
Unveiling the Nanoconfinement Effect on Crystallization of Semicrystalline Polymers Using Coarse-Grained Molecular Dynamics Simulations

Abstract
:1. Introduction
2. Materials and Methods
2.1. CG Model of PVA
2.2. Nanoconfinement Representation
2.3. Simulation Procedures
2.4. Method to Characterize
2.5. Nematic Order Method to Characterize
3. Results and Discussion
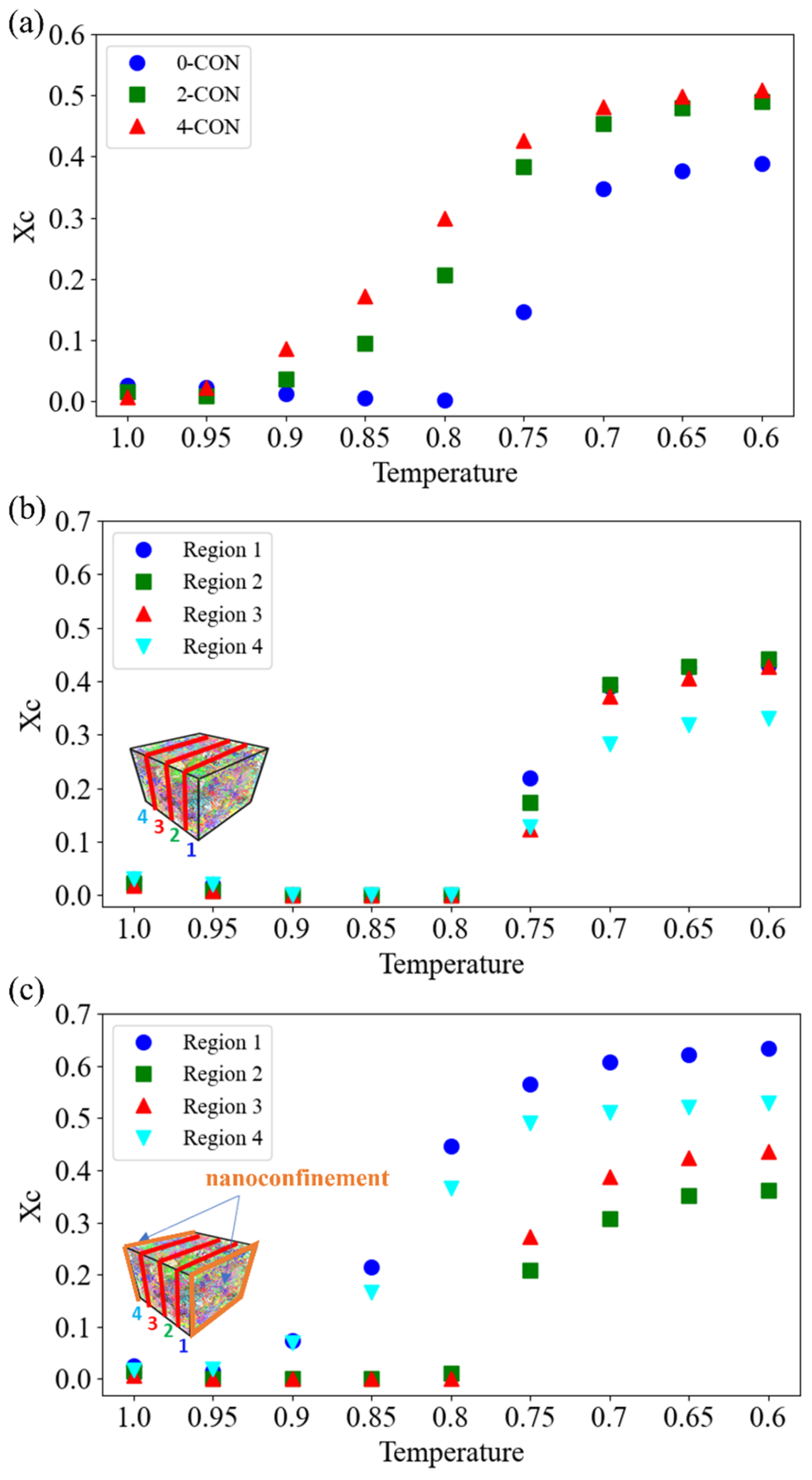
3.1. Effect of Nanoconfinement on and Crystallization Kinetics
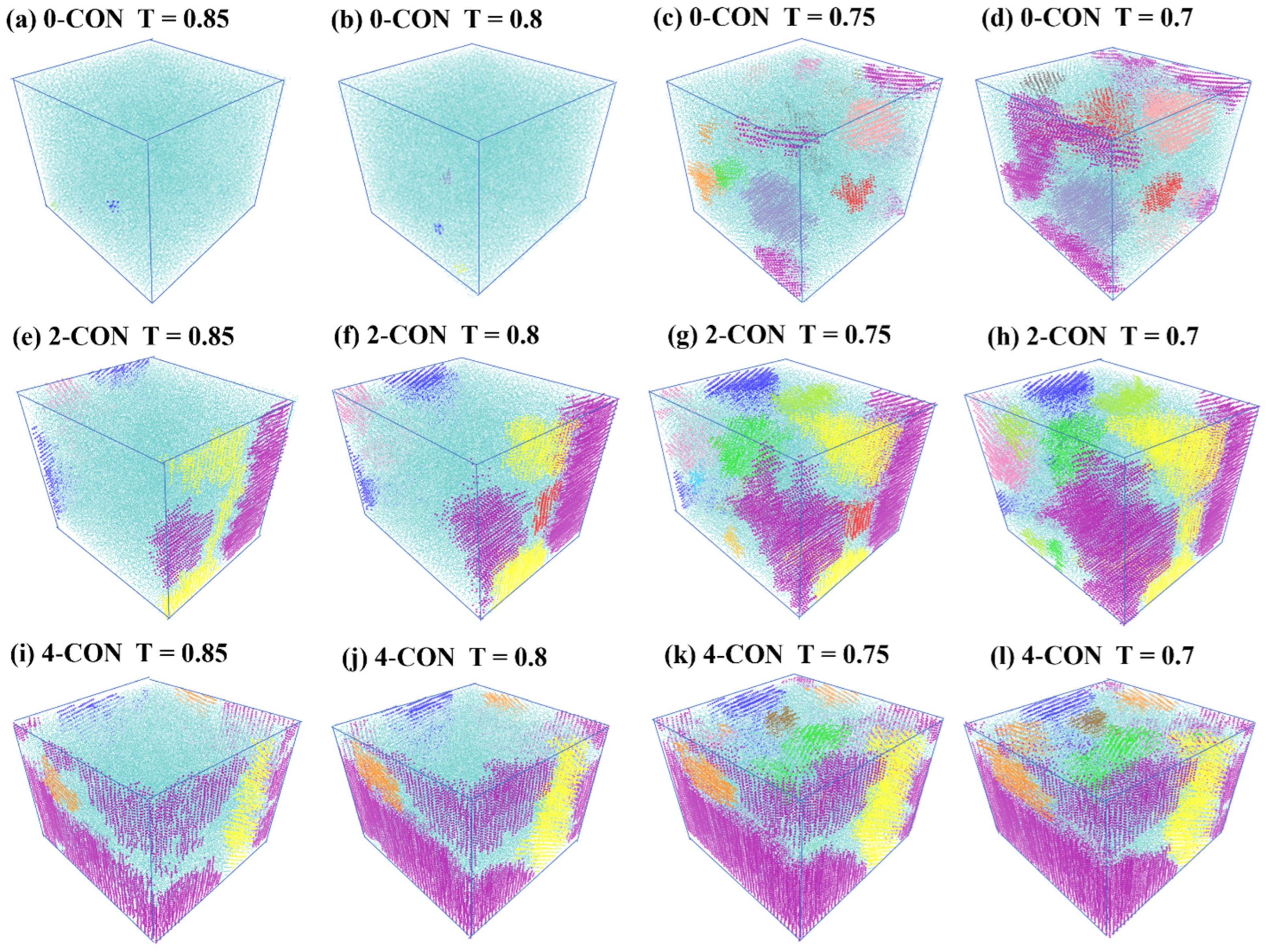
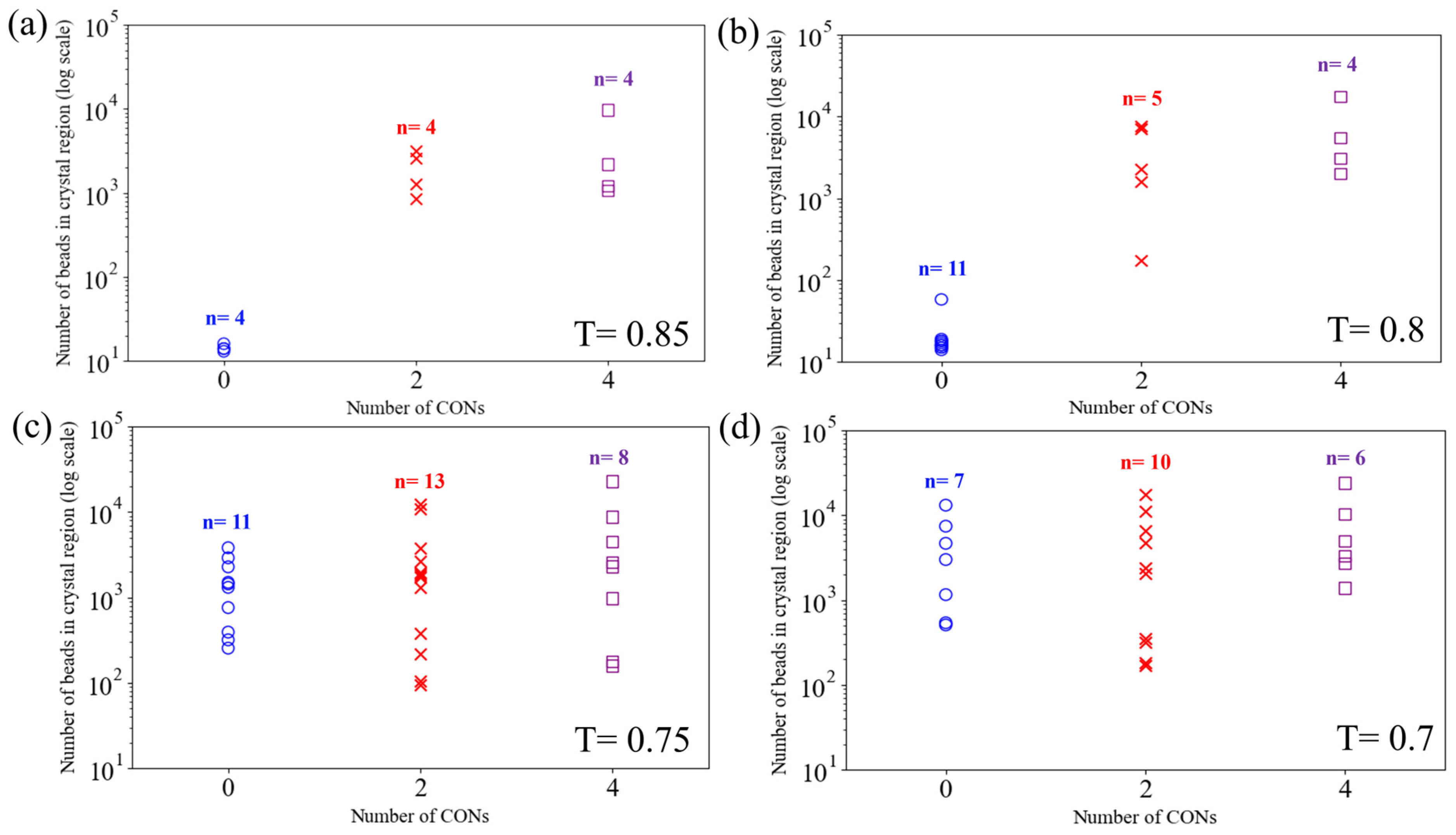
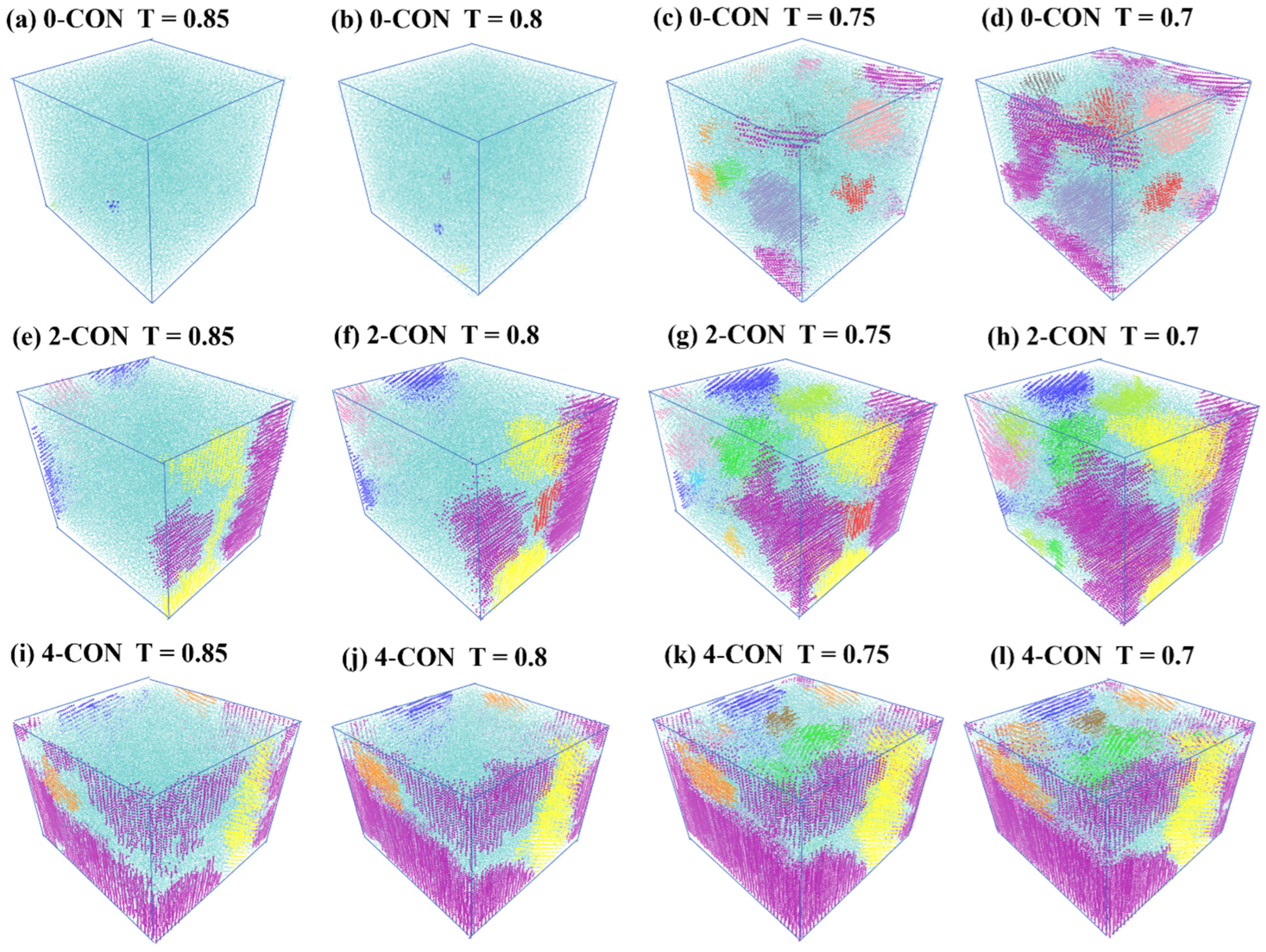
3.2. Effect of Nanoconfinement on Nucleation and Growth of Crystallites
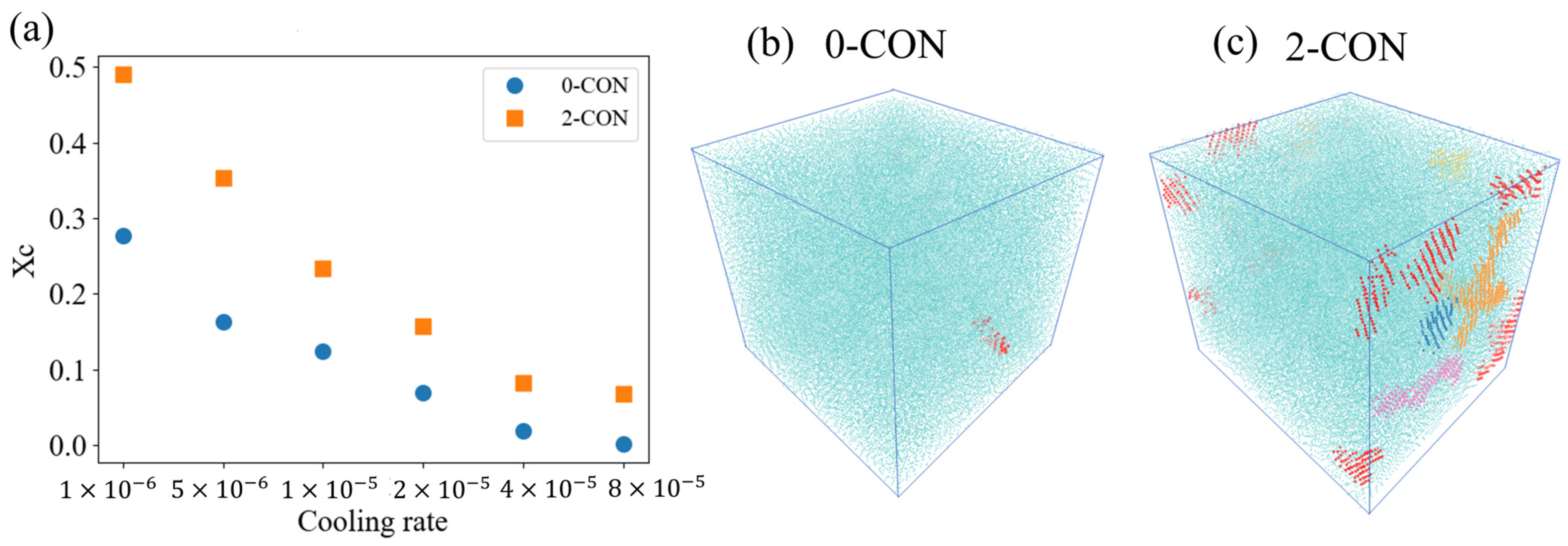
3.3. Effect of Nanoconfinement on Crystallization under Higher Cooling Rate
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, Use, and fate of all plastic ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Q.; Zhao, J.; Li, L. Molecular and thermodynamics descriptions of flow-induced crystallization in semi-crystalline polymers. J. Appl. Phys. 2020, 127, 241101. [Google Scholar] [CrossRef]
- Horn, T.D.; Heidrich, D.; Wulf, H.; Gehde, M.; Ihlemann, J. Multiscale Simulation of Semi-Crystalline Polymers to Predict Mechanical Properties. Polymers 2021, 13, 3233. [Google Scholar] [CrossRef] [PubMed]
- Nile, R.; Feng, S.; Cabello, M.; Wang, D.; Yang, S.; Jin, K. Crystallization Behavior of Semicrystalline Polymers Characterized by an In Situ Fluorescence Technique and Its Sensing Mechanism. Macromol. Chem. Phys. 2023, 224, 2300230. [Google Scholar] [CrossRef]
- Sangroniz, L.; Wang, B.; Su, Y.; Liu, G.; Cavallo, D.; Wang, D.; Muller, A.J. Fractionated crystallization in semicrystalline polymers. Prog. Polym. Sci. 2021, 115, 101376. [Google Scholar] [CrossRef]
- Hu, Y.; Liao, Y.; Zheng, Y.; Ikeda, K.; Okabe, R.; Wu, R.; Ozaki, R.; Xu, J.; Xu, Q. Influence of Cooling Rate on Crystallization Behavior of Semi-Crystalline Polypropylene: Experiments and Mathematical Modeling. Polymers 2022, 14, 3646. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Li, J.; Fan, X.; Wang, Y.; Xiao, Z.; Wang, H.; Liang, D.; Xie, Y. Improved mechanical and antibacterial properties of polyvinyl alcohol composite films using quaternized cellulose nanocrystals as nanofillers. Compos. Sci. Technol. 2023, 232, 109885. [Google Scholar] [CrossRef]
