
Wetting Simulations of High-Performance Polymer Resins on Carbon Surfaces as a Function of Temperature Using Molecular Dynamics



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
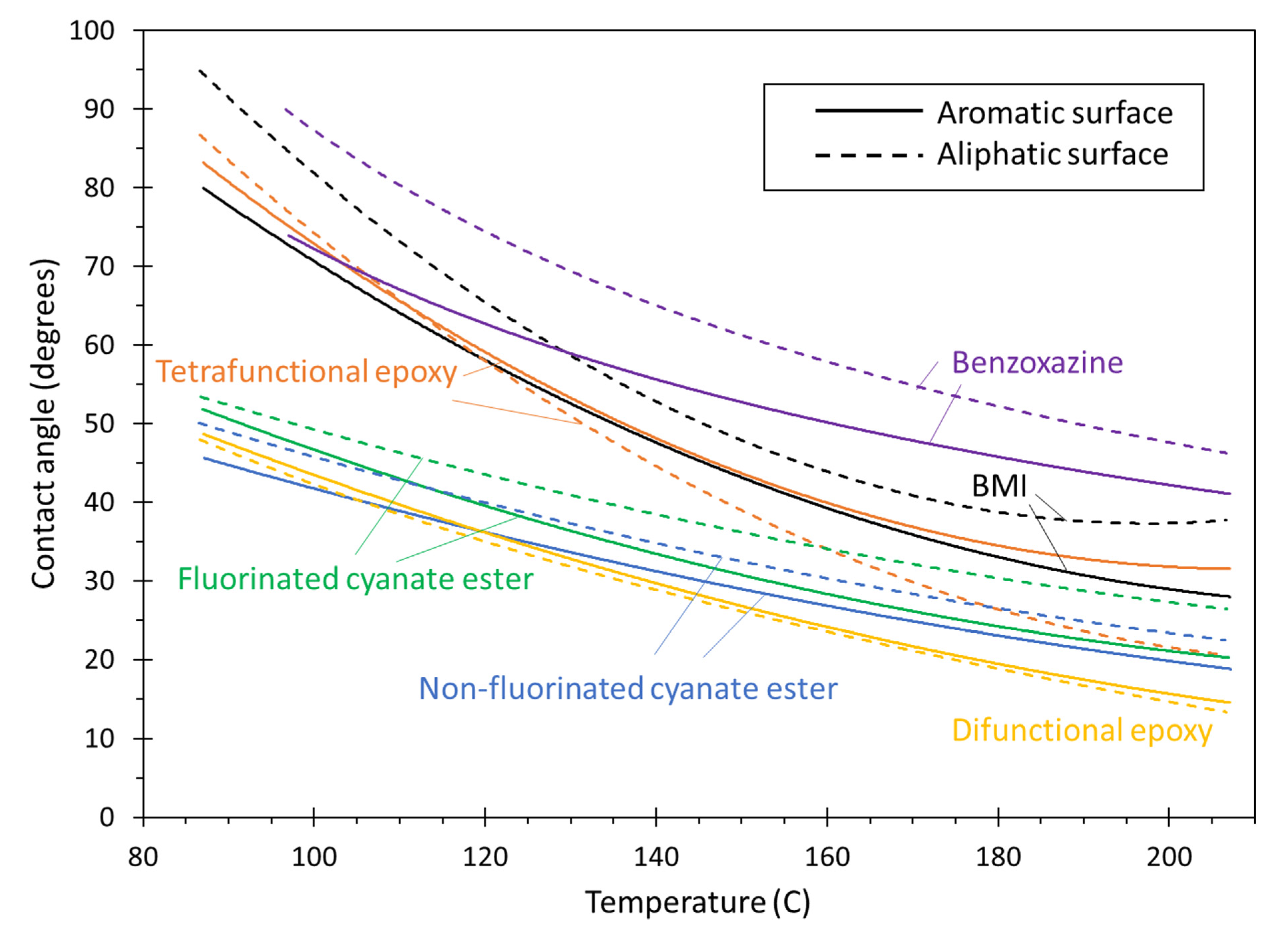
- Bismaleimide (BMI): This is a two-part system consisting of 4,4′-bismaleimidodiphenylmethane (BMPM) and O, O’-diallyl bisphenol A (DABPA), shown in Figure 1. The stochiometric ratio of the two monomers is 1:1, after Huntsman Matrimid 5292.
