Scholar] [CrossRef]
- Dudognon, E.; Bernès, A.; Lacabanne, C. Study by thermostimulated currents of dielectric relaxations through the glass transition in an amorphous polymer: poly(n-butyl methacrylate). Macromolecules 2001, 34, 3988. [Google Scholar] [CrossRef]
- Dudognon, E.; Bernès, A.; Lacabanne, C. Low-frequency chain dynamics of poly(n-hexyl methacrylate) by dielectric spectroscopies. Macromolecules 2002, 35, 5927. [Google Scholar] [CrossRef]
- Beiner, M. Relaxation in poly (alkyl methacrylate)s: Crossover region and nanophase separation. Macromol. Rapid Commun. 2001, 22, 869. [Google Scholar] [CrossRef]
- Beiner, M.; Kabisch, O.; Reichl, S.; Huth, H. Structural and dynamic nanoheterogeneities in higher poly(alkyl methacrylate)s. J. Non Cryst. Sol. 2002, 307-310, 658. [Google Scholar] [CrossRef]
- Hempel, E.; Beiner, M.; Huth, H.; Donth, E. Temperature modulated DSC for the multiple glass transition in poly (n-alkyl methacrylates). Thermochimica Acta 2002, 391, 219. [Google Scholar] [CrossRef]
- Pascui, O.; Beiner, M.; Reichert, D. Identification of slow dynamic processes in poly(n-hexyl methacrylate) by solid-state 1D-MAS exchange NMR. Macromolecules 2003, 36, 3992. [Google Scholar] [CrossRef]
- Beiner, M.; Huth, H. Nanophase separation and hindered glass transition in side-chain polymers. Nat. Mater. 2003, 2, 595. [Google Scholar] [CrossRef]
- Hiller, S.; Pascui, O.; Kabisch, O.; Reichert, D.; Beiner, M. Nanophase separation in side chain polymers: New evidence from structure and dynamics. New J. Phys. 2004, 6, 10. [Google Scholar] [CrossRef]
- Menissez, C.; Sixou, B.; David, L.; Vigier, G. Dielectric and mechanical relaxation behavior in poly(butyl methacrylate) isomers. J. Non Cryst. Sol. 2005, 351, 595. [Google Scholar] [CrossRef]
- Wind, M.; Graf, R.; Renker, S.; Spiess, H.; Steffen, W. Structure of amorphous poly-(ethylmethacrylate): A wide-angle X-ray scattering study. J. Chem. Phys. 2005, 122, 014906. [Google Scholar] [CrossRef]
- Beiner, M. CP832, Flow Dynamics, The Second International Conference on Flow Dynamics; American Institute of Physics: College Park, MD, USA, 2006; p. 134. [Google Scholar]
- Arbe, A.; Genix, A.C.; Arrese-Igor, S.; Colmenero, J.; Richter, D. Dynamics in poly(n-alkyl methacrylates): A neutron scattering, calorimetric, and dielectric study. Macromolecules 2010, 43, 3107. [Google Scholar] [CrossRef] [Green Version]
- Gaborieau, M.; Graf, R.; Kahle, S.; Pakula, T.; Spiess, H.W. Chain dynamics in poly(n-alkyl acrylates) by solid-state NMR, dielectric, and mechanical spectroscopies. Macromolecules 2007, 40, 6249. [Google Scholar] [CrossRef]
- Cowie, J.; Haq, Z.; McEwen, I.; Velickovic, J. Poly (alkyl itaconates): 8. Observations of dual glass transitions and crystallinity in the dialkyl ester series diheptyl to di-eicosyl. Polymer 1981, 22, 327. [Google Scholar] [CrossRef]
- Arrighi, V.; Triolo, A.; McEwen, I.J.; Holmes, P.; Triolo, R.; Amenitsch, H. Observation of local order in poly(di-n-alkyl itaconate)s. Macromolecules 2000, 33, 4989. [Google Scholar] [CrossRef]
- Genix, A.C.; Lauprêtre, F. Subglass and glass transitions of poly(di-n-alkylitaconate)s with various side-chain lengths: Dielectric relaxation investigation. Macromolecules 2005, 38, 2786. [Google Scholar] [CrossRef]
- Gerstl, C.; Schneider, G.J.; Pyckhout-Hintzen, W.; Allgaier, J.; Richter, D.; Alegría, A.; Colmenero, J. Segmental and normal mode relaxation of poly(alkylene oxide)s studied by dielectric spectroscopy and rheology. Macromolecules 2010, 43, 4968. [Google Scholar] [CrossRef]
- Gerstl, C.; Schneider, G.J.; Fuxman, A.; Zamponi, M.; Frick, B.; Seydel, T.; Koza, M.; Genix, A.C.; Allgaier, J.; Richter, D.; et al. Quasielastic neutron scattering study on the dynamics of poly(alkylene oxide)s. Macromolecules 2012, 45, 4394–4405. [Google Scholar] [CrossRef]
- Gerstl, C.; Brodeck, M.; Schneider, G.J.; Su, Y.; Allgaier, J.; Arbe, A.; Colmenero, J.; Richter, D. Short and intermediate range order in poly(alkylene oxide)s. A neutron diffraction and molecular dynamics simulations study. Macromolecules 2012, 45, 7293–7303. [Google Scholar] [CrossRef]
- Colmenero, J.; Brodeck, M.; Arbe, A.; Richter, D. Dynamics of poly(butylene oxide) well above the glass transition. A fully atomistic molecular dynamics simulation study. Macromolecules 2013, 46, 1678–1685. [Google Scholar] [CrossRef]
- Moreno, A.; Arbe, A.; Colmenero, J. Structure and dynamics of self-assembled comb copolymers: Comparison between simulations of a generic model and neutron scattering experiments. Macromolecules 2011, 44, 1695. [Google Scholar] [CrossRef] [Green Version]
- Busselez, R.; Arbe, A.; Alvarez, F.; Colmenero, J.; Frick, B. Study of the structure and dynamics of poly(vinyl pyrrolidone) by molecular dynamics simulations validated by quasielastic neutron scattering and X-ray diffraction experiments. J. Chem. Phys. 2011, 134, 054904. [Google Scholar] [CrossRef] [Green Version]
