
Mechanochemical Encapsulation of Caffeine in UiO-66 and UiO-66-NH2 to Obtain Polymeric Composites by Extrusion with Recycled Polyamide 6 or Polylactic Acid Biopolymer
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. MOFs Synthesis
2.3. Mechanochemical Encapsulation Method
2.4. Preparation of Polymer Composites with UiO-66 Capsules with Caffeine
2.5. Characterization
3. Results and Discussion
3.1. MOF Characterization
3.2. Caffeine-Loaded Capsules
3.3. Characterization of Textile Composites
3.4. Caffeine-Release
3.4.1. CAF@MOF
3.4.2. Polymer Composites
3.5. Benchmark
4. Conclusions
- As indicated by the different characterization techniques (XRD, FTIR, SEM, TGA, NMR, and nitrogen adsorption), UiO-66 has been prepared through a solvent-free synthesis with textural properties (specific surface area and pore size distribution) and a particle size that are different from those of commercially purchased UiO-66.
- With the same characterization techniques indicated in the previous point, it has been demonstrated that UiO-66-NH2 has been prepared through a synthesis with ethanol, avoiding the use of toxic solvents, such as DMF.
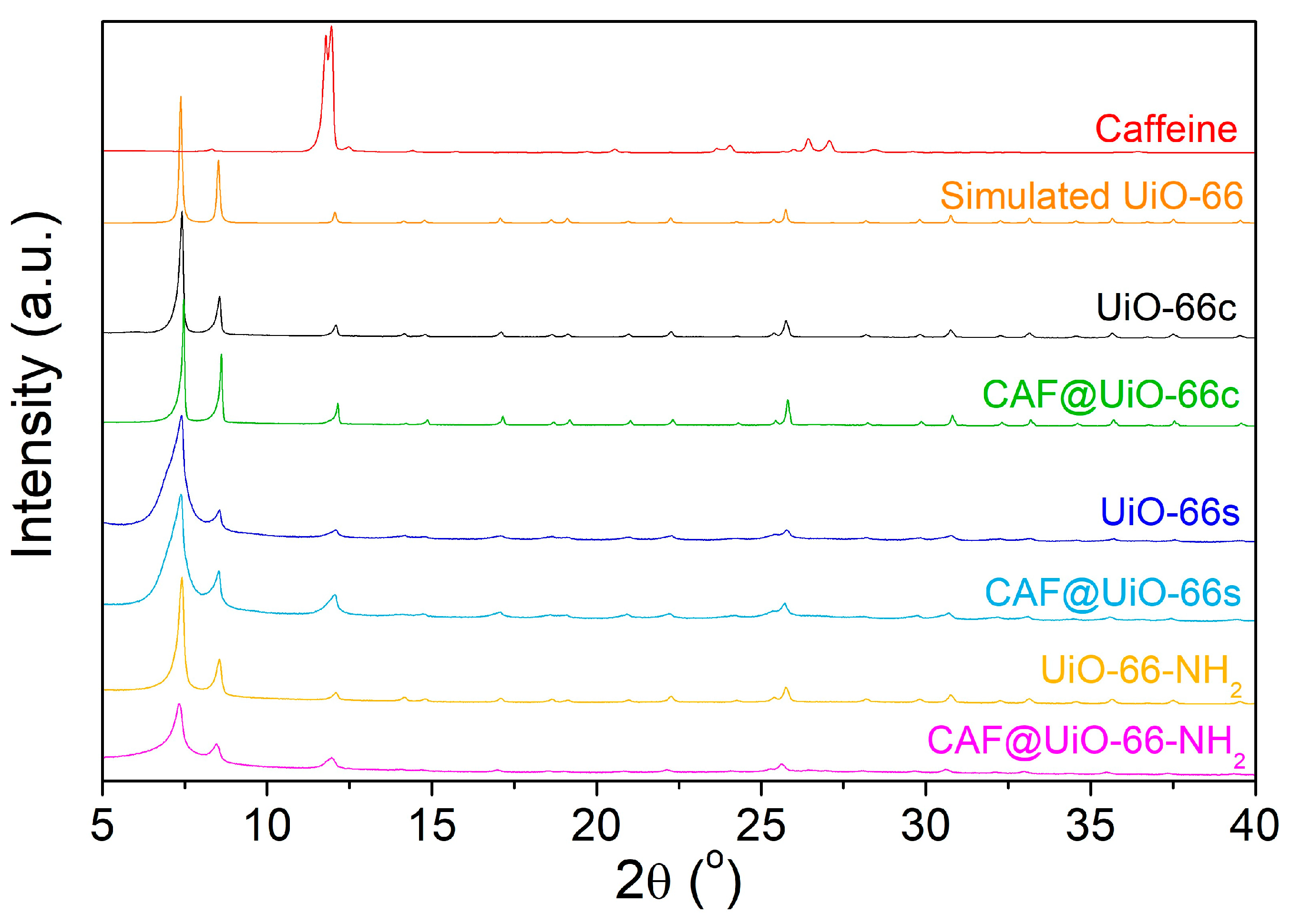
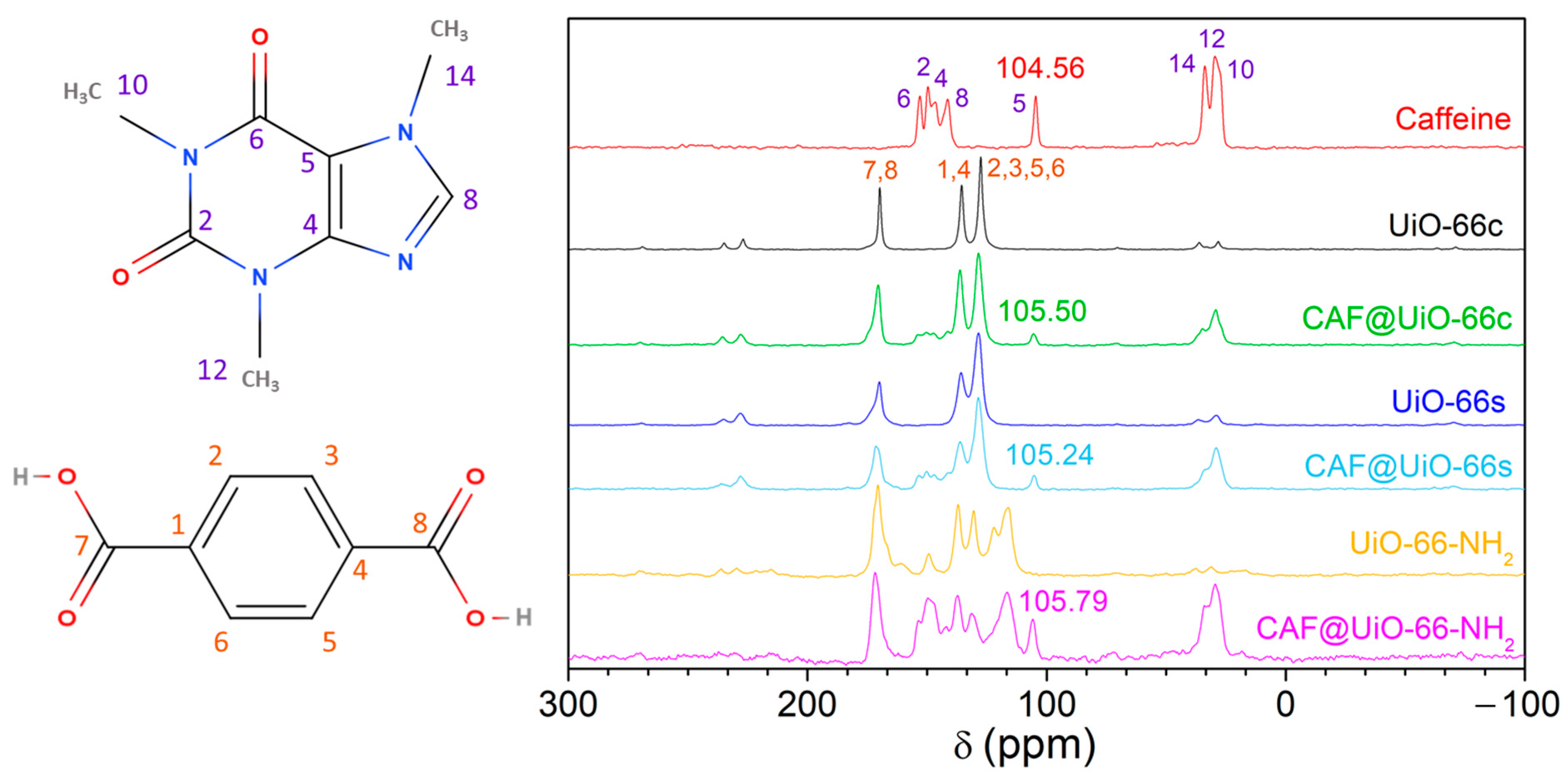
- The encapsulation of 25 wt% of caffeine by milling assisted by a small amount of water in the capsules (MOF@CAF) has been carried out correctly with the available UiO-66 and also with UiO-66-NH2. This assertion is supported by the comparison of MOF and caffeine with these capsules using various techniques, such as XRD, FTIR, SEM, TGA, NMR, and nitrogen adsorption.
- The caffeine release study of the MOF@CAF samples shows a much slower release compared to the physical mixture of caffeine with the MOF. The release is especially slow in sample UiO-66c.