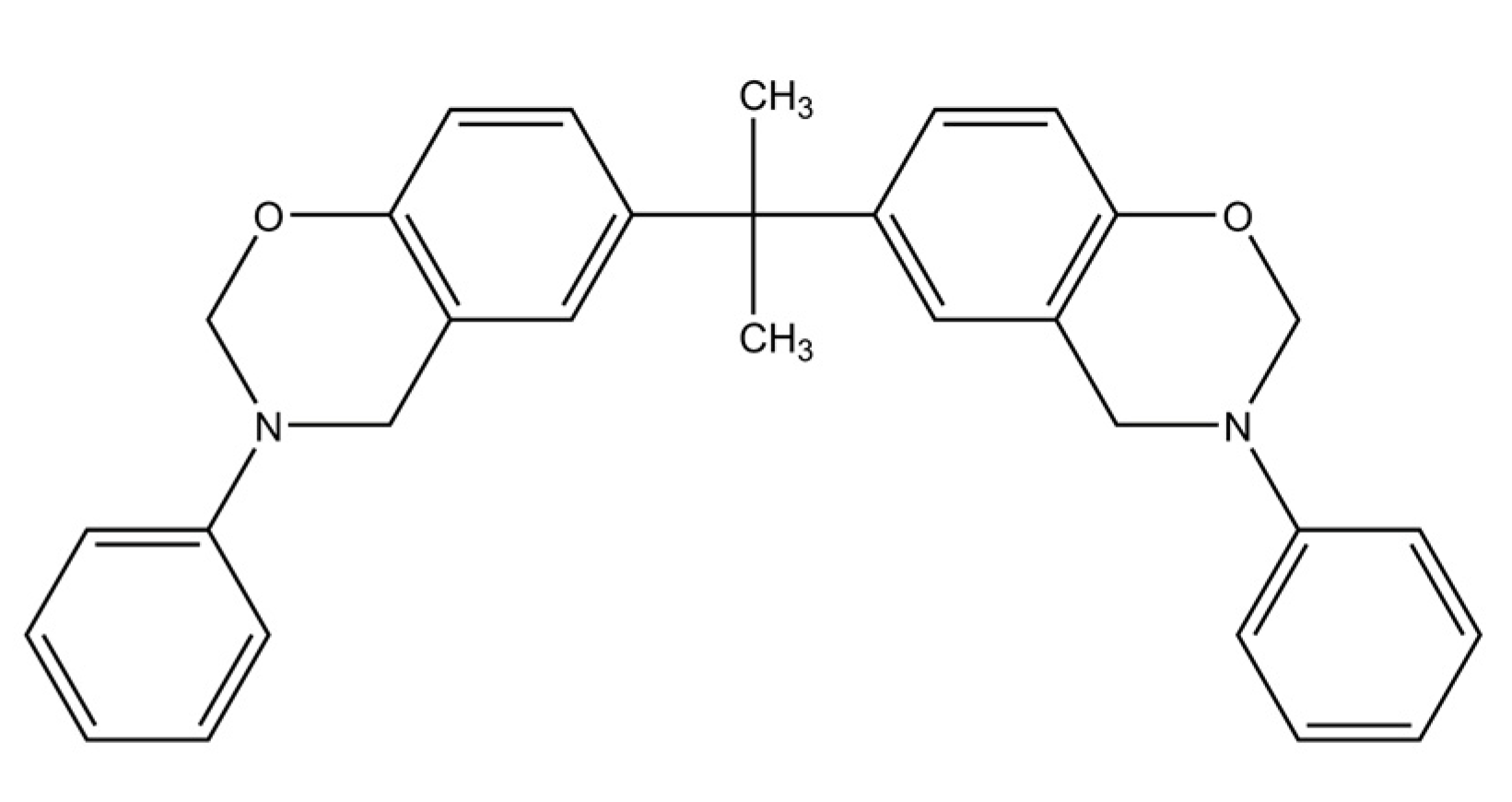
- Benzoxazine: The Bisphenol-A Benzoxazine monomer is shown in Figure 2. This system is modeled after Huntsman Araldite MT 35610.
- Difunctional epoxy: This is a two-part system consisting of diglycidyl ether bisphenol F (DGEBF) and diethyltoluenediamine (DETDA) monomers, as shown in Figure 3. The stochiometric molar ratio of resin to hardener is 2:1. This system is modeled after EPON 862/EPIKURE W.
- Tetrafunctional epoxy: This is a two-part system consisting of tetraglycidyl methylene dianiline (TGMDA) and diaminodiphenyl sulfone (DDS) monomers, as shown in Figure 4. The stochiometric ratio of resin to hardener is 1:1. This system is modeled after Solvay CYCOM 977-3.
- Fluorinated cyanate ester: The hexafluorobisphenol A cyanate ester monomer is shown in Figure 5. This system is modeled after AroCy F-10.
- Non-Fluorinated cyanate ester: The phenol novolac cyanate ester monomer is shown in Figure 6. This system is modeled after Primaset PT 30.
