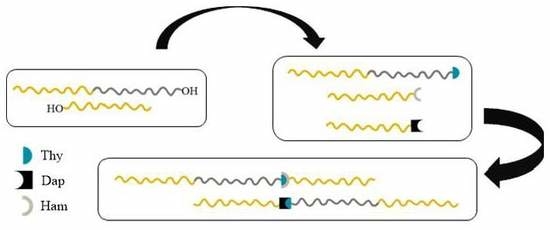
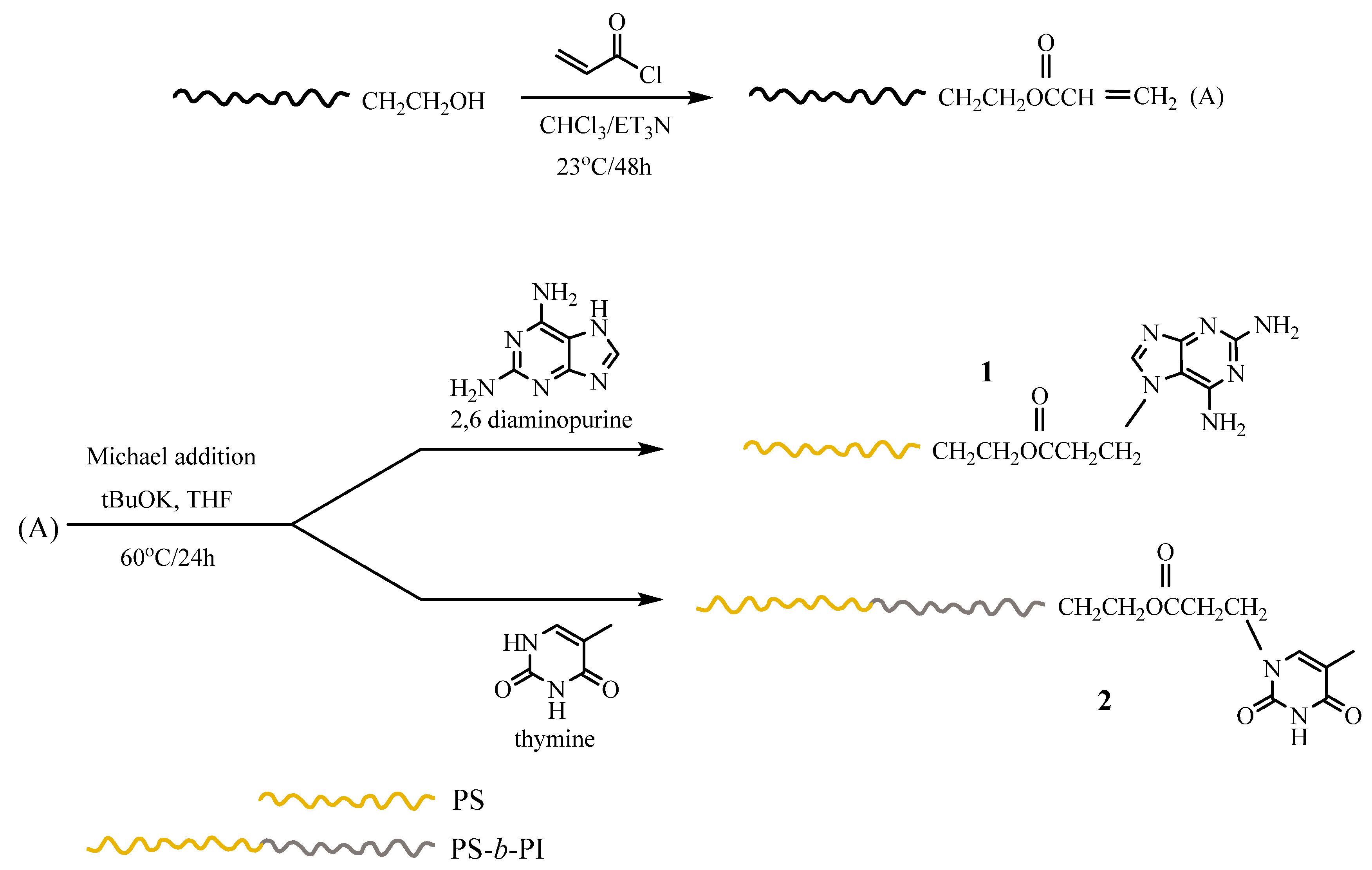
3.3.1. Size-Exclusion Chromatography (SEC)
SEC is neither the crucial nor the major characterization technique for evaluating the formation of supramolecular structures through the presence of hydrogen bonds. These are delicate systems that can be influenced by changes in temperature, the shear forces applied in the separating columns, and the possible existence of aggregation phenomena. Under these circumstances, SEC cannot efficiently resolve the structures that are formed in solution and, therefore, it is not a suitable technique to provide quantitative information in supramolecular polymer chemistry. It is very rare in the literature to find SEC data supporting the synthesis of supramolecular polymers. NMR methodologies are the main tools to verify the existence of hydrogen bonds, which was shown in our case as well. However, in order to strengthen the conclusions regarding the presence of the hydrogen bond interactions, SEC experiments were performed for Blends #1 and #2. In addition, a different blend was created as well, which consisted of the hydroxyl functionalized precursors of the polymers that were used in the previous blends. Specifically, Blend #0 is a mixture of PS-OH and PS-
b-PI-OH, prepared in the same way as Blends #1 and #2. To clarify the exact behaviour of these hydrogen bonded polymers, a variety of characterization methods were employed, starting with SEC in CHCl
3. The SEC trace of Blend #0, shown in
Figure 8, reveals the presence of two different narrow molecular weight distributed peaks. The one at the low elution time is attributed to the PS-OH and the other one, at higher elution time, and is attributed to PS-
