Photo-Induced Micellization of Block Copolymers

1
Department of Environmental and Life Sciences, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku-cho, Toyohashi 441-8580, Japan
2
Department of Materials Science, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku-cho, Toyohashi 441-8580, Japan
*
Author to whom correspondence should be addressed.
Polymers 2010, 2(4), 623-648; https://doi.org/10.3390/polym2040623
Submission received: 30 September 2010
/
Revised: 16 November 2010
/
Accepted: 25 November 2010
/
Published: 26 November 2010
(This article belongs to the Special Issue Advanced Polymer Architectures)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
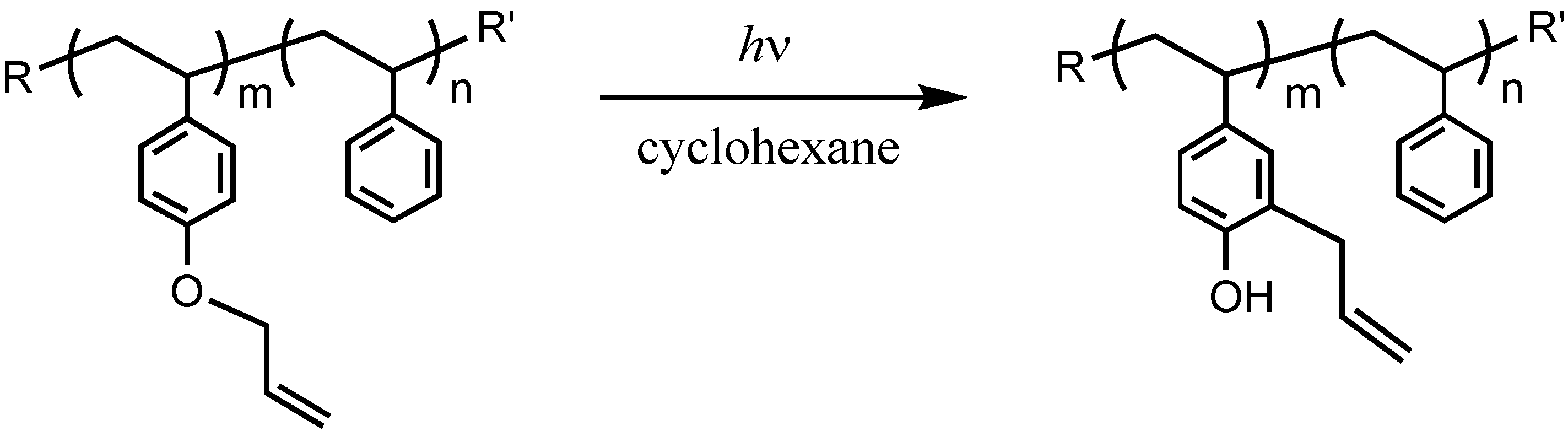{kind=link}
Abstract
:We found novel photo-induced micellizations through photolysis, photoelectron transfer, and photo-Claisen rearrangement. The photolysis-induced micellization was attained using poly(4-tert-butoxystyrene)-block-polystyrene diblock copolymer (PBSt-b-PSt). BSt-b-PSt showed no self-assembly in dichloromethane and existed as isolated copolymers. Dynamic light scattering demonstrated that the copolymer produced spherical micelles in this solvent due to irradiation with a high-pressure mercury lamp in the presence of photo-acid generators, such as bis(alkylphenyl)iodonium hexafluorophosphate, diphenyliodonium hexafluorophosphate, and triphenylsulfonium triflate. The 1H NMR analysis confirmed that PBSt-b-PSt was converted into poly(4-vinylphenol)-block-PSt by the irradiation, resulting in self-assembly into micelles. The irradiation in the presence of the photo-acid generator also induced the micellization of poly(4-pyridinemethoxymethylstyrene)-block-polystyrene diblock copolymer (PPySt-b-PSt). Micellization occurred by electron transfer from the pyridine to the photo-acid generator in their excited states and provided monodispersed spherical micelles with cores of PPySt blocks. Further, the photo-Claisen rearrangement caused the micellization of poly(4-allyloxystyrene)-block-polystyrene diblock copolymer (PASt-b-PSt). Micellization was promoted in cyclohexane at room temperature without a catalyst. During micellization, the elimination of the allyl groups competitively occurred along with the photorearrangement of the 4-allyloxystyrene units into the 3-allyl-4-hydroxystyrene units.
1. Introduction
Stimuli-responsive polymers have attracted considerable attention because these polymers have many industrial applications, such as sensors [1,2,3], drug carriers [4,5,6], artificial muscles [7,8], optical data storage [9], and electric devices with molecular switches [10,11]. The polymers change their spatial structure in response to stimuli, for example, aggregating [12], aligning [13], bending, [14], and spiraling [15,16]. Molecular self-assembly is also caused by the polymers responding to stimuli, resulting in the construction of high-dimensional structures of the polymers. Stimuli inducing self-assembly of the polymers include pH [17,18], temperature [19,20,21,22], and pressure [23,24,25,26]. Recently, it has been reported that chemical reactions inducing self‑assembly of the polymers; the oxidation [27], reduction [28,29,30], and disproportionation [31,32] of stable nitroxyl radicals supported on the side chains of a block copolymer, promote the micellization of the copolymer to provide micelles that function as oxidants and reductants.
Photochemical reaction is a green-sustainable and energy-saving process for manufactures and is an important trigger for the reversible molecular self-assembly. The polymers reversibly change their structure through cis-trans isomerization, dimerization, and conformational changes of photochromic compounds such as azobenzene [33,34,35], spiropyran [36,37], stilbene [38,39,40], cinnamate [39], and triphenylmethane leuco residues [41]. This reversible behavior is manipulated by UV wavelength of the compounds or sometimes temperature. While these reversible reactions function as the on-off switches, irreversible reactions fix the spatial structure changed by photo irradiation. The structure change by the irreversible photoreaction has been investigated on the photolysis of diazosulfonates [42,43,44], 1-iminopyridinium ylides [45], [4(4'-alkoxybenzoyl)phenylmethyl]phosphonic acids [46], and didecyl-2-methoxy-5-nitrophenyl phosphate [47]. The former three kinds of surfactants lose their surface-active ability by photolysis, resulting in destruction of the micelles and vesicles. On the other hand, didecyl-2-methoxy-5-nitrophenyl phosphate formed vesicles by the photolysis.
We focused on the self-assembly of polymers by the irreversible photoreaction with the aim of creating new photo-based materials for use in optical data storage and optical devices. We chose three kinds of the irreversible photoreactions of the photolysis, photoelectron transfer, and photorearrangement. The photolysis and photorearrangement of pendant groups attached to polymers are important methods often used in the field of photoresist [48,49], while the photoelectron transfer is envisaged to be a new technique, without mass transfer and volume shrinkage, for optical data storage. To attain the self-assembly induced by these photoirreversible reactions, three different block copolymers supporting photoreactive groups were prepared; poly(4-tert-butoxystyrene)-block-polystyrene diblock copolymer (PBSt-b-PSt) for the photolysis-induced micellization, poly(4-pyridinemethoxymethylstyrene)-block-polystyrene (PPySt-b-PSt) for the photoelectron transfer-induced micellization, and poly(4-allyloxystyrene)-block-polystyrene (PASt-b-PSt) for the photorearrangement-induced micellization. This review describes the novel micellizations promoting the irreversible reactions of these photoreactive groups attached to the block copolymers.
2. Results and Discussion
2.1. Photolysis-Induced Micellization [50,51]
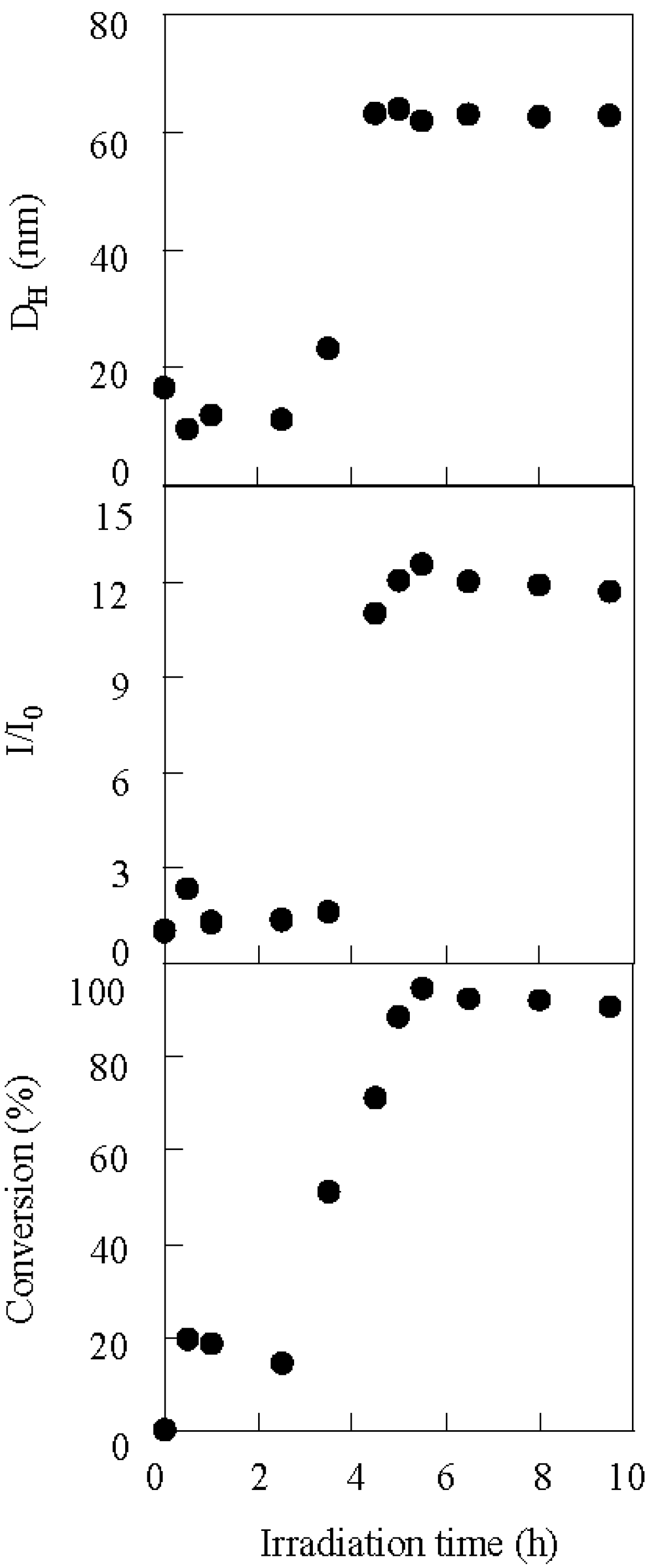
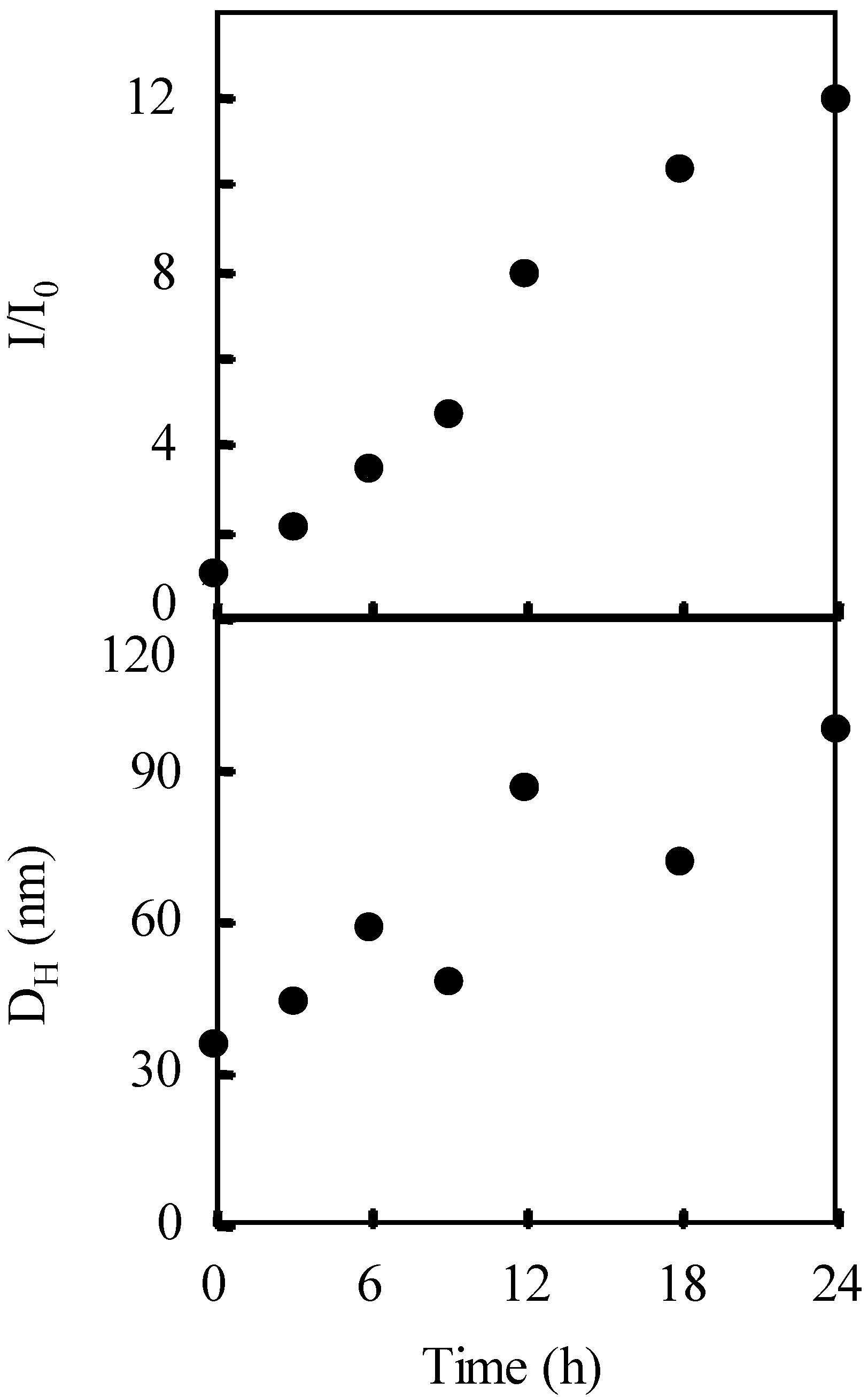
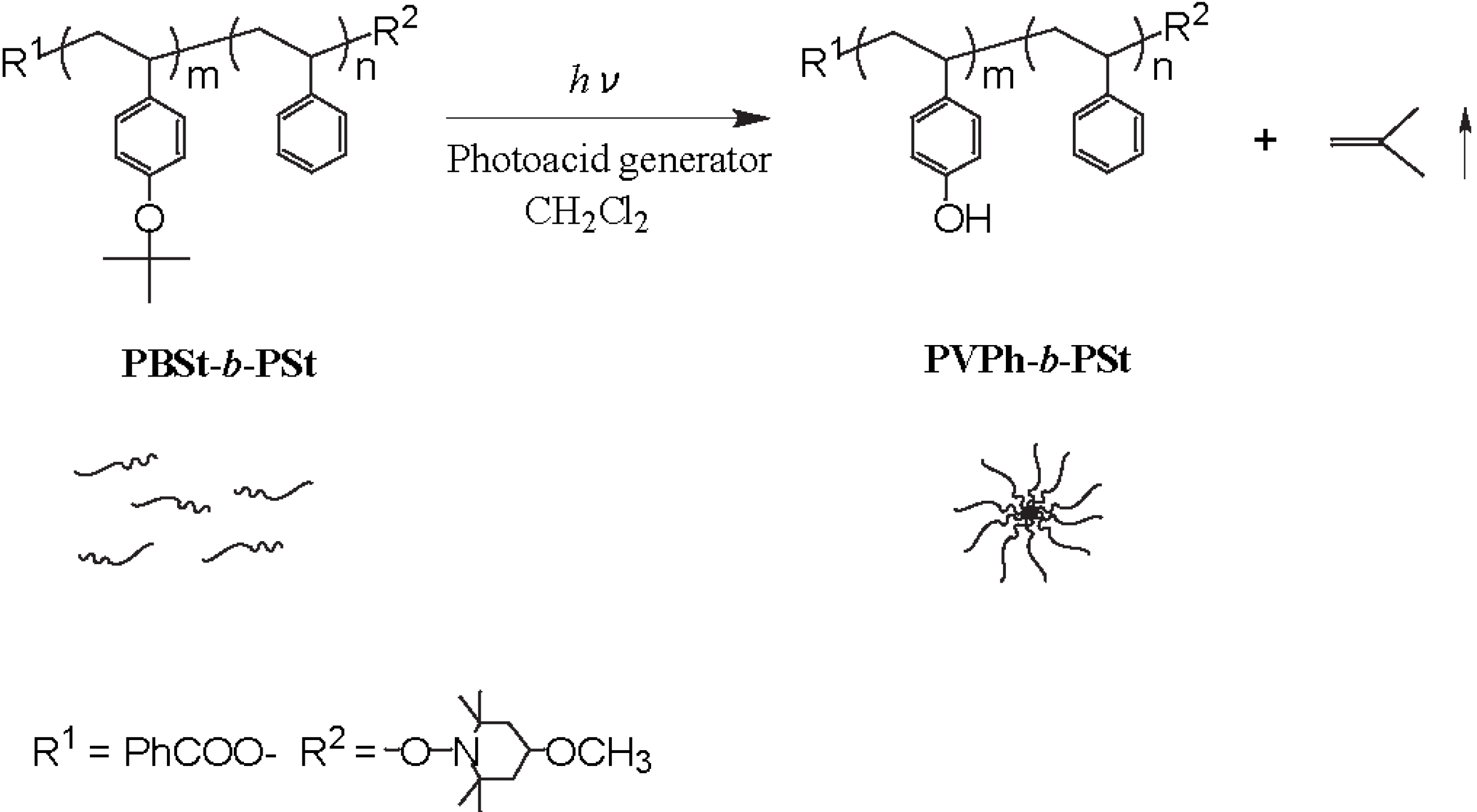
The photolysis-induced micellization was determined for a PBSt-b-PSt diblock copolymer (Scheme 1). The PBSt-b-PSt diblock copolymer shows no self-assembly in dichloromethane since the PBSt and PSt blocks are solvophilic to it. Light scattering studies demonstrated that the copolymer self-assembled into micelles in dichloromethane, by irradiation, in the presence of a photo-acid generator. Figure 1 shows the variation in the hydrodynamic diameter (DH) and the relative scattering intensity (I/I0) of the copolymer with a molecular weight of Mn(PBSt-b-PSt) = 15,000-b-97,000 during irradiation using bis(alkylphenyl)iodonium hexafluorophosphate (BAI) as a photo-acid generator. The molar ratio of BAI to the BSt unit was 0.38. The relative scattering intensity can be regarded as an apparent aggregation number of micelles when it is assumed that change of the functional groups by irradiation has no effect on the scattering intensity. This is because the copolymer concentration is immutable during micellization. The hydrodynamic diameter and scattering intensity showed a good correlation. They increased at 4.5 h and became constant after 5 h, indicating that micellization was then complete. The hydrodynamic diameter of the micelles averaged 63.0 nm, while that of the isolated copolymer, that is a unimer, was 16.6 nm based on the cumulant analysis. The observation of the jump and the constant state within a short time period suggests rapid micellization due to the strong aggregation force.
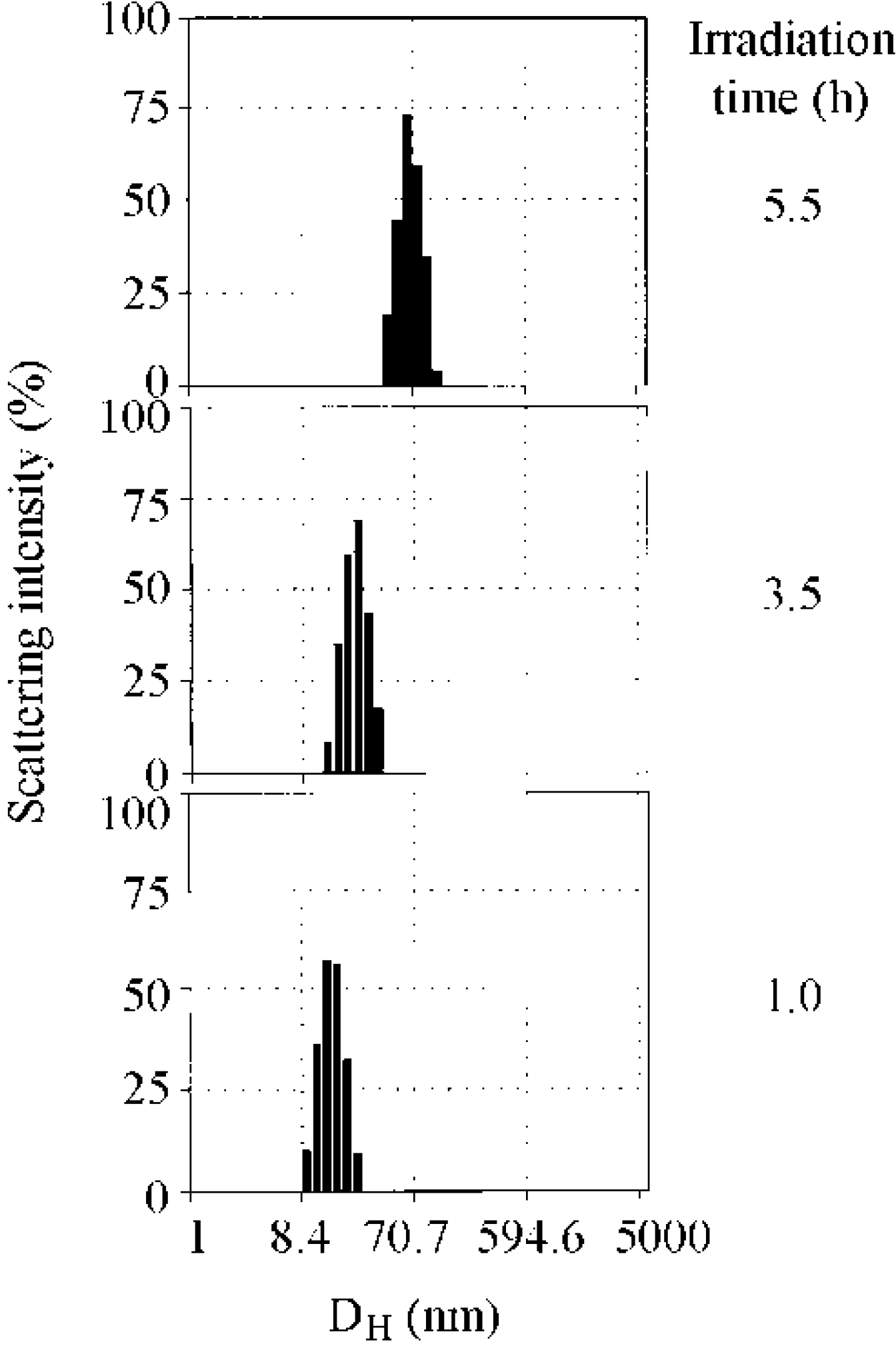
The variation in the scattering intensity distribution of the hydrodynamic diameter also supported the formation of micelles by rapid association. Figure 2 shows the scattering intensity distribution obtained by the Marquadt analysis [52]. The distribution was shifted to the higher side of the hydrodynamic diameter over time by irradiation. The slight shift in distribution at 3.5 h implies that aggregates with a lower aggregation number were formed during the first stage and those associated into micelles, rather than the unimers incorporating step by step into micelles.
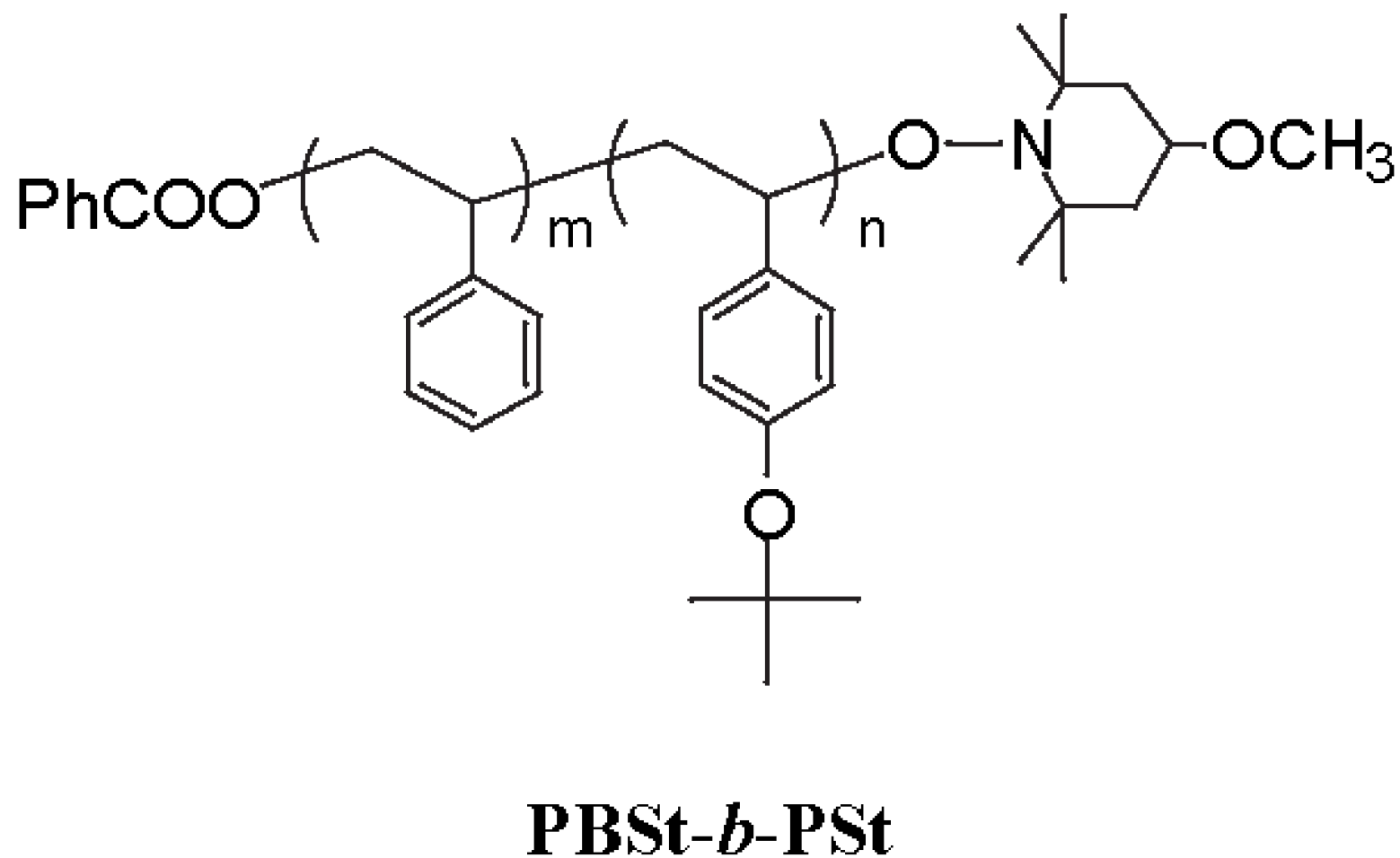
Scheme 1.
The PBSt-b-PSt diblock copolymer.