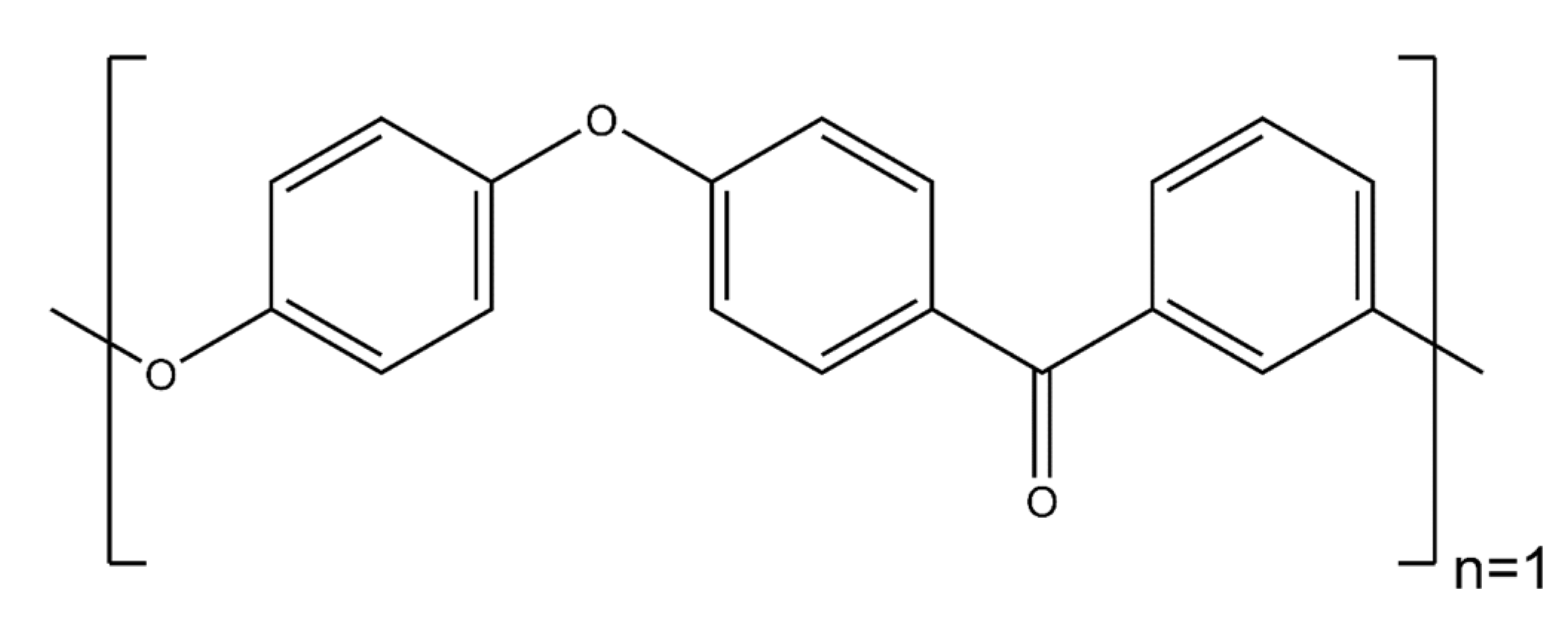
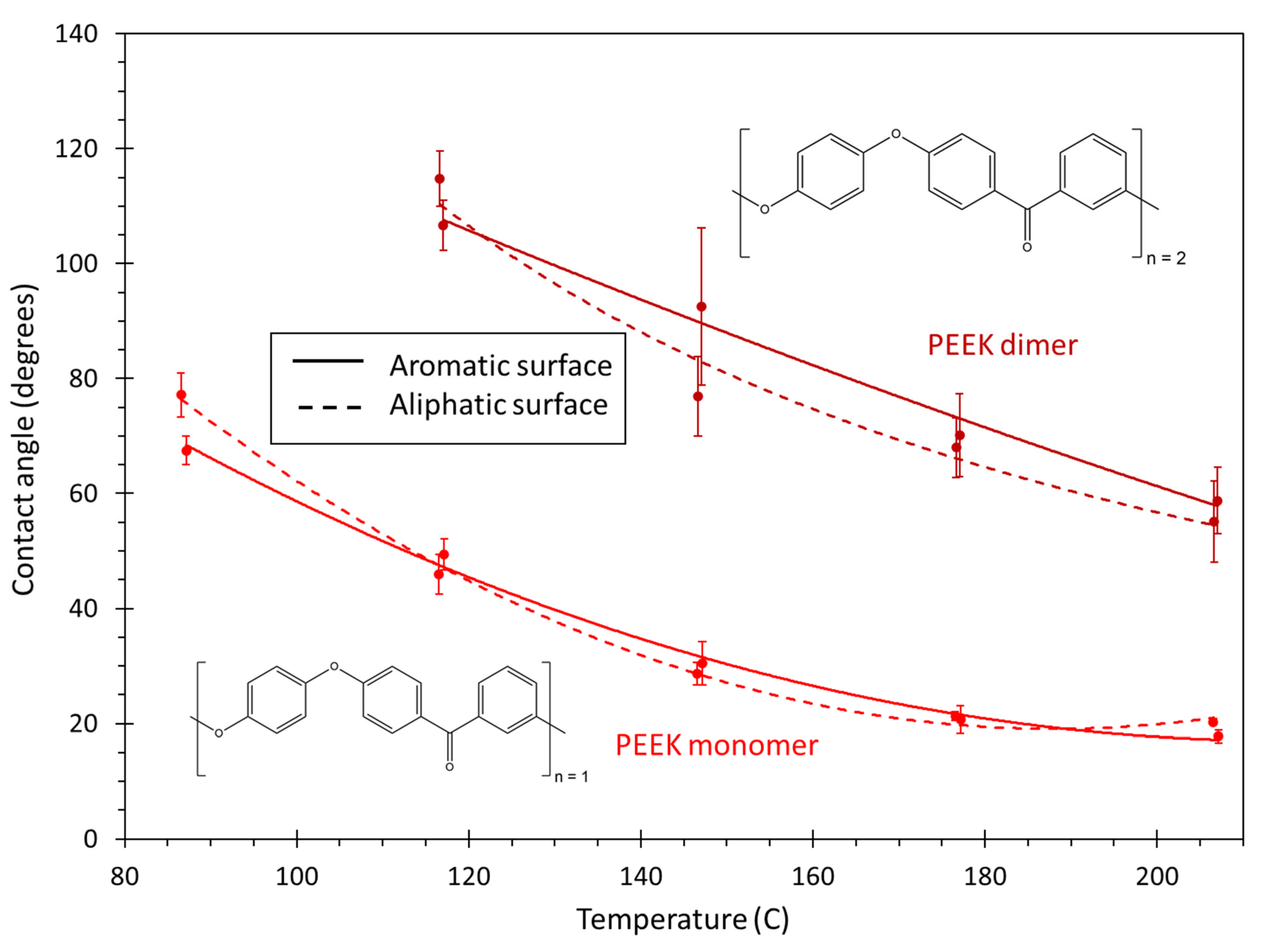
- PEEK monomer: The polyether ether ketone monomer