
Phosphine-Functionalized Core-Crosslinked Micelles and Nanogels with an Anionic Poly(styrenesulfonate) Shell: Synthesis, Rhodium(I) Coordination and Aqueous Biphasic Hydrogenation Catalysis
 ,
,  , and
, and
Abstract
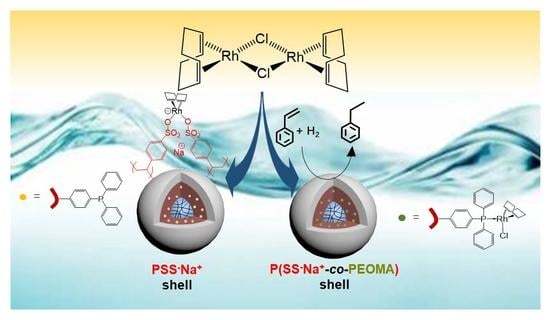
:
1. Introduction
2. Materials and Methods
2.1. Characterization Techniques
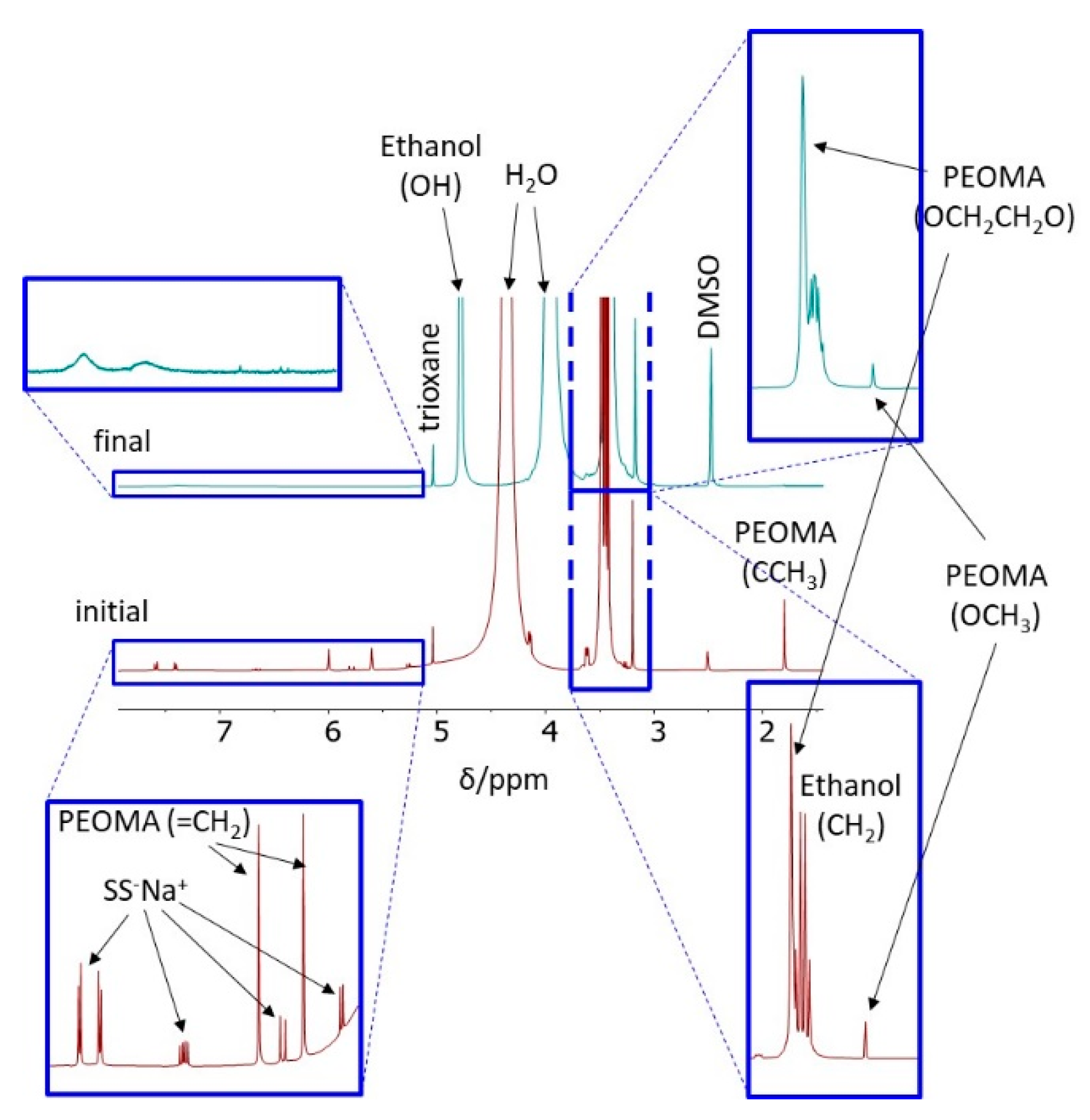
2.1.1. NMR Spectroscopy
2.1.2. Size Exclusion Chromatography (SEC)
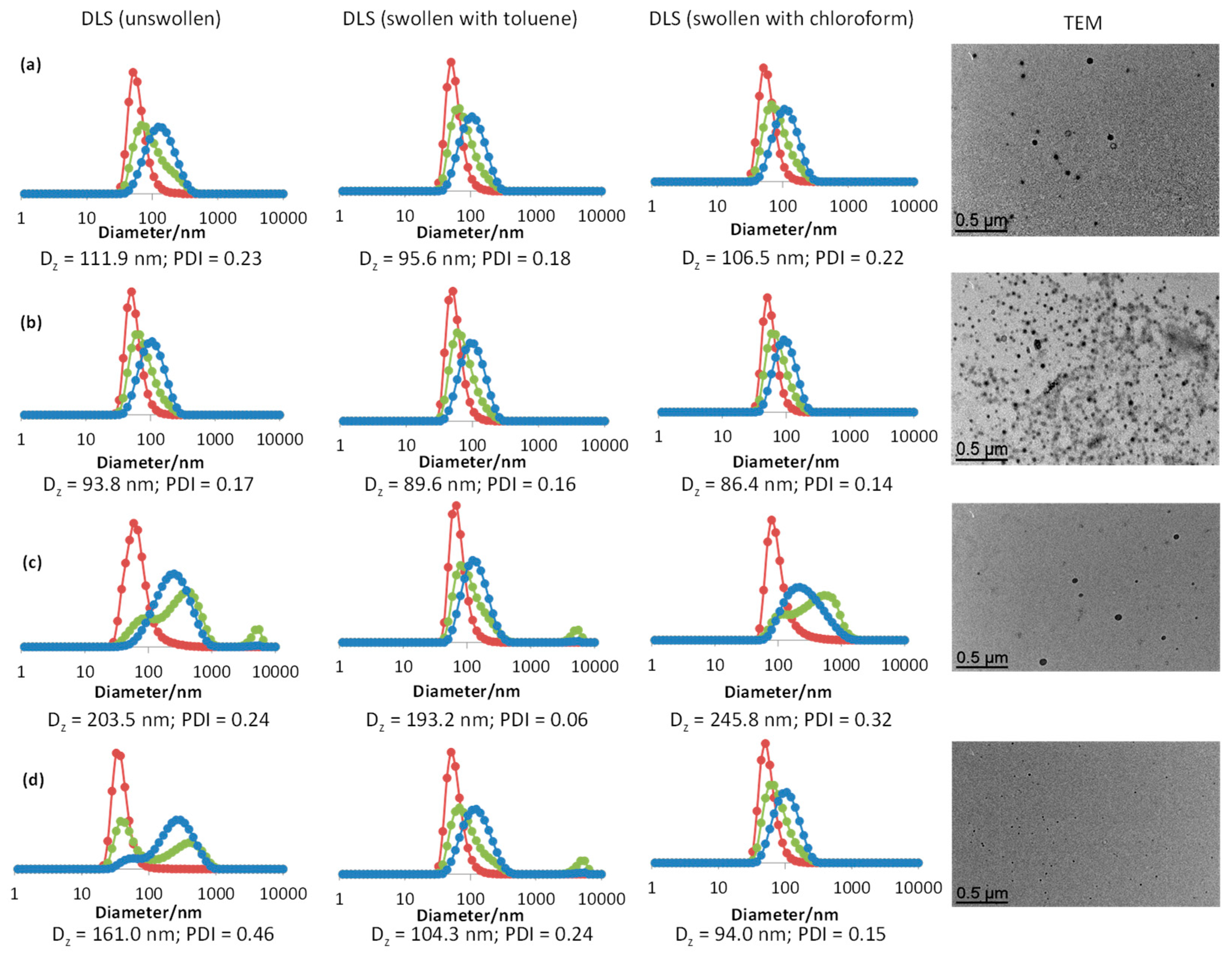
2.1.3. Dynamic Light Scattering (DLS)
2.1.4. Transmission Electron Microscopy (TEM)
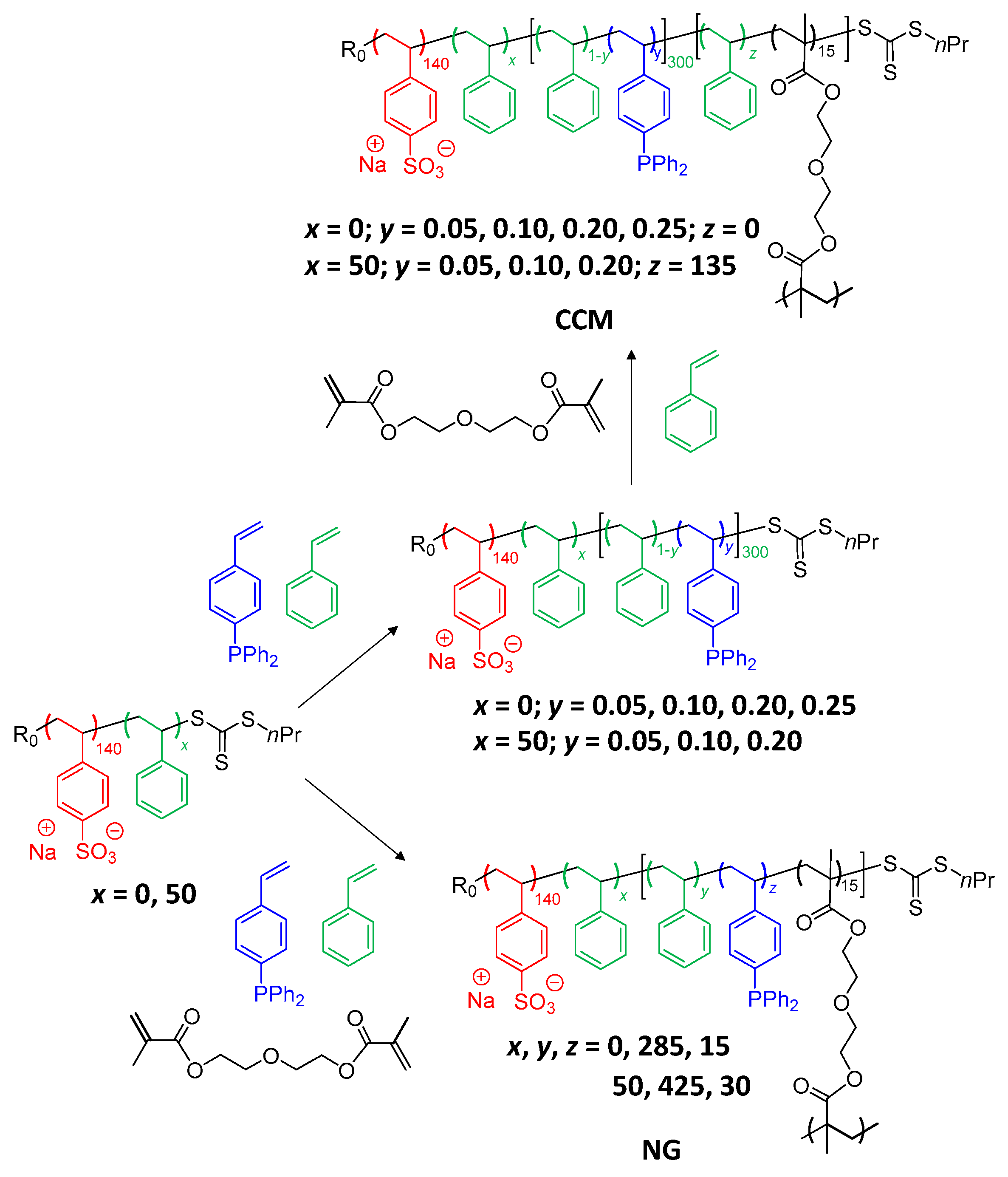
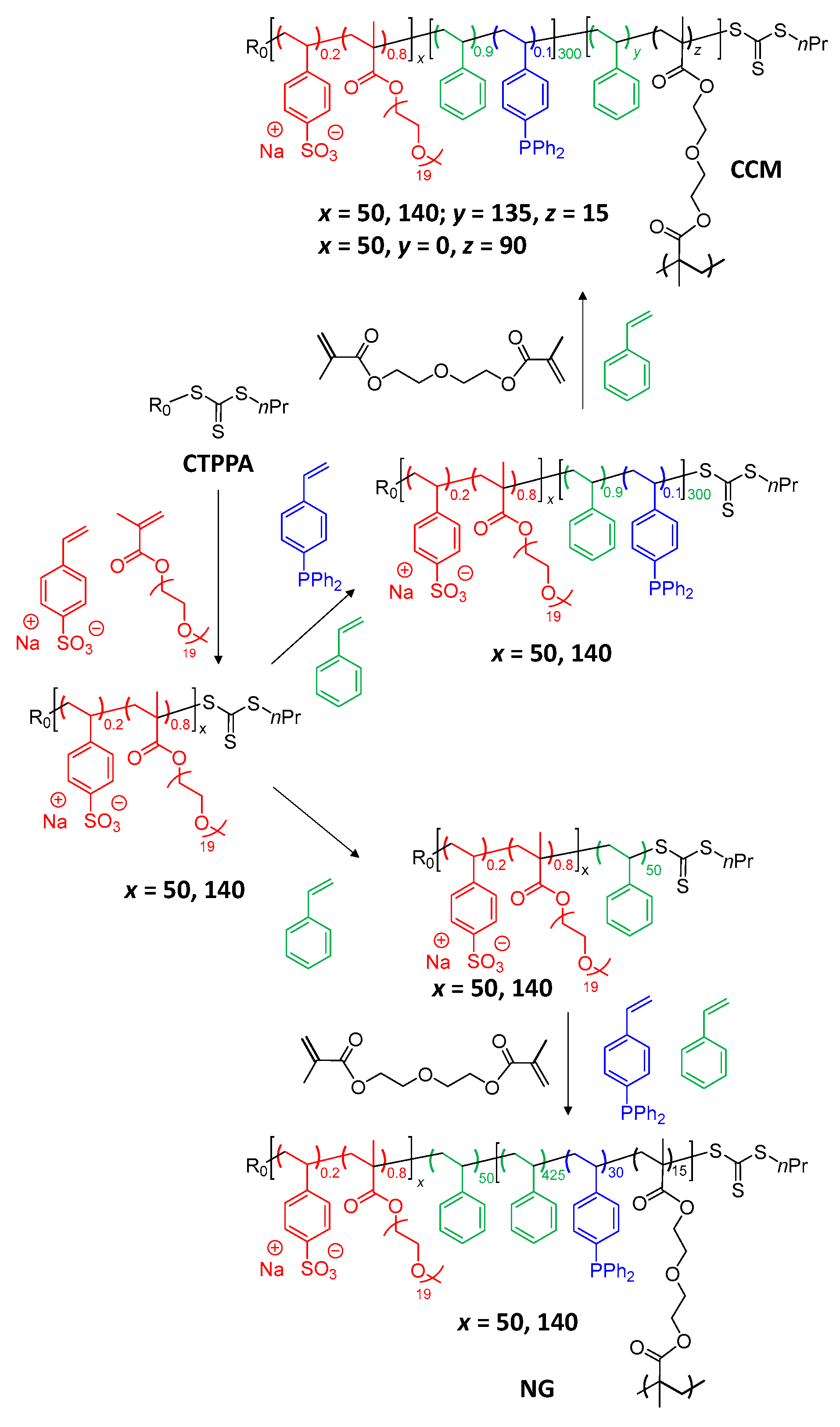
2.2. Synthesis of Phosphine-Functionalized Copolymer Nanoreactors with an Anionic P(SS−Na+) Shell
2.3. Synthesis of Phosphine-Functionalized Copolymer Nanoreactors with an Anionic P(SS−Na+-co-PEOMA) Shell
2.4. General Procedure for Rh Complexation to the Phosphine Ligand within CCM or NG Core
2.5. General Procedure for the Catalyzed Hydrogenation
3. Results
3.1. TPP-Functionalized CCMs and NGs with Hydrophilic P(SS−Na+) Homopolymer Blocks
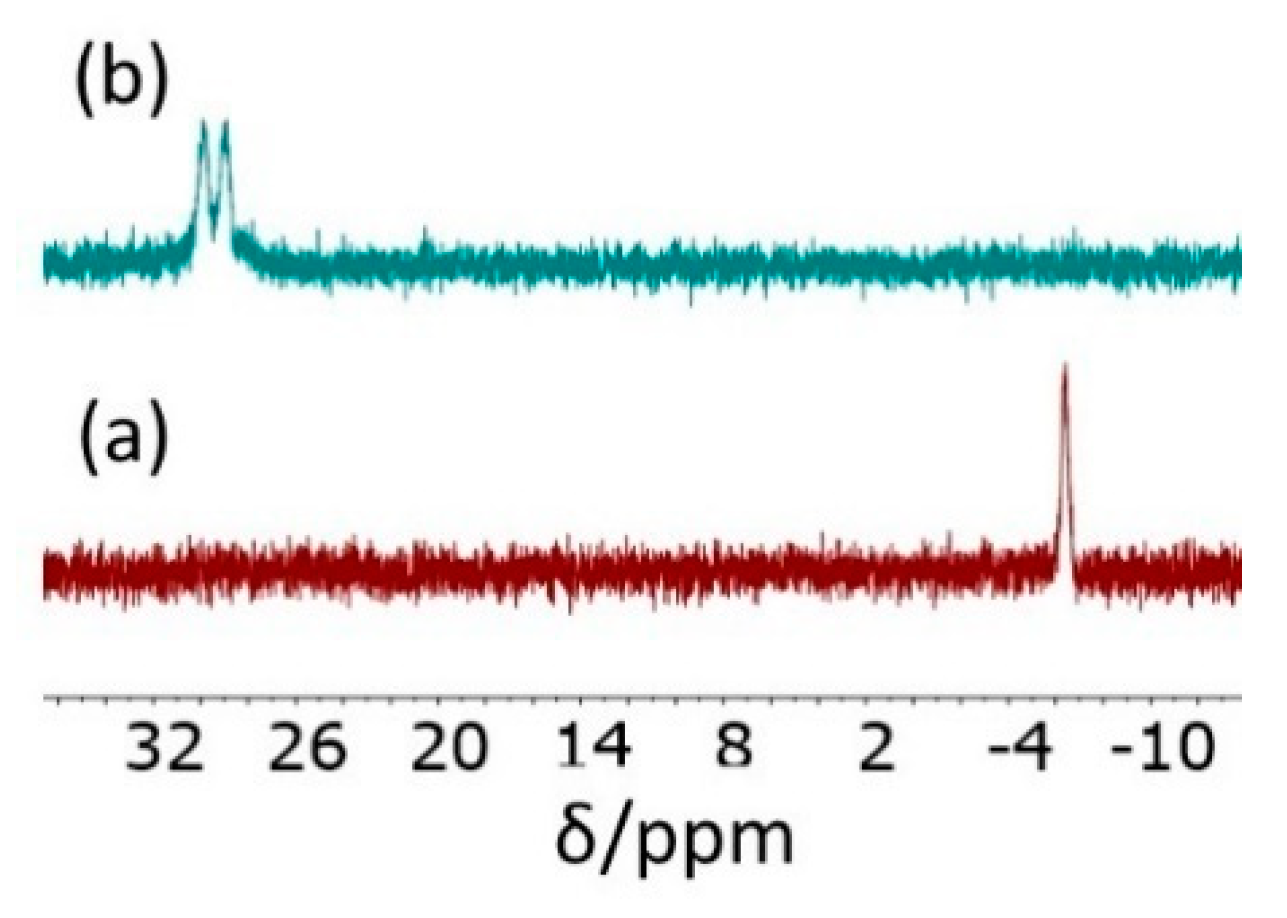
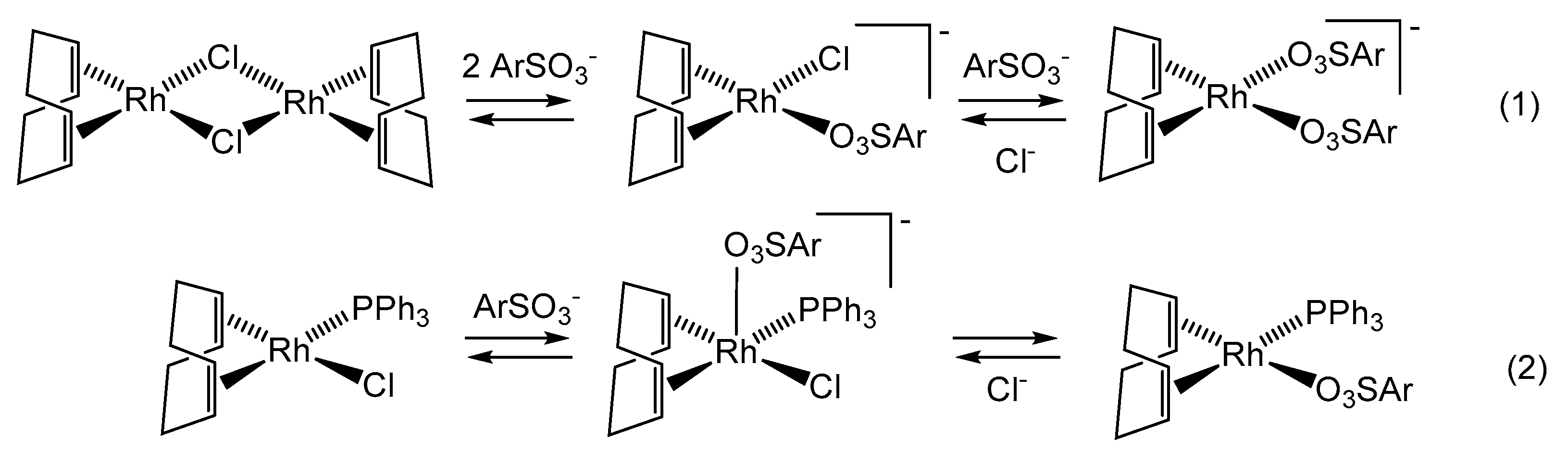
3.2. Treatment with [RhCl(COD)]2 and Model Studies
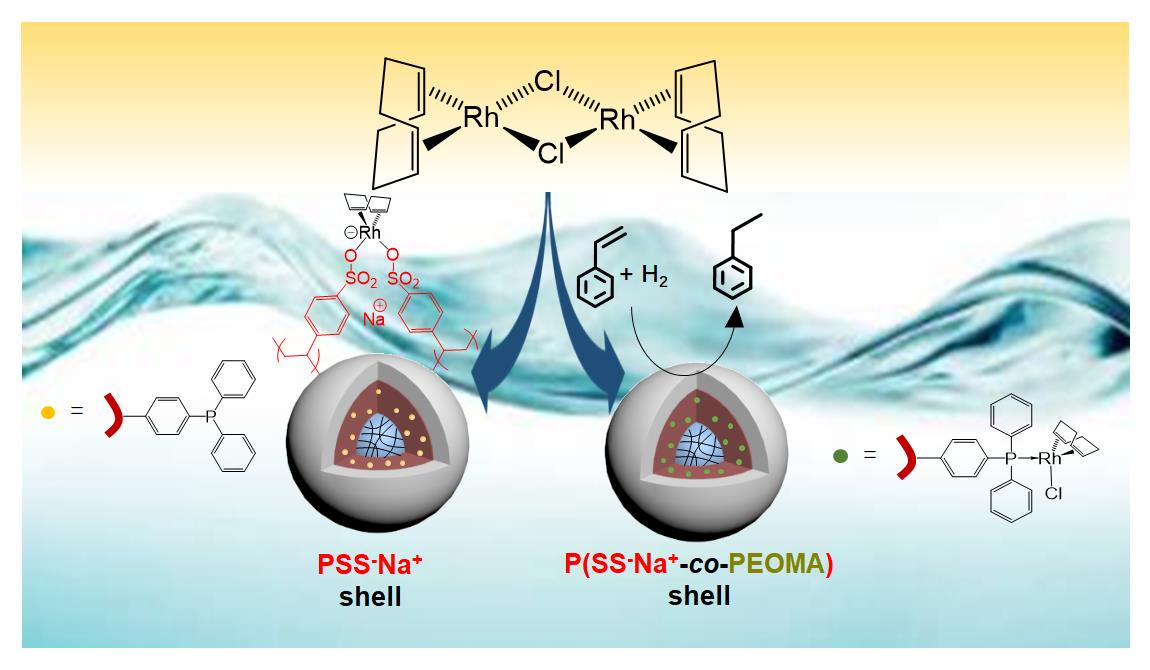
3.3. Synthesis of CCMs and NGs with a P(SS−Na+-co-PEOMA)-Based Shell
3.4. [RhCl(COD)]2 Loading in the CCM and NG with the P(SS−Na+-co-PEOMA)-Based Shell
3.5. Hydrogenation Catalysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornils, B.; Herrmann, W.A. Aqueous Phase Organometallic Catalysis; Wiley: Weinheim, Germany, 1997. [Google Scholar]