Figure 1.
The variation in the hydrodynamic diameter (DH), relative scattering intensity (I/I0), and conversion of the copolymer during irradiation using BAI. Mn(PBSt-b-PSt) = 15,000-b-97,000, [copolymer]0 = 3.30 g/L.
Figure 1.
The variation in the hydrodynamic diameter (DH), relative scattering intensity (I/I0), and conversion of the copolymer during irradiation using BAI. Mn(PBSt-b-PSt) = 15,000-b-97,000, [copolymer]0 = 3.30 g/L.

Figure 2.
Scattering intensity distributions of the hydrodynamic diameter of the copolymer. [copolymer]0 = 3.30 g/L.
Figure 2.
Scattering intensity distributions of the hydrodynamic diameter of the copolymer. [copolymer]0 = 3.30 g/L.

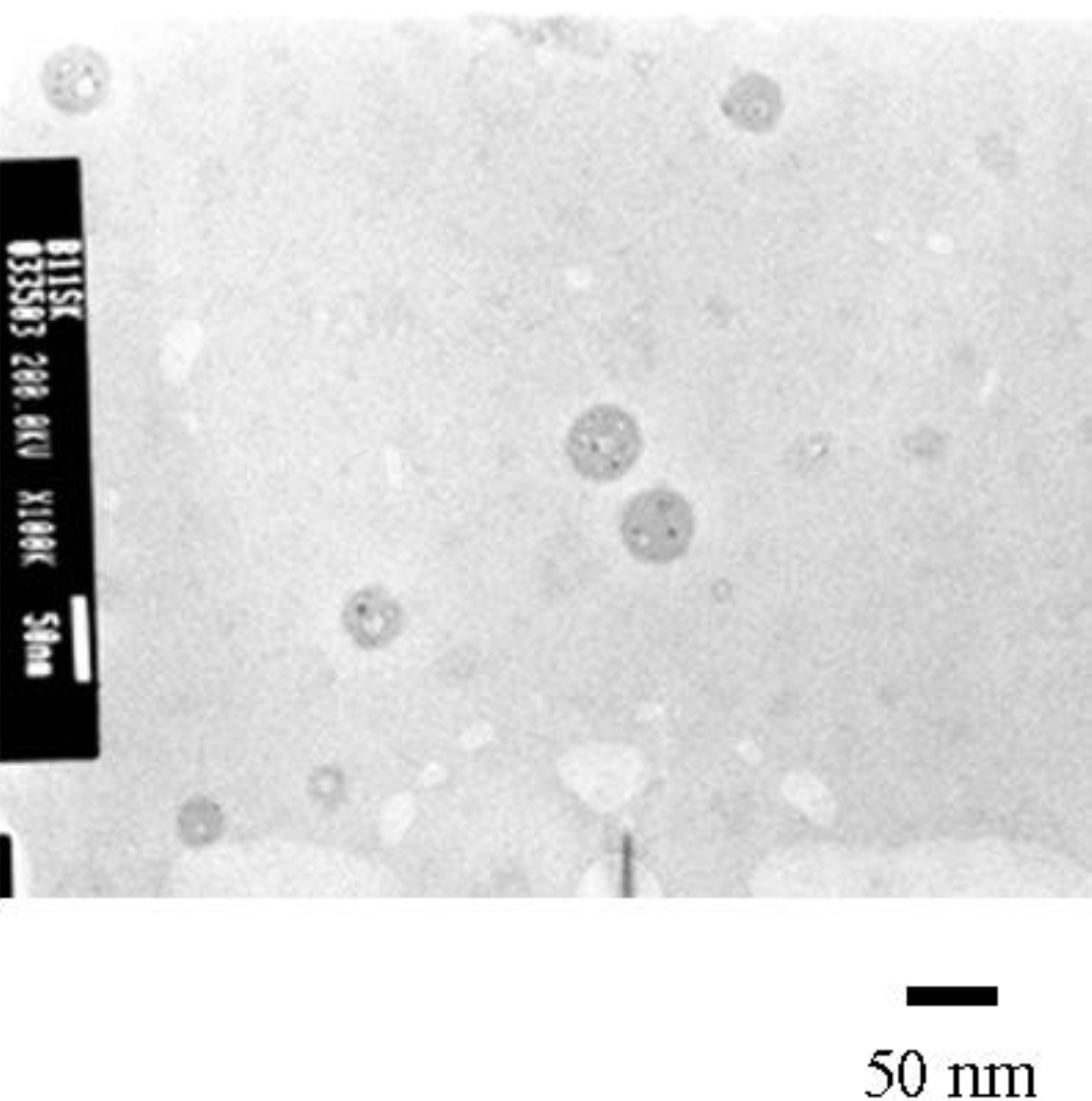
TEM observation confirmed the formation of spherical micelles through irradiation. The TEM image of the micelles is shown in Figure 3. The diameter of the micelles was estimated to average 40.6 nm based on the TEM. Compared to the micellar size determined by the cumulant analysis, the TEM exhibited a smaller diameter of the micelles than the dynamic light scattering. The estimation of the smaller micelles size, can be accounted for by the fact that the micelles in the solution contracted when isolated in air.
Figure 3.
A TEM image of the micelles. Mn(PBSt-b-PSt) = 15,000-b-97,000.