is shown in Figure 7.
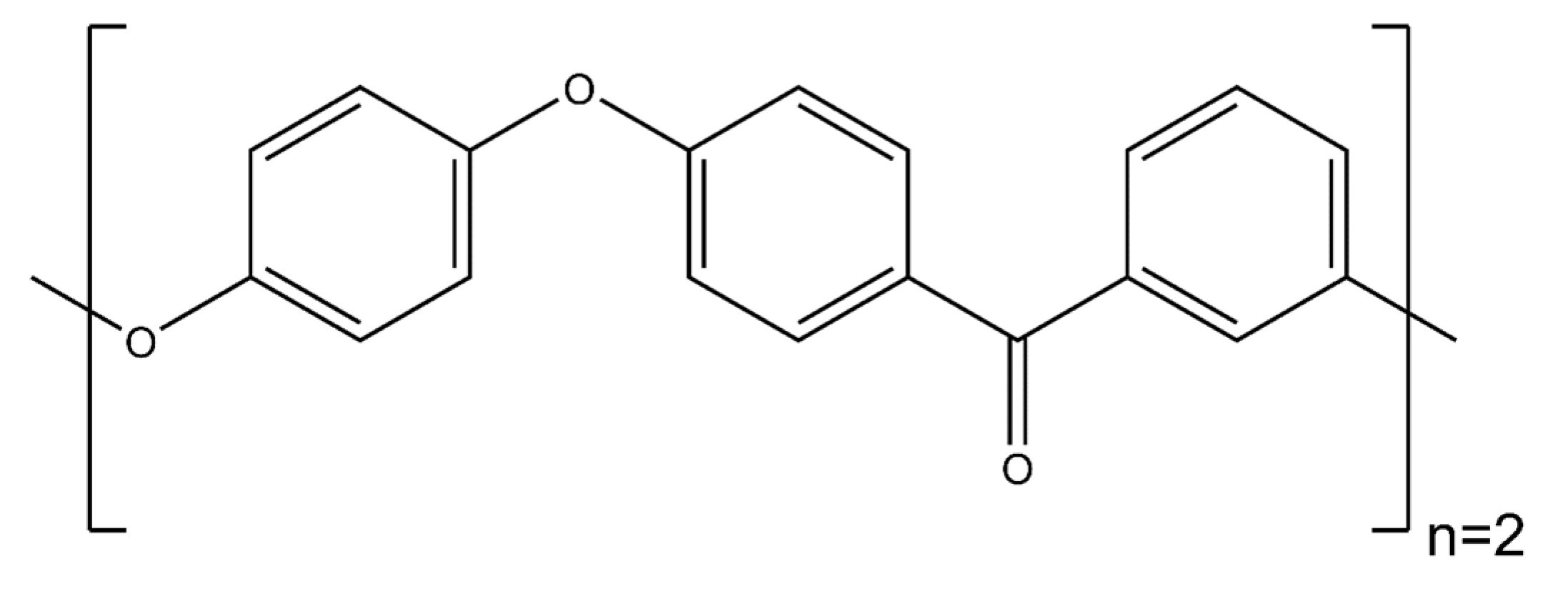
- PEEK dimer: The polyether ether ketone dimer is shown in Figure 8.