- Colmenero, J.; Arbe, A. Segmental dynamics in miscible polymer blends: Recent results and open questions. Soft Matter 2007, 3, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Colmenero, J.; Alvarez, F.; Khairy, Y.; Arbe, A. Modeling the collective relaxation time of glass-forming polymers at intermediate length scales: Application to polyisobutylene. J. Chem. Phys. 2013, 139, 044906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmenero, J.; Alvarez, F.; Arbe, A. Collective dynamics of glass-forming polymers at intermediate length scales—A synergetic combination of neutron scattering, atomistic simulations and theoretical modelling. EPJ Web Conf. 2015, 83, 01001. [Google Scholar] [CrossRef] [Green Version]
- De Gennes, P.G. Liquid dynamics and inelastic scattering of neutrons. Physica 1959, 25, 825. [Google Scholar] [CrossRef]
- Sköld, K. Small energy transfer scattering of cold neutrons from liquid argon. Phys. Rev. Lett. 1967, 19, 1023–1025. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J. Characterization of the “simple-liquid” state in a polymeric system: Coherent and incoherent scattering functions. Phys. Rev. E 2009, 80, 041805. [Google Scholar] [CrossRef]
- Tyagi, M.; Arbe, A.; Alvarez, F.; Colmenero, J.; González, M.A. Short-range order and collective dynamics of poly(vinyl acetate): A combined study by neutron scattering and molecular dynamics simulations. J. Chem. Phys. 2008, 129, 224903. [Google Scholar] [CrossRef]
- Novikov, V.N.; Schweizer, K.S.; Sokolov, A.P. Coherent neutron scattering and collective dynamics on mesoscale. J. Chem.Phys. 2013, 138, 164508. [Google Scholar] [CrossRef] [Green Version]
- Heil, W.; Andersen, K.H.; Cywinski, R.; Humblot, H.; Ritter, C.; Roberts, T.W.; Stewart, J.R. Large solid-angle neutron polarization analysis at thermal neutron wavelengths using a 3He spin-filter. Nucl. Inst. Meth. Phys. Res. A 2002, 485, 551–570. [Google Scholar] [CrossRef]
- Stewart, J.R.; Deen, P.P.; Andersen, K.H.; Schober, H.; Barthélémy, J.F.; Hillier, J.M.; Murani, A.P.; Hayes, T.; Lindenau, B. Disordered materials studied using neutron polarization analysis on the multi-detector spectrometer, D7. J. Appl. Cryst. 2009, 42, 69–84. [Google Scholar] [CrossRef]
- Burankova, T.; Hempelmann, R.; Wildes, A.R.; Embs, J.P. Collective ion diffusion and localized single particle dynamics in pyridinium-based ionic liquids. J. Phys. Chem. B 2014, 118, 14452–14460. [Google Scholar] [CrossRef] [PubMed]
- Casella, G.; Stewart, J.R.; Paternò, G.M.; García-Sakai, V.; Devonport, M.; Galsworthy, P.J.; Bewley, R.I.; Voneshen, D.J.; Raspino, D.; Nilsen, G.J. Polarization analysis on the LET cold neutron spectrometer using a 3He spin-filter: First results. J. Phys. Conf. Ser. 2019, 1316, 012007. [Google Scholar] [CrossRef]
- Chen, W.; Watson, S.; Qiu, Y.; Rodriguez-Rivera, J.A.; Faraone, A. Wide-angle polarization analysis on the multi-axis crystal spectrometer for the study of collective and single particle dynamics of methanol at its prepeak. Phys. B Cond. Matter 2019, 564, 166–171. [Google Scholar] [CrossRef]
- Arbe, A.; Malo de Molina, P.; Alvarez, F.; Frick, B.; Colmenero, J. Dielectric susceptibility of liquid water: Microscopic insights from coherent and incoherent neutron scattering. Phys. Rev. Lett. 2016, 117, 185501. [Google Scholar] [CrossRef] [PubMed]
- Arbe, A.; Colmenero, J.; Richter, D. Broadband Dielectric Spectroscopy; Kremer, F., Schönhals, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; p. 685. [Google Scholar]
- Qvist, J.; Schober, H.; Halle, B. Structural dynamics of supercooled water from quasielastic neutron scattering and molecular simulations. J. Chem. Phys. 2011, 134, 144508. [Google Scholar] [CrossRef]
- Arbe, A.; Moreno, A.J.; Allgaier, J.; Ivanova, O.; Fouquet, P.; Colmenero, J.; Richter, D. Role of dynamic asymmetry on the collective dynamics of comblike polymers: Insights from neutron spin-echo experiments and coarse-grained molecular dynamics simulations. Macromolecules 2016, 49, 4989–5000. [Google Scholar] [CrossRef]
- Moreno, A.J.; Lo Verso, F.; Sanchez-Sanchez, A.; Arbe, A.; Colmenero, J.; Pomposo, J.A. Advantages of orthogonal folding of single polymer chains to soft nanoparticles. Macromolecules 2013, 46, 9748–9759. [Google Scholar] [CrossRef]
- Pomposo, J.A.; Perez-Baena, I.; Lo Verso, F.; Moreno, A.J.; Arbe, A.; Colmenero, J. How far are single-chain polymer nanoparticles in solution from the globular state? ACS Macro Lett. 2014, 3, 767–772. [Google Scholar] [CrossRef]
- Moreno, A.J.; Lo Verso, F.; Arbe, A.; Pomposo, J.A.; Colmenero, J. Concentrated solutions of single-chain nanoparticles: A simple model for intrinsically disordered proteins under crowding conditions. J. Phys. Chem. Lett. 2016, 7, 838–844. [Google Scholar] [CrossRef] [Green Version]
- González-Burgos, M.; Arbe, A.; Moreno, A.J.; Pomposo, J.A.; Radulescu, A.; Colmenero, J. Crowding the environment of single-chain nanoparticles: A combined study by SANS and simulations. Macromolecules 2018, 51, 1573–1585. [Google Scholar] [CrossRef] [Green Version]
- Arbe, A.; Pomposo, J.; Moreno, A.; LoVerso, F.; González-Burgos, M.; Asenjo-Sanz, I.; Iturrospe, A.; Radulescu, A.; Ivanova, O.; Colmenero, J. Structure and dynamics of single-chain nano-particles in solution. Polymer 2016, 105, 532–544. [Google Scholar] [CrossRef]
Figure 1.