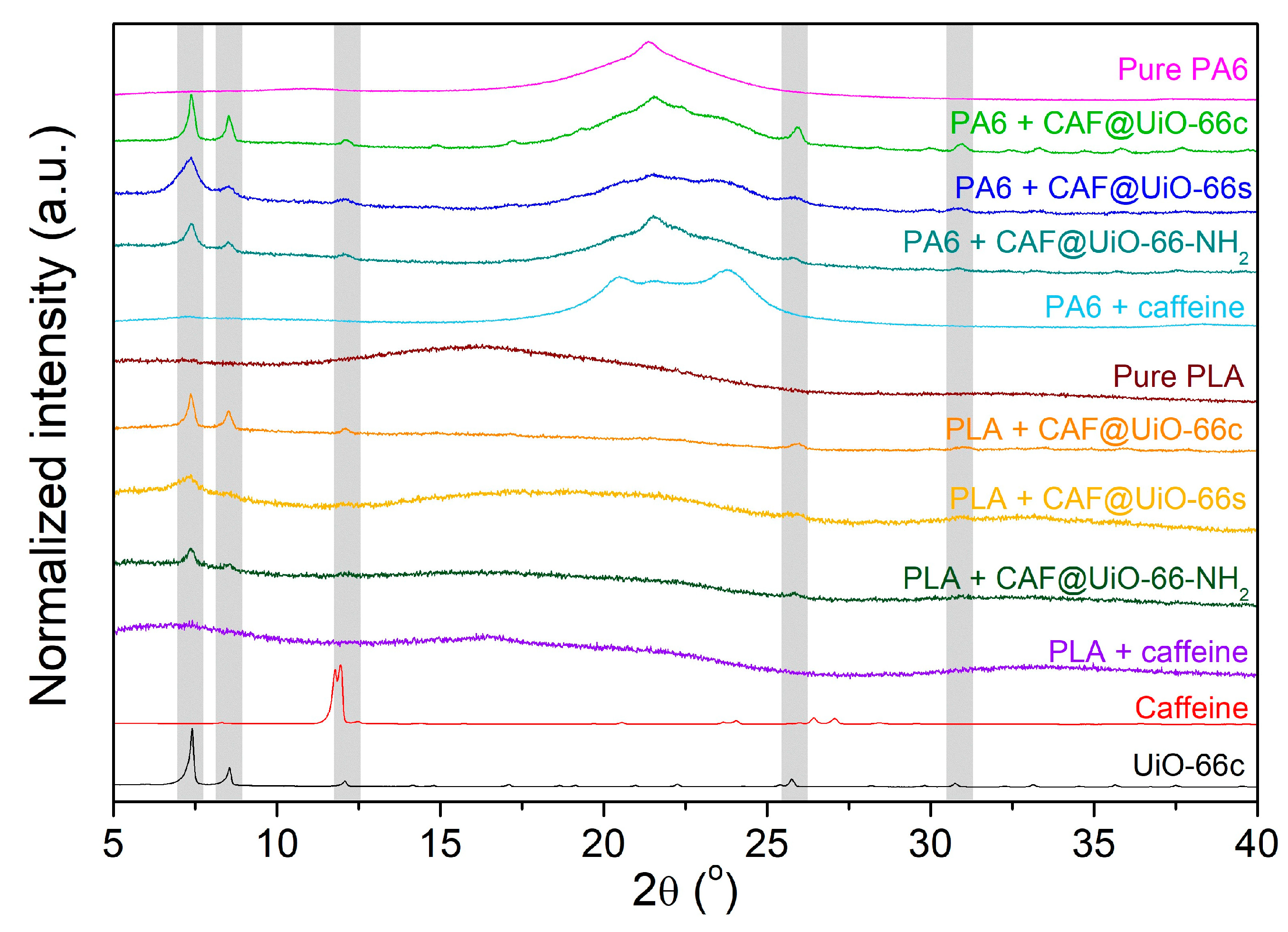
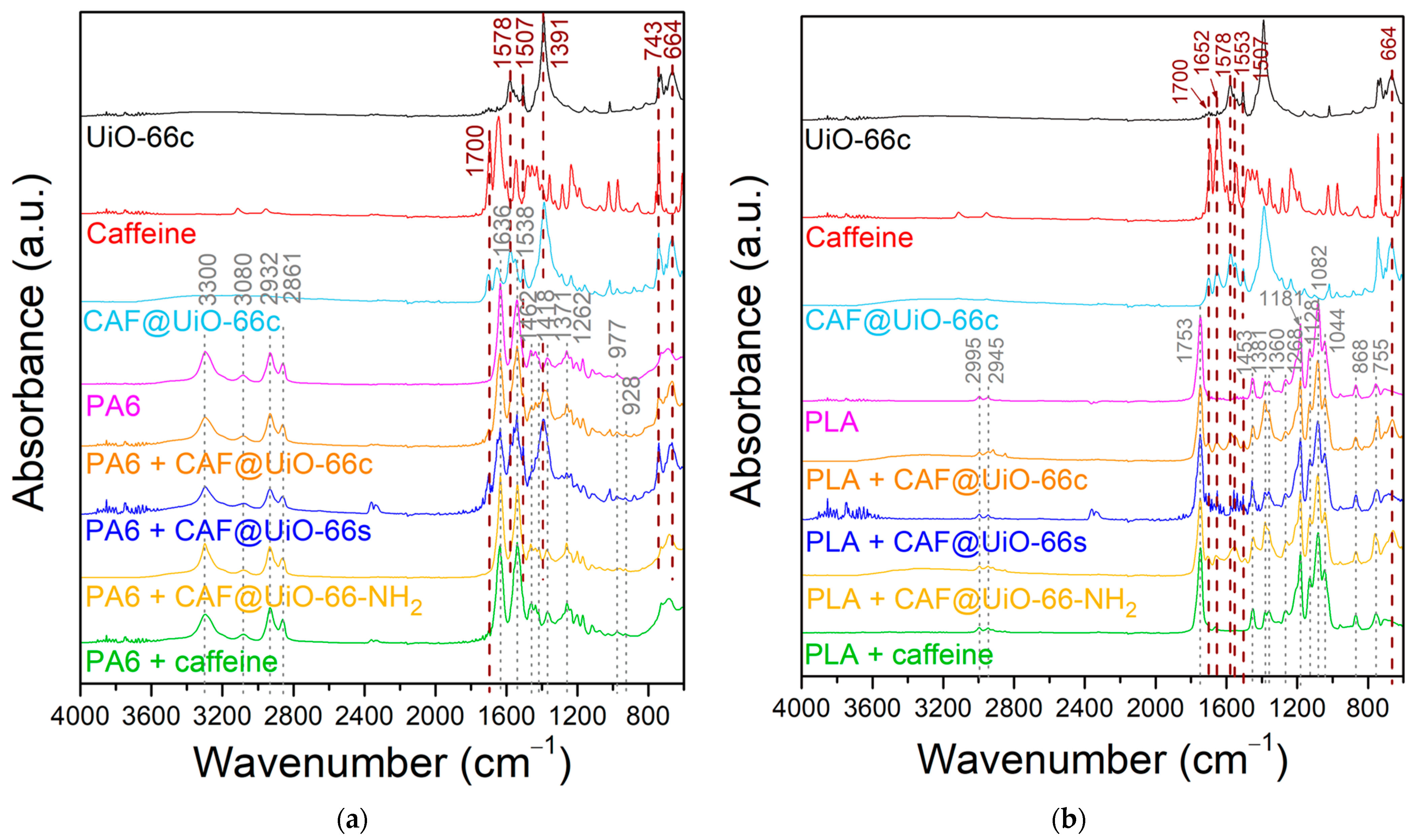
- The PA6 + MOF@CAF and PLA + MOF@CAF composites have been prepared by extrusion with a theoretical caffeine load of 2.5 wt% and, despite the high processing temperatures (190 °C and 260 °C for PLA and PA6, respectively), the capsules retain their fundamental characteristics, as indicated by their characterization (XRD, FTIR, SEM, EDX, and TGA).