b-PI-OH, as a result of the lack of interaction between them. On the contrary, the SEC traces for Blend #1 and #2 are completely different from those of Blend #0, but are very similar to each other.
SEC chromatography of the PS-
b-PI-Thy (
Figure 9) gives a trace at a higher elution volume when compared to its precursor. The presence of Thy leads to the formation of small and compact aggregates having smaller hydrodynamic volume, which elutes later than the –OH functionalized precursor. Except for the main peak, there are also small tails at lower elution volumes. Therefore, it can be assumed that this behavior is due to a dynamic equilibrium in solution between single chains, dimers, and a small population of higher aggregates, which are not stable enough against the external forces of the chromatography. The broad molecular weight distribution of the SEC trace reveals the presence of a mixture of products in solution that cannot be fully separated in the SEC columns. This result is only qualitative since aggregation phenomena cannot be quantitatively resolved by SEC.
PS-Ham and PS-Dap demonstrate similar behaviour, which is relatively different than that of PS-
b-PI-Thy. The main peaks of PS-OH, PS-Dap, and PS-Ham, as shown in
Figure 10, are approximately at the same elution volume. The difference exists when the end groups are attached to the polymers. The bulkiness of the end-groups and their chemical nature prevents the formation of large aggregates. In addition to the main peak, there are only minor shoulders at higher molecular weights, which can be explained as a small tendency for dimerization. This phenomenon is more evident in the PS-Dap, considering the tendency of the Dap end group to self-assemble more intensely than the Ham group, which forms more stable dimeric structures.
