
Synthesis of a New Molecularly Imprinted Polymer and Optimisation of Phenylglyoxylic Acid Extraction from Human Urine Samples Using a Central Composite Design within the Response Surface Methodology
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Analytical Instruments
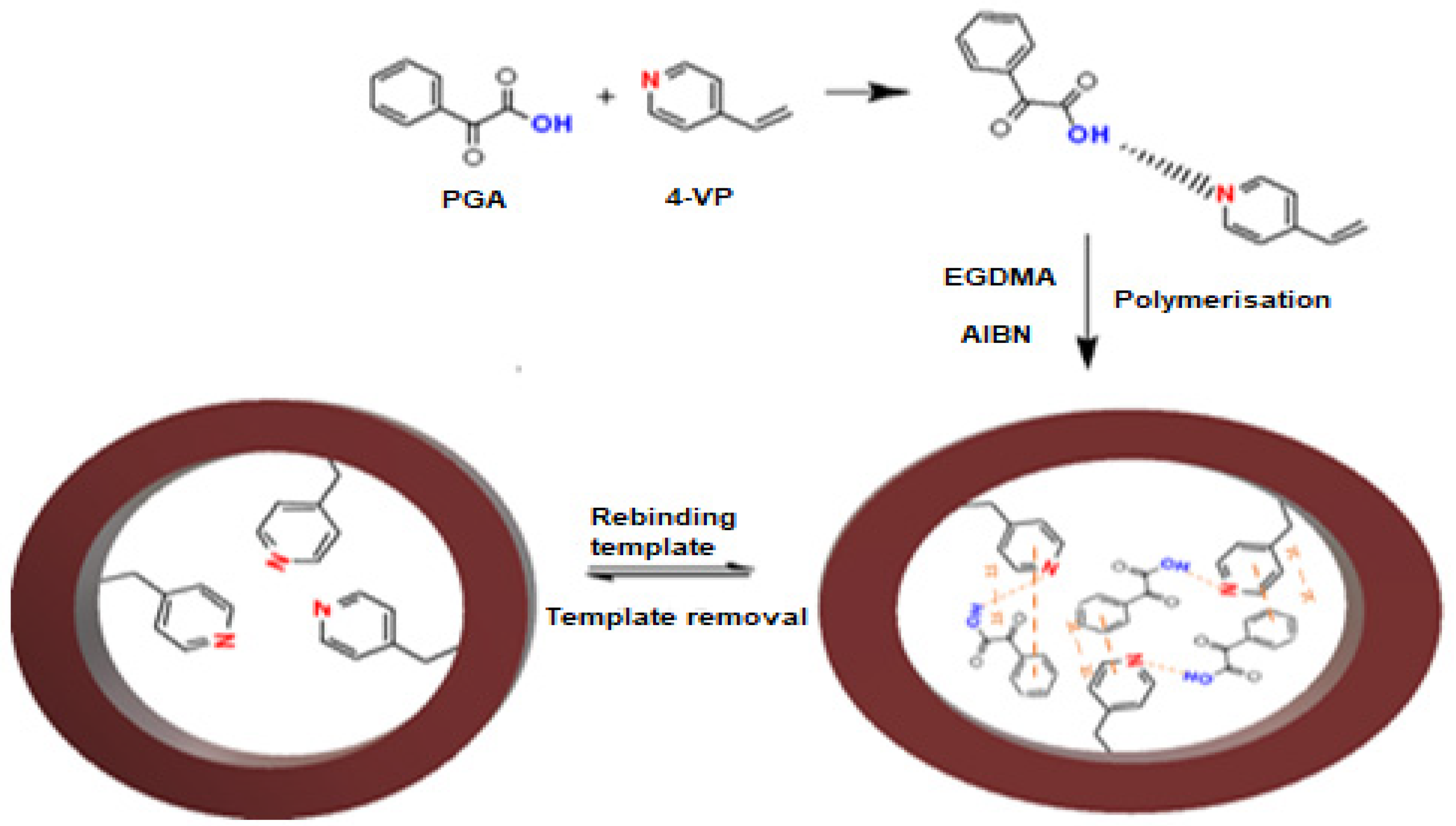
2.3. Preparation of Imprinted Polymer PGA–MIP and NIP
2.4. Polymer Characterisation
2.5. Adsorption Capacities (MIP/NIP)
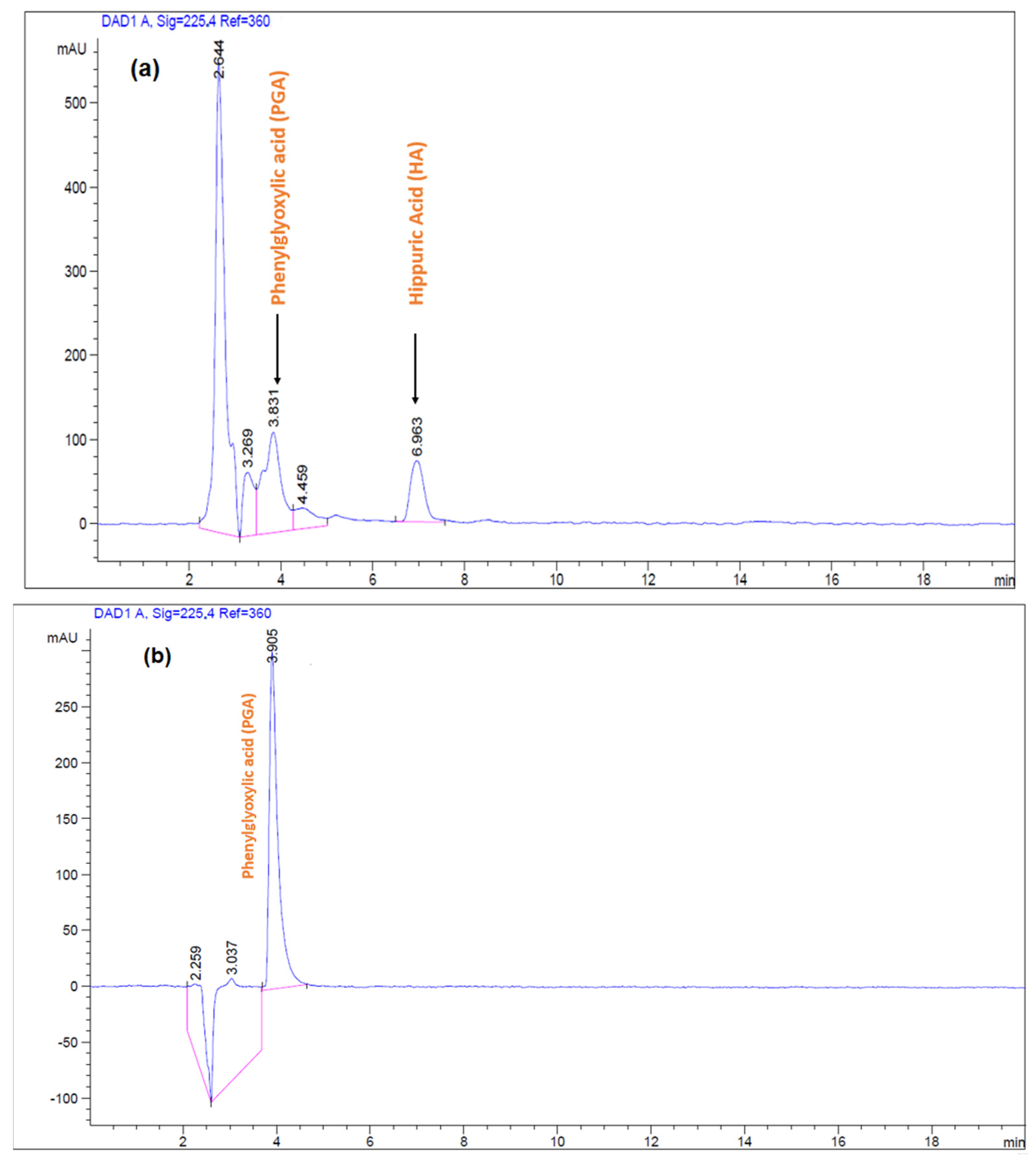
2.6. HPLC–DAD-Analysis Conditions
2.7. Analytical Performance of MIP Extraction
2.8. Optimisation of PGA Adsorption and Desorption by MIP Using Experimental Design Approach
2.9. Selectivity Studies
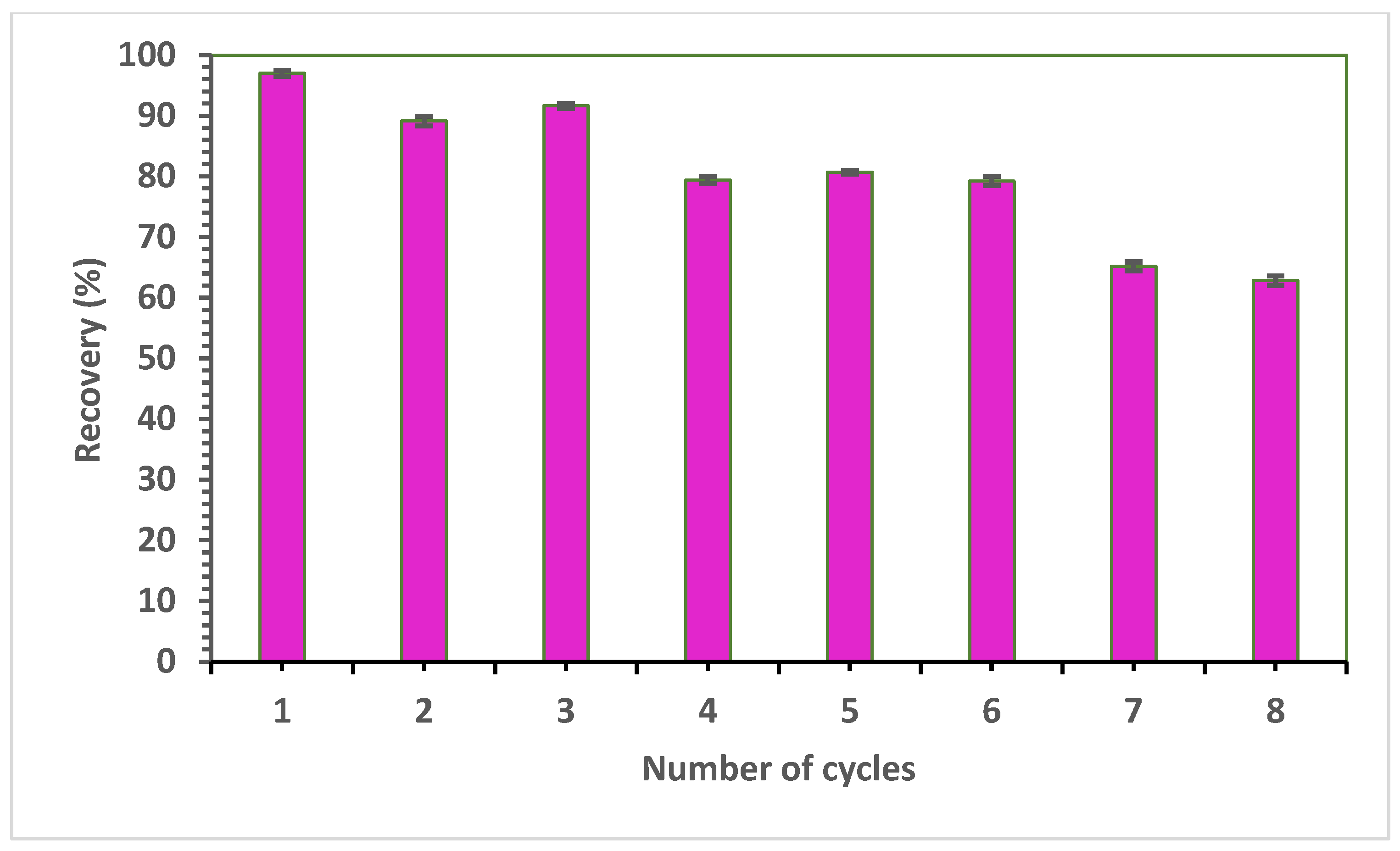
2.10. Reusability Studies
3. Results and Discussion
3.1. Characterisation of Molecular Imprinted Polymer
3.2. FT-IR Spectroscopy
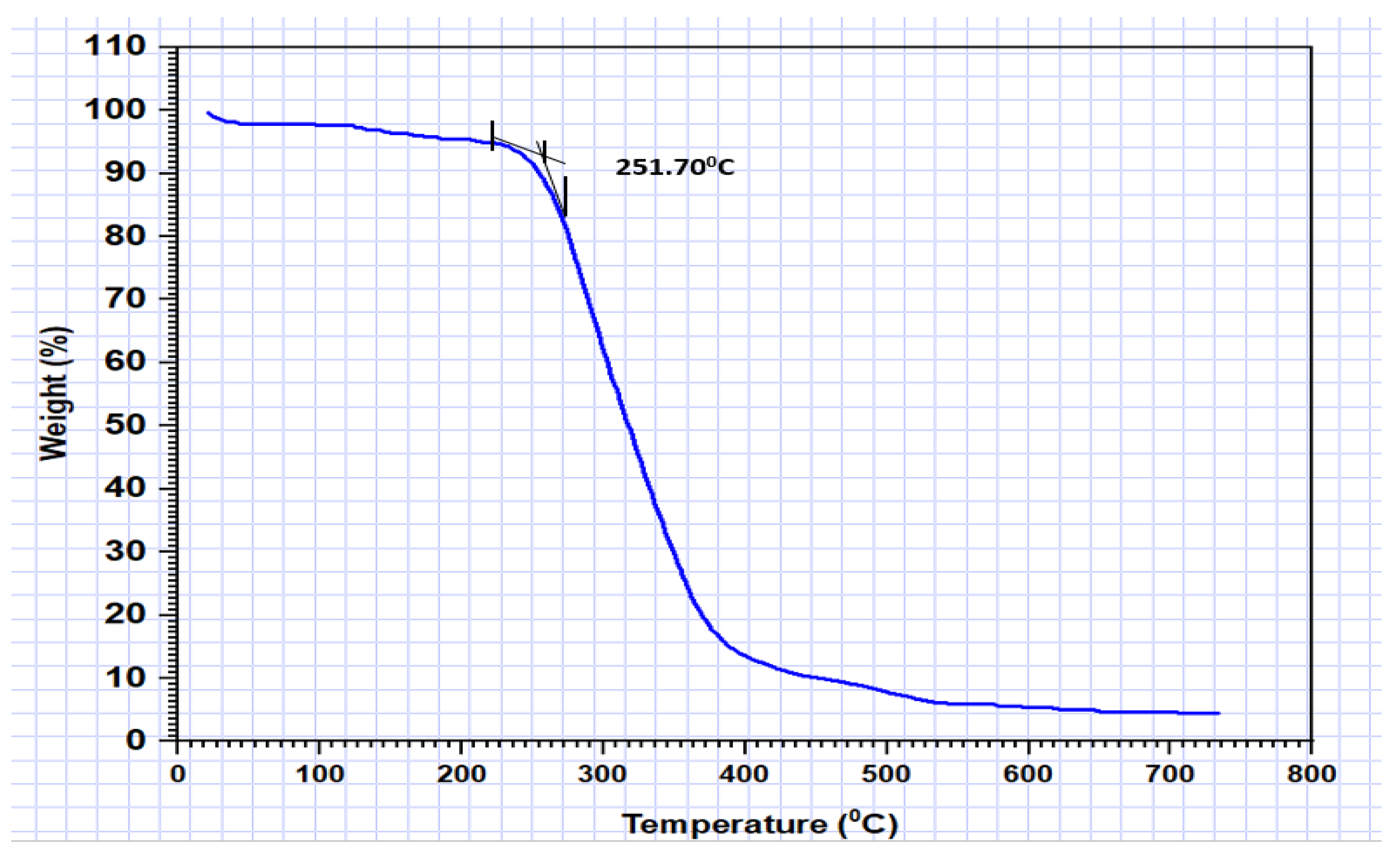
3.3. Thermal Properties
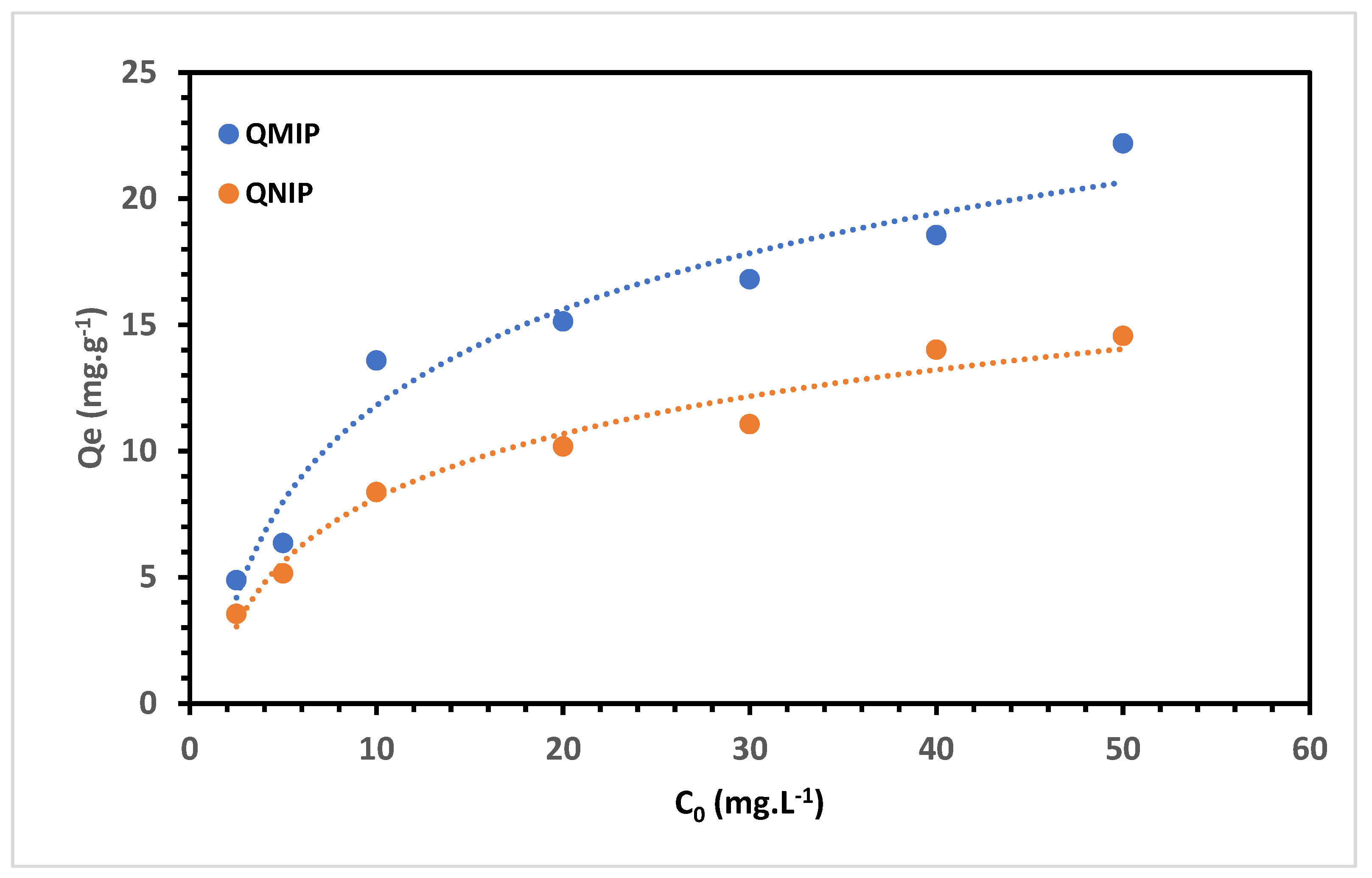
3.4. Adsorption Capacity
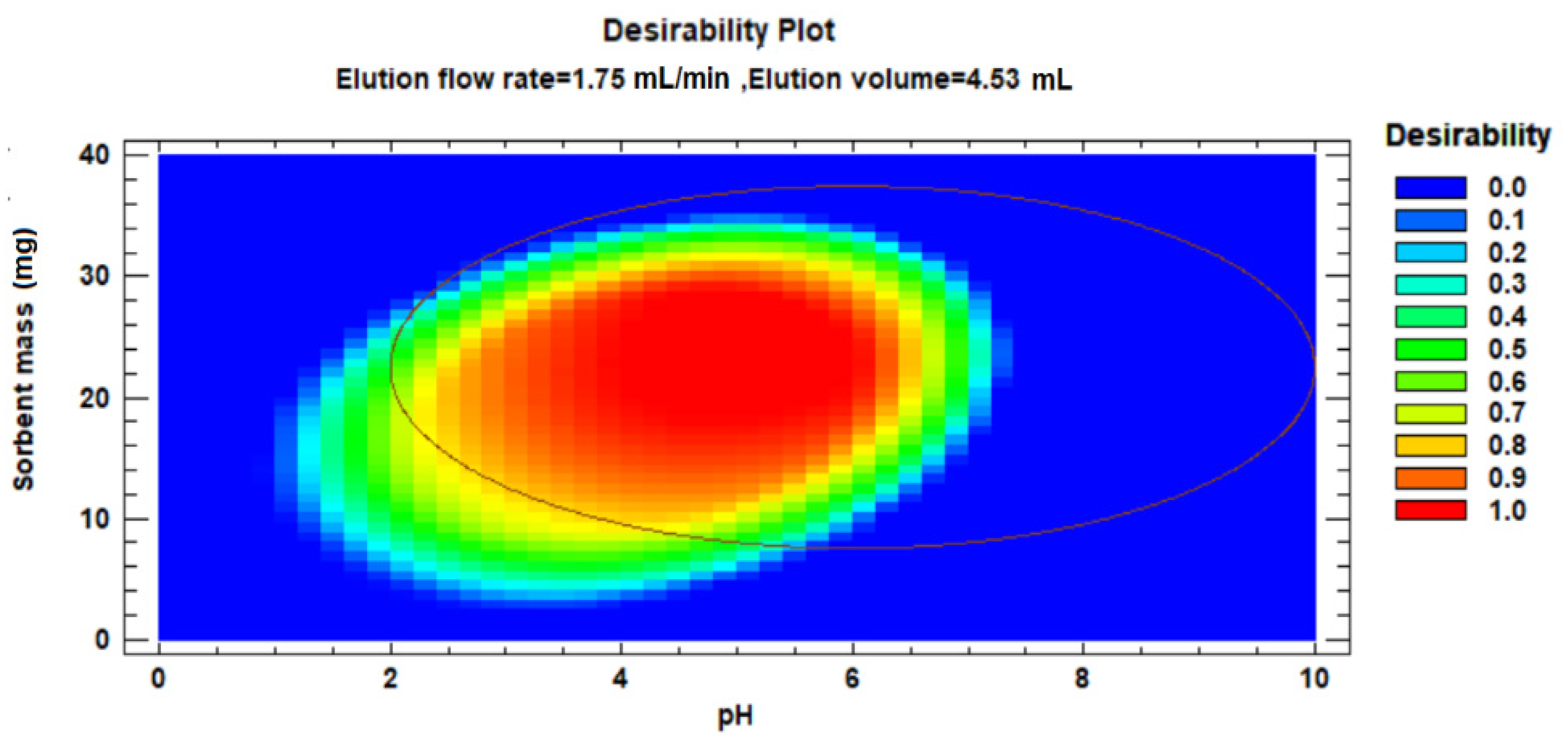
3.5. Optimisation of MIP(4-VP)SPE Procedure by Experimental Design Approach
3.6. Adsorptive Selectivity
3.7. Analytical Performance and Applicability of MIP(-4VP)SPE to Real Urine Samples
3.8. Reusability of SPE-4-VPMIP
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shahtaheri, S.; Abdollahi, M.; Golbabaei, F.; Rahimi, F.A.; Ghamari, F. Monitoring of Mandelic Acid as a Biomarker of Environmental and Occupational Exposures to Styrene. Int. J. Environ. 2008, 2, 169–176. [Google Scholar]
- Soleimani, E.; Bahrami, A.; Afkhami, A.; Shahna, F.G. Selective determination of mandelic acid in urine using molecularly imprinted polymer in microextraction by packed sorbent. Arch. Toxicol. J. 2018, 92, 213–222. [Google Scholar]
- Habibagahi, A.; Alderman, N.; Kubwabo, C. A review of the analysis of biomarkers of exposure to tobacco and vaping products. Anal. Methods 2020, 12, 4276–4302. [Google Scholar] [PubMed]
- Capella, K.M.; Roland, K.; Geldner, N.; deCastro, B.R.; De Jesús, V.R.; van Bemmel, D.; Blount, B.C. Ethylbenzene and styrene exposure in the United States based on urinary mandelic acid and phenylglyoxylic acid: NHANES 2005–2006 and 2011–2012. Environ. Res. 2019, 171, 101–110. [Google Scholar] [PubMed]
- Hajizadeh, Y.; Teiri, H.; Nazmara, S.; Parseh, I. Environmental and biological monitoring of exposures to VOCs in a petrochemical complex in Iran. Environ. Sci. Pollut. Res. 2018, 25, 6656–6667. [Google Scholar]
