
Functionalized Biodegradable Polymers via Termination of Ring-Opening Polymerization by Acyl Chlorides

 ,
,
Abstract
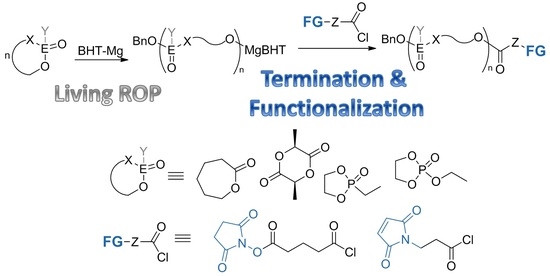
:
1. Introduction
2. Materials and Methods
2.1. General Experimental Remarks
2.2. Synthesis of NHS- and MI-Functionalized Acyl Chlorides
2.2.1. Synthesis of 2,5-dioxopyrrolidin-1-yl 5-chloro-5-oxopentanoate 1
2.2.2. Synthesis of 2,5-dioxopyrrolidin-1-yl 2-(2-chloro-2-oxoethoxy)acetate 2
2.2.3. Synthesis of 2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)acetyl chloride 3
2.2.4. Synthesis of 3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)propanoyl chloride 4
2.3. Synthesis of NHS- and MI-Functionalized Polymers
2.3.1. Polymerization of εCL with Termination by DSC
2.3.2. Polymerization of εCL with Termination by SA Followed by the Reaction with N-hydroxysuccinimide/DCC
2.3.3. Polymerization of εCL with Termination by 1–4
2.3.4. Polymerization of l-LA with Termination by 1 and 4
2.3.5. Polymerization of EtEP with Termination by 1 and 4
2.3.6. Synthesis of εCL/EtEP Block Copolymers with Termination by 1 and 4
2.3.7. Polymerization of EtOEP with Termination by 1, BHT-Mg Catalyst
2.3.8. Polymerization of EtOEP with Termination by 1, TBD/BnOH Catalyst
2.3.9. Reactions of poly(εCL) and poly(EtOEP) with 1
2.4. Reactions of NHS-Functionalized Polymers with iBuNH2
2.4.1. Reactions in CH2Cl2, Synthesis of poly(εCL)-1-N and poly(l-LA)-1-N
2.4.2. Reactions in Aqueous Media, Synthesis of poly(EtEP)-1-N and poly(EtOEP)-1-N
2.4.3. NMR Data
2.5. Reactions of MI-Functionalized Polymers with HSCH2COOMe
2.5.1. Reactions in Organic Solvent, Synthesis of poly(εCL)-4-S and poly(l-LA)-4-S
2.5.2. Reaction in Aqueous Media, Synthesis of poly(EtEP)-4-S
2.5.3. NMR Data
3. Results and Discussion
3.1. Functionalization of the Polymers by Termination of Living Polymerization
3.2. Functionalization by the Reaction of the Polymers with Acyl Chloride 1
3.3. Reactions of NHS-Functionalized Polymers with Amine-Containing Compounds
3.4. Reactions of MI-Functionalized Polymers with Thiol-Containing Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [Green Version]
- Vroman, I.; Tighzert, L. Biodegradable Polymers. Materials 2009, 2, 307–344. [Google Scholar] [CrossRef]
- Englert, C.; Brendel, J.C.; Majdanski, T.C.; Yildirim, T.; Schubert, S.; Gottschaldt, M.; Windhab, N.; Schubert, U.S. Pharmapolymers in the 21st century: Synthetic polymers in drug delivery applications. Prog. Polym. Sci. 2018, 87, 107–164. [Google Scholar] [CrossRef]
- Agarwal, S. Biodegradable Polymers: Present Opportunities and Challenges in Providing a Microplastic-Free Environment. Macromol. Chem. Phys. 2020, 221, 2000017. [Google Scholar] [CrossRef] [Green Version]
- Iha, R.K.; Wooley, K.L.; Nyström, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of Orthogonal “Click” Chemistries in the Synthesis of Functional Soft Materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, M.A.; Klok, H.-A. Peptide/protein–polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. 2008, 2591–2611. [Google Scholar] [CrossRef] [PubMed]
- Theato, P. Synthesis of well-defined polymeric activated esters. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 6677–6687. [Google Scholar] [CrossRef]