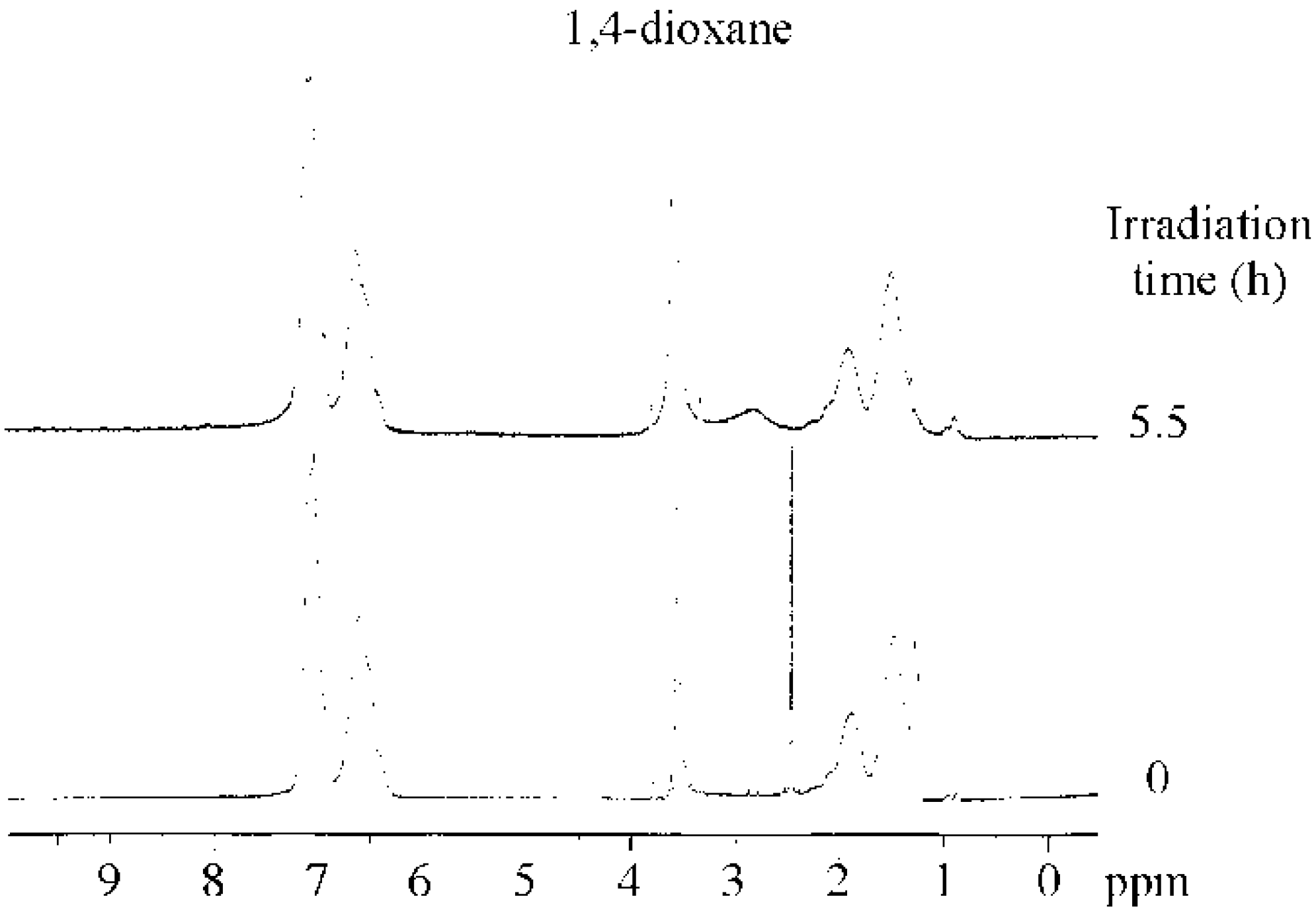
Irradiation of the copolymer in the absence of BAI, and the dark reaction in its presence, produced no changes to the hydrodynamic diameter and scattering intensity. These two control experiments suggest that the structure of PBSt-b-PSt was changed by the irradiation on BAI. The 1H NMR confirmed that micellization was caused by the elimination of the tert-butyl groups in the copolymer. Figure 4 shows the 1H NMR spectra of the copolymer before and after irradiation. The 1H NMR measurements were performed in 1,4-dioxane-d8. Signals at 1.29 ppm, based on the tert-butyl groups were hardly observed after the irradiation. The disappearance of the signals implies that the tert-butyl groups, were eliminated from the copolymer. PBSt-b-PSt should have been converted into poly(4-vinylphenol)-block-PSt (PVPh-b-PSt) by the hydrolysis of the tert-butoxy groups with the photo-acid generator as a catalyst (Scheme 2), based on the mechanism of the hydrolysis of poly(4-tert-butoxystyrene) [53]. A signal, based on the hydroxyl groups of the PVPh blocks, could not be discerned due to the fact that it overlapped with the signals of the aromatic protons and had too low an intensity. In addition, it is clear that the disappearance of the butyl proton signals and no observation of the hydroxyl signal were not based on the self-assembly of the copolymer into micelles. This is because PVPh-b-PSt showed no self-assembly in 1,4-dioxane-d8 and existed as unimers. The conversion of the BSt units into the VPh units was estimated based on the signal intensity of the tert-butyl protons to that of the aromatic protons at 6.3–7.7 ppm. The time-conversion plots are shown in Figure 1. The conversion started increasing at an earlier stage than the scattering intensity. The scattering intensity jumped when the conversion reached 50%, indicating that micellization was dependent on the degree of the VPh unit formation.
Figure 4.
1H NMR spectra of the copolymer before (bottom) and after irradiation (upper, irradiation time = 5.5 h). Solvent: 1,4-dioxane-d8.
Figure 4.
1H NMR spectra of the copolymer before (bottom) and after irradiation (upper, irradiation time = 5.5 h). Solvent: 1,4-dioxane-d8.

Scheme 2.
Micellization induced by photolysis of PBSt-b-PSt.

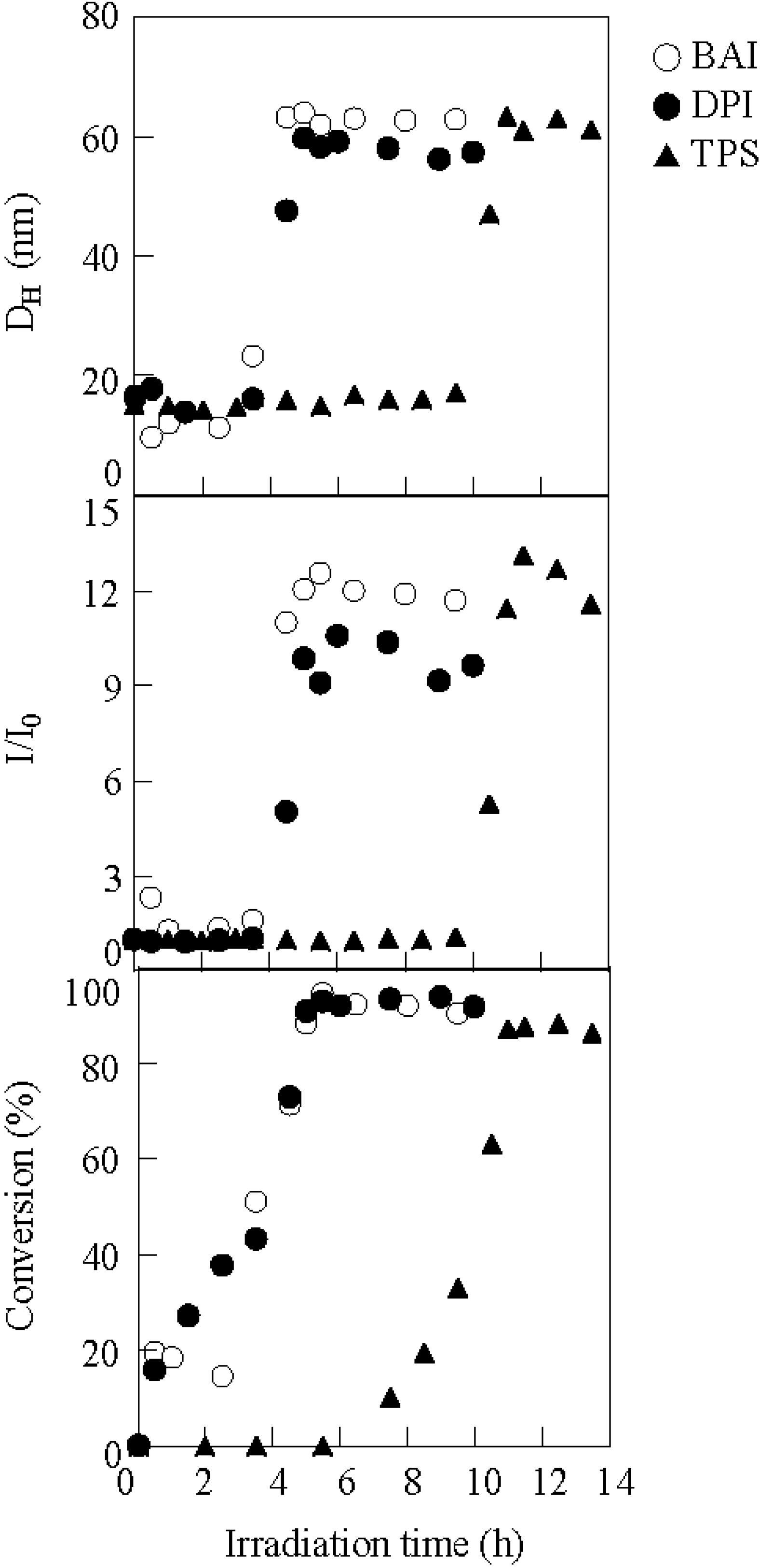
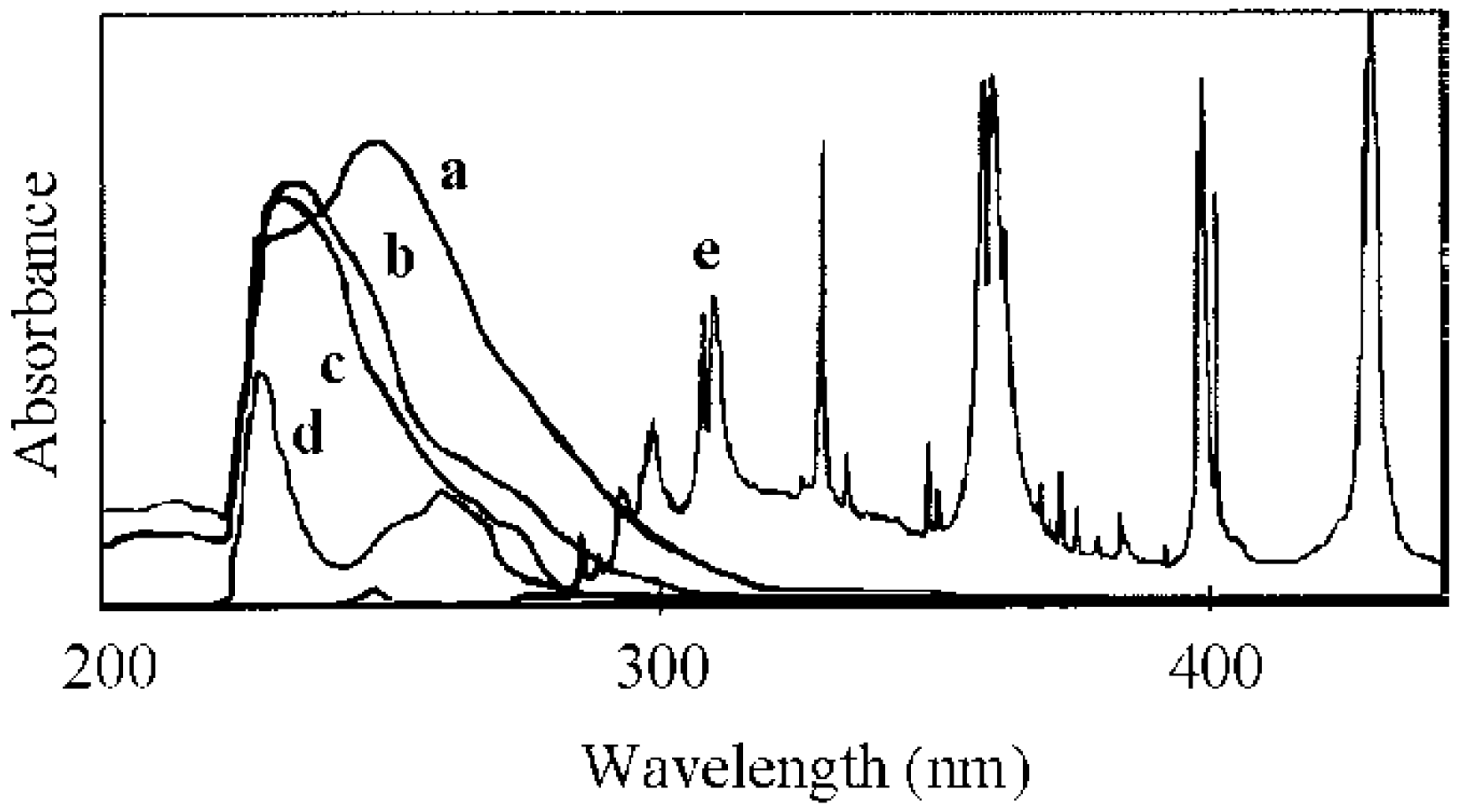
The study of micellization using different kinds of photo-acid generators demonstrated that micellization, coupled with the conversion, were dependent on the ability of the photo-acid generator. Micellization by irradiation was evaluated using diphenyliodonium hexaflurophosphate (DPI) and triphenylsulfonium triflate (TPS). Figure 5 shows the variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation by BAI, DPI, and TPS. The conversion for DPI started increasing slightly earlier than that for BAI, although there is a negligible difference in the transition of the scattering intensity and hydrodynamic diameter. On the other hand, TPS needed a longer irradiation time to promote micellization as compared to BAI and DPI. This difference in promoting micellization was clarified on the basis of the UV analysis of the photoacid generators. Figure 6 shows the UV spectra of the photo-acid generators and the PBSt-b-PSt copolymer, coupled with the illumination intensity of the irradiation, versus the wavelength for the high-pressure mercury lamp. It is considered that the irradiation reaction of the photo-acid generators occurred around 290 nm, because, at this wavelength, the absorption of the photo-acid generator overlapped at its highest proportion with the illumination intensity of the lamp without any obstruction from the copolymer. The absorbance of the photo-acid generators decreased in the order of BAI > DPI > TPS. In particular, TPS had a slight absorption at 290 nm. It can be deduced that the difference in the absorption intensity among the photo-acid generators was reflected in the irradiation time needed to initiate micellization.
Figure 5.
The variation in the hydrodynamic diameter, scattering intensity, and conversion of the copolymer during irradiation in the presence of BAI (○), DPI (●), and TPS (▲). [copolymer]0 = 3.30 g/L. Photo-acid generator/BSt unit = 0.38.
Figure 5.
The variation in the hydrodynamic diameter, scattering intensity, and conversion of the copolymer during irradiation in the presence of BAI (○), DPI (●), and TPS (▲). [copolymer]0 = 3.30 g/L. Photo-acid generator/BSt unit = 0.38.

Figure 6.
UV spectra of BAI (a), DPI (b), TPS (c), and PBSt-b-PSt (d) with the illumination intensity of irradiation of the high-pressure mercury lamp (e). Solvent: dichloromethane. Mn(PBSt-b-PSt) = 15,000-b-97,000.
Figure 6.
UV spectra of BAI (a), DPI (b), TPS (c), and PBSt-b-PSt (d) with the illumination intensity of irradiation of the high-pressure mercury lamp (e). Solvent: dichloromethane. Mn(PBSt-b-PSt) = 15,000-b-97,000.

