


Assessing the In Vivo Biocompatibility of Molecularly Imprinted Polymer Nanoparticles
 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solid Phase Preparation: Trypsin
2.3. NanoMIP Synthesis
2.4. Animal Handling—Rats
2.5. Treatment of Rats with NanoMIPs
2.6. Fluorescence Imaging of Tissue
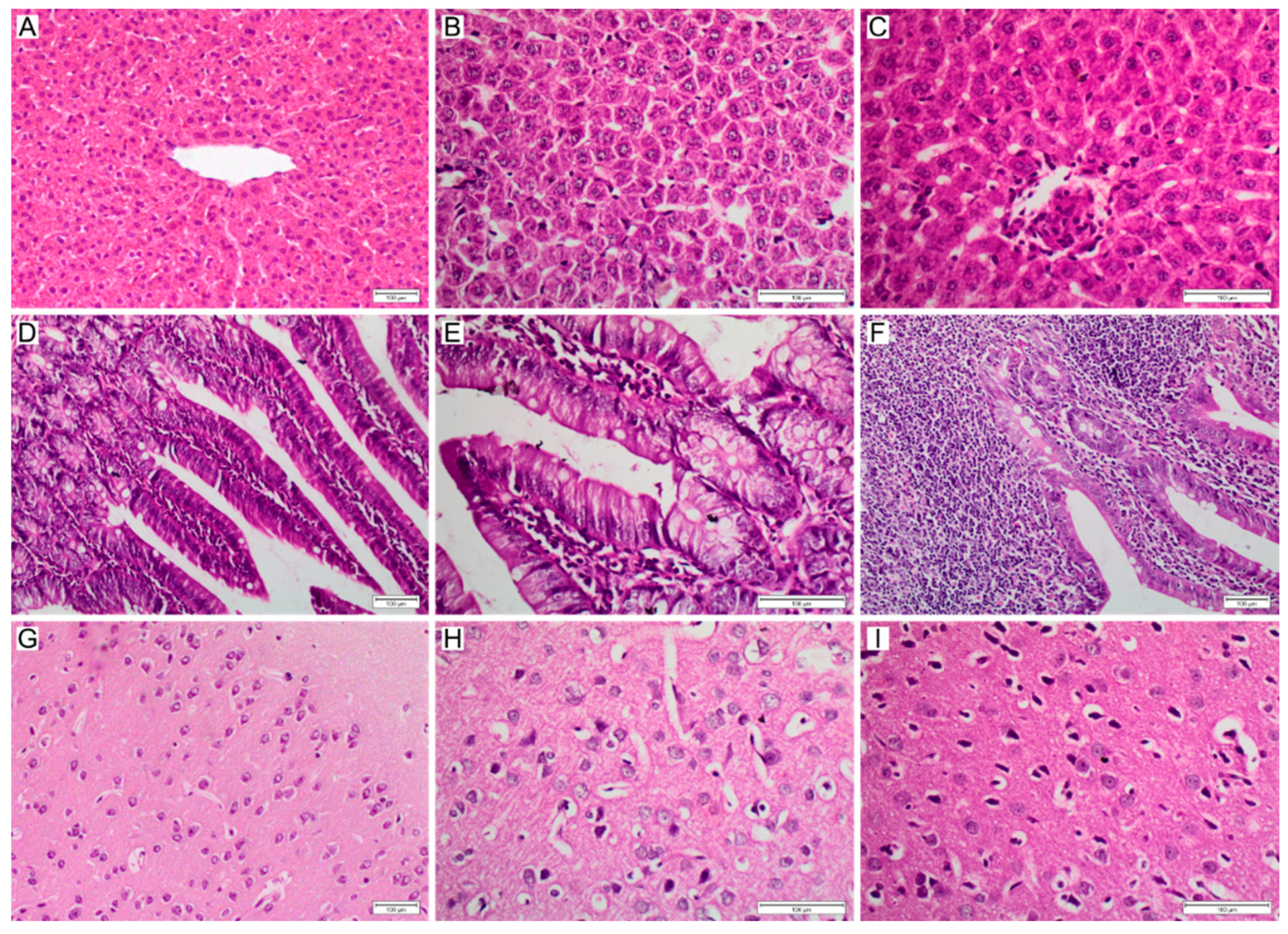
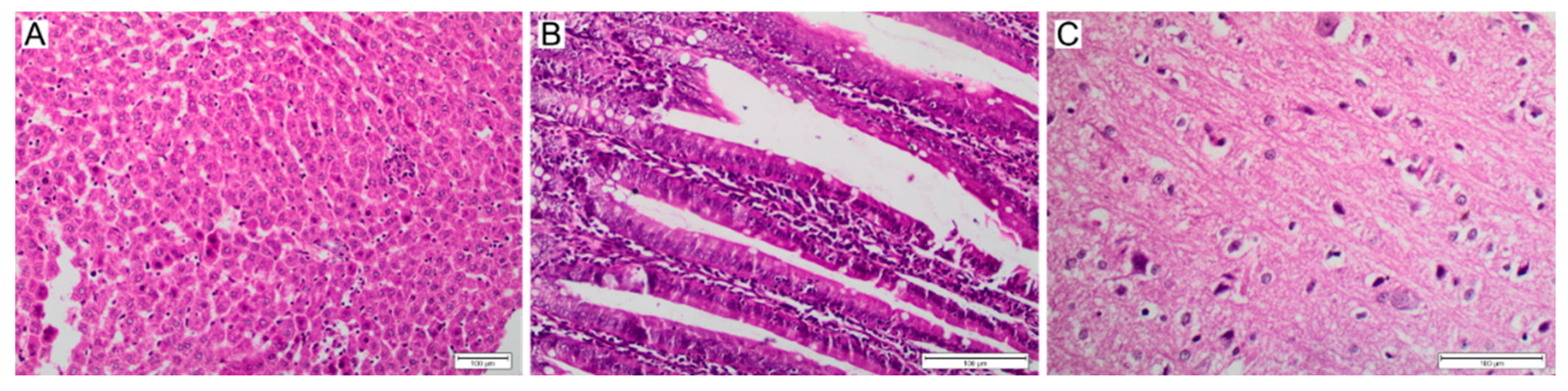
2.7. Histopathological Studies
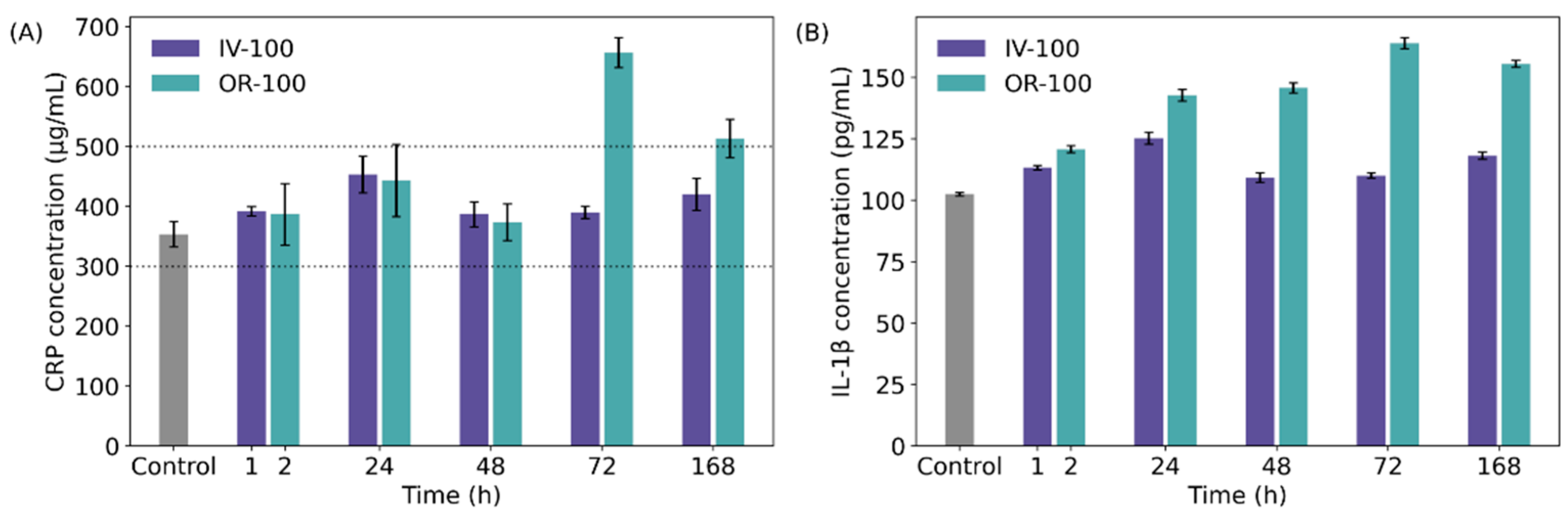
2.8. Detection of C-Reactive Protein (CRP), Interleukin 1-β (Il-1β)
2.9. Quantitative Analysis of NPs in Urine and Faeces
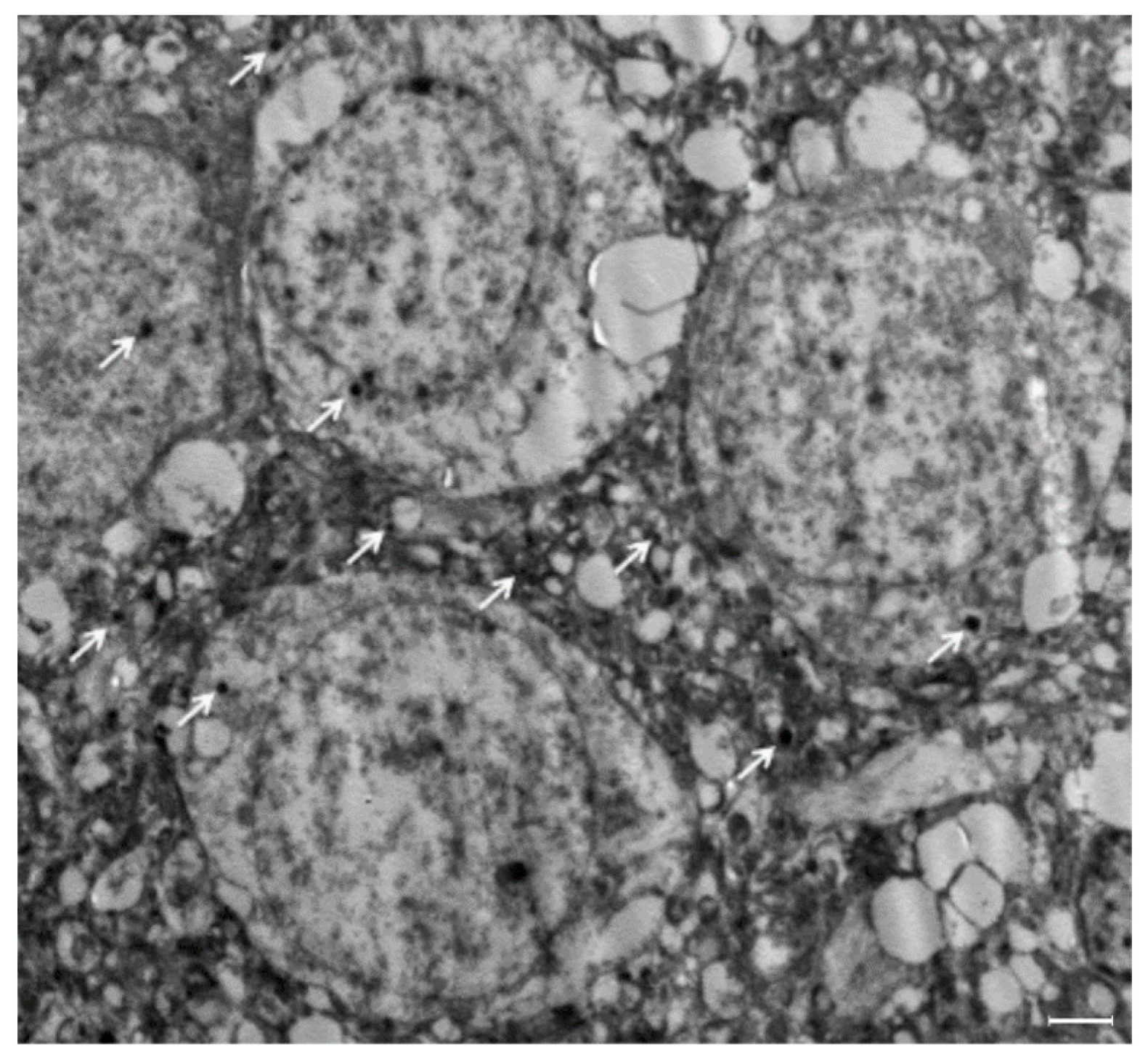
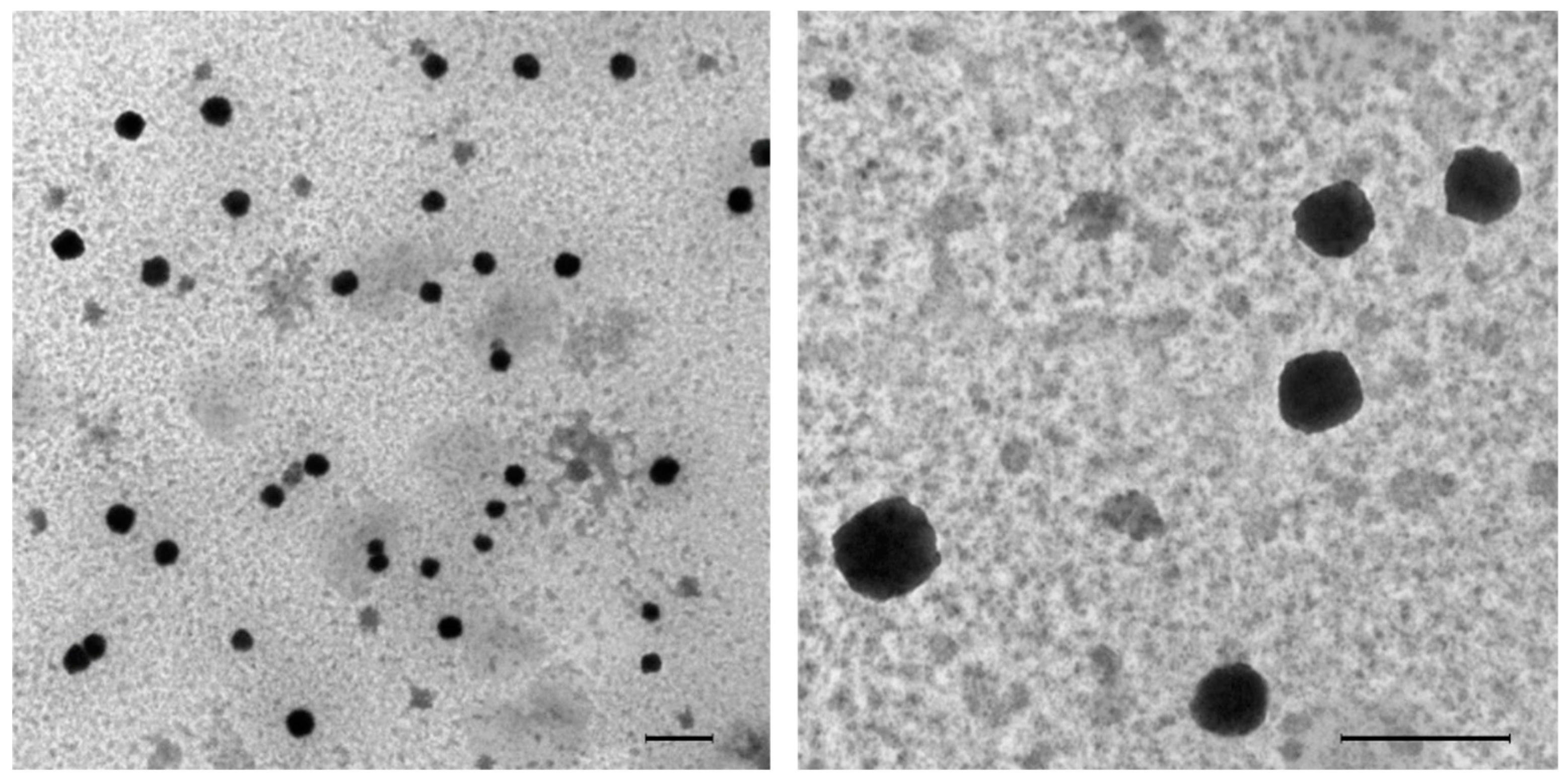
2.10. Detection of MIPs in Urine and Brain Tissue by TEM
2.11. Solid Phase Preparation: EGFR Peptides
2.12. Animal Handling—Mice
2.13. Immunisation of Mice
2.14. Quantification of Ovalbumin-Specific IgG Levels in Mouse Serum
3. Results and Discussion
3.1. Biodistribution of NanoMIPs
3.2. In Vivo Clearance of NanoMIPs
3.3. Cytotoxicity of NanoMIPs
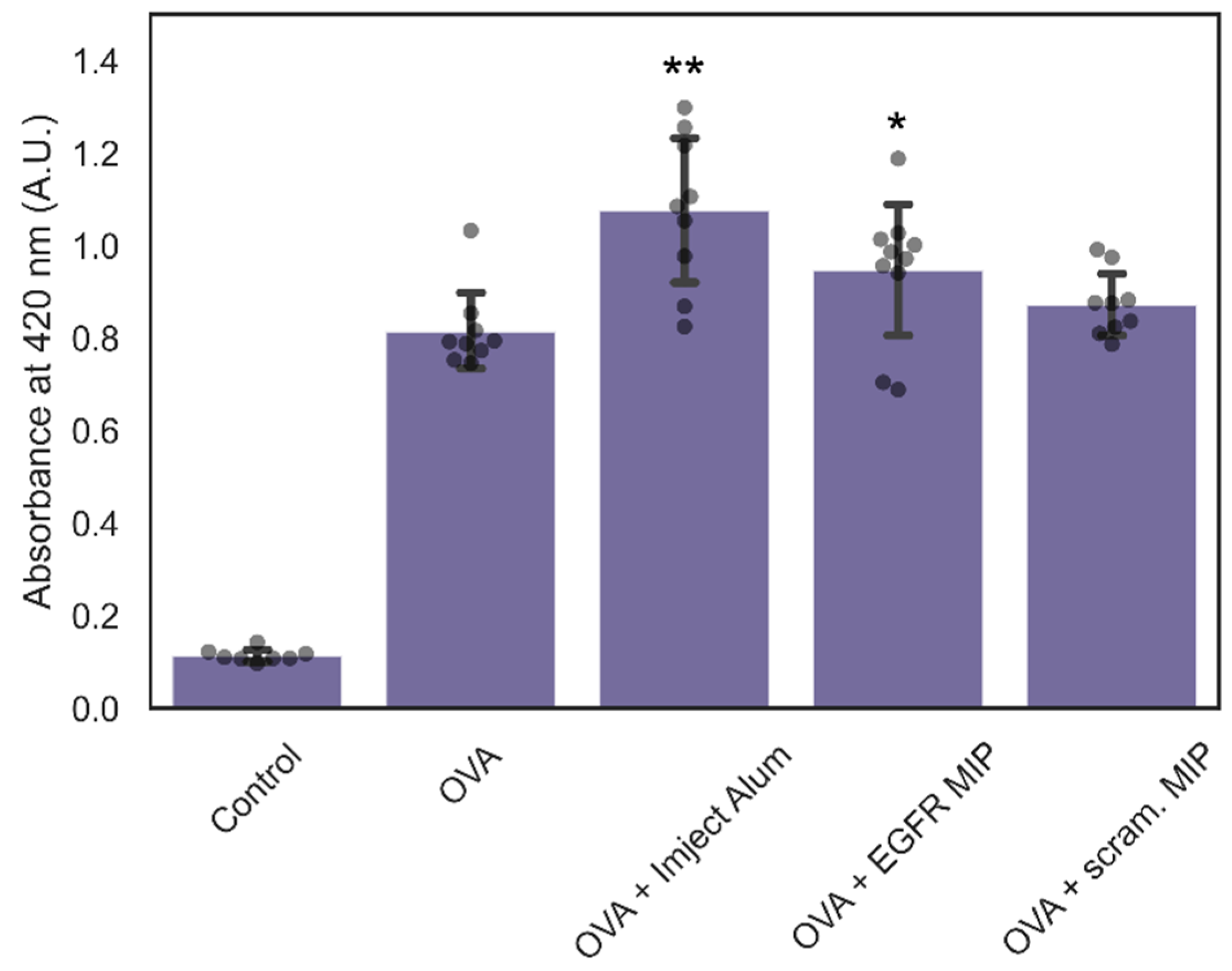
3.4. Immunogenicity of NanoMIPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, J.; Feng, X.; Jiang, Z.; Xu, W.; Guo, H.; Zhuang, X.; Chen, X. Polymer-Mediated Penetration-Independent Cancer Therapy. Biomacromolecules 2019, 20, 4258–4271. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–Drug Conjugate Therapeutics: Advances, Insights and Prospects. Nat. Rev. Drug Discov. 2018, 18, 273–294. [Google Scholar] [CrossRef]