- Seyednejad, H.; Ghassemi, A.H.; van Nostrum, C.F.; Vermonden, T.; Hennink, W.E. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J. Control. Release 2011, 152, 168–176. [Google Scholar] [CrossRef]
- Arslan, M.; Gevrek, T.; Sanyal, A.; Shunmugam, R. Maleimide Containing Thiol-Reactive Polymers: Synthesis and Functionalization. In Functional Polymers; CRC Press: Boca Raton, FL, USA, 2017; pp. 265–293. [Google Scholar]
- Sadaba, N.; Salsamendi, M.; Casado, N.; Zuza, E.; Muñoz, J.; Sarasua, J.-R.; Mecerreyes, D.; Mantione, D.; Detrembleur, C.; Sardon, H. Catechol End-Functionalized Polylactide by Organocatalyzed Ring-Opening Polymerization. Polymers 2018, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J. Control. Release 2017, 260, 46–60. [Google Scholar] [CrossRef]
- Ravasco, J.M.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chem. - A Eur. J. 2019, 25, 43–59. [Google Scholar] [CrossRef]
- Spicer, C.D.; Pashuck, E.T.; Stevens, M.M. Achieving Controlled Biomolecule–Biomaterial Conjugation. Chem. Rev. 2018, 118, 7702–7743. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, H.Y.; Park, M.J. End-Group Chemistry and Junction Chemistry in Polymer Science: Past, Present, and Future. Macromolecules 2020, 53, 746–763. [Google Scholar] [CrossRef] [Green Version]
- Northrop, B.H.; Frayne, S.H.; Choudhary, U. Thiol–maleimide “click” chemistry: Evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity. Polym. Chem. 2015, 6, 3415–3430. [Google Scholar] [CrossRef]
- Lim, C.Y.; Owens, N.A.; Wampler, R.D.; Ying, Y.; Granger, J.H.; Porter, M.D.; Takahashi, M.; Shimazu, K. Succinimidyl Ester Surface Chemistry: Implications of the Competition between Aminolysis and Hydrolysis on Covalent Protein Immobilization. Langmuir 2014, 30, 12868–12878. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Pounder, R.J.; Stanford, M.J.; Brooks, P.; Richards, S.P.; Dove, A.P. Metal free thiol–maleimide ‘Click’ reaction as a mild functionalisation strategy for degradable polymers. Chem. Commun. 2008, 5158–5160. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Bednarek, M. Coupling reaction with thiols as the efficient method of functionalization of ‘‘clickable” polylactide. React. Funct. Polym. 2013, 73, 1130–1136. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiolene “click” reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol–ene “click” reactions and recent applications in polymer and materials synthesis: A first update. Polym. Chem. 2014, 5, 4820–4870. [Google Scholar] [CrossRef]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef] [Green Version]
- Albertsson, A.-C.; Varma, I.K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef] [PubMed]
- Jérôme, C.; LeComte, P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv. Drug Deliv. Rev. 2008, 60, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- LeComte, P.; Jérôme, C. Recent Developments in Ring-Opening Polymerization of Lactones. Adv. Polym. Sci. 2011, 245, 173–217. [Google Scholar] [CrossRef]
- Dove, A.P. Controlled ring-opening polymerisation of cyclic esters: Polymer blocks in self-assembled nanostructures. Chem. Commun. 2008, 6446–6470. [Google Scholar] [CrossRef] [PubMed]
- Olsén, P.; Odelius, K.; Albertsson, A.-C. Thermodynamic Presynthetic Considerations for Ring-Opening Polymerization. Biomacromolecules 2016, 17, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Nifant’Ev, I.; Ivchenko, P. Coordination Ring-Opening Polymerization of Cyclic Esters: A Critical Overview of DFT Modeling and Visualization of the Reaction Mechanisms. Molecules 2019, 24, 4117. [Google Scholar] [CrossRef] [Green Version]