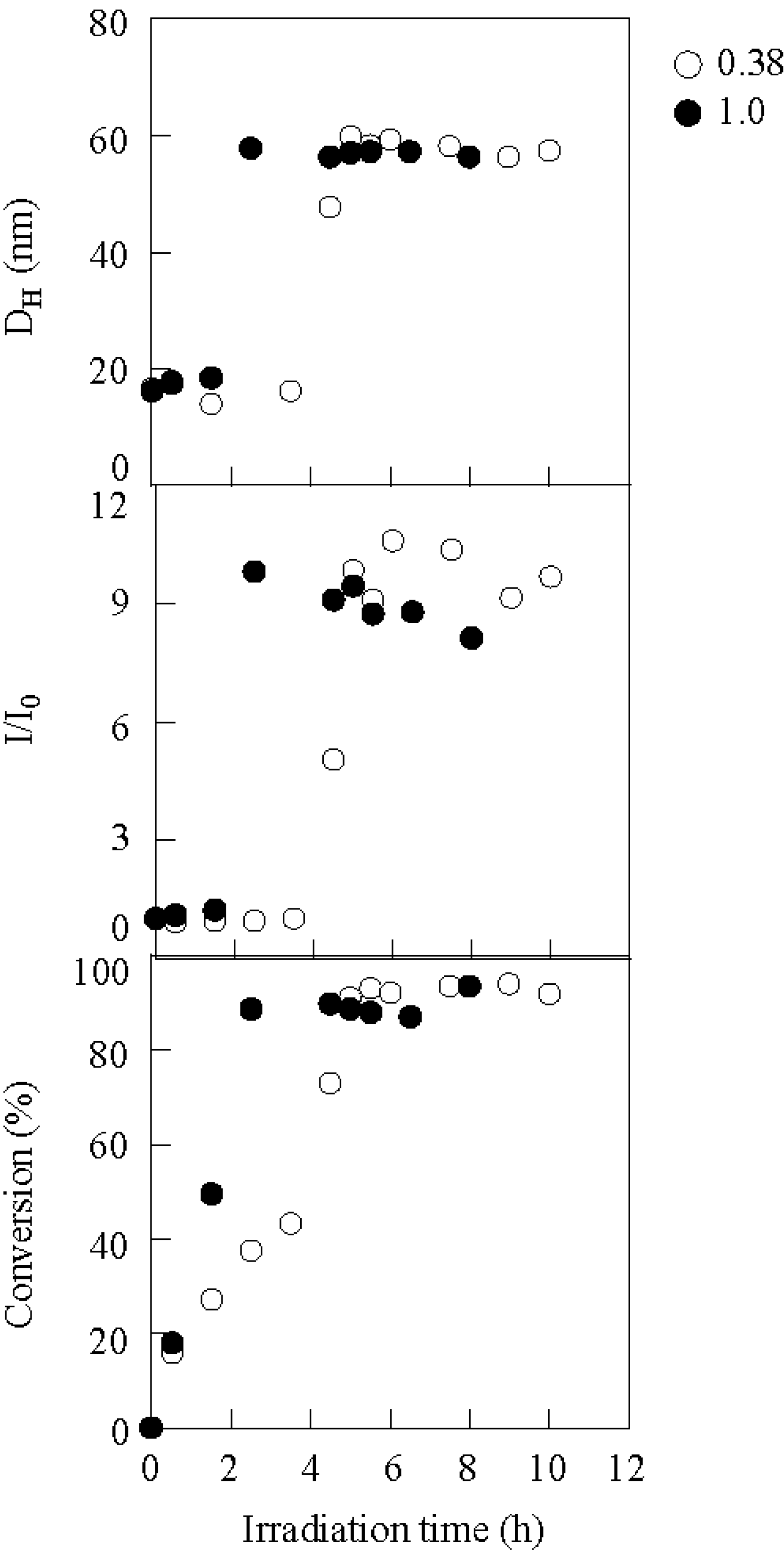
The efficiency of micellization was also dependent on the concentration of the photo-acid generator. Figure 7 shows the variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation, in the presence of DPI at the DPI/BSt molar ratios of 0.38 and 1.00. More sharply and earlier jumping was observed at 1.00, indicating that micellization was promoted more effectively at 1.00. Consequently, the irradiation time needed for micellization was manipulated by the concentration of the photo-acid generator.
Figure 7.
The variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation at 0.38 (○) and 1.00 (●) of DPI/BSt. [copolymer]0 = 3.30 g/L, Mn(PBSt-b-PSt) = 15,000-b-97,000.
Figure 7.
The variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation at 0.38 (○) and 1.00 (●) of DPI/BSt. [copolymer]0 = 3.30 g/L, Mn(PBSt-b-PSt) = 15,000-b-97,000.

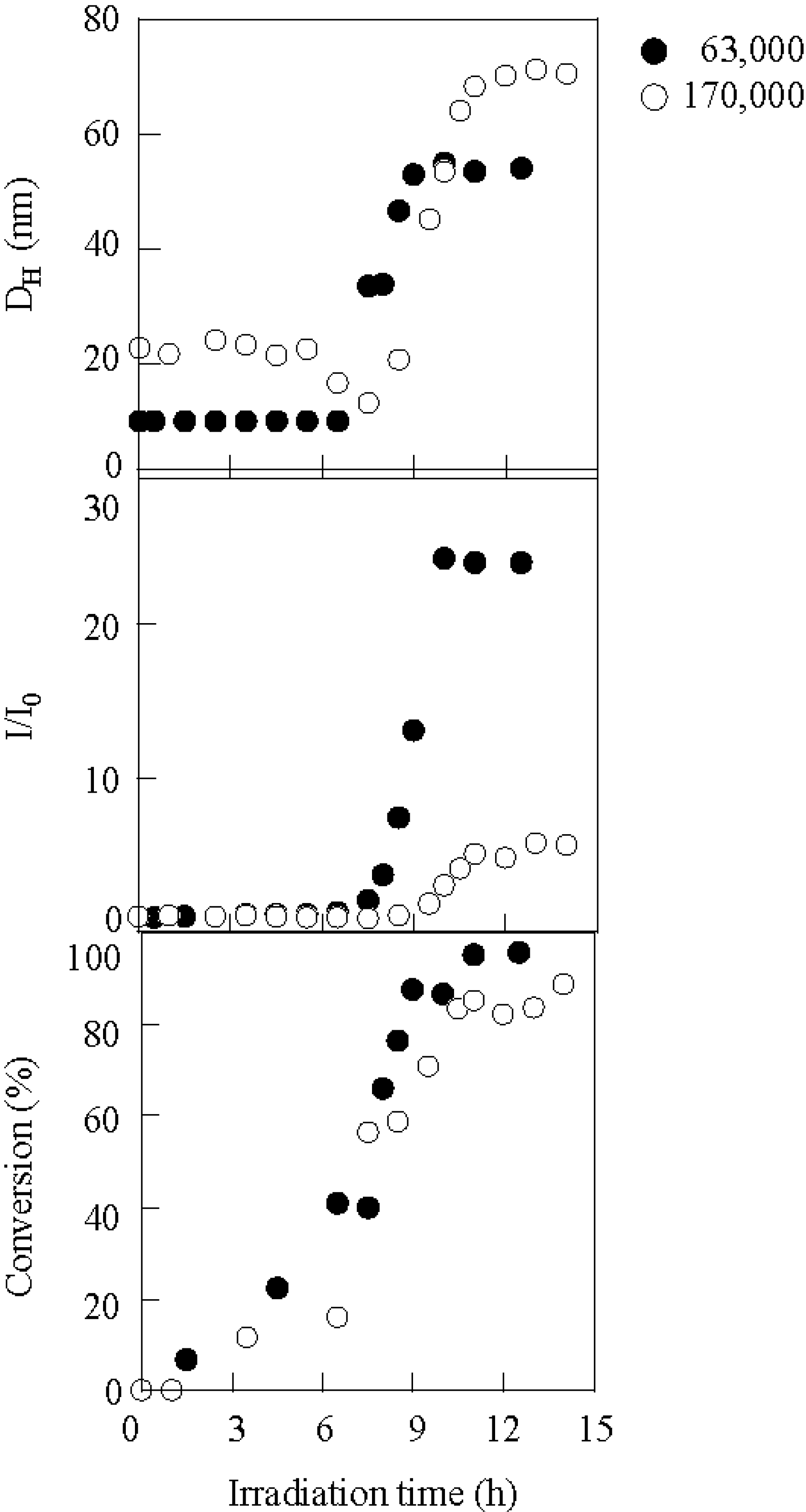
The block length of the copolymer had an effect not only on the micellar size and scattering intensity, but also on the conversion. For the identical PBSt block length (Mn = 15,000), the effect of the PSt block length on micellization was explored. Figure 8 shows the variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation using copolymers with PSt block lengths of Mn = 63,000 and 170,000, respectively. Regarding the hydrodynamic diameter and the scattering intensity, it was observed that the copolymer with the shorter PSt block exhibited transition at an earlier stage than with the longer PSt. The copolymer with the shorter PSt more easily aggregated into micelles. The shorter PSt sample produced smaller micelles with a higher aggregation number due to the shorter length of the PSt blocks. The conversion of the shorter PSt sample also started increasing slightly earlier than for the longer sample. The reason why the shorter PSt sample initiated micellization earlier is also based on the faster conversion.
Figure 8.
The variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation using copolymers with Mn(PSt block) = 63,000 (●) and 170,000 (O). Mn(PBSt block) = 15,000, [copolymer]0 = 3.30 g/L, DPI/BSt = 0.38.
Figure 8.
The variation in the hydrodynamic diameter, scattering intensity, and conversion during irradiation using copolymers with Mn(PSt block) = 63,000 (●) and 170,000 (O). Mn(PBSt block) = 15,000, [copolymer]0 = 3.30 g/L, DPI/BSt = 0.38.

