
Polyurea–Graphene Nanocomposites—The Influence of Hard-Segment Content and Nanoparticle Loading on Mechanical Properties

Abstract
:1. Introduction
2. Materials and Methods
2.1. Polymer Synthesis
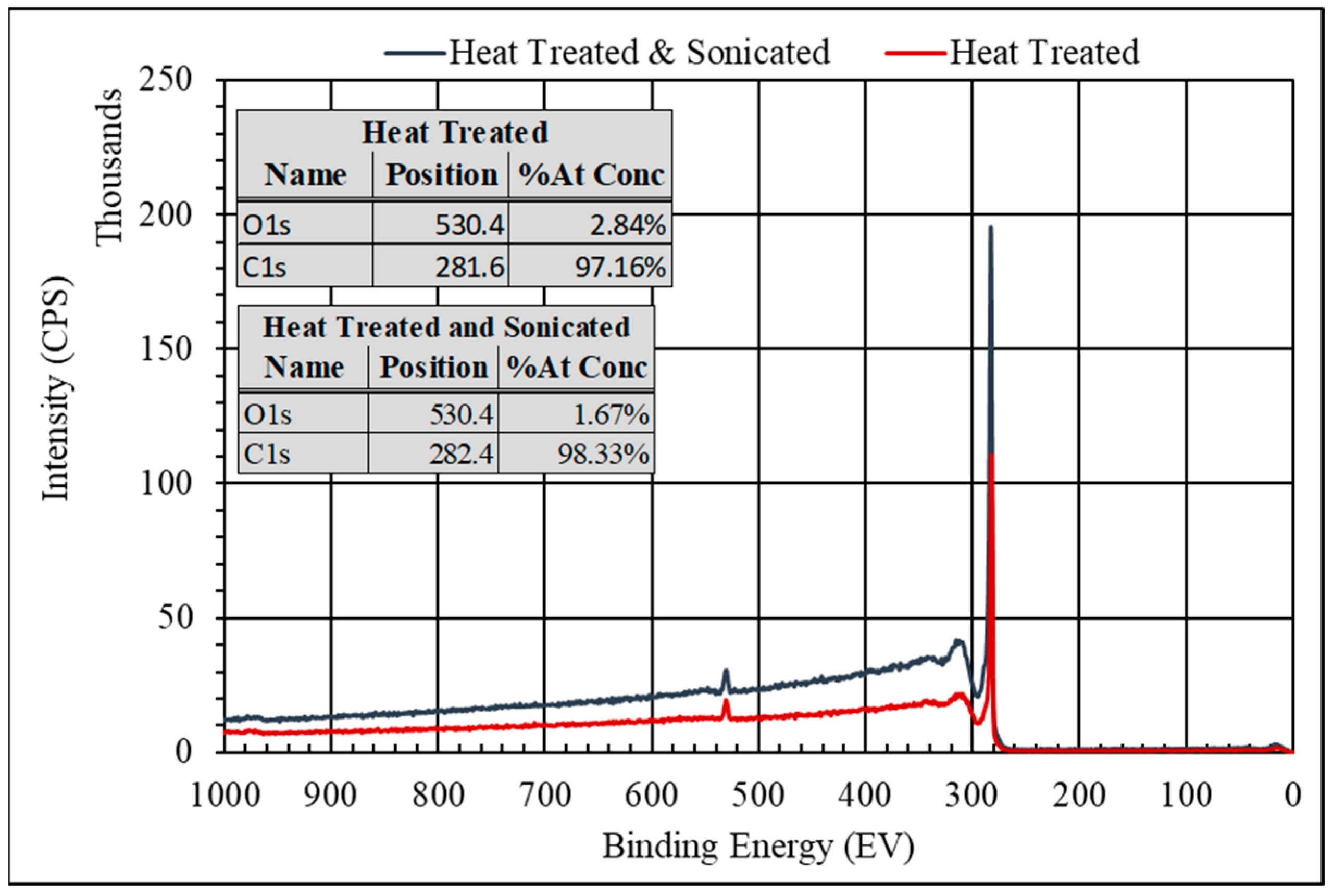
2.2. PUa–xGnP Characterization
2.3. Modeling
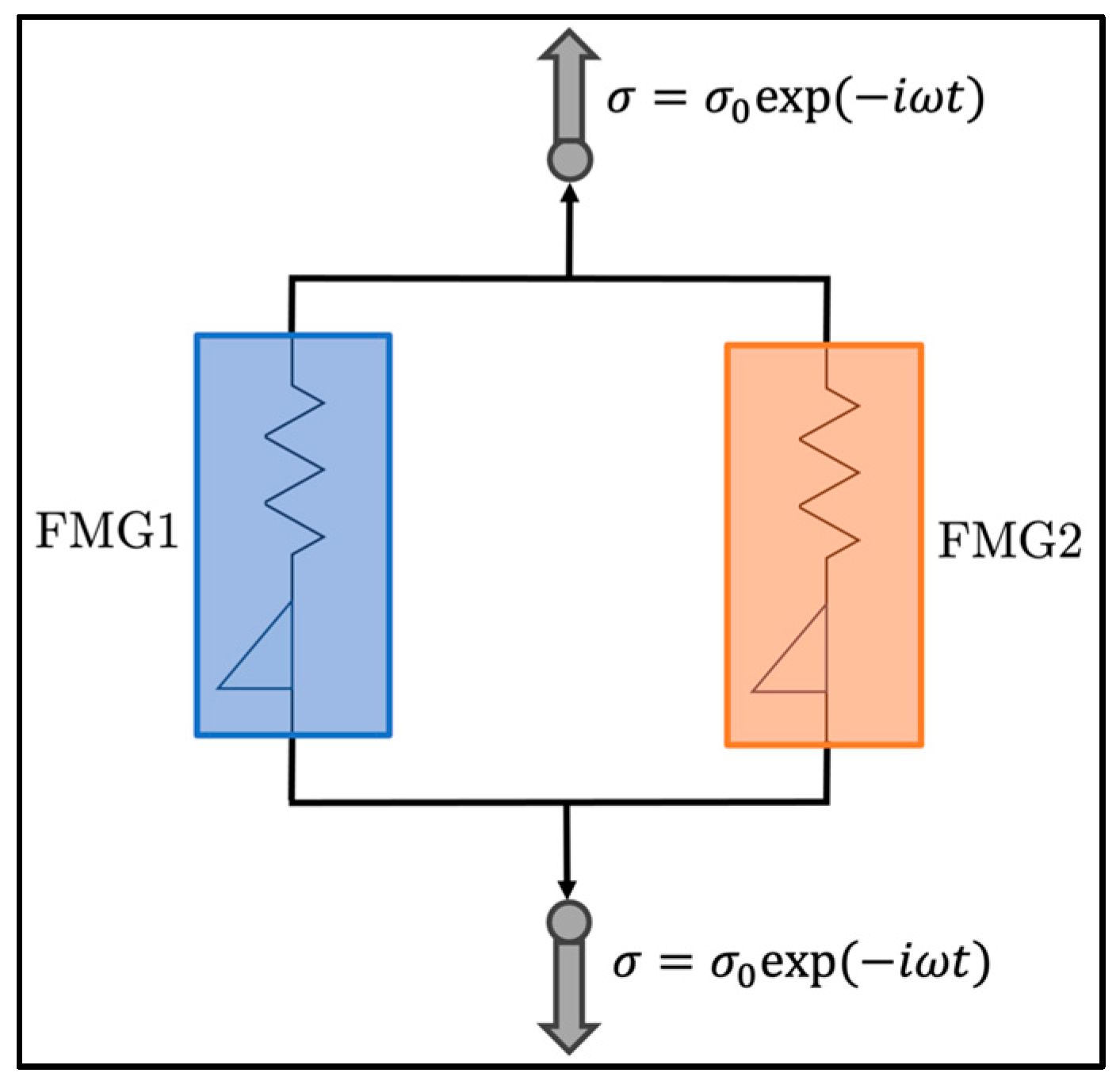
2.3.1. Fractional-Order Maxwell Gel Model
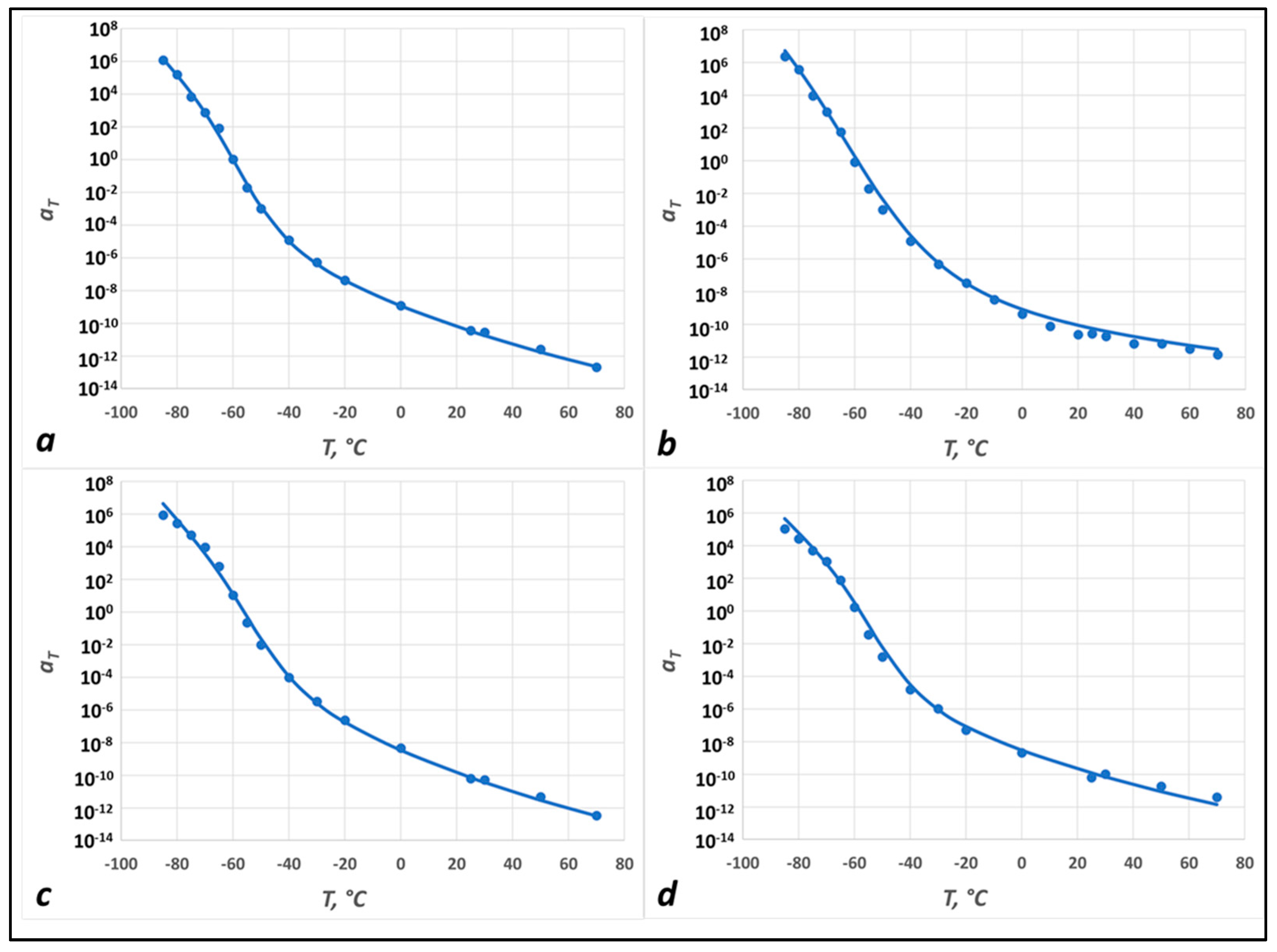
2.3.2. The Shift Factor and the Two State, Two (Time) Scale (TS2) Model
2.3.3. Optimization of the FMG Parameters
3. Results
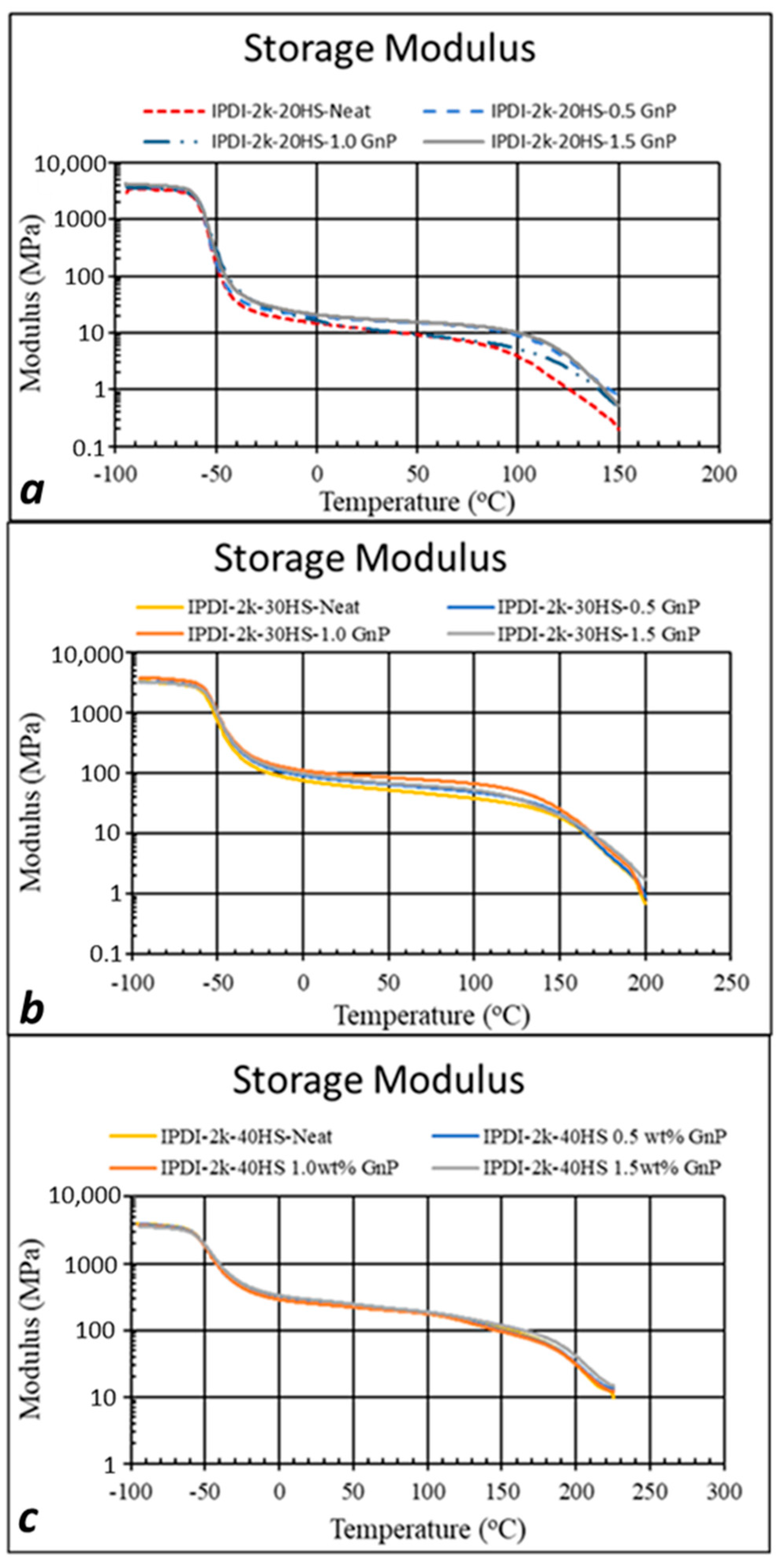
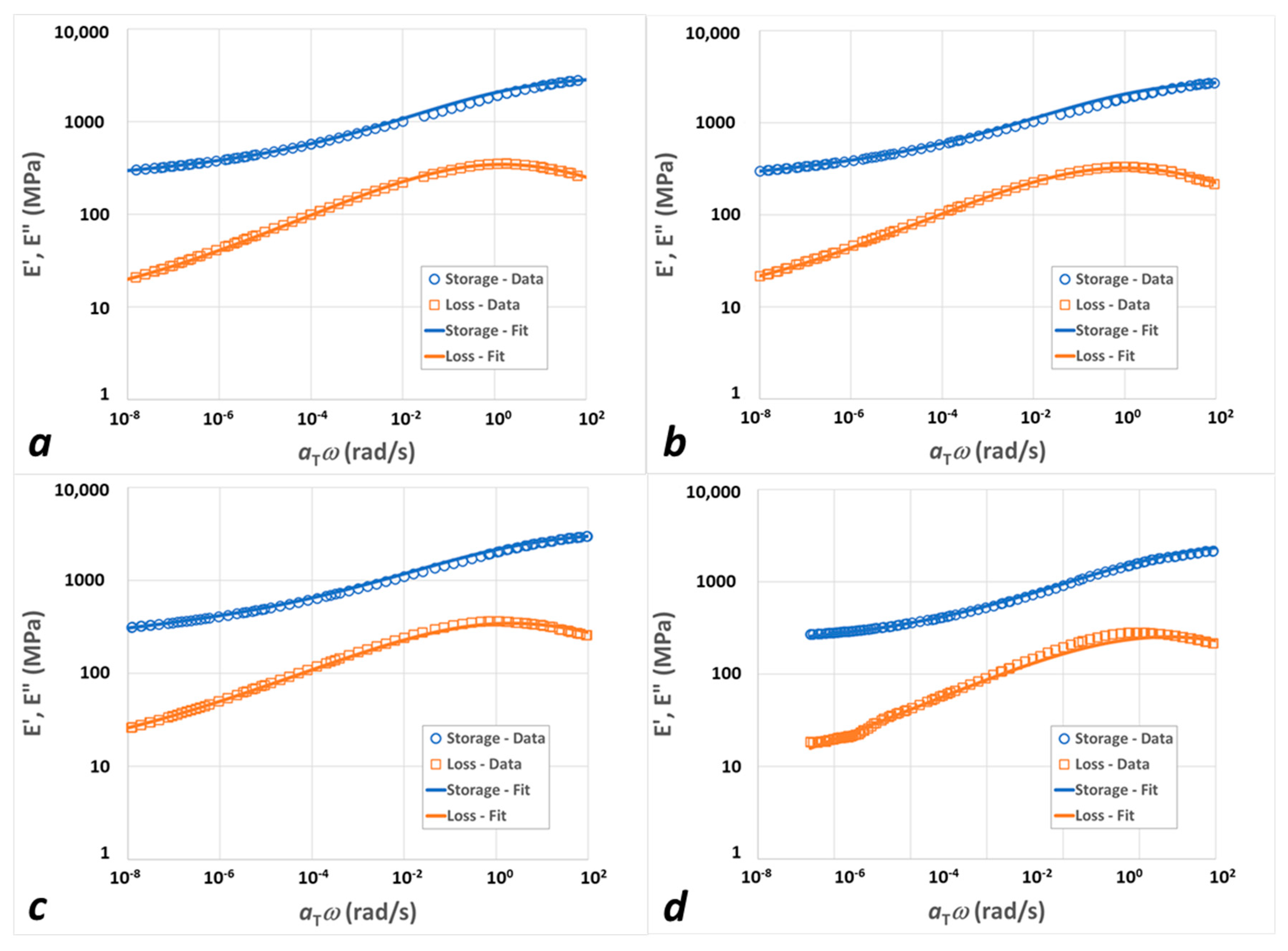
3.1. Experimental Results
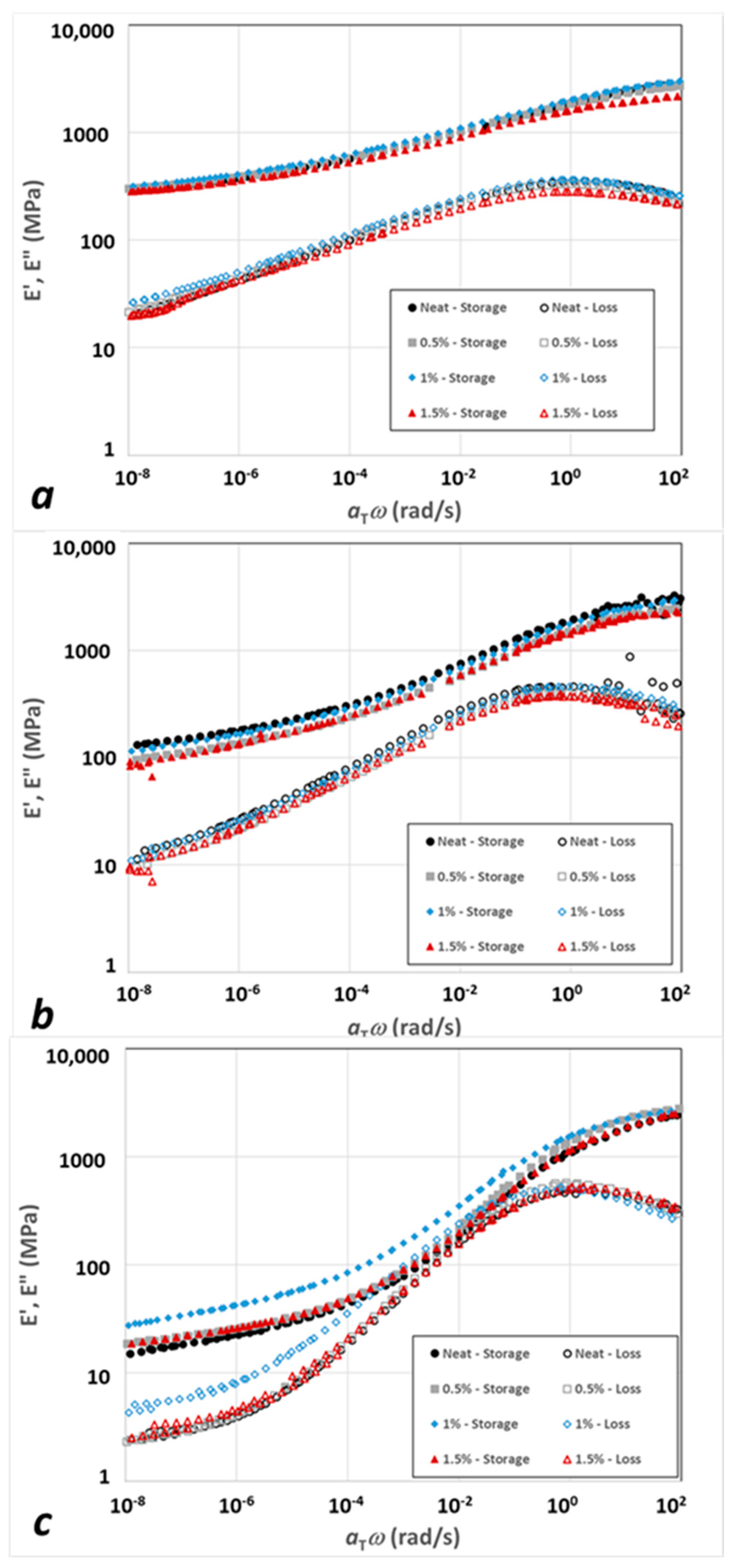
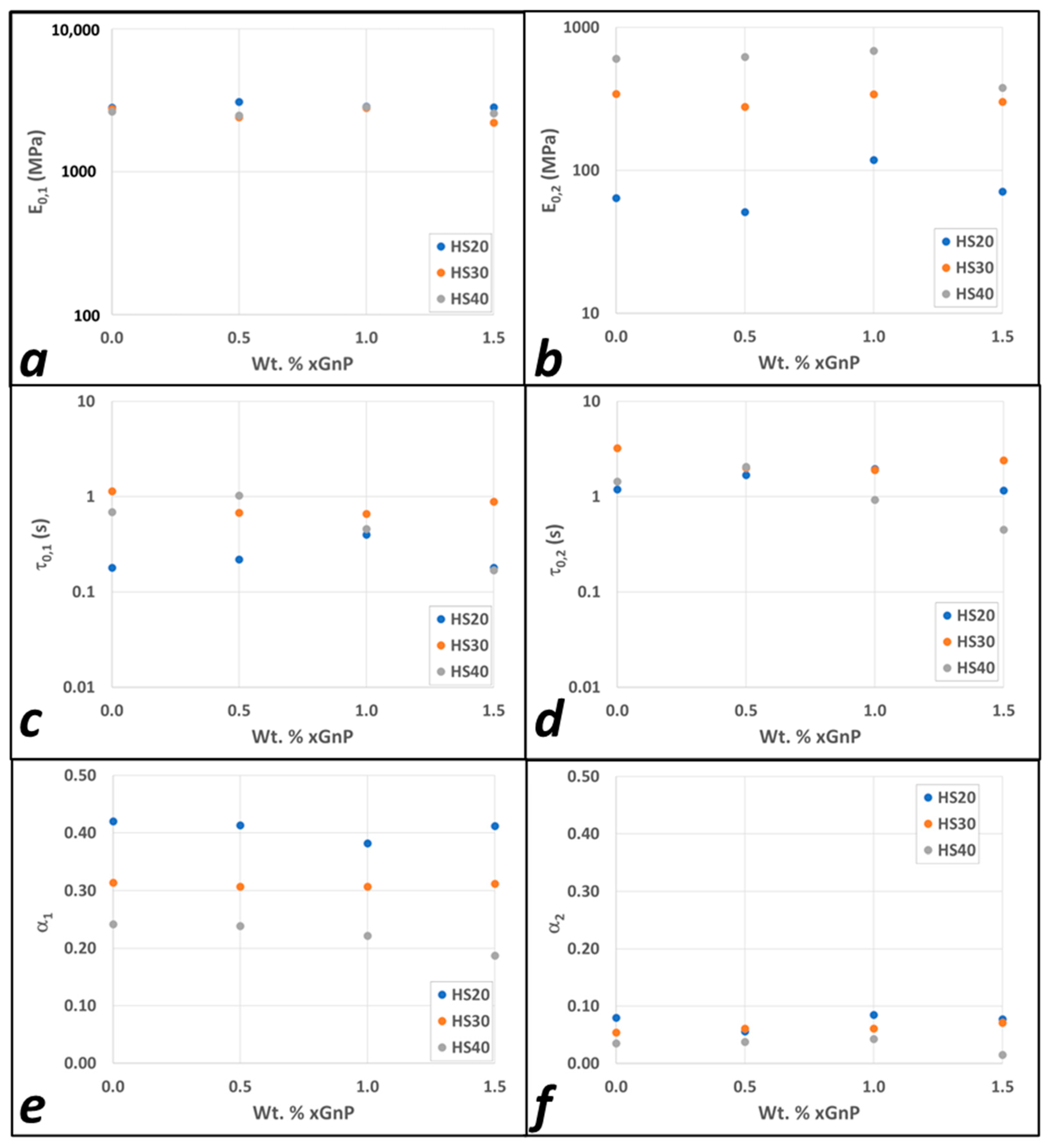
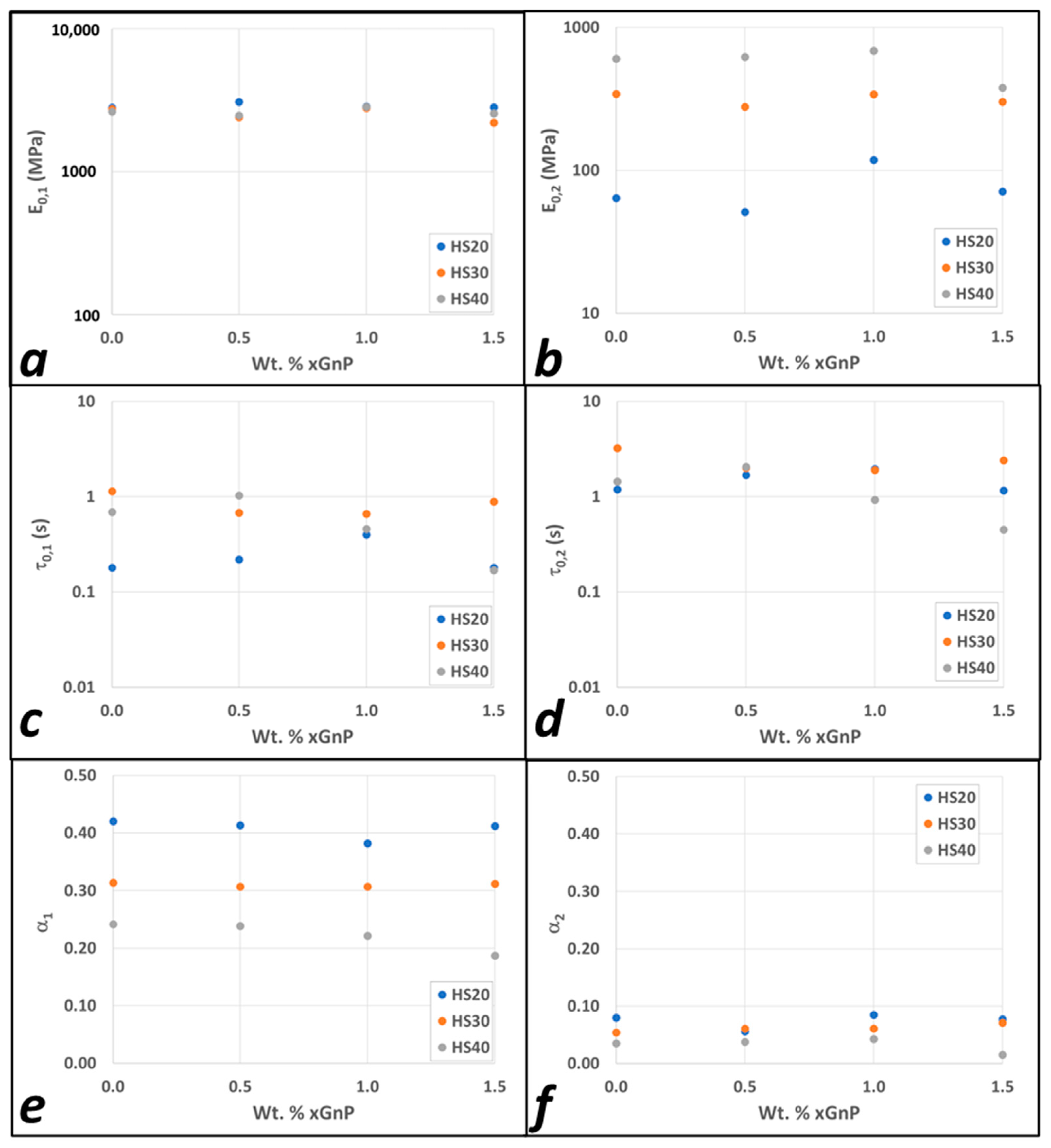
3.2. Modeling Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonnenschein, M.F. Polyurethanes: Science, Technology, Markets, and Trends; Wiley Series on Polymer Engineering and Technology; Wiley: Hoboken, NJ, USA, 2020; ISBN 9781119669463. [Google Scholar]
- Szycher, M. Szycher’s Handbook of Polyurethanes, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 9781439863138/143986313X/9781523108022/1523108029. [Google Scholar]
- Akindoyo, J.O.; Beg, M.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane Types, Synthesis and Applications–a Review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Petrović, Z.S.; Ferguson, J. Polyurethane Elastomers. Prog. Polym. Sci. 1991, 16, 695–836. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Stein, R.S. Small-angle X-ray Scattering Studies of Microdomain Structure in Segmented Polyurethane Elastomers. J. Polym. Sci. Polym. Phys. Ed. 1983, 21, 1439–1472. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Galambos, A.F. Multiple Melting in Segmented Polyurethane Block Copolymers. Macromolecules 1992, 25, 5618–5624. [Google Scholar] [CrossRef]
- Leung, L.M.; Koberstein, J.T. Small-angle Scattering Analysis of Hard-microdomain Structure and Microphase Mixing in Polyurethane Elastomers. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 1883–1913. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Leung, L.M. Compression-Molded Polyurethane Block Copolymers. 2. Evaluation of Microphase Compositions. Macromolecules 1992, 25, 6205–6213. [Google Scholar] [CrossRef]
- Christenson, C.P.; Harthcock, M.A.; Meadows, M.D.; Spell, H.L.; Howard, W.L.; Creswick, M.W.; Guerra, R.E.; Turner, R.B. Model MDI/Butanediol Polyurethanes: Molecular Structure, Morphology, Physical and Mechanical Properties. J. Polym. Sci. B Polym. Phys. 1986, 24, 1401–1439. [Google Scholar] [CrossRef]
- Ginzburg, V.; Bicerano, J.; Christenson, C.P.; Schrock, A.K.; Patashinski, A.Z. Theoretical Modeling of the Relationship between Young’s Modulus and Formulation Variables for Segmented Polyurethanes. J. Polym. Sci. B Polym. Phys. 2007, 45, 2123–2135. [Google Scholar] [CrossRef]
- Garrett, J.T.; Siedlecki, C.A.; Runt, J. Microdomain Morphology of Poly (Urethane Urea) Multiblock Copolymers. Macromolecules 2001, 34, 7066–7070. [Google Scholar] [CrossRef]