- Mohamadyan, M.; Moosazadeh, M.; Borji, A.; Khanjani, N.; Moghadam, S.R. Occupational exposure to styrene and its relation with urine mandelic acid, in plastic injection workers. Environ. Monit. Assess. J. 2019, 191, 62. [Google Scholar]
- Bahrami, A.; Ghamari, F.; Yamini, Y.; Ghorbani Shahna, F.; Koolivand, A. Ion-pair-based hollow-fiber liquid-phase microextraction combined with high-performance liquid chromatography for the simultaneous determination of urinary benzene, toluene, and styrene metabolites. J. Sep. Sci. 2017, 41, 501–508. [Google Scholar]
- Li, A.J.; Pal, V.K.; Kannan, K. A review of environmental occurrence, toxicity, biotransformation, and biomonitoring of volatile organic compounds. Toxicol. Environ. Chem. 2021, 3, 91–116. [Google Scholar]
- Moore, M.M.; Pottenger, L.H.; House-Knight, T. Critical review of styrene genotoxicity focused on the mutagenicity/clastogenicity literature and using current organization of economic cooperation and development guidance. Environ. Mol. Mutagen. 2019, 60, 624–663. [Google Scholar]
- Chua, S.C.; Lee, B.L.; Liau, L.S.; Ong, C.N. Determination of mandelic acid and phenylglyoxylic acid in the urine and its use in monitoring of styrene exposure. J. Anal. Toxicol. 1993, 17, 129–132. [Google Scholar]
- Paci, E.; Pigini, D.; Caporossi, L.; De Rosa, M.; Santoro, A.; Sisto, R.; Papaleo, B.; Tranfo, G. Matrix Effect in the Quantitative Determination of Mandelic and Phenylglyoxylic Acid in Urine Samples by HPLC-MS/MS with Isotopic Dilution. Curr. Anal. Chem. 2013, 9, 439–446. [Google Scholar]
- Pratiwi, R.; Megantara, S.; Rahayu, D.; Pitaloka, I.; Hasanah, A.N. Comparison of Bulk and Precipitation Polymerization Method of Synthesis Molecular Imprinted Solid Phase Extraction for Atenolol using Methacrylic Acid. J. Young Pharm. 2019, 11, 12–16. [Google Scholar]
- Javanbakht, M.; Namjumanesh, M.H.; Akbari-Adergani, B. Molecularly imprinted solid-phase extraction for the selective determination of bromhexine in human serum and urine with high performance liquid chromatography. Talanta 2009, 80, 133–138. [Google Scholar] [PubMed]
- Jia, P.; Gao, L.; Zheng, Y.; Zheng, X.; Wang, C.; Yang, C.; Li, Y.; Zhao, Y. Ultrastable Tb-Organic Framework as a Selective Sensor of Phenylglyoxylic Acid in Urine. ACS Appl. Mater. Interfaces 2021, 13, 33546–33556. [Google Scholar]
- Liu, R.; Ko, C.-C. Molecularly Imprinted Polymer-Based Luminescent Chemosensors. Biosensors 2023, 13, 295. [Google Scholar]
- Akgönüllü, S.; Kılıç, S.; Esen, C.; Denizli, A. Molecularly Imprinted Polymer-Based Sensors for Protein Detection. Polymers 2023, 15, 629. [Google Scholar]