- The caffeine release study of the PA6 + MOF@CAF composites shows a much slower release compared to incorporating pure caffeine into the polymer. Therefore, the encapsulation of caffeine has a determining effect on delaying its release in PA6 polymers.
- Among the MOFs, UiO-66c, with its greater surface area, microporosity, and larger particle size, shows the slowest caffeine release in the polymeric composites, which is also in accordance with the release in the capsules.
- The amino group in the UiO-66 does not appear to play a prominent role in the release of caffeine.
- Both in the CAF@MOF capsules and in pure caffeine, the PLA polymer establishes an interaction with caffeine that delays its release markedly compared to the PA6 polymer. In the PLA composites, the effect of encapsulation is observed at high temperatures.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Size (nm) | |||
|---|---|---|---|
| 2·θ | 7.4° | 8.5° | 25.7° |
| UiO-66c | 49.5 | 56.9 | 53.6 |
| UiO-66s | 13.4 | 21.9 | 23.5 |
| UiO-66-NH2 | 40.2 | 42.4 | 43.6 |






References
- Peng, X.; Umer, M.; Pervez, M.N.; Hasan, K.M.F.; Habib, M.A.; Islam, M.S.; Lin, L.; Xiong, X.; Naddeo, V.; Cai, Y. Biopolymers-based microencapsulation technology for sustainable textiles development: A short review. Case Stud. Chem. Environ. Eng. 2023, 7, 100349. [Google Scholar] [CrossRef]
- García, A.; Ramos, M.; Sanahuja, A.; Garrigós, M. Recent Trends in Microencapsulation for Smart and Active Innovative Textile Products. Curr. Org. Chem. 2018, 22, 1237–1248. [Google Scholar]
- Timilsena, Y.P.; Akanbi, T.O.; Khalid, N.; Adhikari, B.; Barrow, C.J. Complex coacervation: Principles, mechanisms and applications in microencapsulation. Int. J. Biol. Macromol. 2019, 121, 1276–1286. [Google Scholar] [CrossRef]
- Paseta, L.; Simón-Gaudó, E.; Gracia-Gorría, F.; Coronas, J. Encapsulation of essential oils in porous silica and MOFs for trichloroisocyanuric acid tablets used for water treatment in swimming pools. Chem. Eng. J. 2016, 292, 28–34. [Google Scholar] [CrossRef]
- Shi, M.; Lu, B.; Li, X.; Jin, Y.; Ge, M. Thermochromic luminescent fiber based on yellow thermochromic microcapsules: Preparation, properties, and potential application areas. Cellulose 2021, 28, 5005–5018. [Google Scholar] [CrossRef]
- Tariq, Z.; Izhar, F.; Malik, M.H.; Oneeb, M.; Anwar, F.; Abbas, M.; Khan, A. Development of functional textile via microencapsulation of peppermint oils: A novel approach in textile finishing. Res. J. Text. Apparel, 2022; ahead-of-print. [Google Scholar]
- García-Viñuales, S.; Rubio, C.; Martínez-Izquierdo, L.; Zornoza, B.; Piera, E.; Caballero, M.Á.; Téllez, C. Study of Melamine-Formaldehyde/Phase Change Material Microcapsules for the Preparation of Polymer Films by Extrusion. Membranes 2022, 12, 266. [Google Scholar] [CrossRef]
- Julaeha, E.; Puspita, S.; Eddy, D.R.; Wahyudi, T.; Nurzaman, M.; Nugraha, J.; Herlina, T.; Al Anshori, J. Microencapsulation of lime (Citrus aurantifolia) oil for antibacterial finishing of cotton fabric. RSC Adv. 2021, 11, 1743–1749. [Google Scholar] [CrossRef]
- Yaman Turan, N.; Turker, E.; Insaatci, Ö. Microparticles loaded with propolis to make antibacterial cotton. Cellulose 2021, 28, 4469–4483. [Google Scholar] [CrossRef]
- Abdelhameed, R.M.; Rehan, M.; Emam, H.E. Figuration of Zr-based MOF@cotton fabric composite for potential kidney application. Carbohydr. Polym. 2018, 195, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Azizi, N.; Chevalier, Y.; Majdoub, M. Isosorbide-based microcapsules for cosmeto-textiles. Ind. Crops Prod. 2014, 52, 150–157. [Google Scholar] [CrossRef]
- Ghaheh, F.S.; Khoddami, A.; Alihosseini, F.; Jing, S.; Ribeiro, A.; Cavaco-Paulo, A.; Silva, C. Antioxidant cosmetotextiles: Cotton coating with nanoparticles containing vitamin E. Process Biochem. 2017, 59, 46–51. [Google Scholar] [CrossRef]
- Ma, J.; Fan, J.; Xia, Y.; Kou, X.; Ke, Q.; Zhao, Y. Preparation of aromatic β-cyclodextrin nano/microcapsules and corresponding aromatic textiles: A review. Carbohydr. Polym. 2023, 308, 120661. [Google Scholar] [CrossRef] [PubMed]