- Aromatic surface with pure sp2 carbon bonding;
- Aliphatic carbon surface with sp3 carbon bonding near the surface and hydrogen terminations at the surface.
2.2. Molecular Modeling
2.2.1. Carbon Surfaces
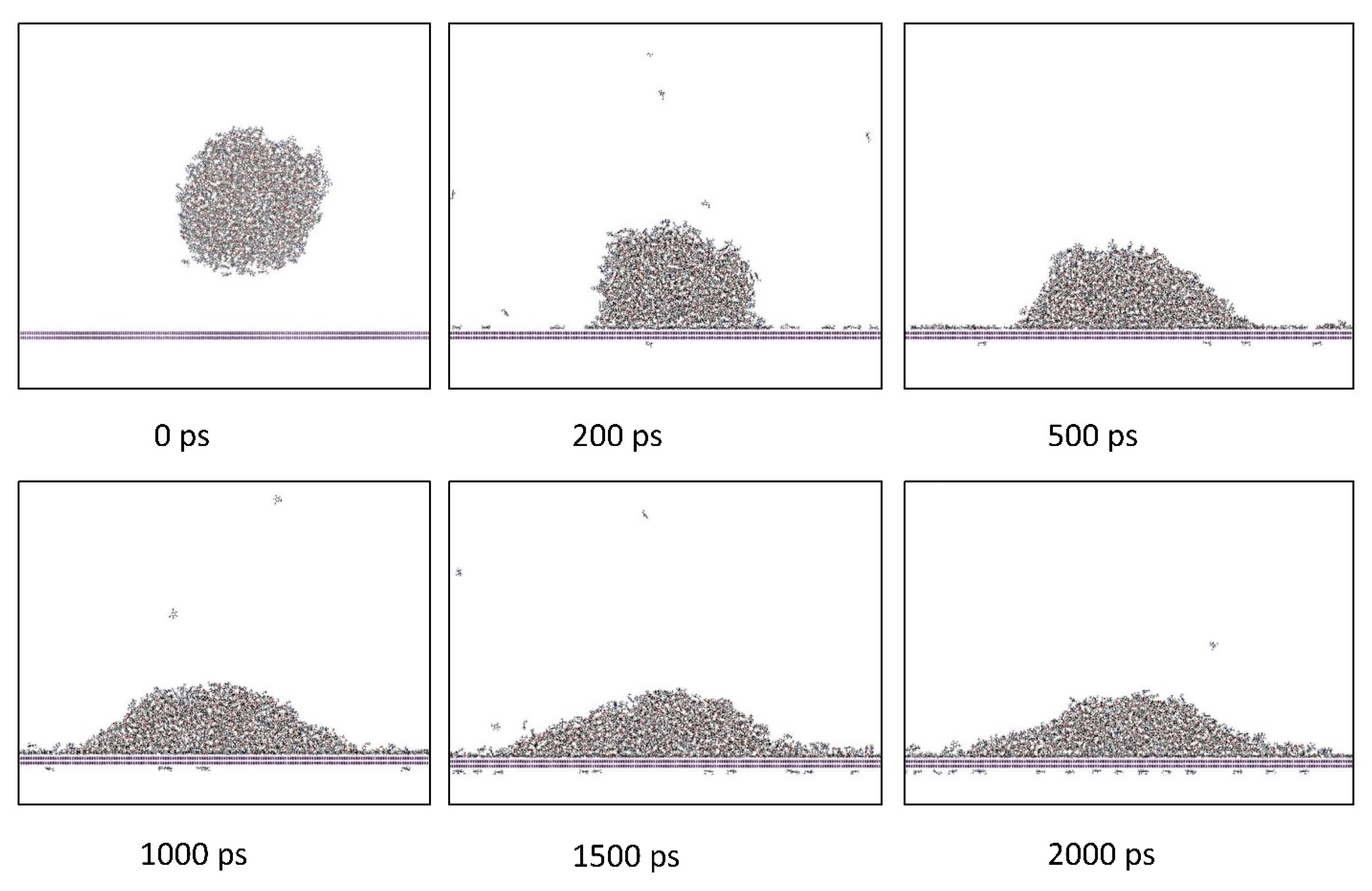
2.2.2. Wetting Simulations
2.2.3. Evaluation of Contact Angle Value
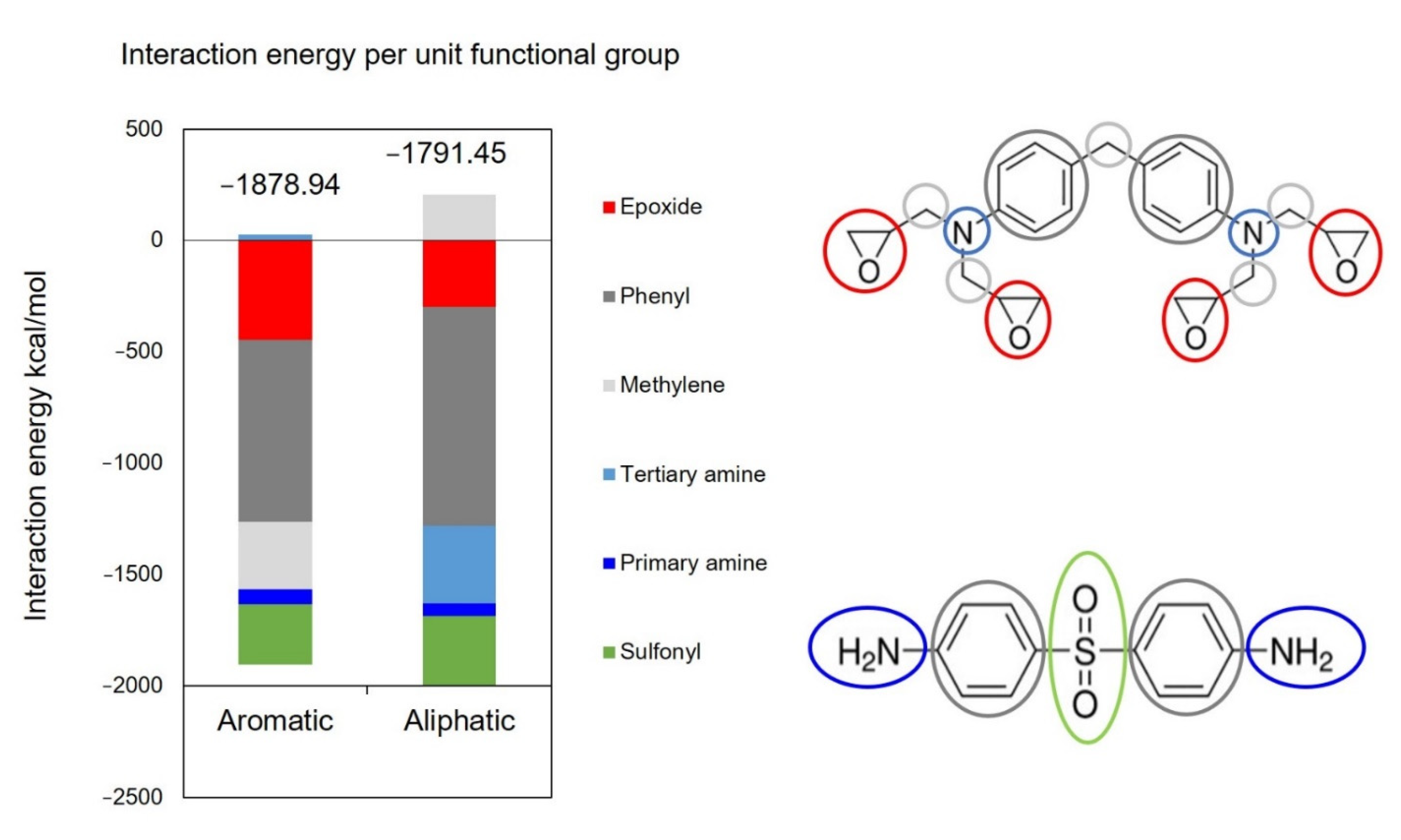
2.2.4. Calculation of Interaction Energy
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Downes, R.D.; Hao, A.; Park, J.G.; Su, Y.-F.; Liang, R.; Jensen, B.D.; Siochi, E.J.; Wise, K.E. Geometrically constrained self-assembly and crystal packing of flattened and aligned carbon nanotubes. Carbon 2015, 93, 953–966. [Google Scholar] [CrossRef]
- Jensen, B.D.; Kim, J.-W.; Sauti, G.; Wise, K.E.; Dong, L.; Wadley, H.N.G.; Park, J.G.; Liang, R.; Siochi, E.J. Toward ultralight high-strength structural materials via collapsed carbon nanotube bonding. Carbon 2020, 156, 538–548. [Google Scholar] [CrossRef]
- Bhattacharya, M. Polymer nanocomposites—A comparison between carbon nanotubes, graphene, and clay as nanofillers. Materials 2016, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Rahmat, M.; Hubert, P. Carbon nanotube–polymer interactions in nanocomposites: A review. Compos. Sci. Technol. 2011, 72, 72–84. [Google Scholar] [CrossRef]
- Goclon, J.; Panczyk, T.; Winkler, K. Investigation of the interfacial properties of polyurethane/carbon nanotube hybrid composites: A molecular dynamics study. Appl. Surface Sci. 2018, 433, 213–221. [Google Scholar] [CrossRef]
- Hadden, C.M.; Jensen, B.D.; Bandyopadhyay, A.; Odegard, G.M.; Koo, A.; Liang, R. Molecular modeling of EPON-862/graphite composites: Interfacial characteristics for multiple crosslink densities. Compos. Sci. Technol. 2013, 76, 92–99. [Google Scholar] [CrossRef]
- Hadden, C.M.; Klimek-McDonald, D.R.; Pineda, E.J.; King, J.A.; Reichanadter, A.M.; Miskioglu, I.; Gowtham, S.; Odegard, G.M. Mechanical properties of graphene nanoplatelet/carbon fiber/epoxy hybrid composites: Multiscale modeling and experiments. Carbon 2015, 95, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Pisani, W.A.; Radue, M.S.; Patil, S.U.; Odegard, G.M. Interfacial modeling of flattened CNT composites with cyanate ester and PEEK polymers. Compos. Part B Eng. 2021, 211, 108672. [Google Scholar] [CrossRef]
- Al Mahmud, H.; Radue, M.S.; Chinkanjanarot, S.; Odegard, G.M. Multiscale Modeling of Epoxy-Based Nanocomposites Reinforced with Functionalized and Non-Functionalized Graphene Nanoplatelets. Polymers 2021, 13, 1958. [Google Scholar] [CrossRef]