- Kohlpaintner, C.W.; Fischer, R.W.; Cornils, B. Aqueous biphasic catalysis: Ruhrchemie/Rhone-Poulenc oxo process. Appl. Catal. A 2001, 221, 219–225. [Google Scholar]
- Behr, A. Thermomorphic solvent systems. In Multiphase Homogeneous Catalysis; Cornils, B., Herrmann, W.A., Horvath, I.T., Leitner, W., Mecking, S., Olivier-Bourbigou, H., Vogt, D., Eds.; Wiley: Weinheim, Germany, 2005; Volume 1, pp. 327–329. [Google Scholar]
- Leclercq, L.; Bricout, H.; Tilloy, S.; Monflier, E. Biphasic aqueous organometallic catalysis promoted by cyclodextrins: Can surface tension measurements explain the efficiency of chemically modified cyclodextrins? J. Colloid Interface Sci. 2007, 307, 481–487. [Google Scholar] [PubMed]
- Hapiot, F.; Menuel, S.; Ferreira, M.; Leger, B.; Bricout, H.; Tilloy, S.; Monflier, E. Catalysis in Cyclodextrin-Based Unconventional Reaction Media: Recent Developments and Future Opportunities. ACS Sustain. Chem. Eng. 2017, 5, 3598–3606. [Google Scholar]
- Oehme, G. Micellar systems. In Aqueous-Phase Organometallic Catalysis: Concepts and Applications, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley: Weinheim, Germany, 2004; pp. 256–271. [Google Scholar]
- Khan, M.N. Micellar Catalysis; CRC Press: Boca Raton, FL, USA, 2006; p. 464. [Google Scholar]
- Cotanda, P.; Petzetakis, N.; O’Reilly, R.K. Catalytic polymeric nanoreactors: More than a solid supported catalyst. MRS Commun. 2012, 2, 119–126. [Google Scholar]
- Lu, A.; O’Reilly, R.K. Advances in nanoreactor technology using polymeric nanostructures. Curr. Opin. Biotech. 2013, 24, 639–645. [Google Scholar] [PubMed]
- Poli, R. Effects of Nanoconfinement on Catalysis; Springer: New York, NY, USA, 2017. [Google Scholar]