- Nifant’Ev, I.; Ivchenko, P. DFT Modeling of Organocatalytic Ring-Opening Polymerization of Cyclic Esters: A Crucial Role of Proton Exchange and Hydrogen Bonding. Polymers 2019, 11, 2078. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, S.M.; Kirillov, E.; Sarazin, Y.; Carpentier, J.-F. Beyond Stereoselectivity, Switchable Catalysis: Some of the Last Frontier Challenges in Ring-Opening Polymerization of Cyclic Esters. Chem. A Eur. J. 2015, 21, 7988–8003. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, D.; Zhang, W.; Solan, G.A.; Ma, Y.; Sun, W.-H. Recent progress in the application of group 1, 2 & 13 metal complexes as catalysts for the ring opening polymerization of cyclic esters. Inorg. Chem. Front. 2019, 6, 2619–2652. [Google Scholar] [CrossRef]
- Miron, T.; Wilchek, M. A simplified method for the preparation of succinimidyl carbonate polyethylene glycol for coupling to proteins. Bioconjugate Chem. 1993, 4, 568–569. [Google Scholar] [CrossRef]
- Steinbach, T.; Wurm, F.R. Degradable Polyphosphoester-Protein Conjugates: “PPEylation” of Proteins. Biomacromolecules 2016, 17, 3338–3346. [Google Scholar] [CrossRef] [Green Version]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, S.S.V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Shuai, X.; Merdan, T.; Unger, F.; Wittmar, A.M.; Kissel, T. Novel Biodegradable Ternary Copolymershy-PEI-g-PCL-b-PEG: Synthesis, Characterization, and Potential as Efficient Nonviral Gene Delivery Vectors. Macromolecules 2003, 36, 5751–5759. [Google Scholar] [CrossRef]
- Steinbach, T.; Becker, G.; Spiegel, A.; Figueiredo, T.; Russo, D.; Wurm, F.R. Reversible Bioconjugation: Biodegradable Poly(phosphate)-Protein Conjugates. Macromol. Biosci. 2016, 17, 1600377. [Google Scholar] [CrossRef]
- Ji, S.; Zhu, Z.; Hoye, T.R.; Macosko, C.W. Maleimide Functionalized Poly(ε-caprolactone)-block-poly(ethylene glycol) (PCL-PEG-MAL): Synthesis, Nanoparticle Formation, and Thiol Conjugation. Macromol. Chem. Phys. 2009, 210, 823–831. [Google Scholar] [CrossRef]
- Pelosi, C.; Duce, C.; Russo, D.; Tiné, M.R.; Wurm, F.R. PPEylation of proteins: Synthesis, activity, and stability of myoglobin-polyphosphoester conjugates. Eur. Polym. J. 2018, 108, 357–363. [Google Scholar] [CrossRef]
- Pelosi, C.; Tinè, M.R.; Wurm, F.R. Main-chain water-soluble polyphosphoesters: Multi-functional polymers as degradable PEG-alternatives for biomedical applications. Eur. Polym. J. 2020, 141, 110079. [Google Scholar] [CrossRef]
- Russo, D.; De Angelis, A.; Garvey, C.J.; Wurm, F.R.; Appavou, M.-S.; Prevost, S. Effect of Polymer Chain Density on Protein–Polymer Conjugate Conformation. Biomacromolecules 2019, 20, 1944–1955. [Google Scholar] [CrossRef]
- Stanford, M.J.; Dove, A.P. One-Pot Synthesis of α,ω-Chain End Functional, Stereoregular, Star-Shaped Poly(lactide). Macromolecules 2009, 42, 141–147. [Google Scholar] [CrossRef]
- Stanford, M.J.; Pflughaupt, R.L.; Dove, A.P. Synthesis of Stereoregular Cyclic Poly(lactide)s via “Thiol−Ene” Click Chemistry. Macromolecules 2010, 43, 6538–6541. [Google Scholar] [CrossRef]