- Qiu, L.Y.; Bae, Y.H. Polymer Architecture and Drug Delivery. Pharm. Res. 2006, 23, 1–30. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Rossin, R.; Turner, J.L.; Becker, M.L.; Joralemon, M.J.; Welch, M.J.; Wooley, K.L. An Assessment of the Effects of Shell Cross-Linked Nanoparticle Size, Core Composition, and Surface PEGylation on in Vivo Biodistribution. Biomacromolecules 2005, 6, 2541–2554. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR Effect: Unique Features of Tumor Blood Vessels for Drug Delivery, Factors Involved, and Limitations and Augmentation of the Effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Nel, A.; Ruoslahti, E.; Meng, H. New Insights into “Permeability” as in the Enhanced Permeability and Retention Effect of Cancer Nanotherapeutics. ACS Nano 2017, 11, 9567–9569. [Google Scholar] [CrossRef]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of Nanoparticle Size, Shape and Surface Chemistry in Oral Drug Delivery. J. Control. Release 2016, 238, 176. [Google Scholar] [CrossRef] [Green Version]
- Sang, Y.; Miao, P.; Chen, T.; Zhao, Y.; Chen, L.; Tian, Y.; Han, X.; Gao, J. Fabrication and Evaluation of Graphene Oxide/Hydroxypropyl Cellulose/Chitosan Hybrid Aerogel for 5-Fluorouracil Release. Gels 2022, 8, 649. [Google Scholar] [CrossRef]
- Canfarotta, F.; Lezina, L.; Guerreiro, A.; Czulak, J.; Petukhov, A.; Daks, A.; Smolinska-Kempisty, K.; Poma, A.; Piletsky, S.; Barlev, N.A. Specific Drug Delivery to Cancer Cells with Double-Imprinted Nanoparticles against Epidermal Growth Factor Receptor. Nano Lett. 2018, 18, 4641–4646. [Google Scholar] [CrossRef]
- Liu, J.F.; Jang, B.; Issadore, D.; Tsourkas, A. Use of Magnetic Fields and Nanoparticles to Trigger Drug Release and Improve Tumor Targeting. Wiley Interdiscip. Rev. Nanomed. Nanonbiotechnol. 2019, 11, e1571. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kitayama, Y.; Sasao, R.; Yamada, T.; Toh, K.; Matsumoto, Y.; Kataoka, K. Molecularly Imprinted Nanogels Acquire Stealth In Situ by Cloaking Themselves with Native Dysopsonic Proteins. Angew. Chem. 2017, 129, 7194–7198. [Google Scholar] [CrossRef]