Cartoon illustrating the relevant structural features of polymers as function of the length scale of observation and magnitudes accessed in neutron diffraction experiments: the single chain structure factor on labelled samples at low scattering angles (left) and the structure factor on fully deuterated samples (right).
Figure 1.
Cartoon illustrating the relevant structural features of polymers as function of the length scale of observation and magnitudes accessed in neutron diffraction experiments: the single chain structure factor on labelled samples at low scattering angles (left) and the structure factor on fully deuterated samples (right).
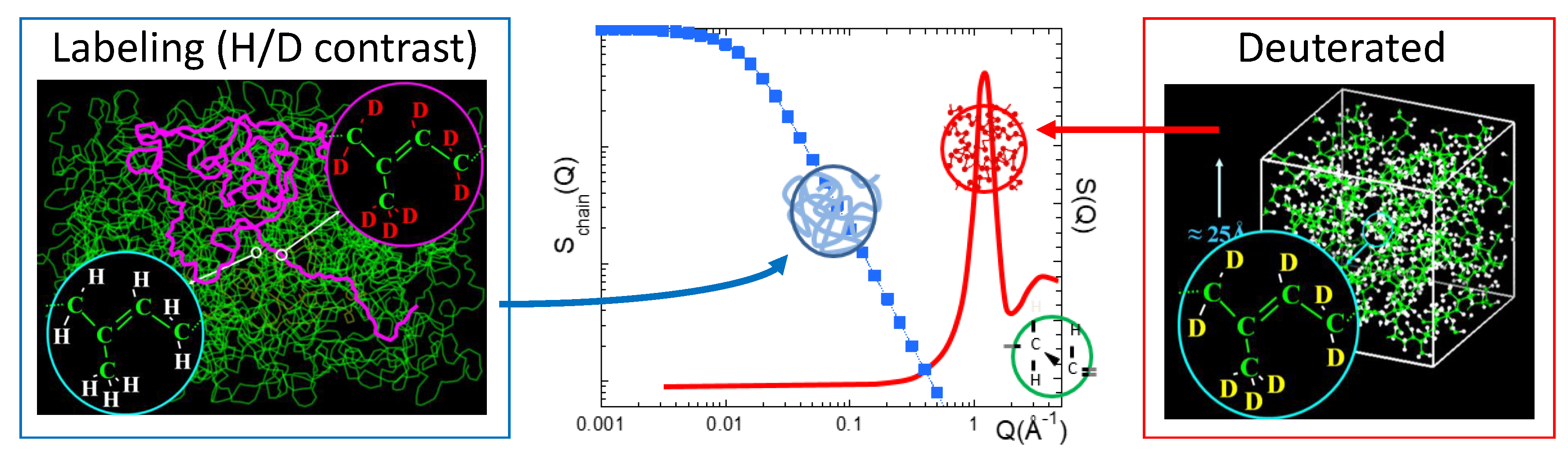
Figure 2.
Illustration of the magnitudes in a scattering experiment and scheme of the functions involved in the different domians in the van Hove formalism of neutron scattering.
Figure 2.
Illustration of the magnitudes in a scattering experiment and scheme of the functions involved in the different domians in the van Hove formalism of neutron scattering.
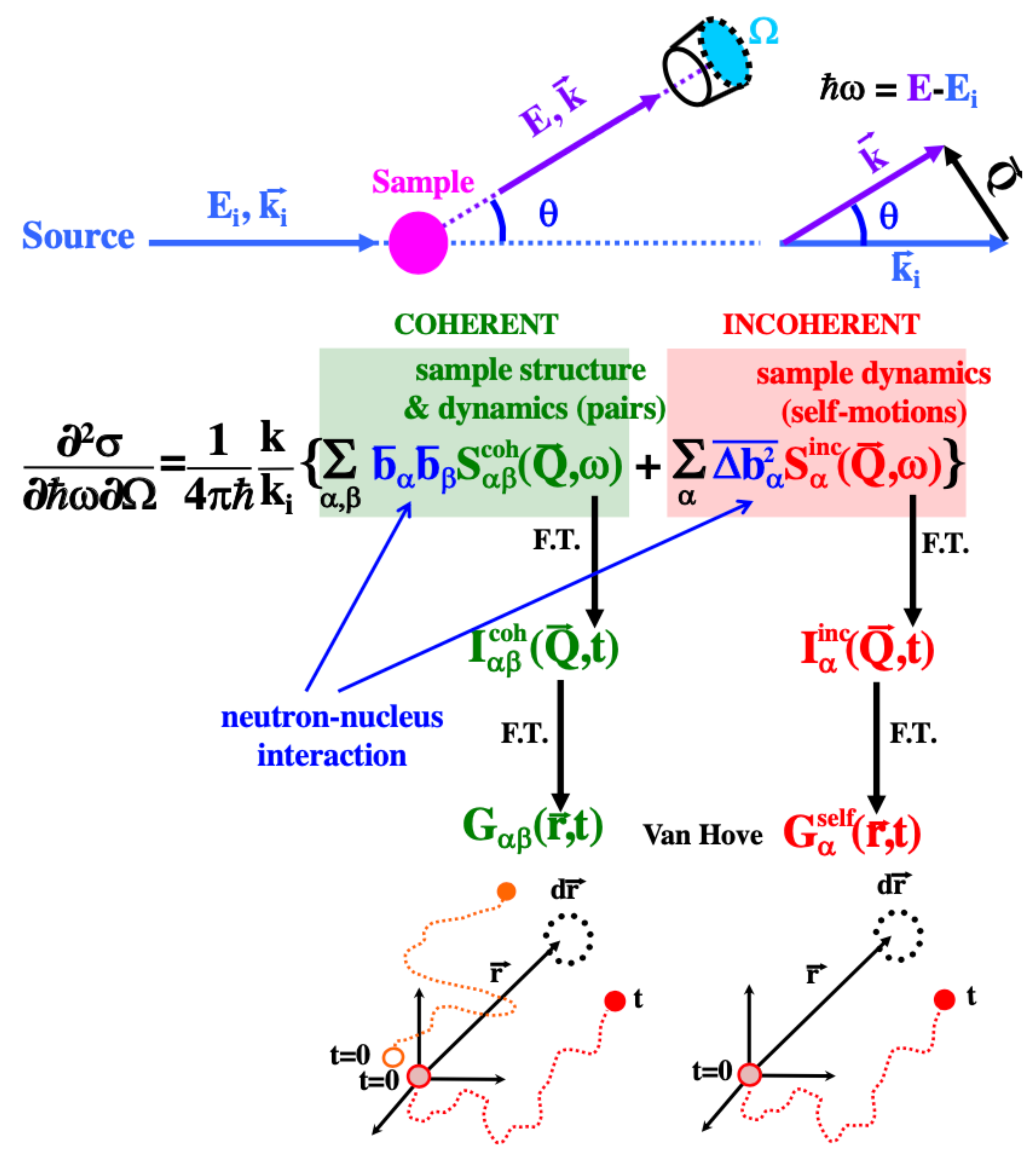
Figure 3.