- Garrett, J.T.; Runt, J.; Lin, J.S. Microphase Separation of Segmented Poly (Urethane Urea) Block Copolymers. Macromolecules 2000, 33, 6353–6359. [Google Scholar] [CrossRef]
- Matsen, M.W.; Bates, F.S. Unifying Weak-and Strong-Segregation Block Copolymer Theories. Macromolecules 1996, 29, 1091–1098. [Google Scholar] [CrossRef]
- Drolet, F.; Fredrickson, G.H. Combinatorial Screening of Complex Block Copolymer Assembly with Self-Consistent Field Theory. Phys. Rev. Lett. 1999, 83, 4317. [Google Scholar] [CrossRef]
- Benoit, H.; Hadziioannou, G. Scattering Theory and Properties of Block Copolymers with Various Architectures in the Homogeneous Bulk State. Macromolecules 1988, 21, 1449–1464. [Google Scholar] [CrossRef]
- Qi, H.J.; Boyce, M.C. Stress–Strain Behavior of Thermoplastic Polyurethanes. Mech. Mater. 2005, 37, 817–839. [Google Scholar] [CrossRef]
- Kolařk, J. Simultaneous Prediction of the Modulus, Tensile Strength and Gas Permeability of Binary Polymer Blends. Eur. Polym. J. 1998, 34, 585–590. [Google Scholar] [CrossRef]
- Bicerano, J. Prediction of Polymer Properties, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2002; ISBN 0203910117. [Google Scholar]
- Tzelepis, D.A.; Suzuki, J.; Su, Y.F.; Wang, Y.; Lim, Y.C.; Zayernouri, M.; Ginzburg, V. V Experimental and Modeling Studies of IPDI-Based Polyurea Elastomers—The Role of Hard Segment Fraction. J. Appl. Polym. Sci. 2023, 140, e53592. [Google Scholar] [CrossRef]
- Velankar, S.; Cooper, S.L. Microphase Separation and Rheological Properties of Polyurethane Melts. 1. Effect of Block Length. Macromolecules 1998, 31, 9181–9192. [Google Scholar] [CrossRef]