- Boh Podgornik, B.; Šandrić, S.; Kert, M. Microencapsulation for Functional Textile Coatings with Emphasis on Biodegradability—A Systematic Review. Coatings 2021, 11, 1371. [Google Scholar] [CrossRef]
- Pérez, E.; Martin, L.; Rubio, C.; Urieta, J.; Piera, E.; Caballero, M.; Téllez, C.; Coronas, J.n. Encapsulation of α-Tocopheryl Acetate into Zeolite Y for Textile Application. Ind. Eng. Chem. Res. 2010, 49, 8495–8500. [Google Scholar] [CrossRef]
- Zornoza, B.; Rubio, C.; Piera, E.; Caballero, M.A.; Julve, D.; Pérez, J.; Téllez, C.; Coronas, J. Caffeine Encapsulation in Metal Organic Framework MIL-53(Al) at Pilot Plant Scale for Preparation of Polyamide Textile Fibers with Cosmetic Properties. ACS Appl. Mater. Interfaces 2022, 14, 22476–22488. [Google Scholar] [CrossRef]
- Liu, X.; Demir, N.K.; Wu, Z.; Li, K. Highly Water-Stable Zirconium Metal–Organic Framework UiO-66 Membranes Supported on Alumina Hollow Fibers for Desalination. J. Am. Chem. Soc. 2015, 137, 6999–7002. [Google Scholar] [CrossRef]
- Ma, D.; Han, G.; Gao, Z.F.; Chen, S.B. Continuous UiO-66-Type Metal–Organic Framework Thin Film on Polymeric Support for Organic Solvent Nanofiltration. ACS Appl. Mater. Interfaces 2019, 11, 45290–45300. [Google Scholar] [CrossRef]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Cirujano, F.G.; Llabrés I Xamena, F.X. Tuning the Catalytic Properties of UiO-66 Metal–Organic Frameworks: From Lewis to Defect-Induced Brønsted Acidity. J. Phys. Chem. Lett. 2020, 11, 4879–4890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Li, C.; Hu, T.; Fan, L. Nanoporous {Co3}-Organic framework for efficiently seperating gases and catalyzing cycloaddition of epoxides with CO2 and Knoevenagel condensation. J. Colloid Interface Sci. 2024, 656, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Pourmadadi, M.; Eshaghi, M.M.; Ostovar, S.; Shamsabadipour, A.; Safakhah, S.; Mousavi, M.S.; Rahdar, A.; Pandey, S. UiO-66 metal-organic framework nanoparticles as gifted MOFs to the biomedical application: A comprehensive review. J. Drug Delivery Sci. Technol. 2022, 76, 103758. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, X.; Noh, H.; Ayoub, G.; Peterson, G.W.; Buru, C.T.; Islamoglu, T.; Farha, O.K. Scalable, room temperature, and water-based synthesis of functionalized zirconium-based metal–organic frameworks for toxic chemical removal. CrystEngComm 2019, 21, 2409–2415. [Google Scholar] [CrossRef]
- Kandiah, M.; Nilsen, M.H.; Usseglio, S.; Jakobsen, S.; Olsbye, U.; Tilset, M.; Larabi, C.; Quadrelli, E.A.; Bonino, F.; Lillerud, K.P. Synthesis and Stability of Tagged UiO-66 Zr-MOFs. Chem. Mater. 2010, 22, 6632–6640. [Google Scholar] [CrossRef]
- Julien, P.A.; Mottillo, C.; Friščić, T. Metal–organic frameworks meet scalable and sustainable synthesis. Green Chem. 2017, 19, 2729–2747. [Google Scholar] [CrossRef]
- Hu, Z.; Peng, Y.; Kang, Z.; Qian, Y.; Zhao, D. A Modulated Hydrothermal (MHT) Approach for the Facile Synthesis of UiO-66-Type MOFs. Inorg. Chem. 2015, 54, 4862–4868. [Google Scholar] [CrossRef]
- Pakamorė, I.; Rousseau, J.; Rousseau, C.; Monflier, E.; Szilágyi, P.Á. An ambient-temperature aqueous synthesis of zirconium-based metal–organic frameworks. Green Chem. 2018, 20, 5292–5298. [Google Scholar] [CrossRef]
- Užarević, K.; Wang, T.C.; Moon, S.-Y.; Fidelli, A.M.; Hupp, J.T.; Farha, O.K.; Friščić, T. Mechanochemical and solvent-free assembly of zirconium-based metal–organic frameworks. Chem. Commun. 2016, 52, 2133–2136. [Google Scholar] [CrossRef]
- D’Amato, R.; Bondi, R.; Moghdad, I.; Marmottini, F.; Mcpherson, M.J.; Naïli, H.; Taddei, M.; Costantino, F. “Shake ‘n Bake” Route to Functionalized Zr-UiO-66 Metal–Organic Frameworks. Inorg. Chem 2021, 60, 14294–14301. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Lo, W.-S.; Kuo, Y.-W.; Chen, W.-J.; Lin, C.-H.; Shieh, F.-K. Green and rapid synthesis of zirconium metal–organic frameworks via mechanochemistry: UiO-66 analog nanocrystals obtained in one hundred seconds. Chem. Commun. 2017, 53, 5818–5821. [Google Scholar] [CrossRef]
- Cunha, D.; Gaudin, C.; Colinet, I.; Horcajada, P.; Maurin, G.; Serre, C. Rationalization of the entrapping of bioactive molecules into a series of functionalized porous zirconium terephthalate MOFs. J. Mater. Chem. B 2013, 1, 1101–1108. [Google Scholar] [CrossRef]