- Nifant’Ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Ivchenko, P.V.; Borisov, R.S.; Churakov, A.V. Monomeric and dimeric magnesium mono-BHT complexes as effective ROP catalysts. Catal. Commun. 2016, 87, 106–111. [Google Scholar] [CrossRef]
- Ivchenko, P.V.; Shlyakhtin, A.V.; Nifant’Ev, I.E. Ring-opening polymerization of glycolide and rac -lactide, catalyzed by aryloxy magnesium complexes: DFT study of reaction profile and stereocontrol mechanism. Mendeleev Commun. 2017, 27, 278–280. [Google Scholar] [CrossRef]
- Nifant’Ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Controlled ring-opening polymerisation of cyclic phosphates, phosphonates and phosphoramidates catalysed by heteroleptic BHT-alkoxy magnesium complexes. Polym. Chem. 2017, 8, 6806–6816. [Google Scholar] [CrossRef]
- Nifant’Ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Minyaev, M.E.; Churakov, A.V.; Karchevsky, S.G.; Birin, K.P.; Ivchenko, P.V. Mono-BHT heteroleptic magnesium complexes: Synthesis, molecular structure and catalytic behavior in the ring-opening polymerization of cyclic esters. Dalton Trans. 2017, 46, 12132–12146. [Google Scholar] [CrossRef] [Green Version]
- Nifant’Ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Synthesis and ring-opening polymerization of glycidyl ethylene phosphate with a formation of linear and branched polyphosphates. Mendeleev Commun. 2018, 28, 155–157. [Google Scholar] [CrossRef]
- Nifant’Ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Tavtorkin, A.N.; Minyaev, M.E.; Kosarev, M.A.; Ivchenko, P.V. Synthesis in aqueous media of poly(ethylene phosphoric acids) by mild thermolysis of homopolymers and block copolymers based on tert -butyl ethylene phosphate. Eur. Polym. J. 2018, 106, 249–256. [Google Scholar] [CrossRef]
- Nifant’Ev, I.; Shlyakhtin, A.; Kosarev, M.; Karchevsky, S.; Ivchenko, P. Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data. Polymers 2018, 10, 1105. [Google Scholar] [CrossRef] [Green Version]
- Nifant’Ev, I.; Shlyakhtin, A.; Kosarev, M.; Gavrilov, D.; Karchevsky, S.; Ivchenko, P. DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates. Polymers 2019, 11, 1641. [Google Scholar] [CrossRef] [Green Version]
- Nifant’Ev, I.; Komarov, P.; Ovchinnikova, V.; Kiselev, A.; Minyaev, M.; Ivchenko, P. Comparative Experimental and Theoretical Study of Mg, Al and Zn Aryloxy Complexes in Copolymerization of Cyclic Esters: The Role of the Metal Coordination in Formation of Random Copolymers. Polymers 2020, 12, 2273. [Google Scholar] [CrossRef]
- Dar’In, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. New Heterocyclic Product Space for the Castagnoli–Cushman Three-Component Reaction. Org. Lett. 2015, 17, 3930–3933. [Google Scholar] [CrossRef] [PubMed]
- Baumhover, N.J.; Anderson, K.; Fernandez, C.A.; Rice, K.G. Synthesis and In Vitro Testing of New Potent Polyacridine−Melittin Gene Delivery Peptides. Bioconjugate Chem. 2009, 21, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, A.J.; Del Amo, V.; Philp, D. Structure–reactivity relationships in a recognition mediated [3+2] dipolar cycloaddition reaction. Org. Biomol. Chem. 2009, 7, 3308–3318. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Steinbach, T.; Wurm, F.R. A Library of Well-Defined and Water-Soluble Poly(alkyl phosphonate)s with Adjustable Hydrolysis. Macromolecules 2015, 48, 3853–3863. [Google Scholar] [CrossRef]