- Turiel, E.; Martín-Esteban, A. Molecularly Imprinted Polymers-Based Microextraction Techniques. Trends Anal. Chem. 2019, 118, 574–586. [Google Scholar]
- Yang, J.; Li, Y.; Wang, J.; Sun, X.; Cao, R.; Sun, H.; Huang, C.; Chen, J. Molecularly imprinted polymer microspheres prepared by Pickering emulsion polymerization for selective solid-phase extraction of eight bisphenols from human urine samples. Anal. Chim. Acta 2015, 872, 35–45. [Google Scholar]
- Wang, S.; Zhang, L.; Zeng, J.; Hu, X.; Wang, X.; Yu, L.; Wang, D.; Ling Cheng, L.; Ahmed, R.; Romanovski, V.; et al. Multi-templates molecularly imprinted polymers for simultaneous recognition of multiple targets: From academy to application. TrAC Trends Anal. Chem. 2023, 166, 117173. [Google Scholar]
- Mulder, H.A.; Halquist, M.S. Growing Trends in the Efficient and Selective Extraction of Compounds in Complex Matrices using Molecularly Imprinted Polymers (MIPs) and their Relevance to Toxicological Analysis. J. Anal. Toxicol. 2020, 45, 312–321. [Google Scholar]
- Wan, Y.; Ma, H.; Lu, B. MIPs in Aqueous Environments. In Advances in Biochemical Engineering/Biotechnology; Springer: Cham, Switzerland, 2015; pp. 131–166. [Google Scholar]
- Vieira, A.C.; Zampieri, R.A.; de Siqueira, M.E.; Martins, I.; Figueiredo, E.C. Molecularly imprinted solid-phase extraction and high-performance liquid chromatography with ultraviolet detection for the determination of urinary trans, trans-muconic acid: A comparison with ionic exchange extraction. Analyst 2012, 137, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, S.; Li, J. Recent advances in molecular imprinting technology: Current status, challenges and highlighted applications. Chem. Soc. Rev. 2011, 40, 2922–2942. [Google Scholar] [CrossRef] [PubMed]
- Pichon, V.; Delaunay, N.; Combes, A. Sample preparation using molecularly imprinted polymers. Anal. Chem. 2020, 92, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Maranata, G.J.; Surya, N.O.; Hasanah, A.N. Optimising factors affecting solid phase extraction performances of molecular imprinted polymer as recent sample preparation technique. Heliyon 2021, 7, e05934. [Google Scholar] [CrossRef]
- Beltran, A.; Borrull, F.; Marcé, R.M.; Cormack, P.A.G. Molecularly-imprinted polymers: Useful sorbents for selective extractions. Trends Anal. Chem. 2010, 29, 1363–1375. [Google Scholar] [CrossRef]