- Thomas, D.; Cebe, P. Self-nucleation and crystallization of polyvinyl alcohol. J. Therm. Anal. Calorim. 2016, 127, 885–894. [Google Scholar] [CrossRef]
- Conte, A.; Buonocore, G.G.; Bevilacqua, A.; Sinigaglia, M.; Nobile, M.A.D. Immobilization of lysozyme on polyvinylalcohol films for active packaging applications. J. Food Prot. 2006, 69, 866–870. [Google Scholar] [CrossRef]
- Kanatt, S.R.; Rao, M.S.; Chawla, S.P.; Sharma, A. Active chitosan–polyvinyl alcohol films with natural extracts. Food Hydrocoll. 2012, 29, 290–297. [Google Scholar] [CrossRef]
- Bonilla, J.; Fortunati, E.; Atares, L.; Chiralt, A.; Kenny, J.M. Physical, structural and antimicrobial properties of poly vinyl alcohol–chitosan biodegradable films. Food Hydrocoll. 2014, 35, 463–470. [Google Scholar] [CrossRef]
- George, G.; Manikandan, H.; Anup Kumar, T.M.; Joshy, S.; Sanju, A.C.; Anandhan, S. Effect of nanofillers on the crystalline and mechanical properties of EVACO polymer nanocomposites. Mater. Today Proc. 2021, 47, 5024–5028. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, L. Structure and property of polyvinyl alcohol/precipitated silica composite hydrogels for microorganism immobilization. Compos. Part B Eng. 2014, 56, 749–755. [Google Scholar] [CrossRef]
- Gong, X.; Tang, C.Y.; Pan, L.; Hao, Z.; Tsui, C.P. Characterization of poly(vinyl alcohol) (PVA)/ZnO nanocomposites prepared by a one-pot method. Compos. Part B Eng. 2014, 60, 144–149. [Google Scholar] [CrossRef]
- Liu, M.; Guo, B.; Du, M.; Jia, D. Drying induced aggregation of halloysite nanotubes in polyvinyl alcohol/halloysite nanotubes solution and its effect on properties of composite film. Appl. Phys. A 2007, 88, 391–395. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, S.; Peng, L.; Wang, Y.; Shang, S.; Miao, D.; Guo, R. AgNps-PVA–coated woven cotton fabric: Preparation, water repellency, shielding properties and antibacterial activity. J. Ind. Text. 2018, 48, 1545–1565. [Google Scholar] [CrossRef]