- Terashima, T.; Kamigaito, M.; Baek, K.-Y.; Ando, T.; Sawamoto, M. Polymer Catalysts from Polymerization Catalysts: Direct Encapsulation of Metal Catalyst into Star Polymer Core during Metal-Catalyzed Living Radical Polymerization. J. Am. Chem. Soc. 2003, 125, 5288–5289. [Google Scholar] [PubMed]
- O’Reilly, R.K.; Hawker, C.J.; Wooley, K.L. Cross-linked block copolymer micelles: Functional nanostructures of great potential and versatility. Chem. Soc. Rev. 2006, 35, 1068–1083. [Google Scholar]
- Liu, Y.; Wang, Y.; Wang, Y.F.; Lu, J.; Pinon, V.; Weck, M. Shell Cross-Linked Micelle-Based Nanoreactors for the Substrate-Selective Hydrolytic Kinetic Resolution of Epoxides. J. Am. Chem. Soc. 2011, 133, 14260–14263. [Google Scholar] [PubMed]
- Terashima, T. Polymer Microgels for Catalysis. In Encyclopedia of Polymer Science and Technology, 4th ed.; Mark, H.F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT Agent Design and Synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self Assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [PubMed] [Green Version]
- Lansalot, M.; Rieger, J.; D’Agosto, F. Polymerization-Induced Self-Assembly: The Contribution of Controlled Radical Polymerization to The Formation of Self-Stabilized Polymer Particles of Various Morphologies. In Macromolecular Self-Assembly; Billon, L., Bourisov, O., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 33–82. [Google Scholar]
- D’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed. 2020, 59, 8368–8392. [Google Scholar]