- Cunha, D.; Ben Yahia, M.; Hall, S.; Miller, S.R.; Chevreau, H.; Elkaïm, E.; Maurin, G.; Horcajada, P.; Serre, C. Rationale of Drug Encapsulation and Release from Biocompatible Porous Metal–Organic Frameworks. Chem. Mater. 2013, 25, 2767–2776. [Google Scholar] [CrossRef]
- Sarker, M.; Jhung, S.H. Zr-MOF with free carboxylic acid for storage and controlled release of caffeine. J. Mol. Liq. 2019, 296, 112060. [Google Scholar] [CrossRef]
- Liédana, N.; Galve, A.; Rubio, C.; Téllez, C.; Coronas, J. CAF@ZIF-8: One-Step Encapsulation of Caffeine in MOF. ACS Appl. Mater. Interfaces 2012, 4, 5016–5021. [Google Scholar] [CrossRef] [PubMed]
- Liédana, N.; Lozano, P.; Galve, A.; Téllez, C.; Coronas, J. The template role of caffeine in its one-step encapsulation in MOF NH2-MIL-88B(Fe). J. Mater. Chem. B 2014, 2, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo-Olivan, R.; Paseta, L.; Potier, G.; López-Ram-de-Viu, P.; Coronas, J. Solvent-Free Encapsulation at High Pressure with Carboxylate-Based MOFs. Eur. J. Inorg. Chem. 2019, 2019, 29–36. [Google Scholar] [CrossRef]
- Wei, T.-H.; Wu, S.-H.; Huang, Y.-D.; Lo, W.-S.; Williams, B.P.; Chen, S.-Y.; Yang, H.-C.; Hsu, Y.-S.; Lin, Z.-Y.; Chen, X.-H.; et al. Rapid mechanochemical encapsulation of biocatalysts into robust metal–organic frameworks. Nat. Commun. 2019, 10, 5002. [Google Scholar] [CrossRef]
- Labay, C.; Canal, J.M.; Navarro, A.; Canal, C. Corona plasma modification of polyamide 66 for the design of textile delivery systems for cosmetic therapy. Appl. Surf. Sci. 2014, 316, 251–258. [Google Scholar] [CrossRef]
- Labay, C.; Canal, J.M.; Canal, C. Relevance of Surface Modification of Polyamide 6.6 Fibers by Air Plasma Treatment on the Release of Caffeine. Plasma Processes Polym. 2012, 9, 165–173. [Google Scholar] [CrossRef]
- Sta, M.; Tada, D.B.; Medeiros, S.F.; Santos, A.M.; Demarquette, N.R. Hydrophilic drug release from electrospun membranes made out of thermo and pH-sensitive polymers. J. Drug Delivery Sci. Technol. 2022, 71, 103284. [Google Scholar] [CrossRef]
- Li, X.; Kanjwal, M.A.; Lin, L.; Chronakis, I.S. Electrospun polyvinyl-alcohol nanofibers as oral fast-dissolving delivery system of caffeine and riboflavin. Colloids Surf. B 2013, 103, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Illangakoon, U.E.; Gill, H.; Shearman, G.C.; Parhizkar, M.; Mahalingam, S.; Chatterton, N.P.; Williams, G.R. Fast dissolving paracetamol/caffeine nanofibers prepared by electrospinning. Int. J. Pharm. 2014, 477, 369–379. [Google Scholar] [CrossRef]
- Li, S.; Girod-Holland, S.; Vert, M. Hydrolytic degradation of poly(dl-lactic acid) in the presence of caffeine base. J. Control. Release 1996, 40, 41–53. [Google Scholar] [CrossRef]
- Tipduangta, P.; Watcharathirawongs, W.; Waritdecha, P.; Sirithunyalug, B.; Leelapornpisid, P.; Chaiyana, W.; Goh, C.F. Electrospun cellulose acetate/polyvinylpyrrolidone fiber mats as potential cosmetic under-eye masks for caffeine delivery. J. Drug Delivery Sci. Technol. 2023, 86, 104732. [Google Scholar] [CrossRef]
- Rezvani Ghomi, E.R.; Khosravi, F.; Saedi Ardahaei, A.S.; Dai, Y.; Neisiany, R.E.; Foroughi, F.; Wu, M.; Das, O.; Ramakrishna, S. The Life Cycle Assessment for Polylactic Acid (PLA) to Make It a Low-Carbon Material. Polymers 2021, 13, 1854. [Google Scholar] [CrossRef]
- Arias, M.J.L.; López, A.; Vilaseca, M.; Vallès, B.; Prieto, R.; Simó, M.; Valle, J.A.B.; Valle, R.D.C.S.C.; Bezerra, F.M.; Bellalta, J.P. Influence of Chitosan Characteristics in the Microencapsulation of Essential Oils. J. Biomed. Sci. Eng. 2021, 14, 119–129. [Google Scholar] [CrossRef]
- Zou, C.; Vagin, S.; Kronast, A.; Rieger, B. Template mediated and solvent-free route to a variety of UiO-66 metal–organic frameworks. RSC Adv. 2016, 6, 102968–102971. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, M.; Han, R. Adsorption of phosphate on UiO-66-NH2 prepared by a green synthesis method. J. Environ. Chem. Eng. 2021, 9, 106672. [Google Scholar] [CrossRef]
- UNE-EN ISO 1133; Plásticos. Determinación del Índice de Fluidez de Materiales Termoplásticos en Masa (IFM) y en Volumen (IFV). AENOR: Madrid, Spain, 2002.