Considering the blends, the formation of the pseudo-tri-blocks is expected to manifest with the appearance of the SEC trace at lower elution volumes. However, this was not the case. An interesting observation regards the identical behavior of the two blends in SEC chromatography, which is a reasonable result due to the use of the same end hydroxyl polymers as the initial reagents for Blend #1 and #2. From
Figure 11 and
Figure 12, it is clear that there is one main peak in Blends #1 and #2, which is an indication that the solutions contain only one molecular species. This is clearly the supramolecular triblock copolymer in contrast to Blend #0, which is a solution of two different, non-interacting species. The main peak of the blends is located between the peaks of PS and PS-
b-PI due to the change of the conformation of the constituting components upon the formation of the supramolecular structures. A more compact structure is present, which will be further conformed by intrinsic viscosity measurements. This indicates that compact structures of low viscosity prevail in solution. Traces in lower elution times are a direct indication of the coexistence of higher aggregates. However, these aggregations are not stable enough, and most of them are disrupted under the employment of the shear forces in the separation columns. This is direct evidence for the formation of the supramolecular desired structures, which was also revealed by NMR spectroscopy.
3.3.2. Dilute Solution Viscometry
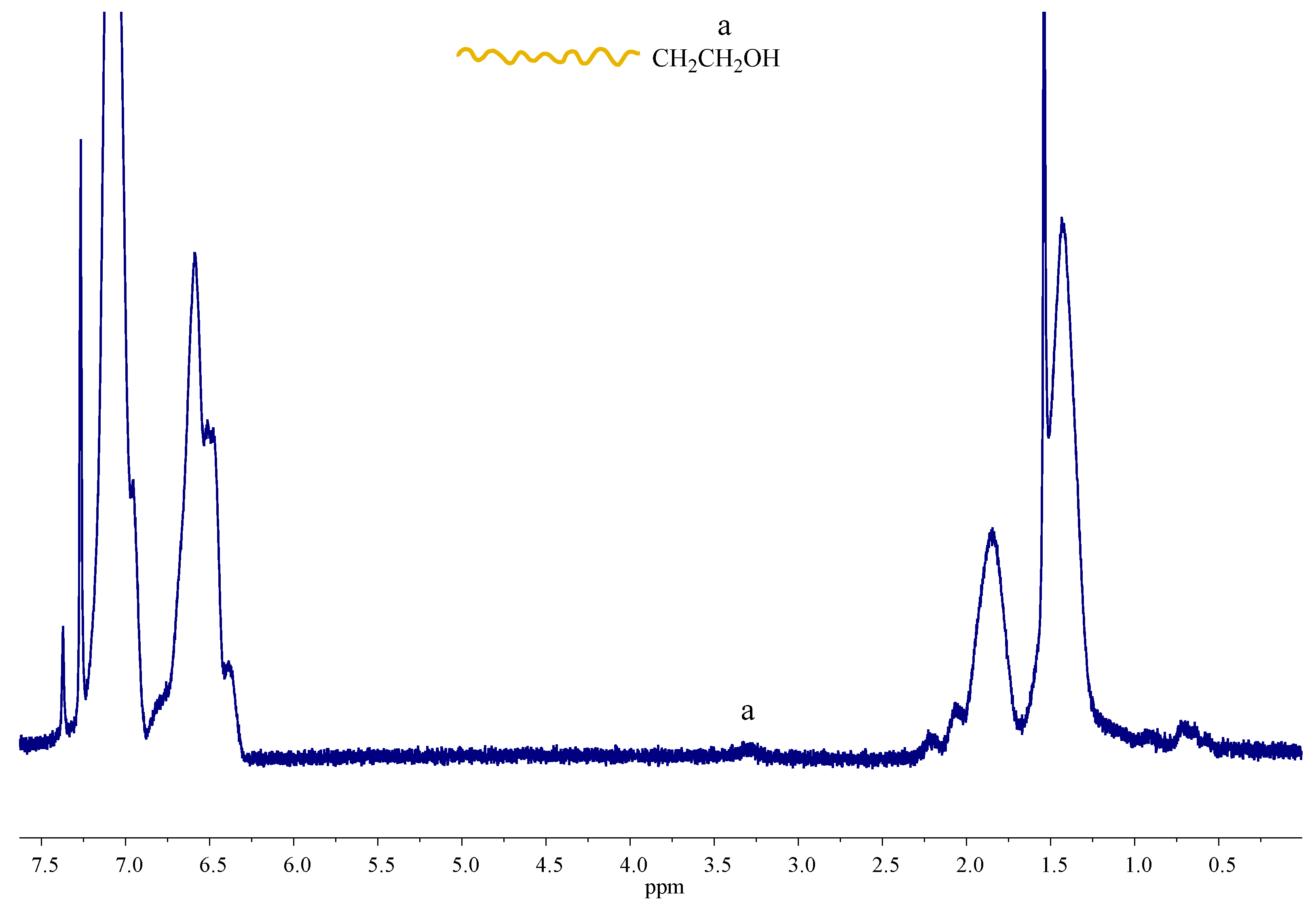
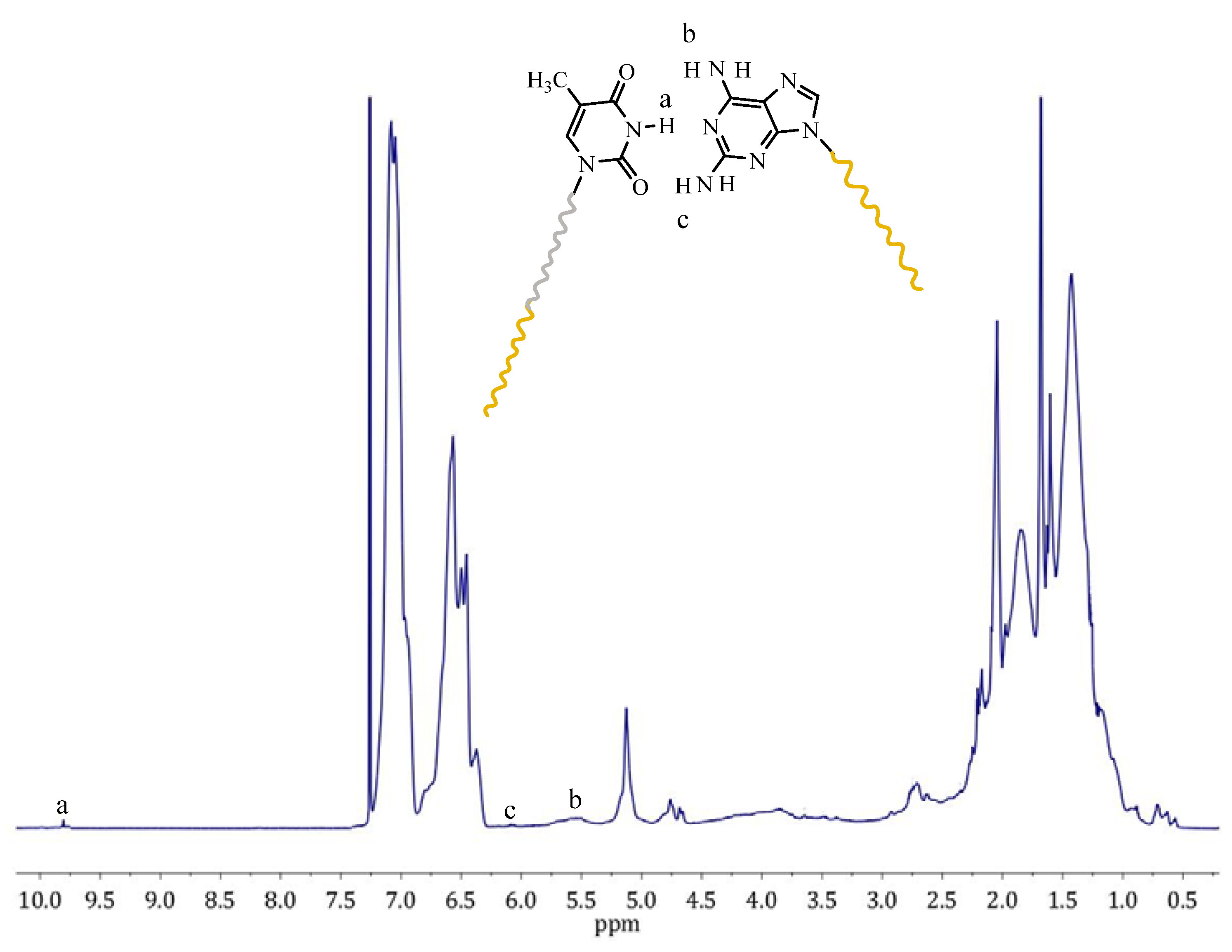
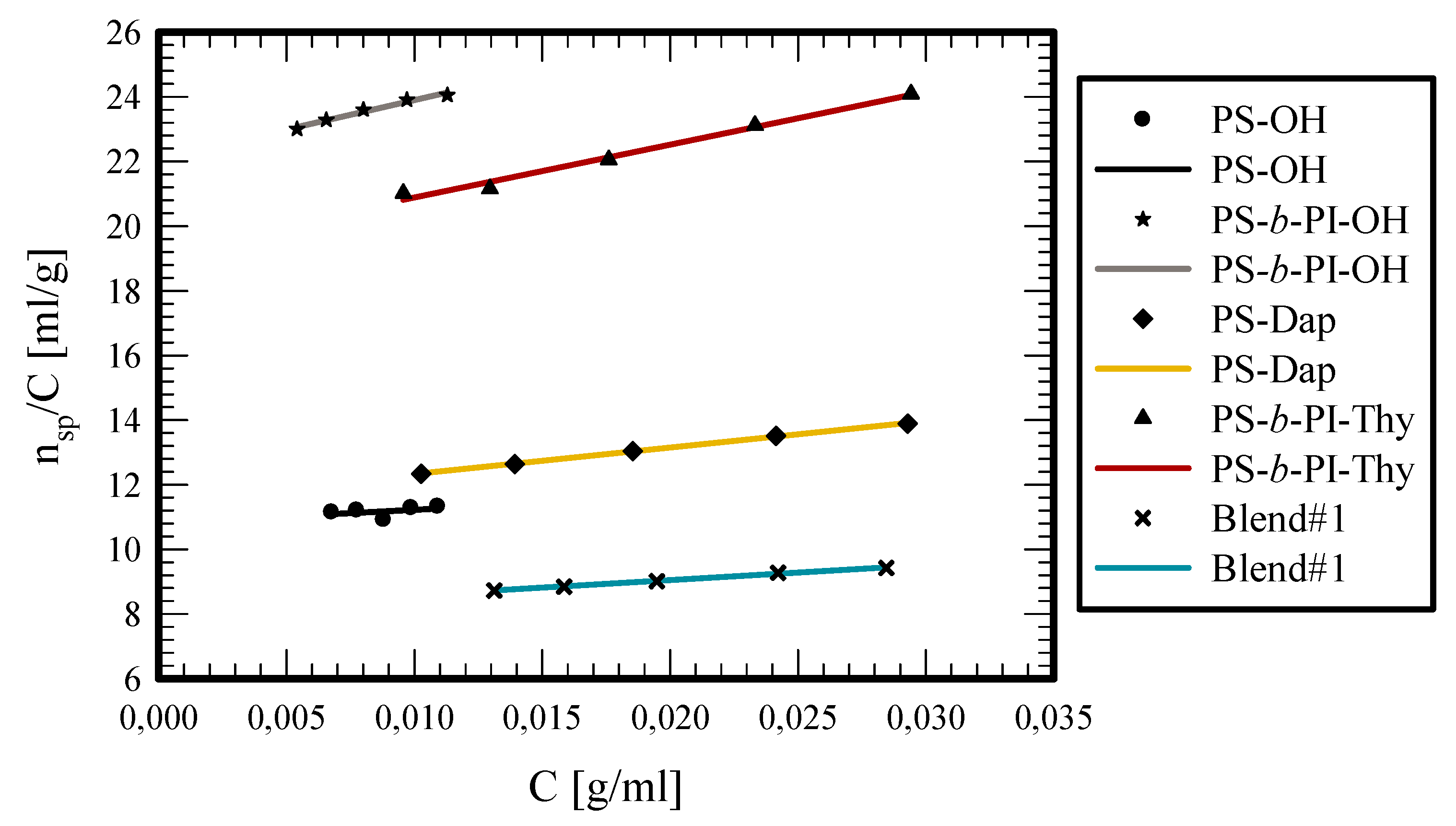
Viscometry measurements were also performed in toluene, which is a non-polar solvent. Thus, this allows for the formation of hydrogen bonds. The results are given in
Table 3, and representative plots are displayed in
Figure 13,
Figure 14 and
Figure S5.