- Zhang, X.; Cardozo, A.F.; Chen, S.; Zhang, W.; Julcour, C.; Lansalot, M.; Blanco, J.-F.; Gayet, F.; Delmas, H.; Charleux, B.; et al. Core-Shell Nanoreactors for Efficient Aqueous Biphasic Catalysis. Chem. Eur. J. 2014, 20, 15505–15517. [Google Scholar] [PubMed] [Green Version]
- Cardozo, A.F.; Julcour, C.; Barthe, L.; Blanco, J.-F.; Chen, S.; Gayet, F.; Manoury, E.; Zhang, X.; Lansalot, M.; Charleux, B.; et al. Aqueous phase homogeneous catalysis using core-shell nano-reactors: Application to rhodium catalyzed hydroformylation of 1-octene. J. Catal. 2015, 324, 1–8. [Google Scholar]
- Chen, S.; Cardozo, A.F.; Julcour, C.; Blanco, J.-F.; Barthe, L.; Gayet, F.; Charleux, B.; Lansalot, M.; D’Agosto, F.; Delmas, H.; et al. Amphiphilic Core-Cross-linked Micelles functionalized with Bis(4-methoxyphenyl)phenylphosphine as catalytic nanoreactors for biphasic hydroformylation. Polymer 2015, 72, 327–335. [Google Scholar]
- Poli, R.; Chen, S.; Zhang, X.; Cardozo, A.; Lansalot, M.; D’Agosto, F.; Charleux, B.; Manoury, E.; Gayet, F.; Julcour, C.; et al. One-Pot RAFT Synthesis of Triphenylphosphine-Functionalized Amphiphilic Core-Shell Polymers and Application as Catalytic Nanoreactors in Aqueous Biphasic Hydroformylation. ACS Symp. Ser. 2015, 1188, 203–220. [Google Scholar]
- Lobry, E.; Cardozo, A.F.; Barthe, L.; Blanco, J.-F.; Delmas, H.; Chen, S.; Gayet, F.; Zhang, X.; Lansalot, M.; D’Agosto, F.; et al. Core phosphine-functionalized amphiphilic nanogels as catalytic nanoreactors for aqueous biphasic hydroformylation. J. Catal. 2016, 342, 164–172. [Google Scholar]
- Chen, S.; Gayet, F.; Manoury, E.; Joumaa, A.; Lansalot, M.; D’Agosto, F.; Poli, R. Coordination chemistry inside polymeric nanoreactors: Interparticle metal exchange processes and ionic compound vectorization in phosphine-functionalized amphiphilic polymer latexes. Chem. Eur. J. 2016, 22, 6302–6313. [Google Scholar] [PubMed]