- Ionita, D.; Cristea, M.; Gaina, C. Prediction of Polyurethane Behaviour via Time-Temperature Superposition: Meanings and Limitations. Polym. Test 2020, 83, 106340. [Google Scholar] [CrossRef]
- Ginzburg, V. A Simple Mean-Field Model of Glassy Dynamics and Glass Transition. Soft Matter 2020, 16, 810–825. [Google Scholar] [CrossRef]
- Jaishankar, A.; McKinley, G.H. A Fractional K-BKZ Constitutive Formulation for Describing the Nonlinear Rheology of Multiscale Complex Fluids. J. Rheol. 2014, 58, 1751–1788. [Google Scholar] [CrossRef]
- Jaishankar, A.; McKinley, G.H. Power-Law Rheology in the Bulk and at the Interface: Quasi-Properties and Fractional Constitutive Equations. Proc. R. Soc. A Math. Phys. Eng. Sci. 2013, 469, 20120284. [Google Scholar] [CrossRef]
- Rathinaraj, J.D.J.; McKinley, G.H.; Keshavarz, B. Incorporating Rheological Nonlinearity into Fractional Calculus Descriptions of Fractal Matter and Multi-Scale Complex Fluids. Fractal Fract. 2021, 5, 174. [Google Scholar] [CrossRef]
- Suzuki, J.; Gulian, M.; Zayernouri, M.; D’Elia, M. Fractional Modeling in Action: A Survey of Nonlocal Models for Subsurface Transport, Turbulent Flows, and Anomalous Materials. J. Peridynamics Nonlocal Model. 2021, 5, 392–459. [Google Scholar] [CrossRef]
- Suzuki, J.L.; Zayernouri, M.; Bittencourt, M.L.; Karniadakis, G.E. Fractional-Order Uniaxial Visco-Elasto-Plastic Models for Structural Analysis. Comput. Methods Appl. Mech. Eng. 2016, 308, 443–467. [Google Scholar] [CrossRef]