- Hasanah, A.N.; Fauzi, D.; Witka, B.Z.; Rahayu, D.; Pratiwi, R. Molecular Imprinted Polymer for Ethylmorphine with Methacrylic Acid and Acrylamide as Functional Monomer in Butanol Using Two Polymerization Method. Mediterr. J. Chem. 2020, 10, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Qronfla, M.M.; Jamoussi, B.; Chakroun, R. Synthesis and Characterization of a New Molecularly Imprinted Polymer for Selective Extraction of Mandelic Acid Metabolite from Human Urine as a Biomarker of Environmental and Occupational Exposures to Styrene. Polymers 2023, 15, 2398. [Google Scholar] [CrossRef]
- Mpupa, A.; Dinc, M.; Mizaikoff, B.; Nomngongo, P.N. Exploration of a Molecularly Imprinted Polymer (MIPs) as an Adsorbent for the Enrichment of Trenbolone in Water. Processes 2021, 9, 186. [Google Scholar] [CrossRef]
- Heravizadeh, O.R.; Khadem, M.; Nabizadeh, R.; Shahtaheri, S.J. Synthesis of molecularly imprinted nanoparticles for selective exposure assessment of permethrin: Optimization by response surface methodology. J. Environ. Health Sci. Eng. 2019, 17, 393–406. [Google Scholar] [CrossRef]
- Elboughdiri, N.; Mahjoubi, A.; Shawabkeh, A.; Khasawneh, H.; Jamoussi, B. Optimization of the Degradation of Hydroquinone, Resorcinol and Catechol Using Response Surface Methodology. Adv. Chem. Eng. Sci. 2015, 5, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Missaoui, I.; Sayedi, L.; Jamoussi, B.; Ben Hassine, B. Response surface optimization for determination of volatile organic compounds in water samples by headspace-gas chromatography-mass spectrometry method. J. Chromatogr. Sci. 2009, 47, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Elboughdiri, N.; Ghernaout, D.; Kriaa, K.; Jamoussi, B. Enhancing the Extraction of Phenolic Compounds from Juniper Berries Using the Box-Behnken Design. ACS Omega 2020, 5, 27990–28000. [Google Scholar] [CrossRef]
- Jablaoui, C.; Besombes, C.; Jamoussi, B.; Rhazi, L.; Allaf, K. Comparison of expander and Instant Controlled Pressure-Drop DIC technologies as thermomechanical pretreatments in enhancing solvent extraction of vegetal soybean oil. Arab. J. Chem. 2020, 13, 7235–7246. [Google Scholar] [CrossRef]
- Bouazizi, S.; Jamoussi, B.; Bousta, D. Application of response surface methodology for optimization of heavy metals biosorption on natural gum of acacia nilotica. Int. J. Eng. Res. 2016, 5, 32–41. [Google Scholar]
- Baizig, M.; Jamoussi, B.; Batis, N. Optimization by RSM of the degradation of three phenolic compounds-Hydroquinone, resorcinol and catechol-On Fe-modified clays. Water Qual. Res. J. Can. 2013, 48, 171–179. [Google Scholar] [CrossRef]