- Patil, S.U.; Radue, M.S.; Pisani, W.A.; Deshpande, P.; Xu, H.; Al Mahmud, H.; Dumitrică, T.; Odegard, G.M. Interfacial characteristics between flattened CNT stacks and polyimides: A molecular dynamics study. Comput. Mater. Sci. 2020, 185, 109970. [Google Scholar] [CrossRef]
- Ambrosi, A.; Pumera, M. Amorphous Carbon Impurities Play an Active Role in Redox Processes of Carbon Nanotubes. J. Phys. Chem. C 2011, 115, 25281–25284. [Google Scholar] [CrossRef]
- Shao, L.; Tobias, G.; Salzmann, C.G.; Ballesteros, B.; Hong, S.Y.; Crossley, A.; Davis, B.G.; Green, M.L.H. Removal of amorphous carbon for the efficient sidewall functionalisation of single-walled carbon nanotubes. Chem. Commun. 2007, 47, 5090–5092. [Google Scholar] [CrossRef]
- Dong, L.; Park, J.G.; Leonhardt, B.E.; Zhang, S.; Liang, R. Continuous Synthesis of Double-Walled Carbon Nanotubes with Water-Assisted Floating Catalyst Chemical Vapor Deposition. Nanomaterials 2020, 10, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surblys, D.; Yamaguchi, Y.; Kuroda, K.; Kagawa, M.; Nakajima, T.; Fujimura, H. Molecular dynamics analysis on wetting and interfacial properties of water-alcohol mixture droplets on a solid surface. J. Chem. Phys. 2014, 140, 034505. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, J.; Hao, X.; Zhang, C.; Wei, N.; Zhang, C. Wetting Properties of Defective Graphene Oxide: A Molecular Simulation Study. Molecules 2018, 23, 1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgren, M.; Allan, N.L.; Cosgrove, T.; George, N. Wetting of Water and Water/Ethanol Droplets on a Non-Polar Surface: A Molecular Dynamics Study. Langmuir 2002, 18, 10462–10466. [Google Scholar] [CrossRef]
- Sergi, D.; Scocchi, G.; Ortona, A. Molecular dynamics simulations of the contact angle between water droplets and graphite surfaces. Fluid Phase Equilibria 2012, 332, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Ohler, B.; Langel, W. Molecular Dynamics Simulations on the Interface between Titanium Dioxide and Water Droplets: A New Model for the Contact Angle. J. Phys. Chem. C 2009, 113, 10189–10197. [Google Scholar] [CrossRef]
- Grossiord, N.; Miltner, H.E.; Loos, J.; Meuldijk, J.; Van Mele, B.; Koning, C.E. On the Crucial Role of Wetting in the Preparation of Conductive Polystyrene−Carbon Nanotube Composites. Chem. Mater. 2007, 19, 3787–3792. [Google Scholar] [CrossRef]