- Joumaa, A.; Chen, S.; Vincendeau, S.; Gayet, F.; Poli, R.; Manoury, E. Rhodium-catalyzed aqueous biphasic hydrogenation of alkenes with amphiphilic phosphine-containing core-shell polymers. Mol. Cat. 2017, 438, 267–271. [Google Scholar]
- Manoury, E.; Gayet, F.; D’Agosto, F.; Lansalot, M.; Delmas, H.; Julcour, C.; Blanco, J.-F.; Barthe, L.; Poli, R. Core-cross-linked micelles and amphiphilic nanogels as unimolecular nanoreactors for micellar-type, metal-based aqueous biphasic catalysis. In Effects of Nanoconfinement on Catalysis, Poli, R., Ed.; Springer: New York, NY, USA, 2017; pp. 147–172. [Google Scholar]
- Joumaa, A.; Gayet, F.; Garcia-Suarez, E.J.; Himmelstrup, J.; Riisager, A.; Poli, R.; Manoury, E. Synthesis of Nixantphos core-functionalized amphiphilic nanoreactors and application to rhodium-catalyzed aqueous biphasic 1-octene hydroformylation. Polymers 2020, 12, 1107. [Google Scholar]
- Sambou, S.S.; Hromov, R.; Ruzhylo, I.; Wang, H.; Allandrieu, A.; Sabatier, C.; Coppel, Y.; Daran, J.-C.; Gayet, F.; Labande, A.; et al. Amphiphilic Polymeric Nanoreactors Containing Rh(I)-NHC Complexes for the Aqueous Biphasic Hydrogenation of Alkenes. Catal. Sci. Technol. 2021, 11, 6811–6824. [Google Scholar]
- Wang, H.; Vendrame, L.; Fliedel, C.; Chen, S.; Gayet, F.; Manoury, E.; Zhang, X.; D’Agosto, F.; Lansalot, M.; Poli, R. Core-cross-linked micelles made by RAFT polymerization with a poly-cationic outer shell based on poly(1-methyl-4-vinylpyridinium). Macromolecules 2020, 53, 2198–2208. [Google Scholar]