2.2. Photoelectron Transfer-Induced Micellization [54]
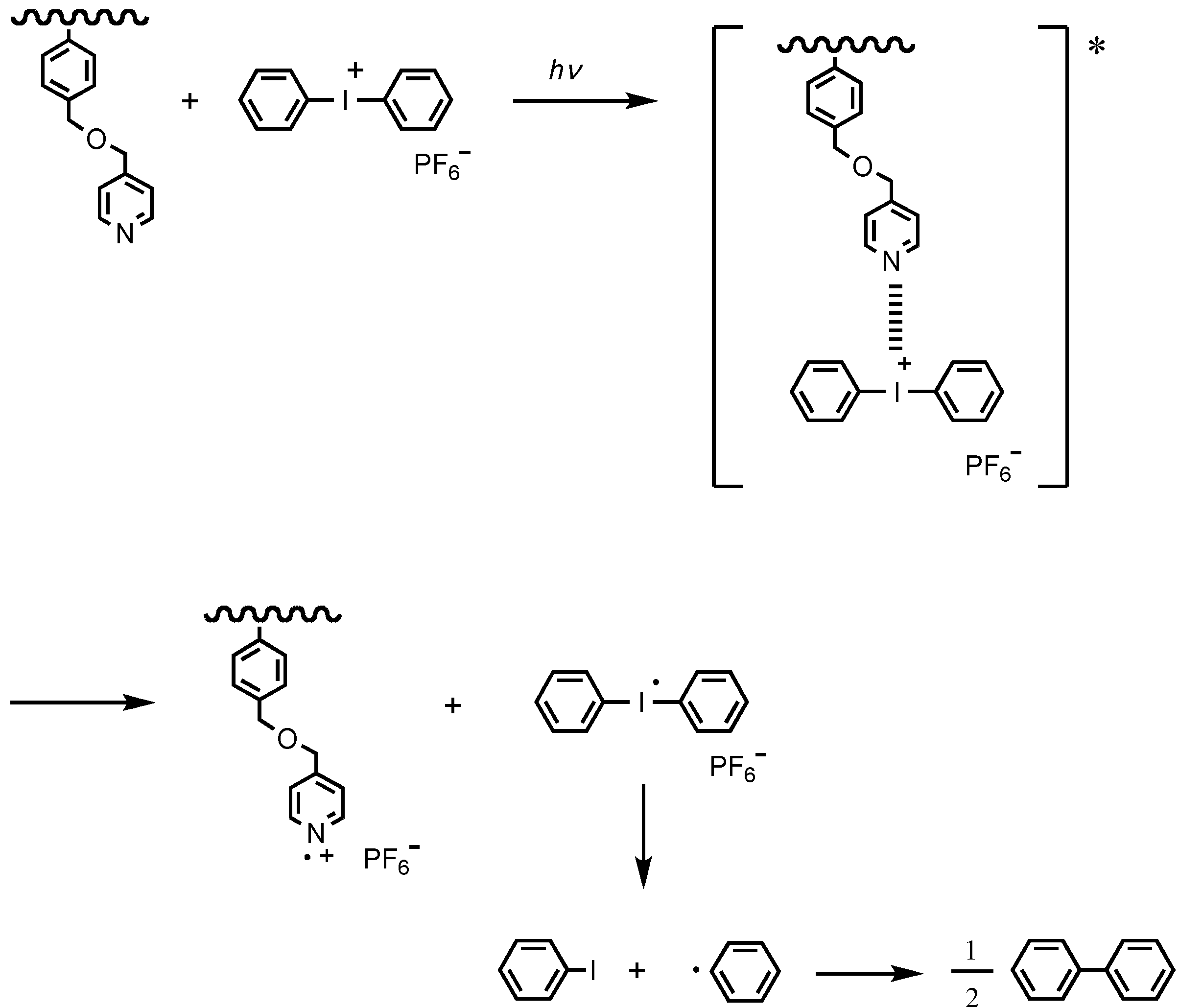
Diaryliodonium salt has been widely used not only as an acid catalyst for the hydrolysis in the excited state, but also as an initiator for photocationic polymerization [55,56] and a promoter for the nitroxide-mediated photo-living radical polymerization [57,58,59,60,61,62,63]. The iodonium salts efficiently undergo photolysis with a quantum yield of 0.7 to 0.9 [64] and have a lower reduction potential than triarylsulfonium salts [65,66]. The mechanism of the photolysis first involves photoexcitation of diarylidodonium salt and then decay of the resulting excited singlet state with both heterolytic and homolytic cleavages of the carbon-iodine bond, resulting in the formation of free-radical, cationic, and cation-radical fragments [64]. The aryl cations and aryliodine cation radicals further react with the solvent, monomers, or impurities to produce photonic acids. In the presence of electron donors, diaryliodonium salt forms an exciplex with the electron donor, followed by a photo-induced electron transfer from the electron donor to the diaryliodonium salt to provide the electron donor cation radical and the diaryliodine radical by irreversible photodecomposition [64,67]. We found a novel photoelectron transfer-induced micellization using the formation of the exciplex by diaryliodonium salt. The irreversible photo-induced self-assembly accompanied by no mass transfer has the potential to provide less volume shrinkage when recording holograms for optical information-storage media.
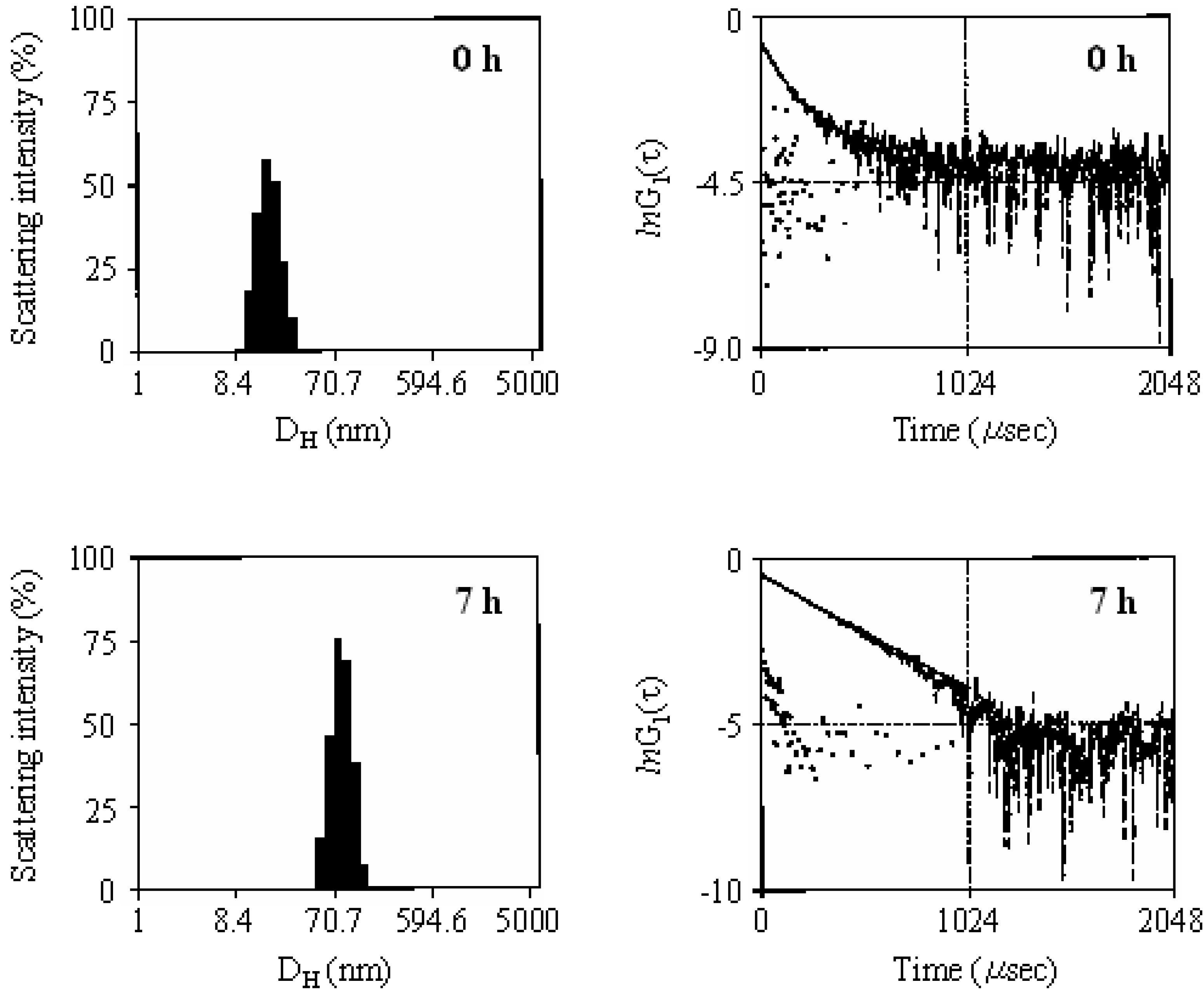
The photoelectron transfer-induced micellization of poly(4-pyridinemethoxymethylstyrene)-block-polystyrene diblock copolymer (PPySt-b-PSt) was performed in 1,4-dioxane using DPI at a 0.3 molar ratio to the PySt unit. The reaction was carried out for 7 h at room temperature by irradiation using a high-pressure mercury lamp. Figure 9 shows the scattering intensity distributions and lnG1(τ) versus τ for the copolymer solution before and after the reaction. The scattering intensity distributions were obtained by Marquardt analysis in the DLS. G1(τ) is the normalized time correlation function of the scattered field and is given by the following equation using G2(τ), the time correlation function of the scattered intensity.
where A is a baseline that corresponds to the square of the average scattering intensity, and β is an efficiency factor. Before the reaction, the distribution of the copolymer was observed at ca. 25 nm hydrodynamic diameter. This distribution shifted to ca. 70 nm by irradiation for 7 h, and no observation was made of the original distribution. All the isolated copolymers self-assembled into micelles. The diameters of the isolated copolymers and micelles were estimated by cumulant analysis to be 27.2 and 68.9 nm, respectively. It was suggested that the copolymer formed monodispersed spherical micelles based on the linear decay of the lnG1(τ) value. For spherical particles with a monodispersity, G1(τ) displays a single exponential decay curve and is represented by Equations (2) and (3)
where Γ is the decay constant and τ is the delay time [67].
G2(τ) = A(1 + β│G1(τ)│2)
G1(τ) = exp(−Γτ)
lnG1(τ) = −Γτ
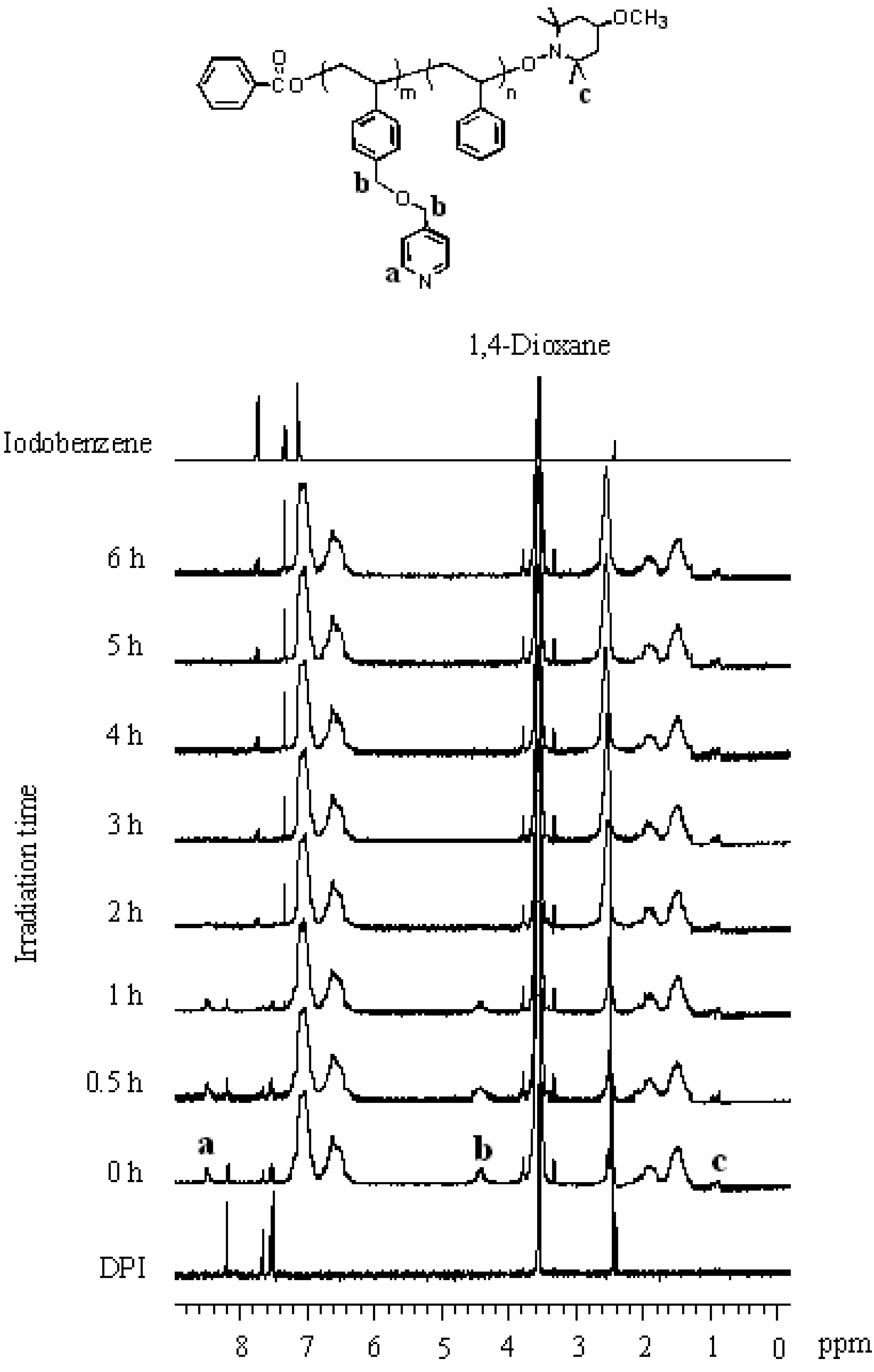
The 1H NMR analysis demonstrated that the micelles had cores formed by the PPySt blocks. The spectra of the copolymer solution at each time step are shown in Figure 10. The copolymer solution before irradiation, exhibited the respective signals based on the copolymer and DPI. Signals of the protons located at the 2-positions on the pyridine were observed at 8.4–8.6 ppm. The signals at 6.0–7.4 ppm were attributed to the aromatic protons. Signals of the methylenes connecting the pyridine with the phenyl were observed at 4.1–4.6 ppm. In addition to the main chain signals at 1.3–2.4 ppm, signals of the tetramethyl protons for the 4-methoxy-TEMPO attached to the polymer chain end were observed at 0.87 and 0.94 ppm. Furthermore, signals originating from DPI were observed at 8.19, 7.66, and 7.52 ppm. These were assigned to the protons at the 2-positions, 4-position, and 3-positions on the phenyls. When the copolymer solution was irradiated, the signal intensity at 8.4–8.6 and 4.1–4.6 ppm decreased with the irradiation time and almost disappeared at 6 h. This disappearance of the signals based on the PPySt blocks indicates that the PPySt blocks were shielded by the self-assembling copolymer, that is, the PPySt blocks formed the cores of the micelles. The signal intensity based on DPI also decreased with time, and finally, the signals were replaced by the new signals observed at 7.75 and 7.33 ppm. These signals were attributed to iodobenzene based on the photodecomposition mechanism of diaryliodonium salts [56,64,69]. The iodobenzene, coupled with biphenyl have been observed in the photodecomposition of DPI [65,70]. This replacement of the signals did not occur when the irradiation of a solution containing only DPI was carried out for 3.5 h. Furthermore, the dark reaction of the copolymer solution in the presence of DPI promoted no micellization. Based on these results, the proposed mechanism of micellization of PPySt-b-PSt by DPI is shown in Scheme 3.
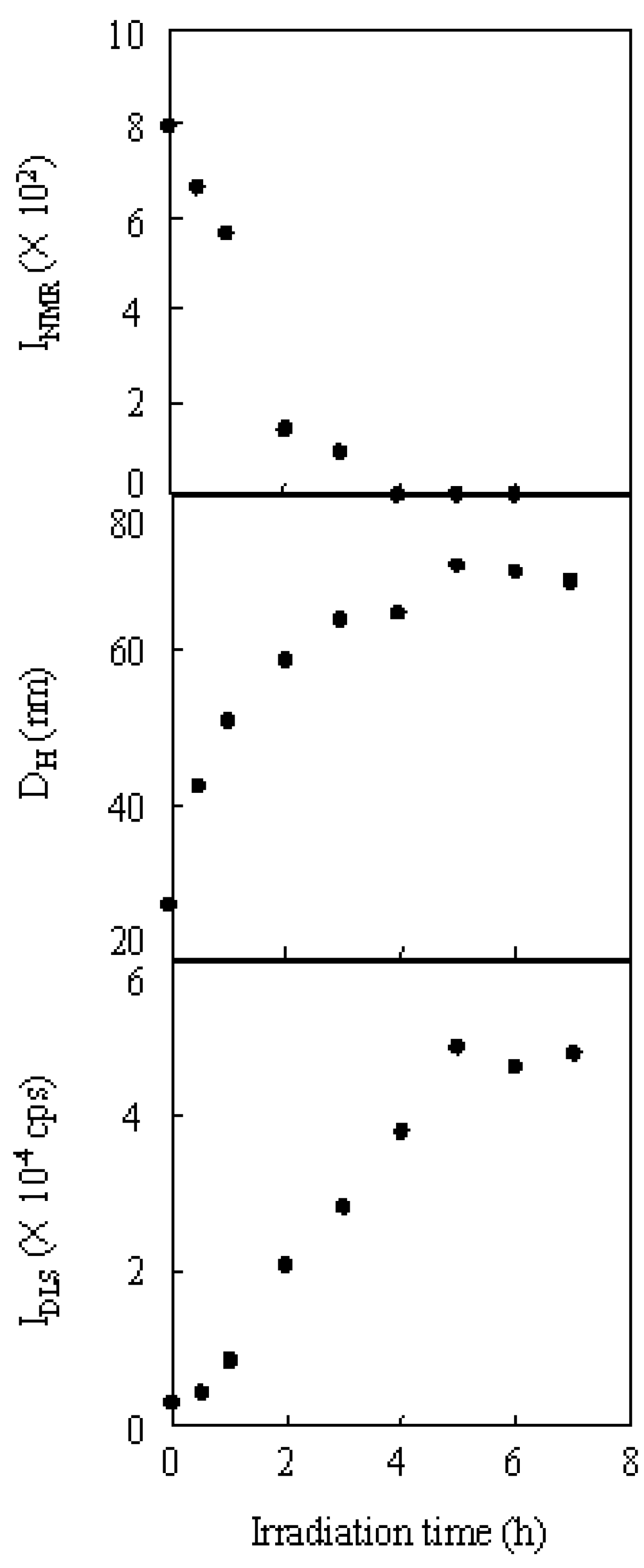
The variations in the hydrodynamic diameter and scattering intensity during micellization, coupled with the signal intensity ratio of the methylene protons to the aromatic protons in the 1H NMR, are shown in Figure 11. While the signal intensity ratio decreased with the irradiation time and reached zero at 4 h, the hydrodynamic diameter and scattering intensity increased and reached a steady state after 5 h. This suggested that the micellization was completed within 5 h.
Figure 9.
The scattering intensity distributions of the hydrodynamic diameter (DH) and plots of lnG1(τ) vs. τ for PPySt-b-PSt before (upper) and after the irradiation for 7 h (lower). [PPySt-b-PSt] = 2.86 g/L.
Figure 9.
The scattering intensity distributions of the hydrodynamic diameter (DH) and plots of lnG1(τ) vs. τ for PPySt-b-PSt before (upper) and after the irradiation for 7 h (lower). [PPySt-b-PSt] = 2.86 g/L.

Figure 10.
1H NMR spectra of the copolymer during irradiation, DPI, and iodobenzene. Solvent: 1,4-dioxane-d8. [PPySt-b-PSt] = 3.00 g/L.
Figure 10.
1H NMR spectra of the copolymer during irradiation, DPI, and iodobenzene. Solvent: 1,4-dioxane-d8. [PPySt-b-PSt] = 3.00 g/L.

Scheme 3.
The photoreaction mechanism of PPySt-b-PSt and DPI.

Figure 11.
The variation in the signal intensity ratio of the methylene protons in 1H NMR (INMR), hydrodynamic diameter (DH), and scattering intensity (IDLS) of the copolymer in DLS during irradiation.
Figure 11.
The variation in the signal intensity ratio of the methylene protons in 1H NMR (INMR), hydrodynamic diameter (DH), and scattering intensity (IDLS) of the copolymer in DLS during irradiation.