- Azeredo, H.M.C.d. Nanocomposites for food packaging applications. Food Res. Int. 2009, 42, 1240–1253. [Google Scholar] [CrossRef]
- Shen, Z.; Rajabi-Abhari, A.; Oh, K.; Yang, G.; Youn, H.J.; Lee, H.L. Improving the Barrier Properties of Packaging Paper by Polyvinyl Alcohol Based Polymer Coating-Effect of the Base Paper and Nanoclay. Polymers 2021, 13, 1334. [Google Scholar] [CrossRef]
- Musetti, A.; Paderni, K.; Fabbri, P.; Pulvirenti, A.; Al-Moghazy, M.; Fava, P. Poly(vinyl alcohol)-based film potentially suitable for antimicrobial packaging applications. J. Food Sci. 2014, 79, E577–E582. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Park, H.-M.; Ha, C.-S. Bio-nanocomposites for food packaging applications. Prog. Polym. Sci. 2013, 38, 1629–1652. [Google Scholar] [CrossRef]
- Xu, J.-Z.; Liang, Y.-Y.; Huang, H.-D.; Zhong, G.-J.; Lei, J.; Chen, C.; Li, Z.-M. Isothermal and nonisothermal crystallization of isotactic polypropylene/graphene oxide nanosheet nanocomposites. J. Polym. Res. 2012, 19, 9975. [Google Scholar] [CrossRef]
- Nagarkar, R.; Patel, J. Polyvinyl Alcohol: A Comprehensive Study. Acta Sci. Pharm. Sci. 2019, 3, 34–44. [Google Scholar]
- Bin-Dahman, O.A.; Shehzad, F.; Al-Harthi, M.A. Influence of graphene on the non-isothermal crystallization kinetics of poly(vinyl alcohol)/starch composite. J. Polym. Res. 2017, 25, 5. [Google Scholar] [CrossRef]
- Lee, K.J.; Lee, J.; Hong, J.Y.; Jang, J. Influence of Amorphous Polymer Nanoparticles on the Crystallization Behavior of Poly(vinyl alcohol) Nanocomposites. Macromol. Res. 2009, 17, 476–482. [Google Scholar] [CrossRef]
- Hassen, A.; El-Sayed, A.M.; Morsi, W.M.; El-Sayed, S. Influence of Cr2O3 nanoparticles on the physical properties of polyvinyl alcohol. J. Appl. Phys. 2012, 112, 093525. [Google Scholar] [CrossRef]
- Lee, J.; Lee, K.J.; Jang, J. Effect of silica nanofillers on isothermal crystallization of poly(vinyl alcohol): In-situ ATR-FTIR study. Polym. Test. 2008, 27, 360–367. [Google Scholar] [CrossRef]
- Bosq, N.; Aht-Ong, D. Nonisothermal crystallization behavior of poly(butylene succinate)/NaY zeolite nanocomposites. Macromol. Res. 2017, 26, 13–21. [Google Scholar] [CrossRef]