- Wang, H.; Vendrame, L.; Fliedel, C.; Chen, S.; Gayet, F.; D’Agosto, F.; Lansalot, M.; Manoury, E.; Poli, R. Triphenylphosphine-functionalized core-cross-linked micelles and nanogels with a polycationic outer shell: Synthesis and application in rhodium-catalyzed biphasic hydrogenations. Chem. Eur. J. 2021, 27, 5205–5214. [Google Scholar] [PubMed]
- Boursier, T.; Chaduc, I.; Rieger, J.; D’Agosto, F.; Lansalot, M.; Charleux, B. Controlled radical polymerization of styrene in miniemulsion mediated by PEO-based trithiocarbonate macromolecular RAFT agents. Polym. Chem. 2011, 2, 355–362. [Google Scholar]
- Wang, H.; Fliedel, C.; Manoury, E.; Poli, R. Core-crosslinked micelles with a poly-anionic poly(styrene sulfonate)-based outer shell made by RAFT polymerization. Polymer 2022, 243, 124640. [Google Scholar]
- Nitti, A.; Carfora, R.; Assanelli, G.; Notari, M.; Pasini, D. Single-Chain Polymer Nanoparticles for Addressing Morphologies and Functions at the Nanoscale: A Review. ACS Appl. Nano Mater. 2022, 5, 13985–13997. [Google Scholar]
- Burk, M.J. C2-Symmetric Bis(phospholanes) and Their Use in Highly Enantioselective Hydrogenation Reactions. J. Am. Chem. Soc. 1991, 113, 8518–8519. [Google Scholar]
- Burk, M.J.; Feaster, J.E.; Nugent, W.A.; Harlow, R.L. Preparation and Use of C2-Symmetric Bis(phospholanes): Production of α-Amino Acid Derivatives via Highly Enantioselective Hydrogenation Reactions. J. Am. Chem. Soc. 1993, 115, 10125–10138. [Google Scholar] [CrossRef]
- Hearley, A.K.; Nowack, R.A.J.; Rieger, B. New single-site palladium catalysts for the nonalternating copolymerization of ethylene and carbon monoxide. Organometallics 2005, 24, 2755–2763. [Google Scholar]