- Heine, D.R.; Grest, G.S.; Webb III, E.B. Spreading dynamics of polymer nanodroplets. Phys. Rev. E 2003, 68, 061603. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Mo, Y.; Zhao, Y.; Jiang, S. Understanding the temperature and size dependence of the contact angle of Cu/Si(1 1 1): A molecular dynamics study. Comput. Mater. Sci. 2018, 150, 222–229. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Q.; Li, X.; Wang, X.; Ke, B.; Li, X.; Shen, Z. Effect of the morphology of adsorbed oleate on the wettability of a collophane surface. Appl. Surface Sci. 2018, 444, 87–96. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. A Math. Phys. Eng. Sci. 2004, 362, 2477–2512. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Lin, T.-J.; Kishore Mishra, R.; Emami, F.S. Thermodynamically Consistent Force Fields for the Assembly of Inorganic, Organic, and Biological Nanostructures: The INTERFACE Force Field. Langmuir 2013, 29, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Suter, U.W. Atomic Charges for Classical Simulations of Polar Systems. J. Phys. Chem. B 2004, 108, 18341–18352. [Google Scholar] [CrossRef]
- Gray, D.; McCaughan, A.; Mookerji, B. Crystal Structure of Graphite, Graphene and Silicon. Phys. Solid State Appl. 2009, 6, 730–732. [Google Scholar]
- Damirchi, B.; Radue, M.; Kanhaiya, K.; Heinz, H.; Odegard, G.M.; van Duin, A.C.T. ReaxFF Reactive Force Field Study of Polymerization of a Polymer Matrix in a Carbon Nanotube-Composite System. J. Phys. Chem. C 2020, 124, 20488–20497. [Google Scholar] [CrossRef]
- Dharmawardhana, C.C.; Kanhaiya, K.; Lin, T.-J.; Garley, A.; Knecht, M.R.; Zhou, J.; Miao, J.; Heinz, H. Reliable computational design of biological-inorganic materials to the large nanometer scale using Interface-FF. Mol. Simul. 2017, 43, 1394–1405. [Google Scholar] [CrossRef]
- LAMMPS Documentation: Pair Styles. Available online: https://lammps.sandia.gov/doc/pair_class2.html (accessed on 20 June 2021).
- LAMMPS Documentation: Pair Style Coul/Cut. Available online: https://lammps.sandia.gov/doc/pair_coul.html (accessed on 20 June 2021).
- Yd, S.; Maroo, S.C. A new algorithm for contact angle estimation in molecular dynamics simulations. In Proceedings of the ASME 2015 13th International Conference on Nanochannels, Microchannels, and Minichannels collocated with the ASME 2015 International Technical Conference and Exhibition on Packaging and Integration of Electronic and Photonic Microsystems, San Francisco, CA, USA, 6–9 July 2015. [Google Scholar]