- Decker, G.E.; Stillman, Z.; Attia, L.; Fromen, C.A.; Bloch, E.D. Controlling Size, Defectiveness, and Fluorescence in Nanoparticle UiO-66 through Water and Ligand Modulation. Chem. Mater. 2019, 31, 4831–4839. [Google Scholar] [CrossRef]
- Liao, X.; Wang, X.; Wang, F.; Yao, Y.; Lu, S. Ligand Modified Metal Organic Framework UiO-66: A Highly Efficient and Stable Catalyst for Oxidative Desulfurization. J. Inorg. Organomet. Polym. Mater. 2021, 31, 756–762. [Google Scholar] [CrossRef]
- Jiang, X.; Li, S.; He, S.; Bai, Y.; Shao, L. Interface manipulation of CO2–philic composite membranes containing designed UiO-66 derivatives towards highly efficient CO2 capture. J. Mater. Chem. A 2018, 6, 15064–15073. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; Mccabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Øien, S.; Wragg, D.; Reinsch, H.; Svelle, S.; Bordiga, S.; Lamberti, C.; Lillerud, K.P. Detailed Structure Analysis of Atomic Positions and Defects in Zirconium Metal–Organic Frameworks. Cryst. Growth Des. 2014, 14, 5370–5372. [Google Scholar] [CrossRef]
- Abid, H.R.; Tian, H.; Ang, H.-M.; Tade, M.O.; Buckley, C.E.; Wang, S. Nanosize Zr-metal organic framework (UiO-66) for hydrogen and carbon dioxide storage. Chem. Eng. J. 2012, 187, 415–420. [Google Scholar] [CrossRef]
- Rodrigues, M.A.; Ribeiro, J.d.S.; Costa, E.d.S.; Miranda, J.L.d.; Ferraz, H.C. Nanostructured membranes containing UiO-66 (Zr) and MIL-101 (Cr) for O2/N2 and CO2/N2 separation. Sep. Purif. Technol. 2018, 192, 491–500. [Google Scholar] [CrossRef]
- Wang, Y.L.; Zhang, S.; Zhao, Y.F.; Bedia, J.; Rodriguez, J.J.; Belver, C. UiO-66-based metal organic frameworks for the photodegradation of acetaminophen under simulated solar irradiation. J. Environ. Chem. Eng. 2021, 9, 106087. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, 1997–2018. Available online: https://imagej.net/ij/ (accessed on 18 February 2024).
- Dong, C.; Wei, F.; Li, J.; Lu, Q.; Han, X. Uniform octahedral ZrO2@C from carbonized UiO-66 for electrocatalytic nitrogen reduction. Mater. Today Energy 2021, 22, 100884. [Google Scholar] [CrossRef]
- Majid, M.F.; Mohd Zaid, H.F.; Abd Shukur, M.F.; Ahmad, A.; Jumbri, K. Physicochemical properties and density functional theory calculation of octahedral UiO-66 with Bis(Trifluoromethanesulfonyl)imide ionic liquids. Heliyon 2023, 9, e20743. [Google Scholar] [CrossRef]
- Enright, G.D.; Terskikh, V.V.; Brouwer, D.H.; Ripmeester, J.A. The Structure of Two Anhydrous Polymorphs of Caffeine from Single-Crystal Diffraction and Ultrahigh-Field Solid-State 13C NMR Spectroscopy. Cryst. Growth Des. 2007, 7, 1406–1410. [Google Scholar] [CrossRef]
- Devautour-Vinot, S.; Maurin, G.; Serre, C.; Horcajada, P.; Paula Da Cunha, D.; Guillerm, V.; De Souza Costa, E.; Taulelle, F.; Martineau, C. Structure and Dynamics of the Functionalized MOF Type UiO-66(Zr): NMR and Dielectric Relaxation Spectroscopies Coupled with DFT Calculations. Chem. Mater. 2012, 24, 2168–2177. [Google Scholar] [CrossRef]
- Vasanthan, N.; Salem, D.R. FTIR spectroscopic characterization of structural changes in polyamide-6 fibers during annealing and drawing. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 536–547. [Google Scholar] [CrossRef]
- Yebra-Rodríguez, A.; Alvarez-Lloret, P.; Rodríguez-Navarro, A.B.; Martín-Ramos, J.D.; Cardell, C. Thermo-XRD and differential scanning calorimetry to trace epitaxial crystallization in PA6/montmorillonite nanocomposites. Mater. Lett. 2009, 63, 1159–1161. [Google Scholar] [CrossRef]