- Papadopoulos, L.; Klonos, P.A.; Terzopoulou, Z.; Psochia, E.; Sanusi, O.M.; Hocine, N.A.; Benelfellah, A.; Giliopoulos, D.; Triantafyllidis, K.; Kyritsis, A.; et al. Comparative study of crystallization, semicrystalline morphology, and molecular mobility in nanocomposites based on polylactide and various inclusions at low filler loadings. Polymer 2021, 217, 123457. [Google Scholar] [CrossRef]
- Shakoor, A.; Thomas, N.L. Talc as a nucleating agent and reinforcing filler in poly(lactic acid) composites. Polym. Eng. Sci. 2014, 54, 64–70. [Google Scholar] [CrossRef]
- Reinsch, V.E.; Rebenfeld, L. Crystallization processes in poly(ethylene terephthalate) as modified by polymer additives and fiber reinforcement. J. Appl. Polym. Sci. 1994, 52, 649–662. [Google Scholar] [CrossRef]
- Jin, S.; McKenna, G.B. Effect of Nanoconfinement on Polymer Chain Dynamics. Macromolecules 2020, 53, 10212–10216. [Google Scholar] [CrossRef]
- Krutyeva, M.; Wischnewski, A.; Monkenbusch, M.; Willner, L.; Maiz, J.; Mijangos, C.; Arbe, A.; Colmenero, J.; Radulescu, A.; Holderer, O.; et al. Effect of nanoconfinement on polymer dynamics: Surface layers and interphases. Phys. Rev. Lett. 2013, 110, 108303. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, J.Y.; Stafford, C.M. Effect of Confinement on Stiffness and Fracture of Thin Amorphous Polymer Films. ACS Macro Lett. 2012, 1, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Alcoutlabi, M.; McKenna, G.B. Effects of confinement on material behaviour at the nanometre size scale. J. Phys. Condens. Matter 2005, 17, R461–R524. [Google Scholar] [CrossRef]
- Shavit, A.; Riggleman, R.A. Influence of Backbone Rigidity on Nanoscale Confinement Effects in Model Glass-Forming Polymers. Macromolecules 2013, 46, 5044–5052. [Google Scholar] [CrossRef]
- Ming, Y.; Zhou, Z.; Zhang, S.; Wei, Y.; Hao, T.; Nie, Y. Molecular simulation of crystallization of polymers confined in cylindrical nanodomain. Polymer 2020, 206, 122818. [Google Scholar] [CrossRef]