- Wang, H.; Fiore, A.M.; Fliedel, C.; Manoury, E.; Philippot, K.; Dell’Anna, M.M.; Mastrorilli, P.; Poli, R. Rhodium nanoparticles inside well-defined unimolecular amphiphilic polymeric nanoreactors: Synthesis and biphasic hydrogenation catalysis. Nanoscale Adv. 2021, 3, 2554–2566. [Google Scholar] [PubMed]
- Narayan, S.; Muldoon, J.; Finn, M.G.; Fokin, V.V.; Kolb, H.C.; Sharpless, K.B. “On water”: Unique reactivity of organic compounds in aqueous suspension. Angew. Chem. Int. Ed. 2005, 44, 3275–3279. [Google Scholar] [CrossRef] [PubMed]
- Kitanosono, T.; Kobayashi, S. Reactions in Water Involving the “On-Water” Mechanism. Chem. Eur. J. 2020, 26, 9408–9429. [Google Scholar] [PubMed]
- Tran, B.L.; Fulton, J.L.; Linehan, J.C.; Lercher, J.A.; Bullock, R.M. Rh(CAAC)-Catalyzed Arene Hydrogenation: Evidence for Nanocatalysis and Sterically Controlled Site-Selective Hydrogenation. ACS Catal. 2018, 8, 8441–8449. [Google Scholar] [CrossRef]
- Ibrahim, M.; Wei, M.M.; Deydier, E.; Manoury, E.; Poli, R.; Lecante, P.; Philippot, K. Rhodium nanoparticles stabilized by ferrocenyl-phosphine ligands: Synthesis and catalytic styrene hydrogenation. Dalton Trans. 2019, 48, 6777–6786. [Google Scholar] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Additive | P/Rh | SS−Na+/Rh | Conv./% | TON (EB) c | TON (EC) c |
|---|---|---|---|---|---|---|
| 1 | R0-[(SS−Na+)0.2-co-PEOMA0.8]140-b-(St0.9-co-DPPS0.1)300-b-(St0.9-co-DEGDMA0.1)150-SC(S)SnPr | 4 | 3.73 | 29.4 | 588 | 0 |
| 2 | R0-[(SS−Na+)0.2-co-PEOMA0.8]50-b-(St0.9-co-DPPS0.1)300-b-DEGDMA90-SC(S)SnPr | 4 | 1.33 | 26.3 | 526 | 0 |
| 3 | R0-[(SS−Na+)0.2-co-PEOMA0.8]140-b-St50-b-(St425-co-DPPS30-co-DEGDMA15)-SC(S)SnPr | 4 | 3.73 | 41.9 | 838 | 0 |