As observed in SEC measurements, the blends had a different behavior in their hydrodynamic volume than expected. A similar behaviour was observed in viscometry. The intrinsic viscosity for the samples PS-OH and PS-b-PI-OH are 10.87 and 22.09 mL/g, respectively. The low Huggins constants of these polymers reveal the absence of any kind of interactions between the –OH groups, which is very reasonable, since they are not polar enough to promote association. When the functional groups (Dap, Ham, and Thy) are located at the polymer chain ends, the intrinsic viscosities are approximately the same as their precursors, considering that the end groups are very small relative to the molecular weight of the polymers. Therefore, their contribution is not pronounced enough to promote any change. However, the Huggins constants were higher than the precursors indicating weaker interactions of the polymers with the solvent. PS-b-PI-Thy shows the smaller change in the KH value since it has the higher molecular weight and, therefore, the effect of the end-group is less pronounced than the other samples. On the other hand, PS-Dap and PS-Ham exhibit much higher Hugging constants. The low molecular weight of these samples reveals that the contribution of the end-groups is much more pronounced, which leads to increased interactions between the end-polar groups. Their interactions are not strong enough to endure the shear forces applied in the capillary tube of the viscometer and, thus, lead to increased intrinsic viscosities.
The experimental results for Blend #0 reveal that there are no interactions between the two polymers. This is manifested by the intrinsic viscosity, which lies between the values of PS-OH and PS-b-PI-OH and the Hugging constant that is almost the same value. A different situation was observed for Blend #1 and #2. Nonetheless, both samples had exactly the same behaviour. For both samples, the intrinsic viscosities’ values are much lower than their precursors. The results are in agreement with the NMR and SEC data confirming that the supramolecular structures behave more or less as hard spheres having smaller hydrodynamic volumes and lower intrinsic viscosities than their linear counterparts. Lastly, it is clear that the Huggins constants for Blends #1 and #2 are very high. This behavior can be attributed to the presence of the hydrogen bonds that form between the end functional groups of each blend. As a result, the structures behave as unimolecular micelles with more compact structures compared to the corresponding linear ones.