- Abdollahi, E.; Khalafi-Nezhad, A.; Mohammadi, A.; Abdouss, M.; Salami-Kalajahi, M. Synthesis of New Molecularly Imprinted Polymer via Reversible Addition Fragmentation Transfer Polymerization as a Drug Delivery System. Polymer 2018, 143, 245–257. [Google Scholar] [CrossRef]
- Canfarotta, F.; Poma, A.; Guerreiro, A.; Piletsky, S. Solid-Phase Synthesis of Molecularly Imprinted Nanoparticles. Nat. Protoc. 2016, 11, 443–455. [Google Scholar] [CrossRef]
- Poma, A.; Guerreiro, A.; Caygill, S.; Moczko, E.; Piletsky, S. Automatic Reactor for Solid-Phase Synthesis of Molecularly Imprinted Polymeric Nanoparticles (MIP NPs) in Water. RSC Adv. 2014, 4, 4203–4206. [Google Scholar] [CrossRef] [Green Version]
- Canfarotta, F.; Waters, A.; Sadler, R.; McGill, P.; Guerreiro, A.; Papkovsky, D.; Haupt, K.; Piletsky, S. Biocompatibility and Internalization of Molecularly Imprinted Nanoparticles. Nano Res. 2016, 9, 3463–3477. [Google Scholar] [CrossRef]
- Qin, Y.T.; Peng, H.; He, X.W.; Li, W.Y.; Zhang, Y.K. Highly Effective Drug Delivery and Cell Imaging Using Fluorescent Double-Imprinted Nanoparticles by Targeting Recognition of the Epitope of Membrane Protein. Anal. Chem. 2019, 91, 12696–12703. [Google Scholar] [CrossRef]
- Neamtu, I.; Rusu, A.G.; Diaconu, A.; Nita, L.E.; Chiriac, A.P. Basic Concepts and Recent Advances in Nanogels as Carriers for Medical Applications. Drug Deliv. 2017, 24, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Safaryan, A.H.M.; Smith, A.M.; Bedwell, T.S.; Piletska, E.V.; Canfarotta, F.; Piletsky, S.A. Optimisation of the Preservation Conditions for Molecularly Imprinted Polymer Nanoparticles Specific for Trypsin. Nanoscale Adv. 2019, 1, 3709–3714. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, N.; Umar, S.; Ashafaq, M.; Akhtar, M.; Iqbal, Z.; Samim, M.; Ahmad, F.J. A Comparative Study of PNIPAM Nanoparticles of Curcumin, Demethoxycurcumin, and Bisdemethoxycurcumin and Their Effects on Oxidative Stress Markers in Experimental Stroke. Protoplasma 2013, 250, 1327–1338. [Google Scholar] [CrossRef]
- Win, Y.Y.; Charoenkanburkang, P.; Limprasutr, V.; Rodsiri, R.; Pan, Y.; Buranasudja, V.; Luckanagul, J.A. In Vivo Biocompatible Self-Assembled Nanogel Based on Hyaluronic Acid for Aqueous Solubility and Stability Enhancement of Asiatic Acid. Polymer 2021, 13, 4071. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.A.; Cagle, P.T. The 100-Year Anniversary of the Description of the Frozen Section Procedure. JAMA 2005, 294, 3135–3137. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, J.D.; Gamble, M. Theory and Practice of Histological Techniques, 6th ed.; Bancroft, J.D., Gamble, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780443102790. [Google Scholar]
- Dinarello, C.A. Interleukin-1 and Interleukin-1 Antagonism. Blood 1991, 77, 1627–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, M.; Tripathi, L.M.; Srivastava, V.M.L.; Puri, A.; Shukla, R. Modulation of Inflammatory Mediators by Ibuprofen and Curcumin Treatment during Chronic Inflammation in Rat. Immunopharmacol. Immunotoxicol. 2003, 25, 213–224. [Google Scholar] [CrossRef]
- Cho, M.; Cho, W.S.; Choi, M.; Kim, S.J.; Han, B.S.; Kim, S.H.; Kim, H.O.; Sheen, Y.Y.; Jeong, J. The Impact of Size on Tissue Distribution and Elimination by Single Intravenous Injection of Silica Nanoparticles. Toxicol. Lett. 2009, 189, 177–183. [Google Scholar] [CrossRef]