- Steinbach, T.; Schröder, R.; Ritz, S.; Wurm, F.R. Microstructure analysis of biocompatible phosphoester copolymers. Polym. Chem. 2013, 4, 4469–4479. [Google Scholar] [CrossRef] [Green Version]
- Nifant’Ev, I.; Shlyakhtin, A.; Bagrov, V.; Lozhkin, B.; Zakirova, G.; Ivchenko, P.; Legon’Kova, O. Theoretical and experimental studies of 1,5,7-triazabicyclo[4.4.0]dec-5-ene-catalyzed ring opening/ring closure reaction mechanism for 5-, 6- and 7-membered cyclic esters and carbonates. React. Kinet. Mech. Catal. 2015, 117, 447–476. [Google Scholar] [CrossRef]
- Liu, X.X.; Melman, A. Templated alkylation of hexahistidine with Baylis–Hillman esters. Chem. Commun. 2013, 49, 9042–9044. [Google Scholar] [CrossRef]
- Corrie, J.E.T.; Trentham, D.R. Synthesis of photoactivatable fluorescein derivatives bearing side chains with varying properties. J. Chem. Soc. Perkin Trans. 1 1995, 1, 1993–2000. [Google Scholar] [CrossRef]
- Camper, N.; Scott, C.J.; Migaud, M.E. Synthesis of an analogue of the bisphosphonate drug Ibandronate for targeted drug-delivery therapeutic strategies. New J. Chem. 2010, 34, 949–955. [Google Scholar] [CrossRef]
- Hoang, M.-D.; Kumar, R.A.; Buisson, D.A.; Ling, W.L.; Gravel, E.; Doris, E.; Ramar, A.K. Self-assembled Polydiacetylene Nanoribbons for Semi-heterogeneous and Enantioselective Organocatalysis of Aldol Reactions in Water. ChemCatChem 2019, 12, 1156–1160. [Google Scholar] [CrossRef]
- Wang, D.; Cao, Y.; Zheng, L.; Lv, D.; Chen, L.; Xing, X.; Zhu, Z.; Li, X.; Chai, Y. Identification of Annexin A2 as a target protein for plant alkaloid matrine. Chem. Commun. 2017, 53, 5020–5023. [Google Scholar] [CrossRef]
- Tian, D.-M.; Qiao, J.; Bao, Y.-Z.; Liu, J.; Zhang, X.-K.; Sun, X.-L.; Zhang, Y.-W.; Yao, X.-S.; Tang, J.-S. Design and synthesis of biotinylated cardiac glycosides for probing Nur77 protein inducting pathway. Bioorganic Med. Chem. Lett. 2019, 29, 707–712. [Google Scholar] [CrossRef]
- Yao, Y.; Yu, L.; Su, X.; Wang, Y.; Li, W.; Wu, Y.; Cheng, X.; Zhang, H.; Wei, X.; Chen, H.; et al. Synthesis, characterization and targeting chemotherapy for ovarian cancer of trastuzumab-SN-38 conjugates. J. Control. Release 2015, 220, 5–17. [Google Scholar] [CrossRef]
- Attatsi, I.K.; Zhu, W.; Liang, X. Noncovalent immobilization of Co(ii)porphyrin through axial coordination as an enhanced electrocatalyst on carbon electrodes for oxygen reduction and evolution. New J. Chem. 2020, 44, 4340–4345. [Google Scholar] [CrossRef]
- Yıldırım, A. Surfactant-Catalyzed Addition of Higher Thiols to N-Aryl Substituted Maleimides. Synth. Commun. 2014, 44, 1137–1141. [Google Scholar] [CrossRef]
- Grin, M.A.; Brusov, S.S.; Shchepelina, E.Y.; Ponomarev, P.V.; Khrenova, M.K.; Smirnov, A.S.; Lebedeva, V.S.; Mironov, A.F. Conjugates of natural chlorins with cyclen as chelators of transition metals. Mendeleev Commun. 2017, 27, 338–340. [Google Scholar] [CrossRef]
- Minyaev, M.E.; Nifant’Ev, I.E.; Shlyakhtin, A.V.; Ivchenko, P.V.; Lyssenko, K.A. Phenoxide and alkoxide complexes of Mg, Al and Zn, and their use for the ring-opening polymerization of ∊-caprolactone with initiators of different natures. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Minyaev, M.E.; Churakov, A.V.; Nifant’Ev, I.E. Structural diversity of polynuclear Mg x O y cores in magnesium phenoxide complexes. Acta Crystallogr. Sect. C Struct. Chem. 2017, 73, 854–861. [Google Scholar] [CrossRef]