3.3.3. Dynamic Light Scattering (DLS)
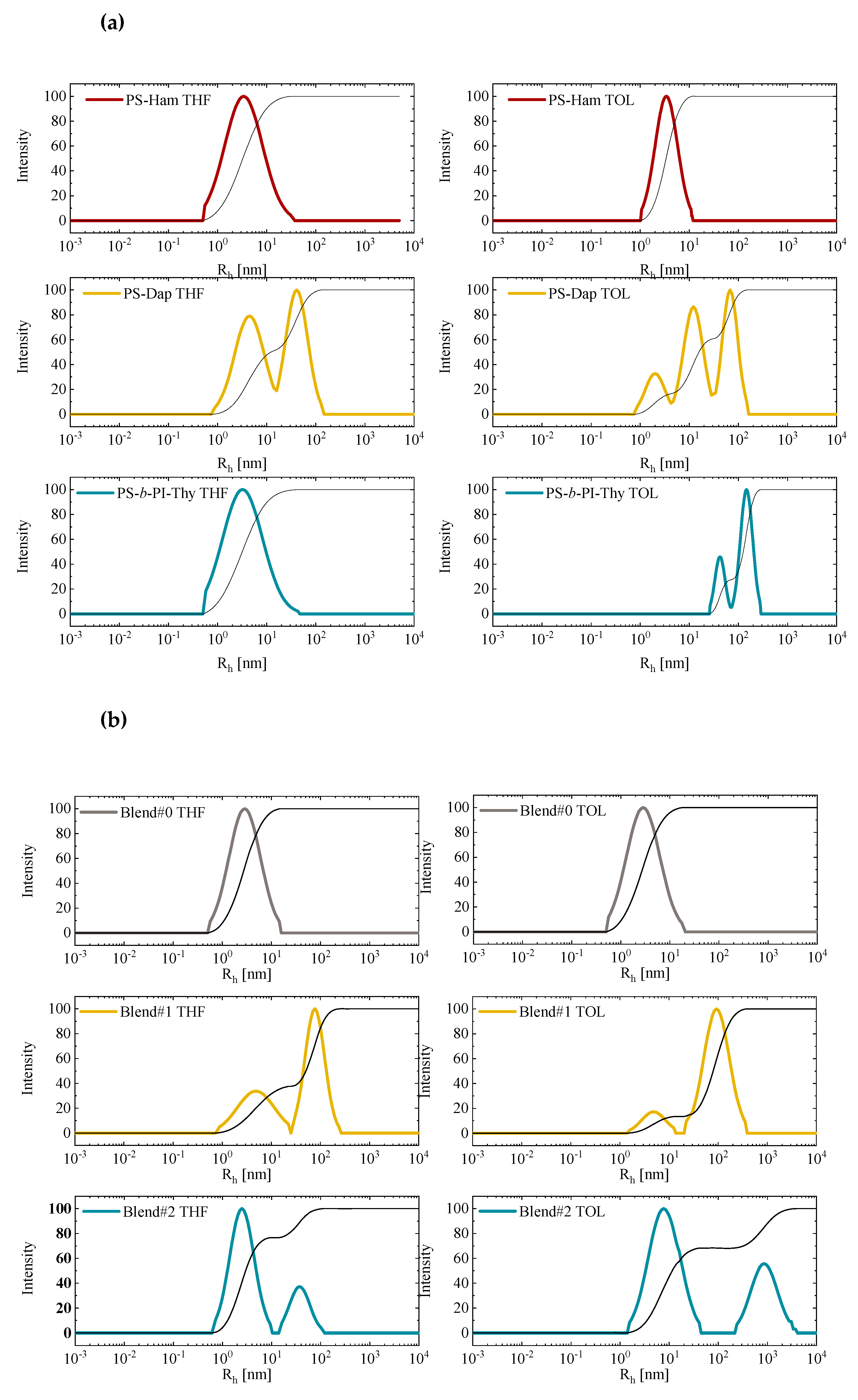
Dynamic light scattering (DLS) studies were conducted in THF, which is a moderately polar solvent. This prevents the formation of hydrogen bonds, and, in toluene, a non-polar solvent, which allows the formation of hydrogen bonds. The measurements were conducted at 25 °C using a 632.8-nm laser source at 90°. Different concentrations were employed for each sample. In all cases, the dependence of the apparent values of Rh did not vary appreciably with concentration. The time-correlation functions were analyzed by the CONTIN program.
The analysis for Blend #0 revealed the absence of any kind of association in either THF or in Toluene. This result is in excellent agreement with the previously reported data from SEC and dilute solution viscometry measurements, which indicate, once more, that the end –OH groups are not polar enough and do not interact strongly to promote association. However, the situation is different when end-polar groups are covalently attached to non-polar polymer chains. These products may behave as amphiphilic systems, which leads to the formation of aggregates in non-polar solvents. The final solution behaviour will depend on the polarity of the end group, the molecular weight of the non-polar macromolecular chain, and the polarity of the solvent. The results from the DLS measurements are provided in
Table 4, whereas characteristic CONTIN plots are provided in
Figure 15.