- Suzuki, J.L.; Naghibolhosseini, M.; Zayernouri, M. A General Return-Mapping Framework for Fractional Visco-Elasto-Plasticity. Fractal Fract. 2022, 6, 715. [Google Scholar] [CrossRef]
- Suzuki, J.L.; Tuttle, T.G.; Roccabianca, S.; Zayernouri, M. A Data-Driven Memory-Dependent Modeling Framework for Anomalous Rheology: Application to Urinary Bladder Tissue. Fractal Fract. 2021, 5, 223. [Google Scholar] [CrossRef]
- Winey, K.I.; Vaia, R.A. Polymer Nanocomposites. MRS Bull. 2007, 32, 314–322. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/Polymer Nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Ray, S.S.; Okamoto, M. Polymer/Layered Silicate Nanocomposites: A Review from Preparation to Processing. Prog. Polym. Sci. 2003, 28, 1539–1641. [Google Scholar]
- Lin, C.-L.; Li, J.-W.; Chen, Y.-F.; Chen, J.-X.; Cheng, C.-C.; Chiu, C.-W. Graphene Nanoplatelet/Multiwalled Carbon Nanotube/Polypyrrole Hybrid Fillers in Polyurethane Nanohybrids with 3D Conductive Networks for EMI Shielding. ACS Omega 2022, 7, 45697–45707. [Google Scholar] [CrossRef]
- Kausar, A. Polyurethane Nanocomposite Coatings: State of the Art and Perspectives. Polym. Int. 2018, 67, 1470–1477. [Google Scholar] [CrossRef]
- Chen, K.; Tian, Q.; Tian, C.; Yan, G.; Cao, F.; Liang, S.; Wang, X. Mechanical Reinforcement in Thermoplastic Polyurethane Nanocomposite Incorporated with Polydopamine Functionalized Graphene Nanoplatelet. Ind. Eng. Chem. Res. 2017, 56, 11827–11838. [Google Scholar] [CrossRef]