| 4 | PPh3 | 3.91 | - | 99.7 ± 0.2 b | 1994 ± 5 b | 2 ± 2 b |
| 5 | PPh3 d | 4.25 | - | 10.4 ± 0.5 b | 208 ± 11 b | 0 |
| 6 | p-CH3C6H4SO3−Na+ | - | 1.33 | 99.9 e,f | 1872 | 126 |
| 7 | R0-[(SS−Na+)0.2-co-PEOMA0.8]50-SC(S)SnPr | - | 1.31 | 99.9 ± 0.1 b,e,f | 1984 ± 12 b | 15 ± 11 b |
| 8 | p-CH3C6H4SO3−Na+ + PPh3 | 3.98 | 3.63 | 79.0 ± 0.4 b | 1580 ± 8 b | 0 |
| 9 | R0-[(SS−Na+)0.2-co-PEOMA0.8]50-SC(S)SnPr + PPh3 | 3.91 | 1.29 | 58 ± 3 b | 1160 ± 60 b | 0 |
| 10 | R0-[(SS−Na+)0.2-co-PEOMA0.8]140-SC(S)SnPr + PPh3 | 4.25 | 3.86 | 85 ± 5 b | 1700 ± 100 b | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Abou-Fayssal, C.J.; Fliedel, C.; Manoury, E.; Poli, R. Phosphine-Functionalized Core-Crosslinked Micelles and Nanogels with an Anionic Poly(styrenesulfonate) Shell: Synthesis, Rhodium(I) Coordination and Aqueous Biphasic Hydrogenation Catalysis. Polymers 2022, 14, 4937. https://doi.org/10.3390/polym14224937
Wang H, Abou-Fayssal CJ, Fliedel C, Manoury E, Poli R. Phosphine-Functionalized Core-Crosslinked Micelles and Nanogels with an Anionic Poly(styrenesulfonate) Shell: Synthesis, Rhodium(I) Coordination and Aqueous Biphasic Hydrogenation Catalysis. Polymers. 2022; 14(22):4937. https://doi.org/10.3390/polym14224937
Chicago/Turabian StyleWang, Hui, Chantal J. Abou-Fayssal, Christophe Fliedel, Eric Manoury, and Rinaldo Poli. 2022. "Phosphine-Functionalized Core-Crosslinked Micelles and Nanogels with an Anionic Poly(styrenesulfonate) Shell: Synthesis, Rhodium(I) Coordination and Aqueous Biphasic Hydrogenation Catalysis" Polymers 14, no. 22: 4937. https://doi.org/10.3390/polym14224937