- Alzahrani, S.A.; Jamoussi, B.; Neamatallah, A.A.; Aloufi, F.A.; Halawani, R.F.; Chakroun, R.; Jablaoui, C. Instant Controlled Pressure Drop (DIC) Processing to Reduce 3-Monochloropropane-1,2-diol Concentration in Palm Oil. Processes 2023, 11, 1085. [Google Scholar] [CrossRef]
- Kivistö, H.; Pekari, K.; Aitio, A. Analysis and stability of phenylglyoxylic and mandelic acids in the urine of styrene-exposed people. Int. Arch. Occup. Environ. Health 1993, 64, 399–403. [Google Scholar] [CrossRef]
- Chakroun, R.; Hedhili, A.; Faidi, A.; Nouaigui, H.; Ben Laiba, M. Simultaneous HPLC Determination of Urinary Metabolites of Toluene, Xylenes, and Ethylbenzene; and Its Application to Biological Monitoring of Tunisian Workers Exposure. Anal. Lett. 2006, 39, 83–97. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, J.; Liu, M.; Han, X.; Peng, Y.; Tian, X.; Liu, J.; Zhang, S. Synthesis of Molecularly Imprinted Polymer via Emulsion Polymerization for Application in Solanesol Separation. Appl. Sci. 2020, 10, 2868. [Google Scholar] [CrossRef] [Green Version]
- Roland, R.M.; Bhawani, S.A. Synthesis and Characterization of Molecular Imprinting Polymer Microspheres of Piperine: Extraction of Piperine from Spiked Urine. J. Anal. Methods Chem. 2016, 2016, 5671507. [Google Scholar] [CrossRef] [Green Version]
- Martins, G.V.; Marques, A.C.; Fortunato, E.; Sales, M.G.F. Paper-based (bio)sensor for label-free detection of 3-nitrotyrosine in human urine samples using molecular imprinted polymer. Sens. Bio-Sens. Res. 2020, 28, 100333. [Google Scholar] [CrossRef]
- Bashir, K.; Guo, P.; Chen, G.; Li, Y.; Ge, Y.; Shu, H.; Fu, Q. Synthesis, characterization, and application of griseofulvin surface molecularly imprinted polymers as the selective solid phase extraction sorbent in rat plasma samples. Arab. J. Chem. 2019, 13, 4082–4091. [Google Scholar] [CrossRef]
- Roushani, M.; Nezhadali, A.; Jalilian, Z.; Azadeh Azadbakht, A. Development of novel electrochemical sensor on the base of molecular imprinted polymer decorated on SiC nanoparticles modified glassy carbon electrode for selective determination of loratadine. Mater. Sci. Eng. C 2017, 17, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Langmuir, I. The constitution and fundamental properties of solids and liquids. II. Liquids. J. Am. Chem. Soc. 1917, 39, 1848–1906. [Google Scholar] [CrossRef] [Green Version]