- Shah, R.; Kausar, A.; Muhammad, B.; Shah, S. Progression from Graphene and Graphene Oxide to High Performance Polymer-Based Nanocomposite: A Review. Polym. Plast. Technol. Eng. 2015, 54, 173–183. [Google Scholar] [CrossRef]
- Albozahid, M.; Naji, H.Z.; Alobad, Z.K.; Wychowaniec, J.K.; Saiani, A. Thermal, Mechanical, and Morphological Characterisations of Graphene Nanoplatelet/Graphene Oxide/High-Hard-Segment Polyurethane Nanocomposite: A Comparative Study. Polymers 2022, 14, 4224. [Google Scholar] [CrossRef] [PubMed]
- Kausar, A. Shape Memory Polyurethane/Graphene Nanocomposites: Structures, Properties, and Applications. J. Plast. Film Sheeting 2020, 36, 151–166. [Google Scholar] [CrossRef]
- Ginzburg, V.V.; Hall, L.M. Theory and Modeling of Polymer Nanocomposites; Springer: Berlin/Heidelberg, Germany, 2021; ISBN 303060442X. [Google Scholar]
- Meng, Q.; Song, X.; Han, S.; Abbassi, F.; Zhou, Z.; Wu, B.; Wang, X.; Araby, S. Mechanical and Functional Properties of Polyamide/Graphene Nanocomposite Prepared by Chemicals Free-Approach and Selective Laser Sintering. Compos. Commun. 2022, 36, 101396. [Google Scholar] [CrossRef]
- Su, X.; Wang, R.; Li, X.; Araby, S.; Kuan, H.-C.; Naeem, M.; Ma, J. A Comparative Study of Polymer Nanocomposites Containing Multi-Walled Carbon Nanotubes and Graphene Nanoplatelets. Nano Mater. Sci. 2022, 4, 185–204. [Google Scholar] [CrossRef]
- Balazs, A.C.; Bicerano, J.; Ginzburg, V.V. Polyolefin/Clay Nanocomposites: Theory and Simulation. In Polyolefin Composites; Wiley: Hoboken, NJ, USA, 2007; pp. 415–448. ISBN 9780471790570. [Google Scholar]