Block-diagram of the strategy followed for combining NS and MD-simulations.

Figure 4.
Ratio between coherent and incoherent differential cross sections measured with diffraction with PA (points) and calculated from the MD-simulations (lines) on PMMA with deuterated ester methyl group (PMMA-d3e) (a) and PMMA with deuterated -methyl group and main chain (PMMA-d5) (b). The respective coherent cross sections calculated from the simulations with the different molecular group correlations, properly weighted by the corresponding neutron scattering lengths, are shown in panels (c,d). Adapted with permission from [57]. Copyright (2006) American Chemical Society.
Figure 4.
Ratio between coherent and incoherent differential cross sections measured with diffraction with PA (points) and calculated from the MD-simulations (lines) on PMMA with deuterated ester methyl group (PMMA-d3e) (a) and PMMA with deuterated -methyl group and main chain (PMMA-d5) (b). The respective coherent cross sections calculated from the simulations with the different molecular group correlations, properly weighted by the corresponding neutron scattering lengths, are shown in panels (c,d). Adapted with permission from [57]. Copyright (2006) American Chemical Society.

Figure 5.
(a) Radial probability distribution function calculated at 200 K ( + 20 K) and t = 1 ns for the different kinds of hydrogens in the 1,4-units of PB. The upper panel displays the schematic representation of the isomeric forms of 1,4-PB monomers and shows with colors the nomenclature for the different types of hydrogens. (b) Radial probability distribution function calculated for H (methyne hydrogen in the trans unit) at the same temperature at the different times indicated. For clarity the origins are shifted to the levels displayed by the horizontal dotted lines. The solid lines show the description obtained by the model proposed in Refs. [75,76].
Figure 5.
(a) Radial probability distribution function calculated at 200 K ( + 20 K) and t = 1 ns for the different kinds of hydrogens in the 1,4-units of PB. The upper panel displays the schematic representation of the isomeric forms of 1,4-PB monomers and shows with colors the nomenclature for the different types of hydrogens. (b) Radial probability distribution function calculated for H (methyne hydrogen in the trans unit) at the same temperature at the different times indicated. For clarity the origins are shifted to the levels displayed by the horizontal dotted lines. The solid lines show the description obtained by the model proposed in Refs. [75,76].
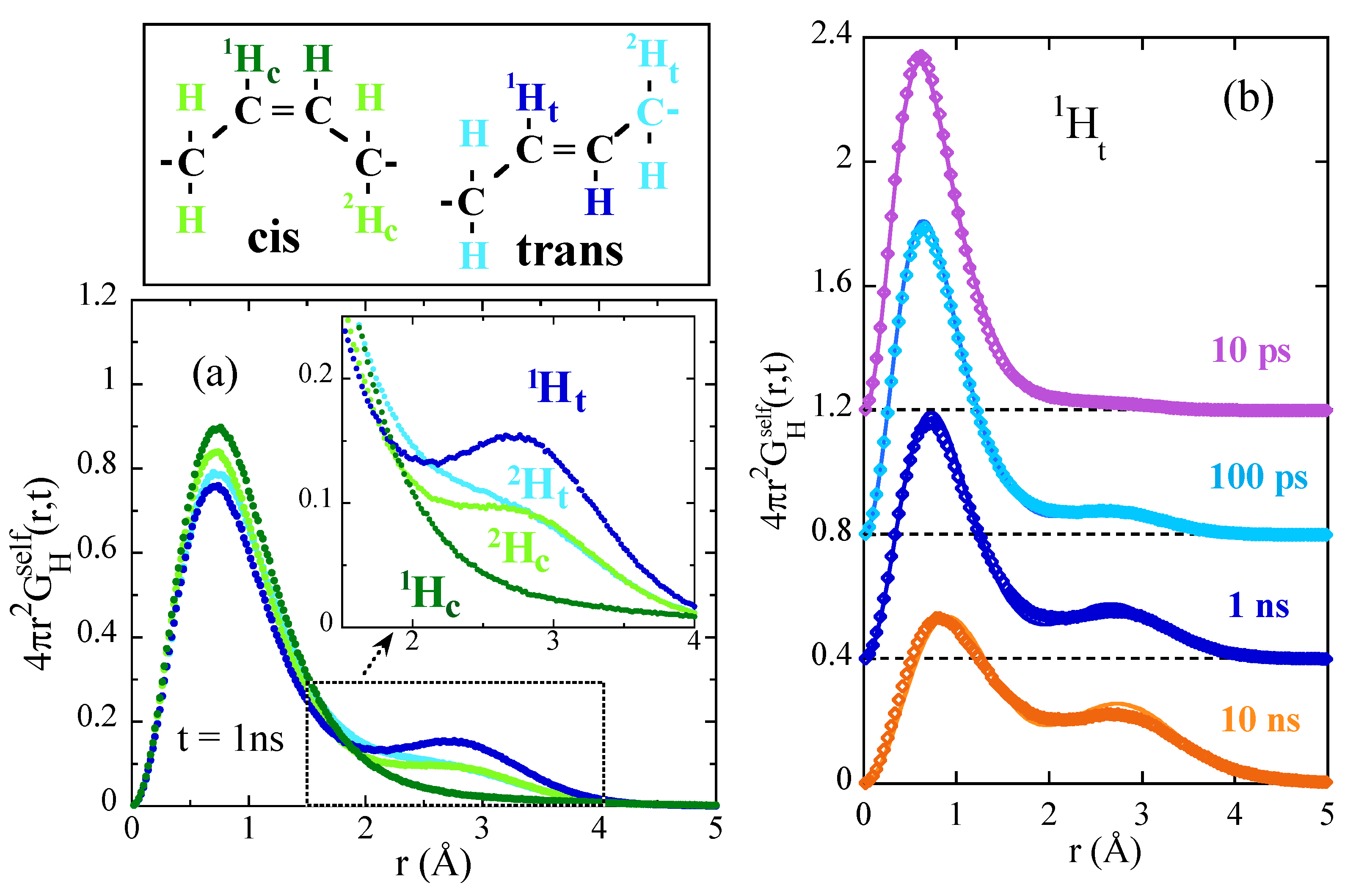
Figure 6.