- Paquet, E.; Viktor, H.L. Molecular dynamics, monte carlo simulations, and langevin dynamics: A computational review. Biomed. Res. Int. 2015, 2015, 183918. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Xia, W.; Keten, S. Interfacial stiffening of polymer thin films under nanoconfinement. Extrem. Mech. Lett. 2015, 4, 89–95. [Google Scholar] [CrossRef]
- Shao, C.; Keten, S. Stiffness Enhancement in Nacre-Inspired Nanocomposites due to Nanoconfinement. Sci. Rep. 2015, 5, 16452. [Google Scholar] [CrossRef]
- Qin, X.; Xia, W.; Sinko, R.; Keten, S. Tuning Glass Transition in Polymer Nanocomposites with Functionalized Cellulose Nanocrystals through Nanoconfinement. Nano Lett. 2015, 15, 6738–6744. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T. Computer modeling of polymer crystallization—Toward computer-assisted materials’ design. Polymer 2009, 50, 1975–1985. [Google Scholar] [CrossRef]
- Xiao, H.; Luo, C.; Yan, D.; Sommer, J. Molecular Dynamics Simulation of Crystallization Cyclic Polymer Melts as Compared to Their Linear Counterparts. Macromolecules 2017, 50, 9796–9806. [Google Scholar] [CrossRef]
- Jamornsuriya, S.; Vao-Soongnern, V. Molecular simulation of an initial stage of the ordered-structure formation of linear and ring polymers upon cooling from the melts. J. Mol. Liq. 2022, 363, 119833. [Google Scholar] [CrossRef]
- Jabbarzadeh, A. The Origins of Enhanced and Retarded Crystallization in Nanocomposite Polymers. Nanomaterials 2019, 9, 1472. [Google Scholar] [CrossRef]
- Han, Z.; Dong, Y.; Tan, Z.; Ma, J.; Yu, P. Study on the crystallization behavior of polyethylene grafted onto carbon nanotubes using molecular dynamics simulation. Mol. Simul. 2020, 46, 1390–1397. [Google Scholar] [CrossRef]
- Yang, Z.; Chiang, C.-C.; Meng, Z. Investigation of dynamic impact responses of layered polymer-graphene nanocomposite films using coarse-grained molecular dynamics simulations. Carbon 2023, 203, 202–210. [Google Scholar] [CrossRef]
- Caviness, C.; Chen, Y.; Yang, Z.; Wang, H.; Wu, Y.; Meng, Z. Improved Ballistic Impact Resistance of Nanofibrillar Cellulose Films with Discontinuous Fibrous Bouligand Architecture. J. Appl. Mech. 2023, 91, 021003. [Google Scholar] [CrossRef]