- Fornes, T.D.; Paul, D.R. Modeling Properties of Nylon 6/Clay Nanocomposites Using Composite Theories. Polymer 2003, 44, 4993–5013. [Google Scholar] [CrossRef]
- Bicerano, J.; Douglas, J.F.; Brune, D.A. Model for the Viscosity of Particle Dispersions. J. Macromol. Sci. Rev. Macromol. Chem. Phys. 1999, 39C, 561–642. [Google Scholar] [CrossRef]
- Pinnavaia, T.J.; Beall, G.W. Polymer-Clay Nanocomposites; John Wiley & Sons, Ltd.: Chichester, UK, 2000. [Google Scholar]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-Forming Liquids. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar] [CrossRef]
- Kennedy, J.; Eberhart, R. Particle Swarm Optimization. In Proceedings of the ICNN’95-International Conference on Neural Networks, Perth, WA, Australia, 27 November–1 December 1995; Volume 4, pp. 1942–1948. [Google Scholar]
- Chen, X.; Wang, X.; Fang, D. A Review on C1s XPS-Spectra for Some Kinds of Carbon Materials. Fuller. Nanotub. Carbon Nanostructures 2020, 28, 1048–1058. [Google Scholar] [CrossRef]
- Kwan, Y.C.G.; Ng, G.M.; Huan, C.H.A. Identification of Functional Groups and Determination of Carboxyl Formation Temperature in Graphene Oxide Using the XPS O 1s Spectrum. Thin. Solid. Film. 2015, 590, 40–48. [Google Scholar] [CrossRef]
- Brune, D.A.; Bicerano, J. Micromechanics of Nanocomposites: Comparison of Tensile and Compressive Elastic Moduli, and Prediction of Effects of Incomplete Exfoliation and Imperfect Alignment on Modulus. Polymer 2002, 43, 369–387. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | IPDI-2k-20HS | IPDI-2k-30HS | IPDI-2k-40HS | |