(a) Master curve giving the Q-dependence of constructed with results from different polymers (see legend; PH:phenoxy), applying polymer-dependent normalizing factors (). The solid line is the Gaussian behavior. (b) Master curve built combining NS from different spectrometers and MD-simulations results for PId3 [85,86], applying T-dependent shift factors . The solid line is the description by the anomalous jump diffusion model with the distribution of jump lengths in the inset. Adapted from [6] with permission from The Royal Society of Chemistry.
Figure 6.
(a) Master curve giving the Q-dependence of constructed with results from different polymers (see legend; PH:phenoxy), applying polymer-dependent normalizing factors (). The solid line is the Gaussian behavior. (b) Master curve built combining NS from different spectrometers and MD-simulations results for PId3 [85,86], applying T-dependent shift factors . The solid line is the description by the anomalous jump diffusion model with the distribution of jump lengths in the inset. Adapted from [6] with permission from The Royal Society of Chemistry.
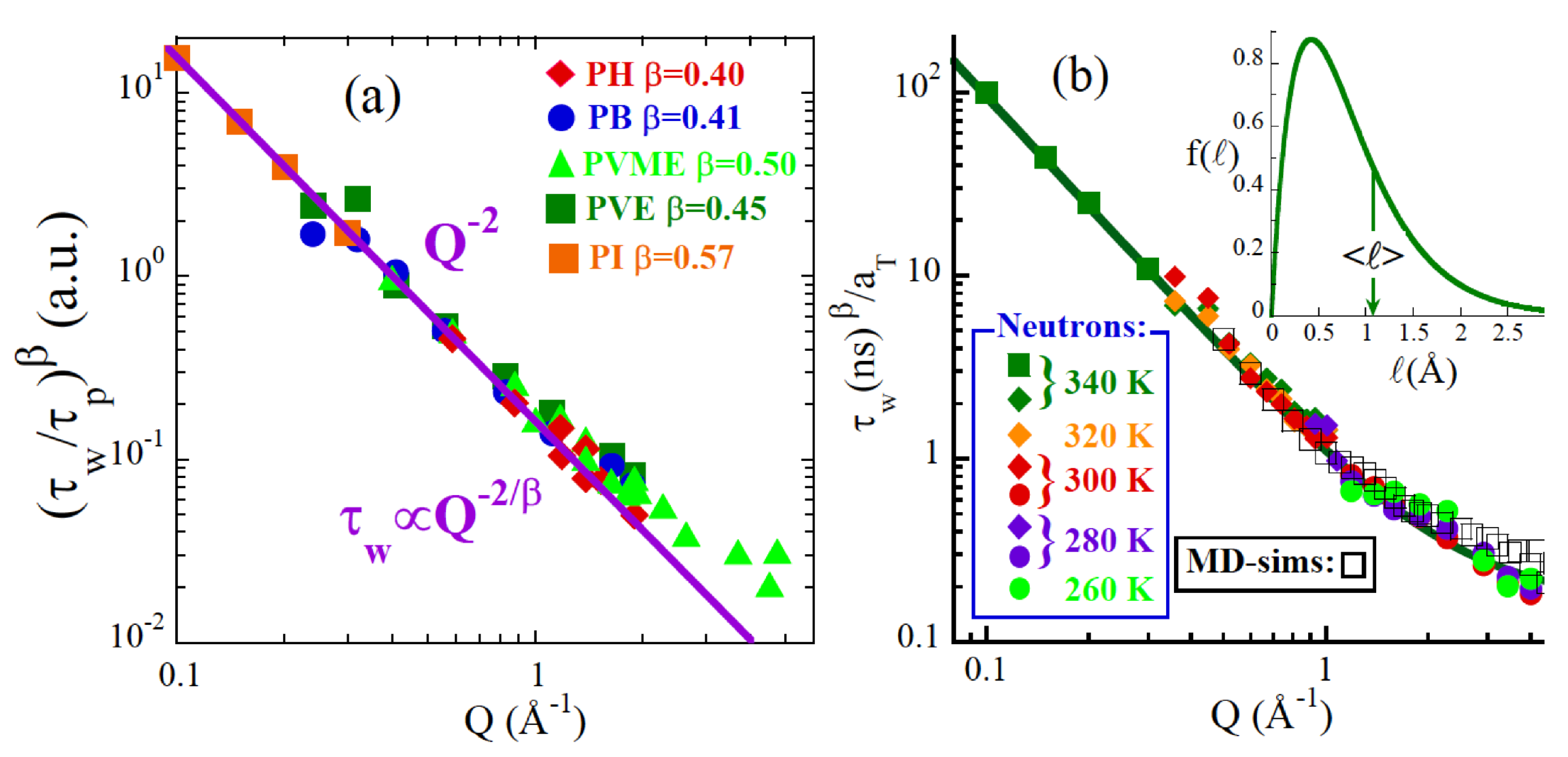
Figure 7.
Comparison of the NSE results on a fully deuterated PIB sample [99] (symbols) and calculated from simulations [65] (solid lines) at 390 K (a) and 335 K (b) at the Q-values indicated in (a). Inset in (b) Q-dependence of the apparent activation energy for collective relaxation obtained from KWW fits of the experimental (solid) and simulated (empty) data. Examples of the fits are shown for . Adapted with permission from [65]. Copyright (2014) American Chemical Society.