- Reith, D.; Meyer, H.; Muller-Plathe, F. Mapping Atomistic to coarse-grained polymer models using automatic simplex optimization to fit structural properties. Macromolecules 2001, 34, 2335–2345. [Google Scholar] [CrossRef]
- Meyer, H.; Müller-Plathe, F. Formation of Chain-Folded Structure in Supercooled Polymer Melts Examined by MD Simulations. Macromolecules 2002, 35, 1241–1252. [Google Scholar] [CrossRef]
- Luo, C.; Sommer, J.-U. Coding coarse grained polymer model for LAMMPS and its application to polymer crystallization. Comput. Phys. Commun. 2009, 180, 1382–1391. [Google Scholar] [CrossRef]
- Jabbari-Farouji, S. Static and dynamic scaling behavior of a polymer melt model with triple-well bending potential. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 1376–1392. [Google Scholar] [CrossRef]
- Jabbari-Farouji, S.; Rottler, J.; Lame, O.; Makke, A.; Perez, M.; Barrat, J.L. Plastic Deformation Mechanisms of Semicrystalline and Amorphous Polymers. ACS Macro Lett. 2015, 4, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Jabbari-Farouji, S.; Rottler, J.; Lame, O.; Makke, A.; Perez, M.; Barrat, J.L. Correlation of structure and mechanical response in solid-like polymers. J. Phys. Condens. Matter 2015, 27, 194131. [Google Scholar] [CrossRef] [PubMed]
- Jabbari-Farouji, S.; Lame, O.; Perez, M.; Rottler, J.; Barrat, J.L. Role of the Intercrystalline Tie Chains Network in the Mechanical Response of Semicrystalline Polymers. Phys. Rev. Lett. 2017, 118, 217802. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Müller-Plathe, F. Formation of chain-folded structures in supercooled polymer melts. J. Chem. Phys. 2001, 115, 7807–7810. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Emamy, H.; Betancourt, B.P.; Vargas-Lara, F.; Starr, F.W.; Douglas, J. The interfacial zone in thin polymer films and around nanoparticles in polymer nanocomposites. J. Chem. Phys. 2019, 151, 124705. [Google Scholar] [CrossRef]
- Starr, F.W.; Schrøder, T.B.; Glotzer, S.C. Molecular Dynamics Simulation of a Polymer Melt with a Nanoscopic Particle. Macromolecules 2002, 35, 4481–4492. [Google Scholar] [CrossRef]