|---|---|---|---|---|
| Isocyanate Prepolymer (A-Side) | IPDI | 30.8 g | 41.6 g | 52.1 g |
| T5000 | 14.5 g | 12.1 g | 10.1 g | |
| D2000 | 57.1 g | 48.5 g | 40.4 g | |
| Toluene | 82.7 g (95 mL) | 165.3 g (190 mL) | 208.8 g (240 mL) | |
| %NCO | 8.7% | 12.9% | 15.3% | |
| Amine Blend (B-Side) | DETDA | 10 g | 19.4 g | 29.3 g |
| D2000 | 90 g | 80.6 g | 70.7 g |
| Model Parameters | 0.0% GnP | 0.5% GnP | 1% GnP | 1.5% GnP | |||||
|---|---|---|---|---|---|---|---|---|---|
| FMG 1 | FMG 2 | FMG 1 | FMG 2 | FMG 1 | FMG 2 | FMG 1 | FMG 2 | ||
| IPDI-2k 20HS | |||||||||
| IPDI-2k 30HS | |||||||||
| 1 | 2 | ||||||||
| IPDI-2k 40HS | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzelepis, D.A.; Khoshnevis, A.; Zayernouri, M.; Ginzburg, V.V. Polyurea–Graphene Nanocomposites—The Influence of Hard-Segment Content and Nanoparticle Loading on Mechanical Properties. Polymers 2023, 15, 4434. https://doi.org/10.3390/polym15224434
Tzelepis DA, Khoshnevis A, Zayernouri M, Ginzburg VV. Polyurea–Graphene Nanocomposites—The Influence of Hard-Segment Content and Nanoparticle Loading on Mechanical Properties. Polymers. 2023; 15(22):4434. https://doi.org/10.3390/polym15224434
Chicago/Turabian StyleTzelepis, Demetrios A., Arman Khoshnevis, Mohsen Zayernouri, and Valeriy V. Ginzburg. 2023. "Polyurea–Graphene Nanocomposites—The Influence of Hard-Segment Content and Nanoparticle Loading on Mechanical Properties" Polymers 15, no. 22: 4434. https://doi.org/10.3390/polym15224434