Figure 7.
Comparison of the NSE results on a fully deuterated PIB sample [99] (symbols) and calculated from simulations [65] (solid lines) at 390 K (a) and 335 K (b) at the Q-values indicated in (a). Inset in (b) Q-dependence of the apparent activation energy for collective relaxation obtained from KWW fits of the experimental (solid) and simulated (empty) data. Examples of the fits are shown for . Adapted with permission from [65]. Copyright (2014) American Chemical Society.
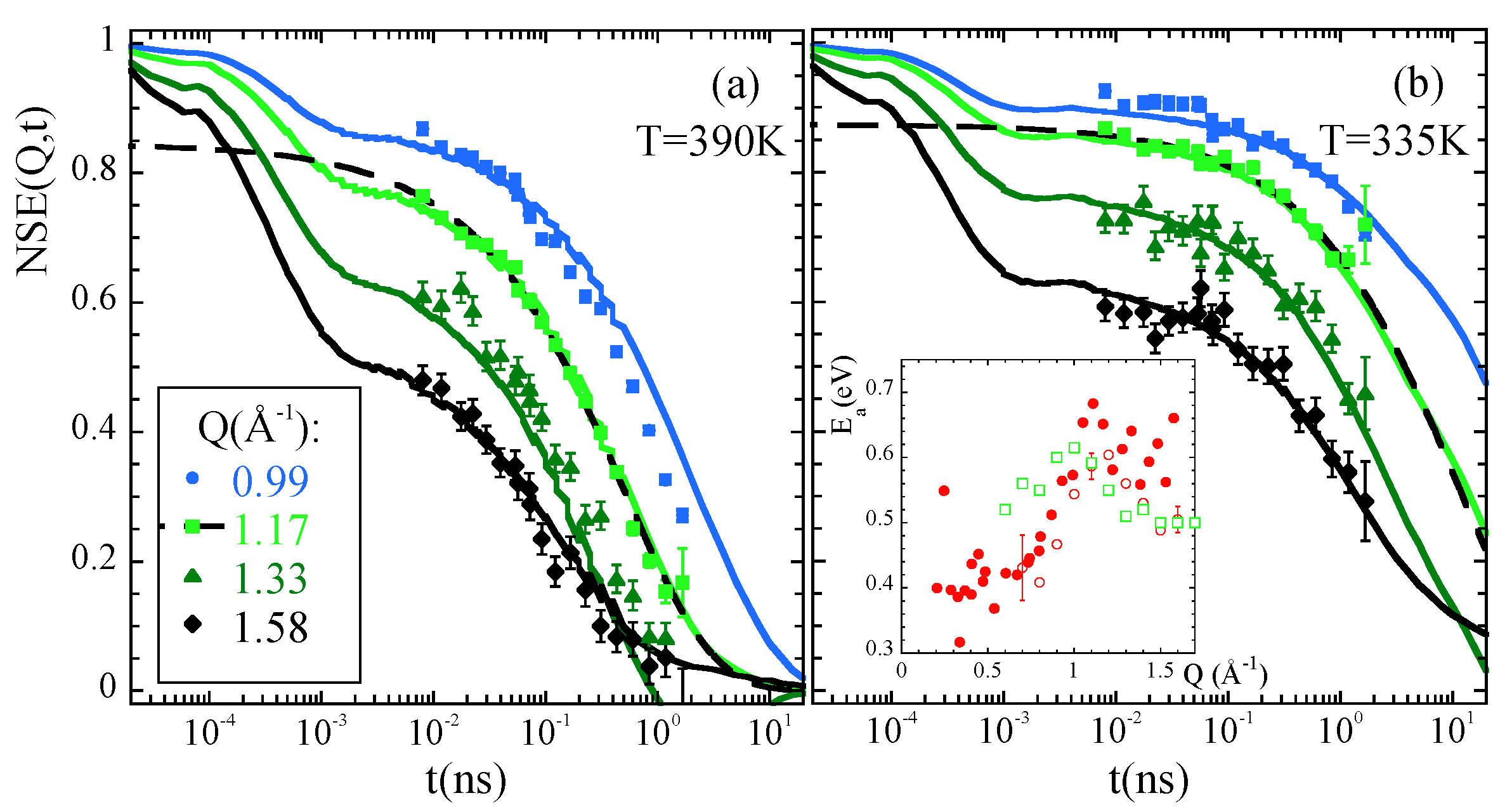
Figure 8.
Normalized partial dynamic structure factors of deuterated PMMA corresponding to the different atomic groups considered [see the insert in (a) for the color code]. The total dynamic structure factor is shown in black. The lines are KWW descriptions for 4 ps. (a) (first structure factor peak); (b) (second structure factor peak). Adapted with permission from [70]. Copyright (2006) American Chemical Society.
Figure 8.
Normalized partial dynamic structure factors of deuterated PMMA corresponding to the different atomic groups considered [see the insert in (a) for the color code]. The total dynamic structure factor is shown in black. The lines are KWW descriptions for 4 ps. (a) (first structure factor peak); (b) (second structure factor peak). Adapted with permission from [70]. Copyright (2006) American Chemical Society.