- Hunter, E.E.; Silver, M. Practical Electron Microscopy: A Beginner’s Illustrated Guide; Cambridge University Press: Cambridge, UK, 1993; ISBN 9780521385398. [Google Scholar]
- Piletsky, S.S.; Piletska, E.; Poblocka, M.; Macip, S.; Jones, D.J.L.; Braga, M.; Cao, T.H.; Singh, R.; Spivey, A.C.; Aboagye, E.O.; et al. Snapshot Imprinting: Rapid Identification of Cancer Cell Surface Proteins and Epitopes Using Molecularly Imprinted Polymers. Nano Today 2021, 41, 101304. [Google Scholar] [CrossRef]
- Piletsky, S.S.; Cruz, A.G.; Piletska, E.; Piletsky, S.A.; Aboagye, E.O.; Spivey, A.C. Iodo Silanes as Superior Substrates for the Solid Phase Synthesis of Molecularly Imprinted Polymer Nanoparticles. Polymers 2022, 14, 1595. [Google Scholar] [CrossRef]
- Guerreiro, A.; Poma, A.; Karim, K.; Moczko, E.; Takarada, J.; de Vargas-Sansalvador, I.P.; Turner, N.; Piletska, E.; de Magalhães, C.S.; Glazova, N.; et al. Influence of Surface-Imprinted Nanoparticles on Trypsin Activity. Adv. Health Mater. 2014, 3, 1426–1429. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, S.; Beyazit, S.; Haupt, K.; Tse Sum Bui, B. Solid-Phase Synthesis of Molecularly Imprinted Nanoparticles for Protein Recognition. Chem. Commun. 2013, 49, 6746–6748. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.I.; Kim, Y.T. Nanotherapeutics Engineered to Cross the Blood-Brain Barrier for Advanced Drug Delivery to the Central Nervous System. J. Ind. Eng. Chem. 2019, 73, 8–18. [Google Scholar] [CrossRef]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRX 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Desai, N. Challenges in Development of Nanoparticle-Based Therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, B.; Zhou, S.; Chen, W.; Chen, H.; Liang, S.; Zheng, L.; Yu, H.; Chu, R.; Wang, M.; et al. Surface Chemistry Governs the Sub-Organ Transfer, Clearance and Toxicity of Functional Gold Nanoparticles in the Liver and Kidney. J. Nanobiotechnol. 2020, 18, 45. [Google Scholar] [CrossRef] [Green Version]
- Park, J.K.; Utsumi, T.; Seo, Y.E.; Deng, Y.; Satoh, A.; Saltzman, W.M.; Iwakiri, Y. Cellular Distribution of Injected PLGA-Nanoparticles in the Liver. Nanomedicine 2016, 12, 1365–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.D.; Smith, S.A.; Thurecht, K.J.; Such, G. Engineered Polymeric Materials for Biological Applications: Overcoming Challenges of the Bio–Nano Interface. Polymers 2019, 11, 1441. [Google Scholar] [CrossRef] [Green Version]
- Armentano, I.; Puglia, D.; Luzi, F.; Arciola, C.R.; Morena, F.; Martino, S.; Torre, L. Nanocomposites Based on Biodegradable Polymers. Materials 2018, 11, 795. [Google Scholar] [CrossRef] [Green Version]
- Missaoui, W.N.; Arnold, R.D.; Cummings, B.S. Toxicological Status of Nanoparticles: What We Know and What We Don’t Know. Chem. Biol. Interact. 2018, 295, 1–12. [Google Scholar] [CrossRef]
- Oberdörster, G.; Oberdörster, E.; Oberdörster, J. Nanotoxicology: An Emerging Discipline Evolving from Studies of Ultrafine Particles. Environ. Health Perspect. 2005, 113, 823–839. [Google Scholar] [CrossRef]
- Wyss, P.P.; Lamichhane, S.P.; Abed, A.; Vonwil, D.; Kretz, O.; Huber, T.B.; Sarem, M.; Shastri, V.P. Renal Clearance of Polymeric Nanoparticles by Mimicry of Glycan Surface of Viruses. Biomaterials 2020, 230, 119643. [Google Scholar] [CrossRef]