- Hunter, C.A.; Sanders, J.K. The nature of. pi.-. pi. interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Zhu, D.; Pignatello, J.J. Characterization of Aromatic Compound Sorptive Interactions with Black Carbon (Charcoal) Assisted by Graphite as a Model. Environ. Sci. Technol. 2005, 39, 2033–2041. [Google Scholar] [CrossRef]
- Björk, J.; Hanke, F.; Palma, C.-A.; Samori, P.; Cecchini, M.; Persson, M. Adsorption of Aromatic and Anti-Aromatic Systems on Graphene through π−π Stacking. J. Phys. Chem. Lett. 2010, 1, 3407–3412. [Google Scholar] [CrossRef]
- Villacañas, F.; Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L. Adsorption of simple aromatic compounds on activated carbons. J. Colloid Interface Sci. 2006, 293, 128–136. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.J.; White, S.R. Cure kinetics, gelation, and glass transition of a bisphenol F epoxide. Polym. Eng. Sci. 2003, 43, 863–874. [Google Scholar] [CrossRef]
- Biolley, N.T.P.; Sillion, B. Polyimide-modified epoxy system: Time-temperature-transformation diagrams, mechanical and thermal properties. Polymer 1994, 35, 558–564. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resin | Number of Atoms in Droplet Model |
|---|---|
| Bismaleimide | 15,488 |
| Benzoxazine | 15,210 |
| Difunctional epoxy | 15,444 |
| Tetrafunctional epoxy | 15,210 |
| Fluorinated cyanate ester | 15,400 |
| Non-fluorinated cyanate ester | 14,960 |
| PEEK monomer | 14,960 |
| PEEK dimer | 14,700 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamane, S.S.; Gaikwad, P.S.; Radue, M.S.; Gowtham, S.; Odegard, G.M. Wetting Simulations of High-Performance Polymer Resins on Carbon Surfaces as a Function of Temperature Using Molecular Dynamics. Polymers 2021, 13, 2162. https://doi.org/10.3390/polym13132162
Bamane SS, Gaikwad PS, Radue MS, Gowtham S, Odegard GM. Wetting Simulations of High-Performance Polymer Resins on Carbon Surfaces as a Function of Temperature Using Molecular Dynamics. Polymers. 2021; 13(13):2162. https://doi.org/10.3390/polym13132162
Chicago/Turabian StyleBamane, Swapnil S., Prashik S. Gaikwad, Matthew S. Radue, S. Gowtham, and Gregory M. Odegard. 2021. "Wetting Simulations of High-Performance Polymer Resins on Carbon Surfaces as a Function of Temperature Using Molecular Dynamics" Polymers 13, no. 13: 2162. https://doi.org/10.3390/polym13132162