- Odinokov, V.N.; Akhmetova, V.R.; Botsman, L.P.; Tolstikov, G.A.; Moiseenkov, A.M. Insect pheromones and their analogs. XI. Synthesis of 2,6-dimethylhepta-1,6-dien-3-ol acetate ? The sex attractant ofPseudococcus comstocki. Chem. Nat. Compd. 1985, 21, 243–244. [Google Scholar] [CrossRef]
- Liu, J.; Ma, D. A Unified Approach for the Assembly of Atisine- and Hetidine-type Diterpenoid Alkaloids: Total Syntheses of Azitine and the Proposed Structure of Navirine C. Angew. Chem. Int. Ed. 2018, 57, 6676–6680. [Google Scholar] [CrossRef] [PubMed]
- An, Y.-L.; Deng, Y.-X.; Zhang, W.; Zhao, S.-Y. Regioselective Hetero-Michael Addition of Oxygen, Sulfur, and Nitrogen Nucleophiles to Maleimides Catalyzed by BF3·OEt2. Synthesis 2015, 47, 1581–1592. [Google Scholar] [CrossRef]
- Lin, Y.-M.; Lu, G.-P.; Cai, C.; Yi, W.-B. An odorless thia-Michael addition using Bunte salts as thiol surrogates. RSC Adv. 2015, 5, 27107–27111. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Mon1 | Mon2 | ROP Termination Agent (TA) | (Mon1)/(Mon2)/(Cat)/(TA) Ratio | Mon1 conv., % 1 | Mon2 conv., % 1 | Mn·103 (theor) 1 | Mn·103 (NMR) 2 | Mn·103 (SEC) 3 | ĐM (SEC) 3 | Comp. of Polymer: Mon1/Mon2/FG (NMR) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | εCL | - | DSC | 50/-/1/5 | >99 | - | 5.96 | 6.0 | 6.4 | 1.19 | 50/-/0.33 |
| 2 | εCL | - | SA, NHS/DCC | 50/-/1/5 | >99 | - | 5.96 | 7.0 | 7.4 | 1.22 | 59/-/0.51 |
| 3 | εCL | - | 1 | 120/-/1/4 | >99 | - | 14.02 | 17.3 | 16.1 | 1.18 | 149/-/1.0 |
| 4 | εCL | - | 2 | 120/-/1/4 | >99 | - | 14.02 | 16.9 | 18.2 | 1.29 | 145/-/0.39 |
| 5 | εCL | - | 3 | 120/-/1/4 | >99 | - | 13.96 | 17.3 | 16.5 | 1.23 | 149/-/0.40 |
| 6 | εCL | - | 4 | 120/-/1/4 | >99 | - | 13.94 | 18.2 | 22.4 | 1.15 | 157/-/0.76 |
| 7 | l-LA | - | 1 | 120/-/1/4 | >99 | - | 17.62 | 18.8 | 21.7 | 1.43 | 128/-/1.0 |
| 8 | l-LA | - | 4 | 120/-/1/4 | >99 | - | 17.56 | 22.8 | 25.6 | 1.55 | 156/-/1.0 |
| 9 | EtEP | - | 1 | 120/-/1/4 | 94 | - | 15.67 | 17.1 | 14.3 | 1.19 | 123/-/1.0 |
| 10 | EtEP | - | 4 | 120/-/1/4 | 95 | - | 15.78 | 17.9 | 14.5 | 1.18 | 130/-/1.0 |
| 11 | εCL | EtEP | 1 | 120/35/1/4 | >99 | 65 | 17.11 | 17.1 | 21.4 | 1.48 | 131/14/1.0 |
| 12 | εCL | EtEP | 4 | 120/35/1/4 | >99 | 79 | 17.72 | 22.9 | 19.5 | 1.46 | 173/21/1.0 |
| 13 | EtOEP | - | 1 | 120/-/1/4 | 96 | - | 17.84 | 22.5 | 16.5 | 1.48 | 146/-/1.0 |
| 14 4 | EtOEP | - | 1 | 50/-/1/4 | >99 | - | 7.85 | 8.3 | 5.2 | 1.86 | 53/-/<0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nifant’ev, I.; Shlyakhtin, A.; Bagrov, V.; Shaputkin, E.; Tavtorkin, A.; Ivchenko, P. Functionalized Biodegradable Polymers via Termination of Ring-Opening Polymerization by Acyl Chlorides. Polymers 2021, 13, 868. https://doi.org/10.3390/polym13060868
Nifant’ev I, Shlyakhtin A, Bagrov V, Shaputkin E, Tavtorkin A, Ivchenko P. Functionalized Biodegradable Polymers via Termination of Ring-Opening Polymerization by Acyl Chlorides. Polymers. 2021; 13(6):868. https://doi.org/10.3390/polym13060868
Chicago/Turabian StyleNifant’ev, Ilya, Andrey Shlyakhtin, Vladimir Bagrov, Evgeny Shaputkin, Alexander Tavtorkin, and Pavel Ivchenko. 2021. "Functionalized Biodegradable Polymers via Termination of Ring-Opening Polymerization by Acyl Chlorides" Polymers 13, no. 6: 868. https://doi.org/10.3390/polym13060868