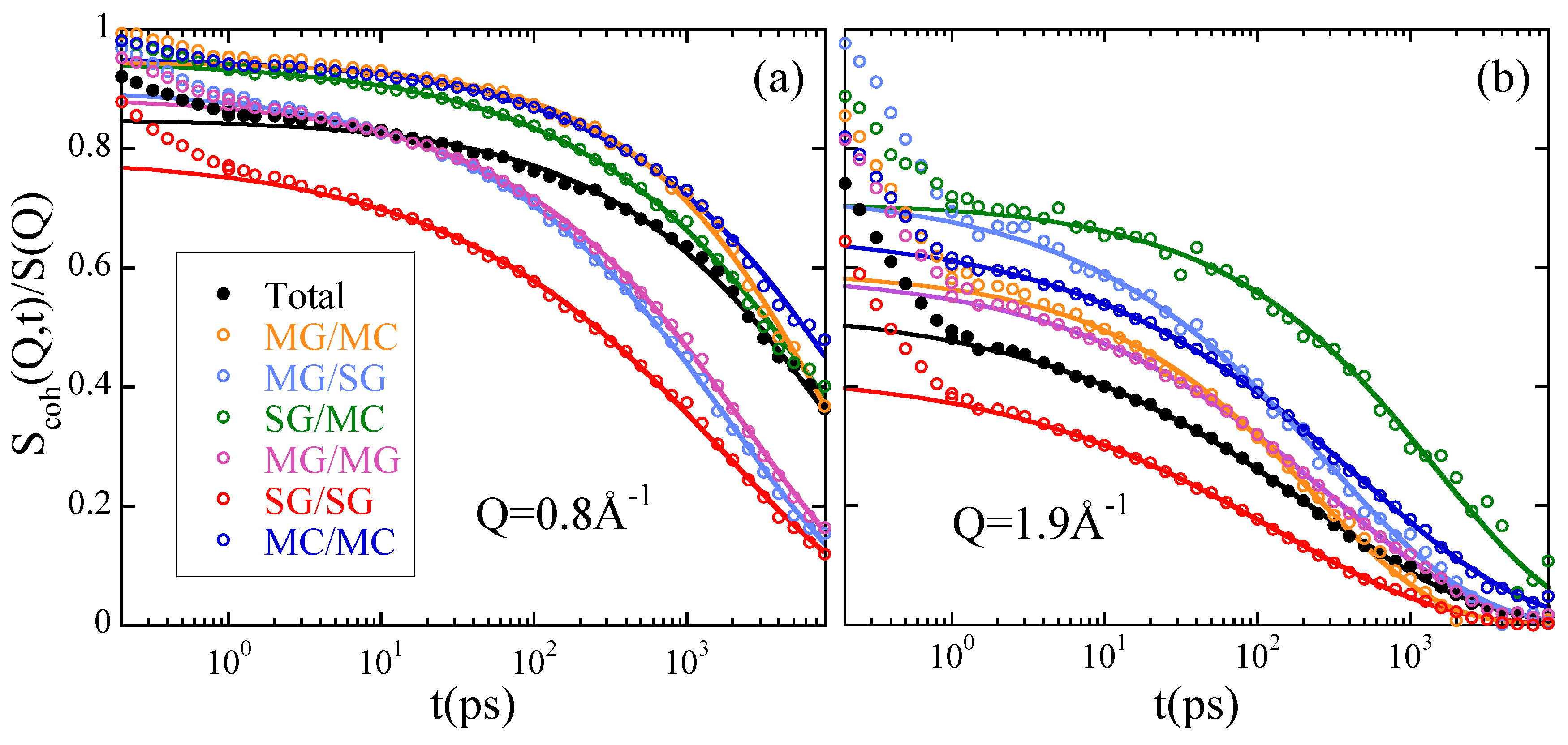
Figure 9.
(a) Normalized Rouse correlators calculated for PEP at + 280 K and the mode-numbers indicated. Lines are fits with stretched exponentials. The obtained stretching parameters and effective friction coefficients (after ARS corrections) are displayed in (b). Rouse behavior corresponds to = 1 and mode-independent friction (solid line). Adapted with permission from [119]. Copyright (2011) American Chemical Society.
Figure 9.
(a) Normalized Rouse correlators calculated for PEP at + 280 K and the mode-numbers indicated. Lines are fits with stretched exponentials. The obtained stretching parameters and effective friction coefficients (after ARS corrections) are displayed in (b). Rouse behavior corresponds to = 1 and mode-independent friction (solid line). Adapted with permission from [119]. Copyright (2011) American Chemical Society.
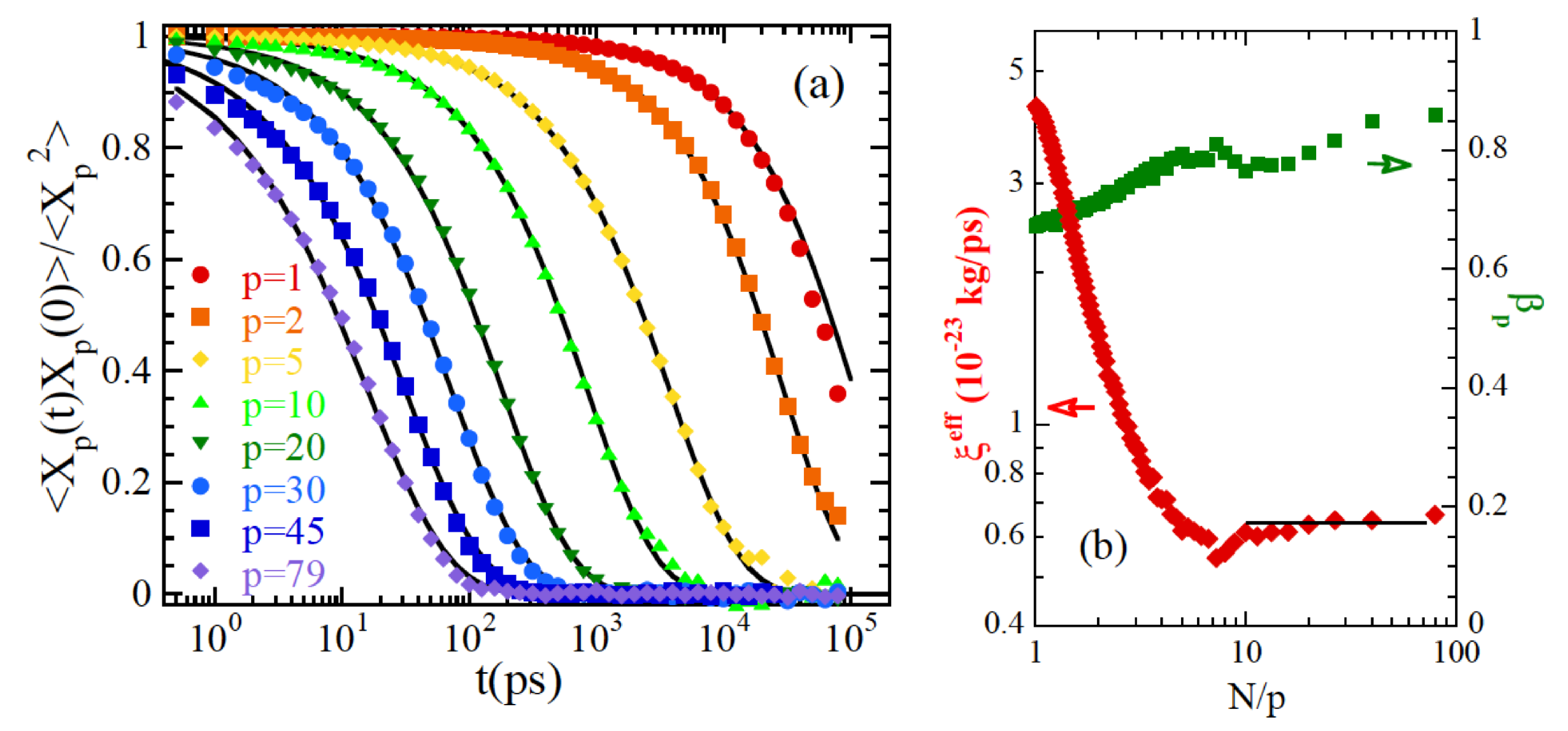
Figure 10.