- Nqombolo, A.; Mpupa, A.; Gugushe, A.S.; Moutloali, R.M.; Nomngongo, P.N. Adsorptive removal of lead from acid mine drainage using cobalt-methylimidazolate framework as an adsorbent: Kinetics, isotherm, and regeneration. Environ. Sci. Pollut. Res. 2019, 26, 3330–3339. [Google Scholar] [CrossRef] [PubMed]
- Gugushe, A.S.; Nqombolo, A.; Nomngongo, P.N. Application of Response Surface Methodology and Desirability Function in the Optimization of Adsorptive Remediation of Arsenic from Acid Mine Drainage Using Magnetic Nanocomposite: Equilibrium Studies and Application to Real Samples. Molecules 2019, 24, 1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.; Ruan, G.; Liang, S.; Xie, F.; Liu, H. Monolithic molecularly imprinted solidphase extraction for the selective determination of trace cytokinins in plant samples with liquid chromatography-electrospray tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 404, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, S.; Jahani, M. Selective solid-phase extraction using molecular imprinted polymer sorbent for the analysis of Florfenicol in food samples. Food Chem. 2013, 141, 1242–1251. [Google Scholar] [CrossRef]
- Urraca, J.L.; Castellari, M.; Barrios, C.A.; Moreno-Bondi, M.C. Multiresidue analysis of fluoroquinolone antimicrobials in chicken meat by molecularly imprinted solid phase extraction and high performance liquid chromatography. J. Chromatogr. A 2014, 1343, 1–9. [Google Scholar] [CrossRef]
- Sorouraddin, S.M.; Afshar Mogaddam, M.R. Development of molecularly imprinted-solid phase extraction combined with dispersive liquid–liquid microextraction for selective extraction and preconcentration of triazine herbicides from aqueous samples. J. Iran. Chem. Soc. 2016, 13, 1093–1104. [Google Scholar] [CrossRef]
- Sanagi, M.M.; Salleh, S.; Ibrahim, W.A.W.; Naim, A.A.; Hermawan, D.; Miskam, M.; Hussain, I.; Aboul-Enein, H.Y. Molecularly imprinted polymer solid-phase extraction for the analysis of organophosphorus pesticides in fruit samples. J. Food Compos. Anal. 2013, 32, 155–161. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Template | Functional Monomer | Cross Linker | Mole Ratio | Progen | Initiator |
|---|---|---|---|---|---|---|
| MIP | PGA | 4-VP | EGDMA | 1:4:20 | ACN | AIBN |
| NIP | - | 4-VP | EGDMA | 1:4:20 | ACN | AIBN |
| MIP(4-VP)SPE Phase | Input Variables | Unit | Symbol | Levels | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| Adsorption Desorption | pH | - | x1 | 4.0 | 9.0 |
| Sorbent mass | mg | x2 | 15.0 | 30.0 | |
| Elution flow rate | mL/min | x3 | 1.0 | 2.5 | |
| Elution solvent volume | mL | x4 | 3.0 | 6.0 | |