2.3. Photo-Claisen Rearrangement-Induced Micellization [69]
Photorearrangements proceed without mass transfer as well as the photoelectron transfer, and are superior to the photoelectron transfer occurring without catalysts. While several photorearrangements have been observed for N-aryl lactams [72,73], the carboxylic acid derivatives of N-aryl amides [74,75], enol esters [76,77], enol lactones [78], enamides [79], and the sulfonic acid derivative of N-aryl amides [80], a number of publications have reported the photo-Claisen rearrangement [81,82,83] and photo-Fries rearrangement [81,84,85,86].
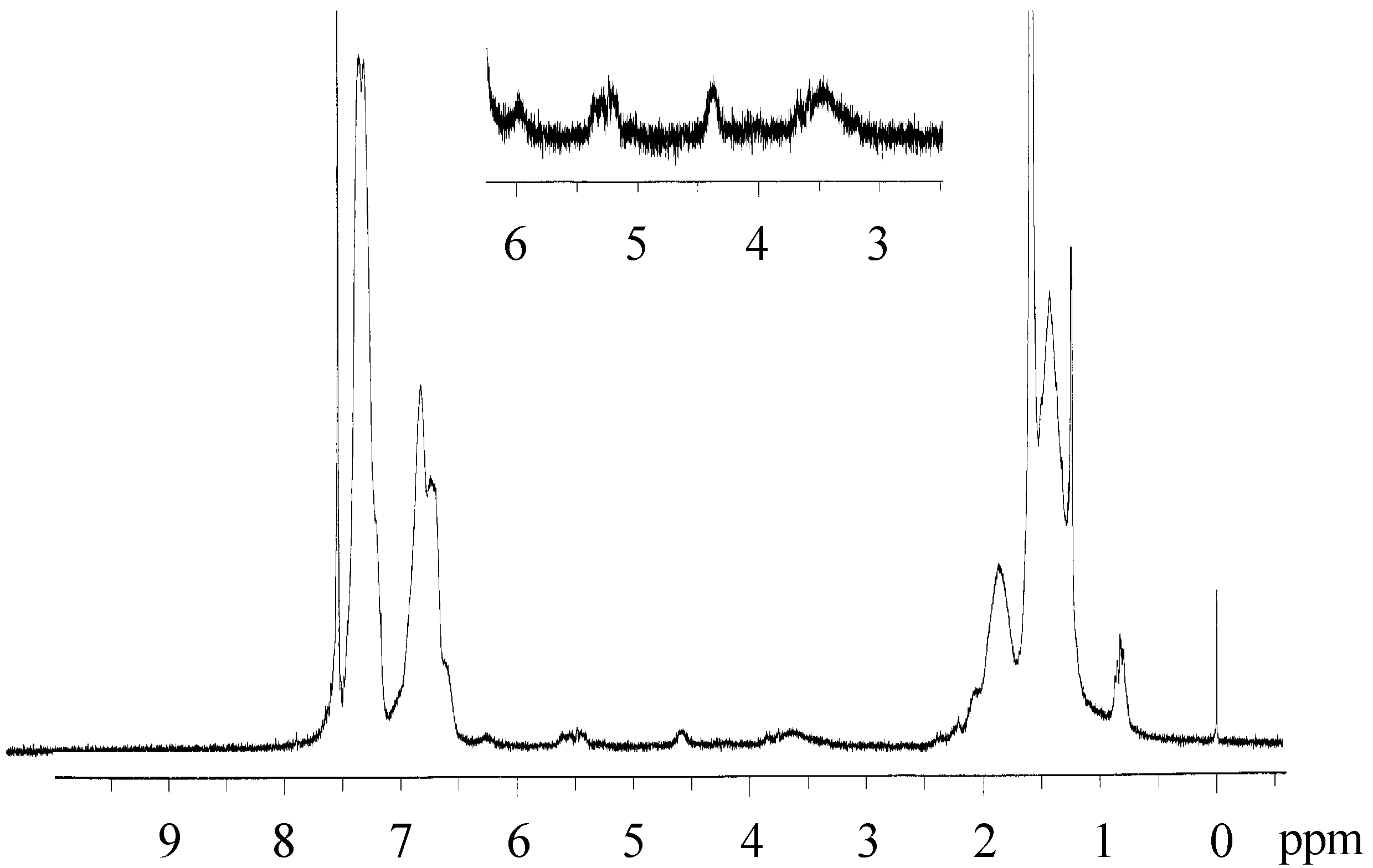
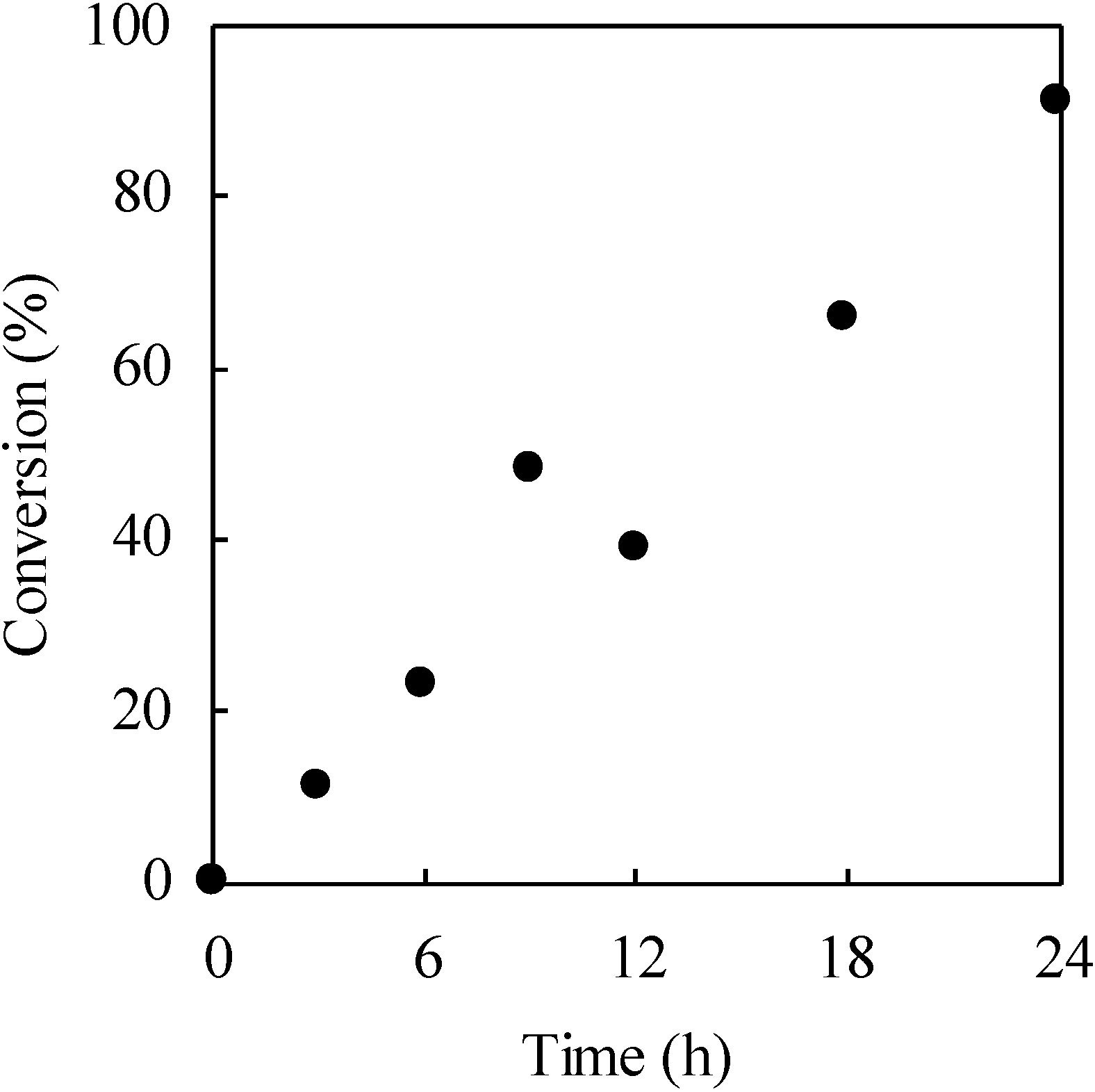
We performed photo-Claisen rearrangement-induced micellization for a poly(4-allyloxystyrene)-block-polystyrene diblock copolymer (PASt-b-PSt). The allyloxystyrene units underwent photo-Claisen rearrangement by irradiation of a cyclohexane solution containing the PASt-b-PSt copolymer (Scheme 4). Figure 12 shows the 1H NMR spectrum of the resulting copolymer by the irradiation of PASt-b-PSt. Irradiation was performed at room temperature for 18 h. In addition to the signals of the allyloxy groups, signals originating from the allylbenzyl groups were observed at 3.1–3.8 ppm. The allylbenzyl groups were attributed to the 3-allyl-4-hydroxystyrene units generated by the photorearrangement of the allyloxystyrene units. A signal for the hydroxyl groups of the 3-allyl-4-hydroxystyrene units was not observed due to its too low intensity and the fact that it overlapped with the signals of the aromatic protons. Accordingly, the conversion of the allyloxystyrene units was estimated on the basis of the proportion of the decrease in the signal intensity of the allyl protons at 4.3–4.7 ppm to that of the aromatic protons at 6.3–7.9 ppm. The conversion was 66% at the 18 h irradiation time. The time-conversion plots of the allyloxystyrene units for the photoreaction are shown in Figure 13. The conversion linearly increased over time. However, the proportion of the increase in the signal intensity of the allylbenzyl protons to the decrease in that of the allyl protons was only 45% at the 18 h irradiation time. This unquantitative proportion of the increase in the signal intensity suggests that part of the allyl groups was eliminated during the reaction. The elimination is considered to produce the vinylphenol units, when taking into account that the photo-Claisen rearrangement of allyl phenyl ether partly caused the formation of phenol [87].
Scheme 4.
The photo-Claisen rearrangement of PASt-b-PSt.

Figure 12.
1H NMR spectrum of the copolymer obtained by irradiation of PASt-b-PSt. Solvent: CDCl3.

Figure 13.
The time-conversion plots of the allyloxystyrene units for the photorearrangement. [copolymer]0 = 1.64 g/L.
Figure 13.
The time-conversion plots of the allyloxystyrene units for the photorearrangement. [copolymer]0 = 1.64 g/L.

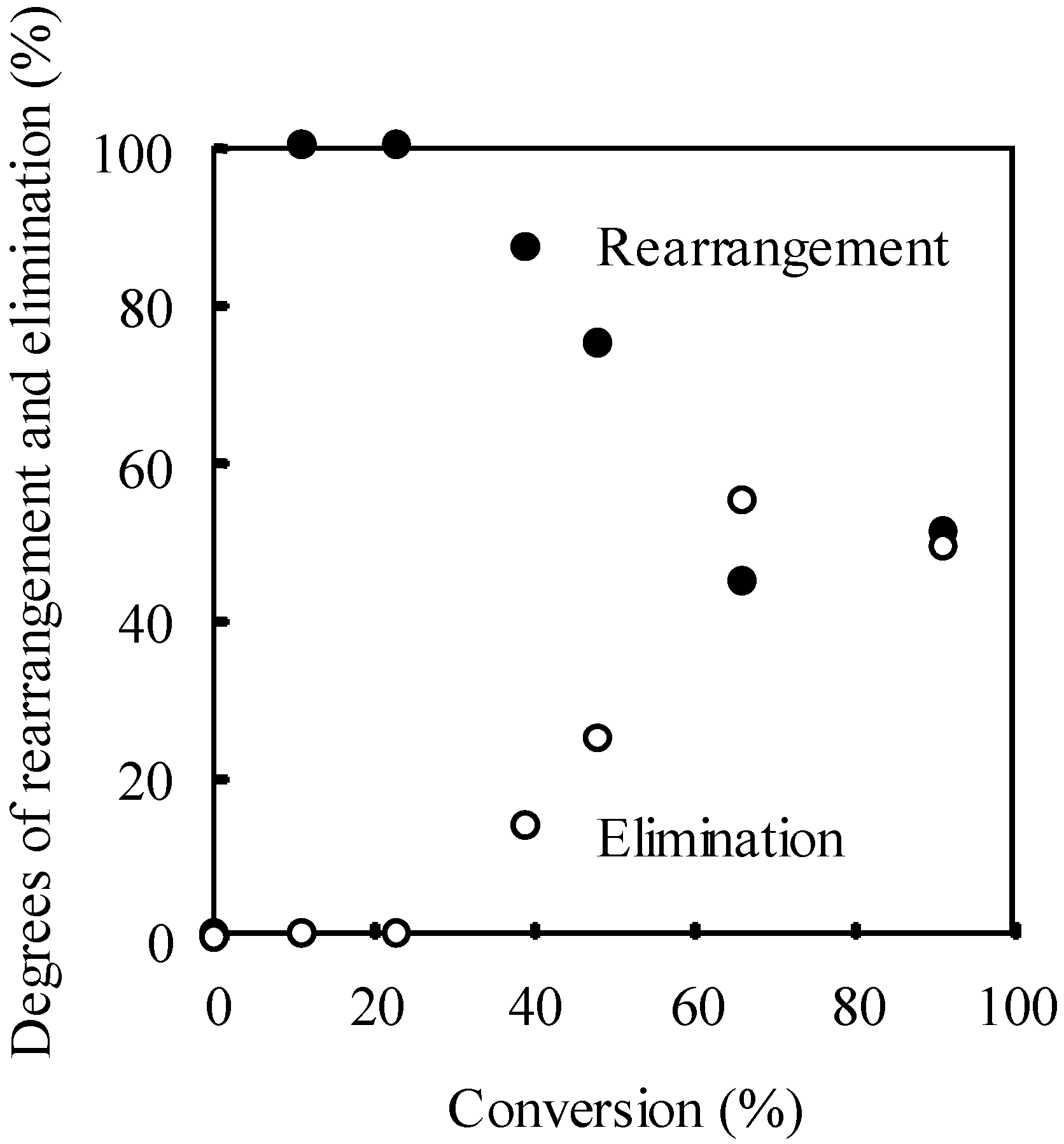
The degree of the rearrangement and elimination of the allyl groups were determined by 1H NMR. Figure 14 shows the plots of the degree of the rearrangement and elimination versus the conversion of the allyloxystyrene units. No elimination occurred up to ca. 20% conversion and the quantitative rearrangement proceeded. However, after the 20% conversion, the degree of rearrangement decreased with the increase in the conversion. Whereas the degree of elimination increased with the increasing conversion. The degree of the rearrangement and elimination were in good agreement regarding their material balance throughout the course of the reaction. Both the rearrangement and elimination finally reached ca. 50% degrees when the conversion was over 60%. This implies that elimination occurs during the course of the allyl rearrangement, rather than after it was completed.
Figure 14.
Plots of the degree of the rearrangement and elimination vs. the conversion of the allyloxystyrene units. [copolymer]0 = 1.64 g/L.
Figure 14.
Plots of the degree of the rearrangement and elimination vs. the conversion of the allyloxystyrene units. [copolymer]0 = 1.64 g/L.

The light-scattering studies demonstrated that the PASt-b-PSt copolymer formed micelles during the photoreaction. The variation in the relative scattering intensity (I/I0) and hydrodynamic diameter (DH) of the copolymer is shown in Figure 15. The light-scattering measurements were performed at 40 °C. The copolymer showed no self-assembly in cyclohexane at 40 °C and existed as a unimer with a 35.3-nm diameter. The presence of the copolymer as unimer was also confirmed by 1H NMR analysis based on the fact that the signal intensity ratios of the protons composing the copolymer were in good agreement with the structure. The scattering intensity increased with time, indicating aggregation of the copolymer. The hydrodynamic diameter of the copolymer also increased with time, although some scattering occurred in the plots. The Marquadt analysis also proved the formation of micelles. Figure 16 shows the scattering intensity distribution for the hydrodynamic diameter of the copolymer before and after irradiation. Before irradiation, the distribution of the hydrodynamic diameter was observed around 35 nm based on the unimers. After irradiation for 24 h, the hydrodynamic diameter distribution shifted to around 100 nm, which is attributed to the micelles. The hydrodynamic diameter of the micelles was estimated to be 98.1 nm. The micelles almost showed a linear decay of lnG1(τ) vs. τ, suggesting a spherical shape. However, not completely reaching linear decay of lnG1(τ) implies micelles were not monodispersed, probably due to the fact that the resulting irradiation blocks contained two kinds of unites of the 3-allyl-4-hydroxystyrene and 4-vinylphenol.
Figure 15.
The variation in the relative scattering intensity and hydrodynamic diameter of the copolymer. [copolymer]0 = 1.64 g/L.
Figure 15.
The variation in the relative scattering intensity and hydrodynamic diameter of the copolymer. [copolymer]0 = 1.64 g/L.

Figure 16.
Scattering intensity distribution for the hydrodynamic diameter of the copolymer before and after irradiation. Irradiation time: 24 h. [copolymer]0 = 1.64 g/L.
Figure 16.
Scattering intensity distribution for the hydrodynamic diameter of the copolymer before and after irradiation. Irradiation time: 24 h. [copolymer]0 = 1.64 g/L.