(a) Direct comparison of the ratio between coherent and incoherent differential cross sections measured at 300 K and calculated from the simulations at 360 K for fully deuterated PAO with = 4. (b) Contributions to the normalized total structure factors for this polymer calculated from the MD-simulations. This panel shows a slice of the simulation cell. Main-chain atoms are represented in blue, side-group atoms in red. The nano-segregation can be appreciated at first sight. The definitions of MC and SG subsystems are shown in the scheme of the monomer in (a) by the green and orange rectangles respectively. Adapted with permission from [150]. Copyright (2012) American Chemical Society.
Figure 10.
(a) Direct comparison of the ratio between coherent and incoherent differential cross sections measured at 300 K and calculated from the simulations at 360 K for fully deuterated PAO with = 4. (b) Contributions to the normalized total structure factors for this polymer calculated from the MD-simulations. This panel shows a slice of the simulation cell. Main-chain atoms are represented in blue, side-group atoms in red. The nano-segregation can be appreciated at first sight. The definitions of MC and SG subsystems are shown in the scheme of the monomer in (a) by the green and orange rectangles respectively. Adapted with permission from [150]. Copyright (2012) American Chemical Society.

Figure 11.
Q-dependence of the average characteristic time for collective (empty squares) and self-correlation (circles) functions of PIB obtained from simulations. Filled squares are the experimental collective times [99]. Solid line: description of the self-correlation times by the anomalous jump diffusion model. Dashed-dotted line: description of the collective times by the proposed model [65,155,156]. The dotted line represents the diffusive contribution and the dashed line the contribution, with the location of the non-diffusive time indicated by the horizontal line. Adapted with permission from [65]. Copyright (2014) American Chemical Society.
Figure 11.
Q-dependence of the average characteristic time for collective (empty squares) and self-correlation (circles) functions of PIB obtained from simulations. Filled squares are the experimental collective times [99]. Solid line: description of the self-correlation times by the anomalous jump diffusion model. Dashed-dotted line: description of the collective times by the proposed model [65,155,156]. The dotted line represents the diffusive contribution and the dashed line the contribution, with the location of the non-diffusive time indicated by the horizontal line. Adapted with permission from [65]. Copyright (2014) American Chemical Society.

Figure 12.
(a) Comparison of the coherent and incoherent dynamical structure factors measured by LET on DO at 295 K at [19]. Vertical dotted lines represent the resolution FWHM. (b) Q-dependence of the characteristic relaxation times: (circles) and (squares; from MD-simulations, squares with crosses). The values have been obtained from incoherent scattering () and from the fit of the coherent scattering results to the model (•). Solid line: fit of to . and -values calculated with the model are shown by the dashed and dashed-dotted lines respectively. The temperature is 295 K [19]. Inset: Ratio between coherent and incoherent differential cross sections of DO at 298 K (hollow dots) as function of Q and calculated from MD-simulations (solid line). Adapted from [19].
Figure 12.
(a) Comparison of the coherent and incoherent dynamical structure factors measured by LET on DO at 295 K at [19]. Vertical dotted lines represent the resolution FWHM. (b) Q-dependence of the characteristic relaxation times: (circles) and (squares; from MD-simulations, squares with crosses). The values have been obtained from incoherent scattering () and from the fit of the coherent scattering results to the model (•). Solid line: fit of to . and -values calculated with the model are shown by the dashed and dashed-dotted lines respectively. The temperature is 295 K [19]. Inset: Ratio between coherent and incoherent differential cross sections of DO at 298 K (hollow dots) as function of Q and calculated from MD-simulations (solid line). Adapted from [19].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Values of the average scattering lengths , their squares and their deviations , as well as the coherent and incoherent scattering cross sections for different isotopes (1 barn = 100 fm).
Table 1.
Values of the average scattering lengths , their squares and their deviations , as well as the coherent and incoherent scattering cross sections for different isotopes (1 barn = 100 fm).
| Isotope | /fm | /fm | /fm | /barns | /barns |
|---|---|---|---|---|---|
| H | −3.7406 | 13.992 | 638.78 | 1.7583 | 80.26 |
| H (D) | 6.6710 | 44.502 | 16.322 | 5.592 | 2.05 |
| C | 6.6511 | 44.237 | 0 | 5.559 | 0 |
| O | 5.8030 | 33.675 | 0 | 4.232 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Arbe, A.; Alvarez, F.; Colmenero, J. Insight into the Structure and Dynamics of Polymers by Neutron Scattering Combined with Atomistic Molecular Dynamics Simulations. Polymers 2020, 12, 3067. https://doi.org/10.3390/polym12123067
AMA Style
Arbe A, Alvarez F, Colmenero J. Insight into the Structure and Dynamics of Polymers by Neutron Scattering Combined with Atomistic Molecular Dynamics Simulations. Polymers. 2020; 12(12):3067. https://doi.org/10.3390/polym12123067
Chicago/Turabian StyleArbe, Arantxa, Fernando Alvarez, and Juan Colmenero. 2020. "Insight into the Structure and Dynamics of Polymers by Neutron Scattering Combined with Atomistic Molecular Dynamics Simulations" Polymers 12, no. 12: 3067. https://doi.org/10.3390/polym12123067
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.