| Run | X1 | X2 | X3 | X4 | Extraction (%) (Mean ± SD) | Retention (%) (Mean ± SD) |
|---|---|---|---|---|---|---|
| 1 | 4 | 15 | 1 | 3 | 88.41 ±0.24 | 81.01 ± 0.76 |
| 2 | 4 | 30 | 1 | 3 | 86.45 ± 0.03 | 79.04 ± 0.85 |
| 3 | 8 | 30 | 1 | 3 | 75.89 ± 0.02 | 27.02 ± 0.13 |
| 4 | 8 | 15 | 2.5 | 3 | 72.02 ± 0.53 | 23.00 ± 0.15 |
| 5 | 4 | 30 | 2.5 | 3 | 88.99 ± 0.25 | 81.05 ± 0.42 |
| 6 | 8 | 30 | 2.5 | 3 | 79.85 ± 0.84 | 26.08 ± 0.18 |
| 7 | 4 | 15 | 1 | 6 | 92.12 ± 0.17 | 85.12 ± 0.06 |
| 8 | 8 | 15 | 1 | 6 | 77.50 ± 0.95 | 29.03 ± 0.12 |
| 9 | 4 | 30 | 1 | 6 | 94.03. ± 0.23 | 64.04 ± 0.73 |
| 10 | 8 | 30 | 1 | 6 | 89.46 ± 0.25 | 41.08 ± 0.06 |
| 11 | 4 | 15 | 2.5 | 6 | 92.80 ± 0.09 | 92.06 ± 0.42 |
| 12 | 8 | 15 | 2.5 | 6 | 79.23 ± 0.05 | 42.02 ± 0.77 |
| 13 | 4 | 30 | 2.5 | 6 | 95.80 ± 0.24 | 62.16 ± 0.10 |
| 14 | 9.73 | 22.5 | 1.75 | 4.5 | 69.02 ± 0.36 | 3.02 ± 0.04 |
| 15 | 6 | 36.5 | 1.75 | 4.5 | 96.04 ± 0.42 | 72.16 ± 0.19 |
| 16 | 6 | 22.5 | 3.15 | 4.5 | 97.22 ± 0.09 | 48.03 ± 0.05 |
| 17 | 6 | 22.5 | 1.75 | 7.3 | 96.71 ± 0.04 | 92.00 ± 0.19 |
| 18 | 6 | 22.5 | 1.75 | 4.5 | 99.69 ± 0.17 | 100.00 ± 1.05 |
| Source | Sum of Squares | df | Mean Square | F-Ratio | p-Value |
|---|---|---|---|---|---|
| x1: pH | 6621.71 | 1 | 6621.71 | 279.40 | 0.0005 |
| x2: Sorbent mass | 36.7358 | 1 | 36.7358 | 1.55 | 0.3015 |
| x3: Elution flow rate | 147.332 | 1 | 147.332 | 6.22 | 0.0882 |
| x4: Elution volume | 67.8588 | 1 | 67.8588 | 2.86 | 0.1892 |
| x1x1 | 1537.69 | 1 | 1537.69 | 64.88 | 0.0040 |
| x1x2 | 438.174 | 1 | 438.174 | 18.49 | 0.0231 |
| x1x3 | 1.25352 | 1 | 1.25352 | 0.05 | 0.8329 |
| x1x4 | 454.533 | 1 | 454.533 | 19.18 | 0.0220 |
| x2x2 | 447.886 | 1 | 447.886 | 18.90 | 0.0225 |
| x2x3 | 134.314 | 1 | 134.314 | 5.67 | 0.0976 |
| x2x4 | 99.793 | 1 | 99.793 | 4.21 | 0.1325 |
| x3x3 | 2145.37 | 1 | 2145.37 | 90.52 | 0.0025 |
| x3x4 | 17.9202 | 1 | 17.9202 | 0.76 | 0.4485 |
| x4x4 | 151.498 | 1 | 151.498 | 6.39 | 0.0856 |
| Total error | 71.1002 | 3 | 23.7001 | ||
| Total (corr.) | 14,272.5 | 17 | |||
| Multiple R2 = 0.9950; Adjusted R2 = 0.9717 | |||||
| Standard Error of Est. = 4.86827; Mean absolute error = 1.77003 | |||||
| Durbin–Watson statistic = 1.74455 (p = 0.1104); Lag 1 residual autocorrelation = 0.107803 | |||||
| Source | Sum of Squares | df | Mean Square | F-Ratio | p-Value |
|---|---|---|---|---|---|
| x1: pH | 328.097 | 1 | 328.097 | 136.38 | 0.0013 |
| x2: Sorbent mass | 87.7088 | 1 | 87.7088 | 36.46 | 0.0091 |
| x3: Elution flow rate | 21.0238 | 1 | 21.0238 | 8.74 | 0.0597 |