3. Experimental Section
Instrumentation 1H NMR measurements were conducted using a Varian 300 FT NMR spectrometer. The size exclusion chromatography (SEC) was performed using a Tosoh GPC-8020 instrument equipped with a DP-8020 dual pump, a CO-8020 column oven, and a RI-8020 refractometer. Two polystyrene gel columns, Tosoh TSK G2000HXL and G4000HXL, were used, with THF as the eluent, at 40 °C. Light scattering measurements were performed with a Photal Otsuka Electronics ELS-8000 electrophoretic light scattering spectrophotometer equipped with a system controller, an ELS controller, and a He-Ne laser operating at λ = 632.8 nm. The irradiation reaction was carried out using a Wacom HX-500 illuminator with a 500 W high-pressure mercury lamp. Transmission electron microscopy (TEM) measurements were performed using a JEOL JEM-2010 electron microscope.
Materials 4-Methoxy-2,2,6,6-tetramethylpiperidine-1-oxyl (MTEMPO) [88] was prepared as reported previously. Benzoyl peroxide (BPO) was precipitated from chloroform and crystallized in methanol at 0 °C. Commercial grade 4-vinylbenzylchloride (VBC) and styrene were washed with aqueous alkaline solution and water and distilled over calcium hydride. The poly(tert-butoxystyrene) terminated with MTEMPO (PBSt-MTEMPO) was prepared as reported previously [89]. The degree of polymerization was DP = 87.3, and the molecular weight was Mn = 15,000, by 1H NMR. SEC estimated the molecular weight and the molecular weight distribution as Mn = 10,000 and Mw/Mn = 1.17, respectively based on polystyrene standards. Commercial grade styrene was washed with aqueous alkaline solution and water and distilled over calcium hydride. Dichloromethane was purified by refluxing on calcium hydride for several hours and distilled over calcium hydride. N,N-Dimethylformamide (DMF) was stirred over calcium hydride at room temperature for 1 h, and was distilled over it, under the reduced pressure. 1,4-Dioxane was purified by refluxing on sodium for several hours and distilled over it. Sodium hydride was washed with hexane and was dried in vacuo for 1 h. Tetrahydrofuran (THF) and cyclohexane were distilled over sodium. Allyl chloride was also distilled over calcium hydride. BAI in 50 wt% propylene carbonate solution was supplied from Wako Pure Chemical Industries Ltd. DPI and TPS were purchased from Sigma-Aldrich. These photoacid generators were used without further purification. 4-Pyridinemethanol was purchased from Tokyo Chemical Industry and was used without further purification.
Synthesis of PBSt-b-PSt with Mn(PBSt-b-PSt) = 15,000-b-97,000 A mixture of the PBSt-MTEMPO (2.00 g) and styrene (10 mL) was placed in an ampule. After degassing the contents, the ampule was sealed in vacuo. Polymerization was carried out at 125 °C for 14 h and terminated by cooling with liquid nitrogen. The reaction mixture was dissolved in dichloromethane and poured into hexane to precipitate the polymer. The polymer was purified by repeated reprecipitation from dichloromethane into hexane. The precipitate was then dried in vacuo for several hours to obtain PBSt-b-PSt (9.78 g). The molecular weight and molecular weight distribution of the copolymer were estimated by SEC as Mn = 58,000 and Mw/Mn = 1.36 based on polystyrene standards. The absolute molecular weight of the copolymer was determined by 1H NMR as Mn(PBSt-b-PSt) = 15,000-b-97,000.
Synthesis of PBSt-b-PSt with Mn(PBSt-b-PSt) = 15,000-b-63,000 A mixture of the PBSt-MTEMPO (1.00 g) and styrene (2.5 mL) was placed in an ampule. After degassing the contents, the ampule was sealed in vacuo. The polymerization was carried out at 125 °C for 35 h and terminated by cooling with liquid nitrogen. The reaction mixture was dissolved in dichloromethane (8 mL) and poured into hexane (2 L). The suspension was subjected to a centrifugal separator to precipitate a polymer. The precipitate was collected, then dried in vacuo for several hours to obtain PBSt-b-PSt (1.61 g). The molecular weight and molecular weight distribution of the copolymer were estimated by SEC as Mn = 40,000 and Mw/Mn = 1.31 based on polystyrene standards. The absolute molecular weight of the copolymer was determined by 1H NMR as Mn(PBSt-b-PSt) = 15,000-b-63,000.
Synthesis of PBSt-b-PSt with Mn(PBSt-b-PSt) = 15,000-b-170,000 A mixture of the PBSt-MTEMPO (0.5 g) and styrene (5 mL) was placed in an ampule. After degassing the contents, the ampule was sealed in vacuo. The polymerization was carried out at 125 °C for 64 h and terminated by cooling with liquid nitrogen. The reaction mixture was dissolved in dichloromethane (30 mL) and poured into hexane (3 L) to precipitate a polymer. The precipitate was dried in vacuo for several hours to obtain PBSt-b-PSt (5.05 g). The molecular weight and molecular weight distribution of the copolymer were estimated by SEC as Mn = 60,000 and Mw/Mn = 1.97 based on polystyrene standards. The absolute molecular weight of the copolymer was determined by 1H NMR as Mn(PBSt-b-PSt) = 15,000-b-170,000.
Synthesis of poly(4-vinylbenzylchloride) (PVBC) VBC (32.5 g, 213 mmol), BPO (0.650 g, 2.68 mmol), and MTEMPO (0.600 g, 3.22 mmol) were placed in an ampule. After degassing the contents, the ampule was sealed in vacuo. The polymerization was carried out at 125 °C for 130 min and terminated by cooling with liquid nitrogen. The product was dissolved in dichloromethane (70 mL) and poured into methanol (10 L) to precipitate a polymer. The resulting polymer was dried in vacuo for 7 h to obtain PVBC (26.8 g). The polymer was purified by reprecipitation from dichloromethane into methanol. The degree of polymerization was DP = 78.6 and the molecular weight was Mn = 12,000 by 1H NMR. The molecular weight distribution was Mw/Mn = 1.29 by SEC based on the polystyrene standards.
Synthesis of poly(4-vinylbenzylchloride-block-polystyrne) diblock copolymer (PVBC-b-PSt) The PVBC (6.00 g) and styrene (27.3 g, 262 mmol) were placed in an ampule. After degassing the contents, the ampule was sealed in vacuo. The polymerization was carried out at 125 °C for 38 h. The product was dissolved in dichloromethane (220 mL) and then poured into methanol (12 L) to precipitate a polymer. The resulting polymer was dried in vacuo for 9 h to obtain PVBC-b-PSt (25.7 g). The polymer was purified by reprecipitation from dichloromethane into methanol. The degree of polymerization of the PSt block was DP = 467.2 and its molecular weight was Mn = 49,000 by 1H NMR. The molecular weight distribution of the copolymer was Mw/Mn = 1.88 by SEC based on the polystyrene standards.
Synthesis of PPySt-b-PSt A solution of 4-pyridinemethanol (1.42 g, 13.0 mmol) in DMF (20 mL) was added to a suspension of sodium hydride (468 mg, 19.5 mmol) in DMF (1 mL) at 0 °C under nitrogen. The mixture was stirred at room temperature for 1 h. A solution of the PVBC-b-PSt (2.00 g, 2.59 mmol as the VBC unit) in DMF (20 mL) was added to the mixture at 0 °C under nitrogen. The mixture was stirred at 0 °C under nitrogen for 2 h and was further stirred at room temperature for 17 h. The resulting mixture was poured into methanol (2 L) to precipitate a polymer. The precipitates were collected and dried in vacuo for 6 h to obtain PPySt-b-PSt (2.00 g). The substitution degree of the chloride by the 4-pyridinemethoxide was confirmed to be 100% by 1H NMR. The molecular weight of the PPySt-b-PSt was determined by 1H NMR to be Mn(PPySt-b-PSt) = 18,000-b-45,000.
Synthesis of a poly(vinylphenol)-block-polystyrene diblock copolymer (PVPh-b-PSt) The PBSt-b-PSt (Mn(PBSt-b-PSt) = 15,000-b-97,000, 2.00 g) was dissolved in THF (70 mL). Concentrated hydrochloric acid (7 mL) was added to the copolymer solution. The mixture was heated at 85 °C for 4.5 h. The resulting solution was concentrated to ca. 30 mL by an evaporator and was poured into water (1 L) to precipitate a polymer. The precipitates were collected by filtration, and then freeze-dried with 1,4-dioxane. PVPh-b-PSt (1.593 g) was obtained. The molecular weight of the PVPh-b-PSt was determined to be Mn(PVPh-b-PSt)= 11,000-b-97,000 by 1H NMR.
Synthesis of PASt-b-PSt The PVPh-b-PSt (0.70 g) was dissolved in DMF (15 mL). Sodium hydride (0.414 g, 17.3 mmol) was added to the copolymer solution at 0 °C under nitrogen atmosphere. The suspension was stirred at 0 °C for 5 min and was further stirred at room temperature for 1 h. Allyl chloride (1.41 g, 18.4 mmol) in DMF (5 mL) was added to the suspension at 0 °C. The mixture was stirred at 0 °C for 5 min and was further stirred at room temperature for 20 h. The resulting solution was poured into methanol (1 L) to precipitate a polymer. The precipitates were collected by filtration and then dried in vacuo for several hours. PASt-b-PSt (0.68 g) was obtained. The molecular weight of the PASt-b-PSt was determined to be Mn(PASt-b-PSt)= 14,000-b-97,000 by 1H NMR.
Irradiation reaction of PBSt-b-PSt: general procedure PBSt-b-PSt (Mn(PBSt-b-PSt) = 15,000-b-97,000, 363 mg) was dissolved in dichloromethane (110 mL). After the solution had stood at room temperature for 1 h, the solution was injected through a microporous filter. BAI in 50 wt% propylene carbonate solution (146 mg) was put in a 100mL-round flask covered with aluminum foil. The PBSt-b-PSt solution (97.3 mL) was poured into the flask containing the photoacid generator. The mixture was irradiated with a high-pressure mercury lamp under a nitrogen atmosphere at room temperature for the specified time. The resulting solution was subjected to light scattering measurement at 20 °C. The solution was concentrated by an evaporator and was poured into hexane to remove the photoacid generator. The precipitated polymer was purified by repeated reprecipitation from dichloromethane into hexane. The precipitate was then dried in vacuo for several hours to quantitatively obtain the resulting polymer.
Irradiation reaction of PPySt-b-PSt The PPySt-b-PSt (286 mg, 0.339 mmol of the PySt unit) and DPI (43 mg, 0.101 mmol) were dissolved in 1,4-dioxane (100 mL). The solution was injected through a microporous filter and was placed in a round-bottom flask. The solution was irradiated using a high-pressure mercury lamp at room temperature for 7 h. The resulting solution (3.5 mL) was subjected to light scattering measurement performed at 20 °C.
Irradiation reaction of PASt-b-PSt: general procedure Cyclohexane was deoxygenated by bubbling with nitrogen for 15 min. The PASt-b-PSt (8.2 mg) was dissolved in deoxygenated cyclohexane (5 mL). This copolymer solution was stirred at room temperature for 30 min under nitrogen atmosphere to deoxygenate it. The solution was irradiated with a high-pressure mercury lamp at room temperature for a specified time. The resulting solution was concentrated with an evaporator and was freeze-dried with 1,4-dioxane. The resulting copolymer was subjected to light scattering measurements at 40 °C and 1H NMR analysis.
Light scattering measurements The light scattering measurements were performed at the angle θ = 90°. The hydrodynamic diameter of the copolymer was estimated by cumulant analysis, while the scattering intensity distribution of hydrodynamic diameter was obtained by Marquadt analysis [52].
TEM measurements A drop of the micellar solution obtained by irradiation was allowed to fall on a Cu grid with a carbon substrate, and the solvent was immediately evacuated using a filter paper. The grid was dried in air for a few hours and then subjected to TEM observations.
1H NMR measurement The PPySt-b-PSt (3.0 mg, 3.56 × 10−3 mmol of the PySt unit) and DPI (0.4 mg, 9.39 × 10−4 mmol) were dissolved in 1,4-dioxane-d8 (1 mL). The solution was placed in an NMR tube and irradiated using a high-pressure mercury lamp at room temperature. The 1H NMR measurement of the resulting solution was performed.
4. Conclusions
The novel photo-induced micellizations were attained through photolysis, photoelectron transfer, and photo-Claisen rearrangement using block copolymers supporting the photoreactive groups at the side chains. The tert-butoxy groups on PBSt-b-PSt were photodecomposed into the hydroxyl groups by irradiation in the presence of the photo-acid generators, causing self‑assembly of the copolymer into micelles. The photoelectron transfer-induced micellization was attained using the pyridine-containing diblock copolymer and the photo-acid generator. The micellization of PPySt-b-PSt was expected to occur by the formation of the exciplex through the electron transfer from the pyridine groups to the photo-acid generator. The copolymer formed monodispersed spherical micelles with cores of PPySt blocks. The photo-Claisen rearrangement also promoted the micellization of the block copolymer. The photorearrangement of the allyloxystyrene units of PASt-b-PSt quantitatively proceeded up to a 20% conversion to produce the 3-allyl-4-hydroxystyrene units, while elimination of the allyl groups competitively occurred after 20% conversion. Photorearrangement and elimination degrees showed good agreement in their material balance throughout the course of the reaction. These studies on micellizations induced by irreversible photoreactions, demonstrate that many photochemical reactions can be employed as driving forces of the self-assembly of polymers to produce new architectures and nanomaterials.
References
- Ahlheim, M.; Hellensleben, M.L. Radikalisch polymerisierbare gallens uren in monoschichten, mizellen und vesikeln. Makromol. Chem. 1992, 193, 779–797. [Google Scholar] [CrossRef]
- Isahara, M.; Nakanishi, K.; Ono, K.; Sato, M.; Kikchi, M.; Sito, Y.; Yura, H.; Matsui, T.; Hattori, H.; Uenoyama, M.; Kurita, A. Photocrosslinkable chitosan as a dressing for wound occlusion and accelerator in healing process. Biomaterials 2002, 23, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Hu, Z.; Li, Y. Polymer gel as thermally responsive attenuator for ultrasonic waves. Appl. Phys. Lett. 1999, 74, 2233–2235. [Google Scholar] [CrossRef]
- Sideratou, Z.; Tsiourvas, D.; Paleos, C.M. Quaternized poly(propylene imine) dendrimers as novel pH-sensitive controlled-release systems. Langmuir 2000, 16, 1766–1769. [Google Scholar] [CrossRef]
- Yoon, X.A.; Burgess, D.J. Effect of cationic surfactant on transport of model drugs in emulsion systems. J. Pharm. Pharmacol. 1997, 49, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.J.; Lawrence, S.M.; Barlow, D.J. Aggregation and surface properties of synthetic double-chain non-ionic surfactants in aqueous solution. J. Pharm. Pharmacol. 1997, 49, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Smela, E. Conjugated polymer actuators for biomedical applications. Adv. Mater. 2003, 15, 481–494. [Google Scholar] [CrossRef]
- Hara, S.; Zama, T.; Takashima, W.; Kaneto, K. Artificial muscles based on polypyrrole actuators with large strain and stress induced electrically. Polym. J. 2004, 36, 151–161. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kawata, Y. Field enhancement by surface plasmon polariton in self-assembling nanopatterned media. Appl. Phys. Lett. 2006, 88, 123113. [Google Scholar] [CrossRef]
- Feringa, B.L.; Jager, W.F.; De Lange, B. Organic materials for reversible optical data storage. Tetrahedron 1993, 49, 8267–8310. [Google Scholar] [CrossRef]
- Liang, L.; Feng, X.D.; Liu, J.; Rieke, P.C.; Fryxell, G.E. Reversible surface properties of glass plate and capillary tube grafted by photopolymerization of N-isopropylacrylamide. Macromolecules 1998, 31, 7845–7850. [Google Scholar] [CrossRef]
- Arotcarena, M.; Heise, B.; Ishaya, S.; Laschewsky, A. Switching the inside and outside of aggregates of water-soluble block copolymers with double thermoresponsivity. J. Am. Chem. Sos. 2002, 124, 3787–3793. [Google Scholar] [CrossRef]