- Emamy, H.; Kumar, S.K.; Starr, F.W. Diminishing Interfacial Effects with Decreasing Nanoparticle Size in Polymer-Nanoparticle Composites. Phys. Rev. Lett. 2018, 121, 207801. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Bessa, M.A.; Xia, W.; Liu, W.K.; Keten, S. Predicting the macroscopic fracture energy of Epoxy Resins from atomistic molecular simulations. Macromolecules 2016, 49, 9474–9483. [Google Scholar] [CrossRef]
- Yoshiharu, N.; Shigenori, K.; Masahisa, W.; Takeshi, O. Cellulose Microcrystal Film of High Uniaxial Orientation. Macromolecules 1997, 30, 6395–6397. [Google Scholar] [CrossRef]
- Yang, J.; Yao, X.; Meng, Z. Investigation of Molecular Mechanisms of Polyvinylidene Fluoride under the Effects of Temperature, Electric Poling, and Mechanical Stretching Using Molecular Dynamics Simulations. Polymer 2022, 245, 124691. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Rutledge, G.C. Molecular simulation of bundle-like crystal nucleation from n-eicosane melts. J. Chem. Phys. 2011, 135, 024903. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Moon, J.; Kim, B.; Cho, M. Multi-scale coarse-grained molecular dynamics simulation to investigate the thermo-mechanical behavior of shape-memory polyurethane copolymers. Polymer 2020, 213, 123228. [Google Scholar] [CrossRef]
- Stewart, I.W. The Static and Dynamic Continuum Theory of Liquid Crystals: A Mathematical Introduction; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Bornani, K.; Rahman, M.A.; Benicewicz, B.; Kumar, S.; Schadler, L. Using Nanofiller Assemblies to Control the Crystallization Kinetics of High-Density Polyethylene. Macromolecules 2021, 54, 5673–5682. [Google Scholar] [CrossRef]
- Xiang, A.; Lv, C.; Zhou, H. Changes in Crystallization Behaviors of Poly(Vinyl Alcohol) Induced by Water Content. J. Vinyl Addit. Technol. 2020, 26, 613–622. [Google Scholar] [CrossRef]
- Lu, W.; Lu, C.; Hu, J.; Wu, J.; Zhou, Q. Effects of the blending time on the properties and non-isothermal crystallization behavior of PA6/EVOH blends. Polym. Eng. Sci. 2021, 61, 1719–1731. [Google Scholar] [CrossRef]
- Qi, X.-d.; Yang, J.; Zhang, N.; Huang, T.; Zhou, Z.-w.; Kuhnert, I.; Potschke, P.; Wang, Y. Selective localization of carbon nanotubes and its effect on the structure and properties of polymer blends. Prog. Polym. Sci. 2021, 123, 101471. [Google Scholar] [CrossRef]