- Hendrickson, G.R.; Andrew Lyon, L. Microgel Translocation through Pores under Confinement. Angew. Chem. Int. Ed. 2010, 49, 2193–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, D.E.; Peppas, N.A. Opsonization, Biodistribution, and Pharmacokinetics of Polymeric Nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Heo, D.; Park, J.; Na, S.; Suh, J.S.; Haam, S.; Wook Park, S.; Huh, Y.M.; Yang, J. Role of Surface Charge in Cytotoxicity of Charged Manganese Ferrite Nanoparticles towards Macrophages. Nanotechnology 2012, 23, 505702. [Google Scholar] [CrossRef] [PubMed]
- Yokel, R.A.; Florence, R.L.; Unrine, J.M.; Tseng, M.T.; Graham, U.M.; Wu, P.; Grulke, E.A.; Sultana, R.; Hardas, S.S.; Butterfield, D.A. Biodistribution and Oxidative Stress Effects of a Systemically-Introduced Commercial Ceria Engineered Nanomaterial. Nanotoxicology 2009, 3, 234–248. [Google Scholar] [CrossRef]
- Boitard, C.; Curcio, A.; Rollet, A.L.; Wilhelm, C.; Ménager, C.; Griffete, N. Biological Fate of Magnetic Protein-Specific Molecularly Imprinted Polymers: Toxicity and Degradation. ACS Appl. Mater. Interfaces 2019, 11, 35556–35565. [Google Scholar] [CrossRef]
- Padilla, N.D.; Bleeker, W.K.; Lubbers, Y.; Rigter, G.M.M.; van Mierlo, G.J.; Daha, M.R.; Hack, C.E. Rat C-Reactive Protein Activates the Autologous Complement System. Immunology 2003, 109, 564. [Google Scholar] [CrossRef]
- Du, Z.; Zhao, D.; Jing, L.; Cui, G.; Jin, M.; Li, Y.; Liu, X.; Liu, Y.; Du, H.; Guo, C.; et al. Cardiovascular Toxicity of Different Sizes Amorphous Silica Nanoparticles in Rats after Intratracheal Instillation. Cardiovasc. Toxicol. 2013, 13, 194–207. [Google Scholar] [CrossRef]