- Farias-Aguilar, J.C.; Ramírez-Moreno, M.J.; Téllez-Jurado, L.; Balmori-Ramírez, H. Low pressure and low temperature synthesis of polyamide-6 (PA6) using Na0 as catalyst. Mater. Lett. 2014, 136, 388–392. [Google Scholar] [CrossRef]
- Riba, J.R.; Cailloux, J.; Cantero, R.; Canals, T.; Maspoch, M.L. Multivariable methods applied to FTIR: A powerful technique to highlight architectural changes in poly(lactic acid). Polym. Test. 2018, 65, 264–269. [Google Scholar] [CrossRef]
- Vanderhart, D.L.; Asano, A.; Gilman, J.W. Solid-State NMR Investigation of Paramagnetic Nylon-6 Clay Nanocomposites. 1. Crystallinity, Morphology, and the Direct Influence of Fe3+ on Nuclear Spins. Chem. Mater. 2001, 13, 3781–3795. [Google Scholar] [CrossRef]
- Devautour-Vinot, S.; Martineau, C.; Diaby, S.; Ben-Yahia, M.; Miller, S.; Serre, C.; Horcajada, P.; Cunha, D.; Taulelle, F.; Maurin, G. Caffeine Confinement into a Series of Functionalized Porous Zirconium MOFs: A Joint Experimental/Modeling Exploration. J. Phys. Chem. C 2013, 117, 11694–11704. [Google Scholar] [CrossRef]
- Proikakis, C.S.; Tarantili, P.A.; Andreopoulos, A.G. The role of polymer/drug interactions on the sustained release from poly(dl-lactic acid) tablets. Eur. Polym. J. 2006, 42, 3269–3276. [Google Scholar] [CrossRef]
- Sitkowski, J.; Stefaniak, L.; Nicol, L.; Martin, M.L.; Martin, G.; Webb, G.A. Complete assignments of the 1H, 13C and 15N NMR spectra of caffeine. Spectrochim. Acta Part A 1995, 51, 839–841. [Google Scholar] [CrossRef]
- Zhou, J.; Guo, M.; Wu, D.; Shen, M.; Liu, D.; Ding, T. Synthesis of UiO-66 loaded-caffeic acid and study of its antibacterial mechanism. Food Chem. 2023, 402, 134248. [Google Scholar] [CrossRef]














| BET Surface Area (m2/g) | Total Pore Volume 1 (cm3/g) | Micropore Volume 2 (cm3/g) | |
|---|---|---|---|
| UiO-66c | 1298 ± 24 | 0.626 | 0.544 |
| UiO-66s | 771 ± 12 | 0.600 | 0.271 |
| UiO-66-NH2 | 650 ± 11 | 0.380 | 0.240 |
| CAF@UiO-66c | 216 ± 3 | 0.130 | 0.078 |
| CAF@UiO-66s | 396 ± 6 | 0.340 | 0.129 |
| CAF@UiO-66-NH2 | 150 ± 2 | 0.120 | 0.044 |
| Polymer | Capsule | wt% Capsules (Experimental) 1 |
|---|---|---|
| PA6 | CAF@UiO-66c | 9.4 ± 5.5 |
| CAF@UiO-66s | 9.0 ± 6.7 | |
| CAF@UiO-66-NH2 | 11.4 ± 8.0 | |
| PLA | CAF@UiO-66c | 5.7 ± 1.5 |
| CAF@UiO-66s | 11 ± 6.0 | |
| CAF@UiO-66-NH2 | 8.9 ± 0.4 |
| Polymer | Capsule | MVR (cm3/10 min) |
|---|---|---|
| PA6 | Pure | 78.78 ± 4.50 |
| CAF@UiO-66c | 73.42 ± 6.37 | |
| CAF@UiO-66s | 60.32 ± 14.17 | |
| CAF@UiO-66-NH2 | 63.88 ± 6.91 | |
| Caffeine | 77.01 ± 4.65 | |
| PLA | Pure | 5.75 ± 0.45 |
| CAF@UiO-66c | 2.40 ± 0.88 | |
| CAF@UiO-66s | 2.96 ± 1.45 | |
| CAF@UiO-66-NH2 | 1.25 ± 0.36 | |
| Caffeine | 3.66 ± 0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pina-Vidal, C.; Berned-Samatán, V.; Piera, E.; Caballero, M.Á.; Téllez, C. Mechanochemical Encapsulation of Caffeine in UiO-66 and UiO-66-NH2 to Obtain Polymeric Composites by Extrusion with Recycled Polyamide 6 or Polylactic Acid Biopolymer. Polymers 2024, 16, 637. https://doi.org/10.3390/polym16050637
Pina-Vidal C, Berned-Samatán V, Piera E, Caballero MÁ, Téllez C. Mechanochemical Encapsulation of Caffeine in UiO-66 and UiO-66-NH2 to Obtain Polymeric Composites by Extrusion with Recycled Polyamide 6 or Polylactic Acid Biopolymer. Polymers. 2024; 16(5):637. https://doi.org/10.3390/polym16050637
Chicago/Turabian StylePina-Vidal, Cristina, Víctor Berned-Samatán, Elena Piera, Miguel Ángel Caballero, and Carlos Téllez. 2024. "Mechanochemical Encapsulation of Caffeine in UiO-66 and UiO-66-NH2 to Obtain Polymeric Composites by Extrusion with Recycled Polyamide 6 or Polylactic Acid Biopolymer" Polymers 16, no. 5: 637. https://doi.org/10.3390/polym16050637