- Leclair, S.; Mathew, L.; Giguere, M.; Motallebi, S.; Zhao, Y. Photoinduced alignment of ferroelectric liquid crystals using azobenzene polymer networks of chiral polyacrylates and polymethacrylates. Macromolecules 2003, 36, 9024–9032. [Google Scholar] [CrossRef]
- Yu, Y.; Nakano, M.; Ikeda, T. Photomechanics: directed bending of a polymer film by light. Nature 2003, 425, 145. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, O.; Fabbri, D.; Fissi, A.; Ciardelli, F. Photomodulated conformational changes of azo-modified poly(L-glutamic acid) in micellar systems. Makromol. Chem. Rapid Commun. 1988, 9, 637–640. [Google Scholar] [CrossRef]
- Fissi, A.; Pieroni, O.; Ciardelli, F.; Ruggeri, G.; Umezawa, K. Photoresponsive polypeptides: photochromism and conformation of poly(L-glutamic acid) containing spiropyran units. Biopolymers 1993, 33, 1505–1517. [Google Scholar] [CrossRef]
- Chen, W.J.; Li, G.Z.; Zhou, G.W.; Zhai, L.M.; Li, Z.M. pH-induced spontaneous vesicle formation from NaDEHP. Chem. Phys. Lett. 2003, 374, 482–486. [Google Scholar] [CrossRef]
- Bergsma, M.; Fielden, M.L.; Engberts, J.B.F.N. pH-Dependent aggregation behavior of a sugar-amine Gemini surfactant in water: Vesicles, micelles, and monolayers of hexane-1,6-bis(hexadecyl-1’-deoxyglucitylamine. J. Colloid Interface Sci. 2001, 243, 491–495. [Google Scholar] [CrossRef]
- Yin, H.Q.; Zhou, Z.K.; Huang, J.B.; Zheng, R.; Zhang, Y.Y. Temperature-induced micelle to vesicle transition in the sodium dodecylsulfate/dodecyltriethylammonium bromide system. Angew. Chem. Int. Ed. 2003, 42, 2188–2191. [Google Scholar] [CrossRef]
- Majhi, P.R.; Blume, A. Thermodynamic characterization of temperature-induced micellization and demicellization of detergents studied by differential scanning calorimetry. Langmuir 2001, 17, 3844–3851. [Google Scholar] [CrossRef]
- Yoshida, E.; Ohta, M.; Terada, Y. Reversible control of micellization induced by hydrogen bond crosslinking for a nonamphiphilic diblock copolymer with an α,ω-diamine. Polym. Adv. Technol. 2005, 16, 183–188. [Google Scholar] [CrossRef]
- Yoshida, E.; Tanaka, M.; Takata, T. Self-assembly control of a pyridine-containing diblock copolymer by perfluorinated counter anions during salt-induced micellization. Colloid Polym. Sci. 2005, 283, 1100–1107. [Google Scholar] [CrossRef]
- McClain, J.B.; Canelas, D.A.; Samulski, E.T.; DeSimone, J.M.; Londono, J.D.; Cochran, H.D.; Wignall, G.D.; Chillura-Martino, G.D.; Triolo, R. Design of nonionic surfactants for supercritical carbon dioxide. Science 1996, 274, 2049–2052. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chu, B. Laser light scattering study of pressure-induced micellization of a diblock copolymer of poly(1,1-dihydroperfluorooctylacrylate) and poly(vinyl acetate) in supercritical carbon dioxide. Macromolecules 1998, 31, 5300–5308. [Google Scholar] [CrossRef]
- Celso, L.; Triolo, A.; Triolo, F.; Donato, D.I.; Steinhart, M.; Kriechbaum, M.; Amenitsch, H.; Triolo, R. Synchrotron SAXS investigation on aggregation phenomena in supercritical carbon dioxide. Eur. Phys. J. Soft Matter 2002, 8, 311–314. [Google Scholar] [CrossRef]
- Yoshida, E.; Nagakubo, A. Convenient synthesis of microspheres by self-assembly of random copolymers in supercritical carbon dioxide. Colloid Polym. Sci. 2007, 285, 441–447. [Google Scholar] [CrossRef]
- Yoshida, E.; Tanaka, T. Oxidation-induced micellization of a diblock copolymer containing stable nitroxyl radicals. Colloid Polym. Sci. 2006, 285, 135–144. [Google Scholar] [CrossRef]
- Yoshida, E.; Tanaka, T. Reduction-induced micellization of a diblock copolymer containing stable nitroxyl radicals. Colloid Polym. Sci. 2008, 286, 827–830. [Google Scholar] [CrossRef]
- Saji, T.; Hoshino, K.; Aoyagi, S. Reversible formation and disruption of micelles by control of the redox state of the surfactant tail group. J. Chem. Soc. Chem. Commun. 1985, 13, 865–866. [Google Scholar] [CrossRef]
- Saji, T.; Ebata, K.; Sugawara, K.; Liu, S.; Kobayashi, K. Electroless plating of organic thin films by reduction of nonionic surfactants containing an azobenzene group. J. Am. Chem. Soc. 1994, 116, 6053–6054. [Google Scholar] [CrossRef]
- Yoshida, E.; Ogawa, H. Micelle formation induced by disproportionation of stable nitroxyl radicals supported on a diblock copolymer. J. Oleo Sci. 2007, 56, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E. Control of micellization induced by disproportionation of 2,2,6,6-tetramethylpiperidine-1-oxyl supported on side chains of a block copolymer. Colloid Polym. Sci. 2009, 287, 1365–1368. [Google Scholar] [CrossRef]
- Lovrien, R. The photoviscosity effect. Proc. Natl Acad. Sci. USA 1967, 57, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Matejka, L.; Dusek, K. Photochromic polymers: Photoinduced conformational changes and effect of polymeric matrix on the isomerization of photochromes. Makromol. Chem. 1981, 182, 3223–3236. [Google Scholar] [CrossRef]
- Pieroni, O.; Fissi, A. Synthetic photochromic polypeptides: possible models for photoregulation in biology. J. Photochem. Photobiol. B. Biol. 1992, 12, 125–140. [Google Scholar] [CrossRef]
- Menju, A.; Hayashi, K.; Irie, M. Photoresponsive polymers. 3. Reversible solution viscosity change of poly(methacrylic acid) having spirobenzopyran pendant groups in methanol. Macromolecules 1981, 14, 755–758. [Google Scholar]
- Taguchi, M.; Li, G.; Gu, Z.; Sato, O.; Einaga, Y. Magnetic vesicles of amphiphilic spiropyran containing iron oxide particles on a solid state substrate. Chem. Mater. 2003, 15, 4756–4760. [Google Scholar] [CrossRef]
- Meier, H. The photochemistry of stilbenoid compounds and their role in materials technology. Angew. Chem. Int. Ed. Engl. 1992, 31, 1399–1420. [Google Scholar] [CrossRef]
- Szczubialka, K.; Nowakoaska, M. Response of micelles formed by smart terpolymers to stimuli studied by dynamic light scattering. Polymer 2003, 44, 5269–5274. [Google Scholar] [CrossRef]
- Eastoe, J.; Sanchez, M.; Dominguez, S.; Wyatt, P.; Beeby, A.; Heenan, R. Properties of a stilibene-containing Gemini photosurfactant: Light-triggered changes in surface tension and aggregation. Langmuir 2002, 18, 7837–7844. [Google Scholar] [CrossRef]
- Irie, M.; Hosoda, M. Photoresponsive polymers. Reversible solution viscosity change of poly(N,N-dimethylacrylamide) with pendant triphenylmethane leucohydroxide residues in methanol. Makromol. Chem. Rapid Commun. 1985, 6, 533–536. [Google Scholar]
- Dunkin, I.R.; Gittinger, A.; Sherrington, D.C.; Whittaker, P. A photodestructible surfactant. J. Chem. Soc. Chem. Commun. 1994. [CrossRef]
- Mezger, T.; Nuyken, O.; Meindl, K.; Wokaun, A. Light decomposable emulsifiers: application of alkyl-substituted aromatic azosulfonates in emulsion polymerization. Prog. Org. Coatings 1996, 29, 147–157. [Google Scholar] [CrossRef]
- Nuyken, O.; Voit, B. The photoactive diazosulfonate group and its role in polymer chemistry. Macromol. Chem. Phys. 1997, 198, 2337–2372. [Google Scholar] [CrossRef]
- Haubs, M.; Ringsdorf, H. Photosensitive monolayers, bilayer membranes and polymers. New J. Chem. 1987, 11, 151–156. [Google Scholar]
- Okamoto, Y.; Yoshida, H.; Takamuku, S. Photochemical reaction of [4(4’-alkoxybenzoyl)phenylmethylphosphonic acids. Application to a photo-degradable surfactant. Chem. Lett. 1988, 17, 569–572. [Google Scholar]
- Veronese, A.; Berclaz, N.; Luisi, P.L. Photoinduced formation of bilayer vesicles. J. Phys. Chem. B 1998, 102, 7078–7080. [Google Scholar] [CrossRef]
- Cohen, S.M.; Young, R.H.; Markhart, A.H. Transparent ultraviolet-barrier coatings. J. Polym. Sci. A 1971, 9, 3263–3299. [Google Scholar] [CrossRef]
- Tessier, T.G.; Frechet, J.M.J. The Photo-fries rearrangement and its use in polymeric imaging systems. ACS Sympo. Ser. 1985, 266, 269–292. [Google Scholar]
- Yoshida, E.; Kuwayama, S. Micelle formation induced by photolysis of a poly(tert-butoxystyrene)-block-polystyrene diblock copolymer. Colloid Polym. Sci. 2007, 285, 1287–1291. [Google Scholar] [CrossRef]
- Yoshida, E.; Kuwayama, S. Photolysis-induced micellization of a poly(4-tert-butoxystyrene)-block-polystyrene diblock copolymer. Colloid Polym. Sci. 2008, 286, 1621–1627. [Google Scholar] [CrossRef]
- Marquardt, D.W. An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Indust. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Conlon, D.A.; Crivello, J.V.; Lee, J.L.; O’Brien, M.J. The synthesis, characterization, and deblocking of poly(4-tert-butoxystyrene) and poly(4-tert-butoxy-.alpha.-methylstyrene). Macromolecules 1989, 22, 509–516. [Google Scholar] [CrossRef]
- Yoshida, E.; Kuwayama, S.; Kawaguchi, S. Photo-induced micellization of poly(4-pyridinemethoxymethylstyrene)-block-polystyrene using a photo-acid generator. Colloid Polym. Sci. 2010, 288, 91–95. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. New photoinitiators for cationic polymerization. J. Polym. Sci. Symp. 1976, 56, 383–395. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W.; Volante, C.N. Photoinitiated cationic polymerization using diaryliodonium salts. J. Radiat. Curing 1977, 4, 2–16. [Google Scholar]
- Crivello, J.V.; Sangermano, M. Visible and long-wavelength photoinitiated cationic polymerization. J. Polym. Sci. A Polym. Chem. 2001, 39, 343–356. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by a nitroxide mediator. Colloid Polym. Sci. 2008, 286, 1663–1666. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by 2,2,6,6-tetramethylpiperidine-1-oxyl in the presence of a photo-acid generator. Colloid Polym. Sci. 2009, 287, 767–772. [Google Scholar] [CrossRef]
- Yoshida, E. Synthesis of poly(methyl methacrylate)-block-poly(tetrahydrofuran) by photo-living radical polymerization using a 2,2,6,6-tetramethylpiperidine-1-oxyl macromediator. Colloid Polym. Sci. 2009, 287, 1417–1424. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate using alkoxyamine as an initiator. Colloid Polym. Sci. 2010, 288, 7–13. [Google Scholar] [CrossRef]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of vinyl acetate. Colloid Polym. Sci. 2010, 288, 73–78. [Google Scholar] [CrossRef]
- Yoshida, E. Effect of azoinitiators on nitroxide-mediated photo-living radical polymerization of methyl methacrylate. Colloid Polym. Sci. 2010, 288, 341–345. [Google Scholar] [CrossRef]
- Crivello, J.V. The discovery and development of onium salt cationic photoinitiators. J. Polym. Sci. A Polym. Chem. 1999, 37, 4241–4254. [Google Scholar] [CrossRef]
- Pappas, S.P.; Gatechair, L.; Jilek, J.H. Photoinitiation of cationic polymerization. III. Photosensitization of diphenyliodonium and triphenylsulfonium salts. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 77–84. [Google Scholar] [CrossRef]
- Kunze, A.; Muller, U.; Tittes, K.; Fouassier, J.P.; Morlet-Savary, F. Triplet quenching by onium salts in polar and nonpolar solvents. J. Photochem. Photobiol. A 1997, 110, 115–122. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Dye-sensitized photoinitiated cationic polymerization. J. Polym. Sci. Polym. Chem. Ed. 1976, 16, 2441–2451. [Google Scholar] [CrossRef]
- Brown, W. Light Scattering Principles and Development; Clarendon Press Oxford: Gloucestershire, UK, 1996; pp. 439–442. [Google Scholar]
- Kellmann, A. Primary photochemical processes of cationic acridine orange in aqueous solution studied by flash photolysis. Photochem. Photobiol. 1974, 20, 103–108. [Google Scholar] [CrossRef]
- Devoe, R.J.; Sahyun, M.R.V.; Serpone, N.; Sharma, D.K. Transient intermediates in the photolysis of iodonium cations. Can. J. Chem. 1987, 65, 2342–2349. [Google Scholar] [CrossRef]
- Yoshida, E.; Kuwayama, S. Micelle formation induced by photo-Claisen rearrangement of poly(4-allyloxystyrene)-block-polystyrene. Colloid Polym. Sci. 2009, 287, 789–793. [Google Scholar] [CrossRef]
- Fischer, M. Photochemische reaktionen, V: Photochemische synthese mittlerer und grosser stickstoffhaltiger ringe. Tetrahedron Lett. 1968, 9, 4295–4298. [Google Scholar] [CrossRef]
- Fischer, M.; Mattheus, A. Photochemische reaktionen, VI. Photoumlagerungen von N-phenyl-lactamen. Chem. Ber. 1969, 102, 342–350. [Google Scholar]
- Bellus, D.; Schaffner, K. Photochemische reaktionen. 43. Mitteilung [1]. UV.-bestrahlung von N-phenylurethan und N-phenylthiourethan. Helv. Chim. Acta 1968, 51, 221–224. [Google Scholar]
- Kan, R.O.; Furey, R.L. Photochemical generation and decomposition of dibenzoylaniline. Tetrahedron Lett. 1966, 7, 2573–2578. [Google Scholar] [CrossRef]
- Finnegan, R.A.; Hagen, A.W. An analogue of the photo-fries rearrangement; the photolysis of vinyl benzoate. Tetrahedron Lett. 1963, 4, 365–368. [Google Scholar] [CrossRef]
- Gorodetsky, M.; Mazur, Y. Photochemistry of enolic systems. II. Irradiation of dienol acetates. J. Am. Chem. Soc. 1964, 86, 5213–5218. [Google Scholar]
- Yogev, A.; Mazur, Y. Irradiation of enol lactones. J. Am. Chem. Soc. 1965, 87, 3520–3521. [Google Scholar] [CrossRef]
- Bertele, E.; Boos, H.; Dunitz, J.D.; Elsinger, F.; Eschenmoser, A.; Felner, I.; Gribi, H.P.; Gschwend, H.; Meyer, E.F.; Pesaro, M.; Scheffold, R. Ein synthetischer Zugang zum Corrinsystem. Angew. Chem. 1964, 76, 393–399. [Google Scholar] [CrossRef]
- Nozaki, H.; Okada, T.; Noyori, R.; Kawanishi, M. Photochemical rearrangement of arenesulphonanilides to p-aminodiarylsulphones. Tetrahedron 1966, 22, 2177–2180. [Google Scholar] [CrossRef]
- Pitchumani, K.; Warri, M.; Ramamurthy, V. Remarkable product selectivity during photo-Fries and photo-Claisen rearrangements within zeolites. J. Am. Chem. Soc. 1996, 118, 9428–9429. [Google Scholar] [CrossRef]
- Pincock, A.L.; Pincock, J.A.; Stefanova, R. Substituent effects on the rate constants for the photo-Claisen rearrangement of allyl aryl ethers. J. Am. Chem. Soc. 2002, 124, 9768–9778. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Veglia, A.V.; Rossi, R.H. β-Cyclodextrin effects on photo-Claisen rearrangement of allyl phenyl ether. Can. J. Chem. 1997, 75, 1151–1155. [Google Scholar] [CrossRef]
- Anderson, J.C.; Reese, C.B. Photo-induced Fries rearrangements. Proc. Chem. Soc. 1960, 217. [Google Scholar]
- Arai, T.; Tobita, S.; Shizuka, H. Direct measurements of the rates of 1,3- and 1,5-sigmatropic hydrogen shifts in the photo-Fries rearrangements of phenyl acetate. Chem. Phys. Lett. 1994, 223, 521–526. [Google Scholar] [CrossRef]
- Magdy, M.; Malik, A.; Mayo, P. Surface photochemistry: The amide photo-Fries rearrangement. Can. J. Chem. 1983, 62, 1275–1278. [Google Scholar]
- Pitchumani, K.; Warrier, M.; Ramamurthy, V. Utility of zeolitic medium in photo-Fries and photo-Claisen rearrangements. Res. Chem. Intermed. 1999, 25, 623–631. [Google Scholar] [CrossRef]
- Miyazawa, T.; Endo, T.; Shiihashi, S.; Ogawara, M. Selective oxidation of alcohols by oxoaminium salts (R2N:O+ X-). J. Org. Chem. 1985, 50, 1332–1334. [Google Scholar] [CrossRef]
- Yoshida, E.; Kunugi, S. Micelle formation of poly(vinyl phenol)-block-polystyrene through hydrogen bond crosslinking by α, ω-diamine. J. Polym. Sci. A Polym. Chem. Ed. 2002, 40, 3063–3067. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Yoshida, E.; Kuwayama, S. Photo-Induced Micellization of Block Copolymers. Polymers 2010, 2, 623-648. https://doi.org/10.3390/polym2040623
AMA Style
Yoshida E, Kuwayama S. Photo-Induced Micellization of Block Copolymers. Polymers. 2010; 2(4):623-648. https://doi.org/10.3390/polym2040623
Chicago/Turabian StyleYoshida, Eri, and Satoshi Kuwayama. 2010. "Photo-Induced Micellization of Block Copolymers" Polymers 2, no. 4: 623-648. https://doi.org/10.3390/polym2040623