The PS-Ham sample, both in THF and in toluene, shows one population of a low Rh value. It seems that the bulky nature of the end group prevents the formation of higher aggregates in both solvents. However, the higher Rh value measured in toluene (4.77 nm against 2.07 nm in THF) and the lower polydispersity value from the CONTIN analysis in toluene indicate the formation of more compact dimeric structures in toluene.
The PS-Dap sample, on the other hand, showed pronounced differences compared to the PS-Ham sample. Strong association effects were obtained both in THF and in toluene. The smaller size and the more polar nature of the Dap end group, due to the presence of primary –NH2 groups, compared to the Ham end group promote much higher degrees of association in solution. Actually, the situation is more complex in toluene since three different populations are observed in this solvent. The Rh values of the various populations are higher in toluene, and, most importantly, the contribution of the large aggregates is much more pronounced in toluene than in THF, which indicates that the relatively polar nature of THF prevents the formation of higher aggregates.
Lastly, the PS-b-PI-Thy sample showed a distinct difference in the two solvents of different polarity. A single population of low Rh value is observed in THF. The relatively polar nature of the solvent along with the high molecular weight of the di-block copolymer prevent the formation of aggregates. On the contrary, from CONTIN analysis, the non-polar toluene promotes strong association with the appearance of two distinct populations with both having large Rh values.
The Blends #1 and #2 were also studied in THF and in toluene. In toluene, supramolecular triblock copolymers exist. The internal hydrogen bonded groups may be able to promote association due to their polar nature, which was already indicated from the DLS studies of the individual polymeric components bearing the end-polar groups Dap and Thy. In THF, the hydrogen bonds are not stable and, therefore, the supramolecular structures are broken down into their constituent components. However, even in THF, association is also expected for the supramolecular structures that still remain in solution and from the end-functionalized polymers that are formed after the disruption of the hydrogen bonds, as discussed earlier. Taking into account these statements, it is possible to understand the DLS measurements and, therefore, the solution behaviour of the blends.
CONTIN analysis of the results coming from Blend #1 in toluene indicates the presence of two distinct populations. The first, with a low Rh value, is attributed to the free supramolecular structures, whereas the second population, with a high Rh value, is attributed to associations of the supramolecular structures. The aggregates prevail over the free supramolecular tri-blocks, as shown by the relative area of the two peaks from CONTIN analysis. In THF, two populations are also obtained from CONTIN analysis. The first population is attributed to possible remaining supramolecular tri-blocks and to the PS-b-PI-Thy components since both have Rh values very close to the Rh value of this peak. The second peak, of high Rh value, is attributed to aggregates from remaining supramolecular structures and to mixed aggregates from PS-Dap and PS-b-PI-Thy linear chains. It should be noted that, in THF, the second population is less pronounced compared to that in toluene and has a lower Rh value due to the less extended association phenomena in THF.
The aggregation effects are more clear in the case of Blend #2 in toluene. Judging from the relative size of the two populations, equilibrium between aggregates and clusters can be observed. The micellar structures prevail over the clusters, which can be concluded from the areas of the two populations by CONTIN analysis. The stronger aggregation phenomena in Blend #2 compared to Blend #1 can be attributed to the stronger interactions between the interacting groups in the first case (Ham against Thy groups) and their larger size, which facilitates the accommodation of additional supramolecular tri-blocks to micellar structures. The association phenomena in THF are much less intense. CONTIN analysis reveals that the major population is of very low Rh value, which corresponds to surviving supramolecular copolymers and their components, whereas the second minor population indicates the presence of micellar structures coming from the remaining intact supramolecular tri-block copolymers.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}