- Hammani, S.; Moulai-Mostefa, N.; Samyn, P.; Bechelany, M.; Dufresne, A.; Barhoum, A. Morphology, Rheology and Crystallization in Relation to the Viscosity Ratio of Polystyrene/Polypropylene Polymer Blends. Materials 2020, 13, 926. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Ferreira, L.A.S.; Manrich, S.; Pessan, L.A. Crystallization behavior of PBT/ABS polymer blends. J. Appl. Polym. Sci. 1999, 71, 423–430. [Google Scholar] [CrossRef]
- Singh, T.; Gangil, B.; Ranakoti, L.; Joshi, A. Effect of silica nanoparticles on physical, mechanical, and wear properties of natural fiber reinforced polymer composites. Polym. Compos. 2021, 42, 2396–2407. [Google Scholar] [CrossRef]
- Shen, R.; Xue, S.; Xu, Y.; Liu, Q.; Feng, Z.; Ren, H.; Zhai, H.; Kong, F. Research Progress and Development Demand of Nanocellulose Reinforced Polymer Composites. Polymers 2020, 12, 2113. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neighbor Domain Id | Unit Orientation Vector | Neighbor Domain Id | Unit Orientation Vector | Neighbor Domain Id | Unit Orientation Vector |
|---|---|---|---|---|---|
| 1 | 6 | 11 | |||
| 2 | 7 | 12 | |||
| 3 | 8 | 13 | |||
| 4 | 9 | 14 | |||
| 5 | 10 |
| Time Step | Cooling Rate |
|---|---|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Chen, Y.; Yang, Z.; Dai, L.; Choi, H.; Meng, Z. Unveiling the Nanoconfinement Effect on Crystallization of Semicrystalline Polymers Using Coarse-Grained Molecular Dynamics Simulations. Polymers 2024, 16, 1155. https://doi.org/10.3390/polym16081155
Yang J, Chen Y, Yang Z, Dai L, Choi H, Meng Z. Unveiling the Nanoconfinement Effect on Crystallization of Semicrystalline Polymers Using Coarse-Grained Molecular Dynamics Simulations. Polymers. 2024; 16(8):1155. https://doi.org/10.3390/polym16081155
Chicago/Turabian StyleYang, Ji, Yitong Chen, Zhangke Yang, Linjiale Dai, Hongseok Choi, and Zhaoxu Meng. 2024. "Unveiling the Nanoconfinement Effect on Crystallization of Semicrystalline Polymers Using Coarse-Grained Molecular Dynamics Simulations" Polymers 16, no. 8: 1155. https://doi.org/10.3390/polym16081155