- Thakur, N.; Thakur, S.; Chatterjee, S.; Das, J.; Sil, P.C. Nanoparticles as Smart Carriers for Enhanced Cancer Immunotherapy. Front. Chem. 2020, 8, 1217. [Google Scholar] [CrossRef]
- da Silva Santos, E.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.d.B.V.; Júnior, I.J.d.S.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR Targeting for Cancer Therapy: Pharmacology and Immunoconjugates with Drugs and Nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Ekpenyong-Akiba, A.E.; Canfarotta, F.; Abd, B.; Poblocka, M.; Casulleras, M.; Castilla-Vallmanya, L.; Kocsis-Fodor, G.; Kelly, M.E.; Janus, J.; Althubiti, M.; et al. Detecting and Targeting Senescent Cells Using Molecularly Imprinted Nanoparticles. Nanoscale Horiz 2019, 4, 757–768. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kassem, S.; Piletsky, S.S.; Yesilkaya, H.; Gazioglu, O.; Habtom, M.; Canfarotta, F.; Piletska, E.; Spivey, A.C.; Aboagye, E.O.; Piletsky, S.A. Assessing the In Vivo Biocompatibility of Molecularly Imprinted Polymer Nanoparticles. Polymers 2022, 14, 4582. https://doi.org/10.3390/polym14214582
Kassem S, Piletsky SS, Yesilkaya H, Gazioglu O, Habtom M, Canfarotta F, Piletska E, Spivey AC, Aboagye EO, Piletsky SA. Assessing the In Vivo Biocompatibility of Molecularly Imprinted Polymer Nanoparticles. Polymers. 2022; 14(21):4582. https://doi.org/10.3390/polym14214582
Chicago/Turabian StyleKassem, Samr, Stanislav S. Piletsky, Hasan Yesilkaya, Ozcan Gazioglu, Medhanie Habtom, Francesco Canfarotta, Elena Piletska, Alan C. Spivey, Eric O. Aboagye, and Sergey A. Piletsky. 2022. "Assessing the In Vivo Biocompatibility of Molecularly Imprinted Polymer Nanoparticles" Polymers 14, no. 21: 4582. https://doi.org/10.3390/polym14214582