| x4: Elution volume | 184.642 | 1 | 184.642 | 76.75 | 0.0031 |
| x1x1 | 253.047 | 1 | 253.047 | 105.18 | 0.0020 |
| x1x2 | 61.9063 | 1 | 61.9063 | 25.73 | 0.0148 |
| x1x3 | 1.91072 | 1 | 1.91072 | 0.79 | 0.4385 |
| x1x4 | 21.4099 | 1 | 21.4099 | 8.90 | 0.0584 |
| x2x2 | 61.9367 | 1 | 61.9367 | 25.74 | 0.0148 |
| x2x3 | 0.863509 | 1 | 0.863509 | 0.36 | 0.5913 |
| x2x4 | 17.1837 | 1 | 17.1837 | 7.14 | 0.0755 |
| x3x3 | 25.4209 | 1 | 25.4209 | 10.57 | 0.0475 |
| x3x4 | 0.369892 | 1 | 0.369892 | 0.15 | 0.7212 |
| x4x4 | 83.7748 | 1 | 83.7748 | 34.82 | 0.0097 |
| Total error | 7.21747 | 3 | 2.40582 | ||
| Total (corr.) | 1506.59 | 17 | |||
| Multiple R2 = 0.9952; Adjusted R2 = 0.9728 | |||||
| Standard Error of Est. = 1.55107; Mean absolute error = 0.507682 | |||||
| Durbin-Watson statistic = 1.29337 (p = 0.0224); Lag 1 residual autocorrelation = 0.335221 | |||||
| Target and Metabolites Solution | Polymer | Kd (mL/g) | IF | α(IFPGA/IFHA) | |
|---|---|---|---|---|---|
| PGA | MIP | 272.57 | 2.34 | 2.79 | |
| NIP | 116.17 | ||||
| HA | MIP | 89.33 | 0.84 | ||
| NIP | 106.37 | ||||
| Metabolite | LOD (mg.L−1) | LOQ (mg.L−1) |
|---|---|---|
| PGA | 0.5 | 1.6 |
| HA | 3.4 | 11.4 |
| Spiked Urine (mg.L−1) | Recovery (%) | RSD (%) (n = 3) |
|---|---|---|
| 5 | 97.32 ± 4.29 | 4.61 |
| 50 | 98.78 ± 3.08 | 3.72 |
| 125 | 99.06 ± 3.25 | 3.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qronfla, M.M.; Jamoussi, B.; Chakroun, R.; Al-Mur, B.A.; Halawani, R.F.; Aloufi, F.A. Synthesis of a New Molecularly Imprinted Polymer and Optimisation of Phenylglyoxylic Acid Extraction from Human Urine Samples Using a Central Composite Design within the Response Surface Methodology. Polymers 2023, 15, 3279. https://doi.org/10.3390/polym15153279
Qronfla MM, Jamoussi B, Chakroun R, Al-Mur BA, Halawani RF, Aloufi FA. Synthesis of a New Molecularly Imprinted Polymer and Optimisation of Phenylglyoxylic Acid Extraction from Human Urine Samples Using a Central Composite Design within the Response Surface Methodology. Polymers. 2023; 15(15):3279. https://doi.org/10.3390/polym15153279
Chicago/Turabian StyleQronfla, Murad. M., Bassem Jamoussi, Radhouane Chakroun, Bandar A. Al-Mur, Riyadh F. Halawani, and Fahed A. Aloufi. 2023. "Synthesis of a New Molecularly Imprinted Polymer and Optimisation of Phenylglyoxylic Acid Extraction from Human Urine Samples Using a Central Composite Design within the Response Surface Methodology" Polymers 15, no. 15: 3279. https://doi.org/10.3390/polym15153279