Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents

Abstract
: The nanostructure of bone has been replicated using a polymer-induced liquid-precursor (PILP) mineralization process. This polymer-mediated crystallization process yields intrafibrillar mineralization of collagen with uniaxially-oriented hydroxyapatite crystals. The process-directing agent, an anionic polymer which we propose mimics the acidic non-collagenous proteins associated with bone formation, sequesters calcium and phosphate ions to form amorphous precursor droplets that can infiltrate the interstices of collagen fibrils. In search of a polymeric agent that produces the highest mineral content in the shortest time, we have studied the influence of various acidic polymers on the in vitro mineralization of collagen scaffolds via the PILP process. Among the polymers investigated were poly-L-aspartic acid (PASP), poly-L-glutamic acid (PGLU), polyvinylphosphonic acid (PVPA), and polyacrylic acid (PAA). Our data indicate that PASP and the combination of PGLU/PASP formed stable mineralization solutions, and yielded nano-structured composites with the highest mineral content. Such studies contribute to our goal of preparing biomimetic bone graft substitutes with composition and structure that mimic bone.
1. Introduction
Bone is a composite material composed of collagen and hydroxyapatite (HA) [Ca10(PO4)6(OH)2], which has been highly studied due to its unique mechanical properties and the possibility of creating synthetic bone substitutes. The structure of bone is arranged in hierarchical levels, described by Weiner and Wagner [1] as starting from the individual constituents of hydroxyapatite and collagen fibrils, to the nanostructure of an interpenetrating mineralized collagen network, to the lamellar microstructure of osteons, to the macrostructure of cancellous and cortical bone. This study focuses on the nanostructural level, which is comprised of collagen fibrils mineralized with intrafibrillar, [001] uniaxially aligned platelets of hydroxyapatite nanocrystals [2-4]. There is great interest in developing a biomimetic method of replicating the bone mineralization process in vitro. One proposed process for recreating bone structure, called the polymer-induced liquid-precursor (PILP) process, focuses on the intrafibrillar mineralization of self-assembled collagen arrays, and the evidence suggests that the mechanism involves an amorphous, liquid-phase mineral precursor. We have proposed that non-collagenous proteins (NCPs) that are close to the mineralization front play a crucial role in the biomineralization process of bone by inducing such a precursor pathway [2,5-11].
The extracellular matrix of bone and dentin contains small quantities of NCPs, such as osteopontin, bone sialoprotein, dentin matrix protein 1, and dentin sialophosphoprotein [12]. These proteins are all highly anionic due to the prevalence of carboxylate groups on the polyaspartic acid and polyglutamic acid residues which comprise the protein backbone. Although their exact roles in the mechanisms of bone and dentin formation are not fully understood, the NCPs are believed to play a crucial role in the mineralization of bone and dentin. This assertion is supported by studies demonstrating that mutations in genes that code for these proteins result in abnormal bone and dentin mineralization [13-16]. Extensive study of the NCPs has also determined that some of their functions are dependent on post-translations modifications (PTMs), such as phosphorylation of the serine residues, which further augments their anionic character [17].
Based on evidence from our in vitro studies, we have proposed that these anionic proteins sequester ions, resulting in liquid-liquid phase separation, as has been observed in our CaCO3 PILP model system [18,19]. The minor phase consists of metastable, amorphous, liquid-phase nanodroplets that are a precursor to the more stable amorphous calcium phosphate, and ultimately the crystalline form of hydroxyapatite. These droplets are referred to as the PILP phase. Due to the fluidic nature of these droplets, it has been proposed that they are drawn into the interstices of the collagen fibrils by capillary forces, where they solidify and leave the fibrils embedded with hydroxyapatite nanocrystals [2,20].
Our current in vitro model uses poly-L-aspartic acid (PASP) as a simple polymer to mimic the role of the acidic proteins present during bone formation. The structure of PASP consists of a carboxylate group attached to an amino acid backbone by one methylene group. It was selected for the PILP model system due to the fact that it is one of the two most prevalent amino acids in the NCPs, and is therefore suitable as a simple mimic of their functions in biomineralization. The discovery that PASP added to bone graft substitutes in animal models results in increased bone regeneration sparked further investigation into its role in the NCPs [21]. Although the mechanism is not fully understood, it was concluded that PASP serves as a nucleating agent and growth mediator for hydroxyapatite [22]. Studies of PASP with various hydroxylation degrees by Chi et al. demonstrated that the carboxylate group performs a crucial role as a crystal growth modifier and in directing the synthesis of calcium phosphate [23]. We offer a different view, that the role of such anionic NCPs in bone formation may be more related to their ability to direct the crystallization process to where it follows an amorphous precursor pathway. For example, the use of PASP has enabled intrafibrillar mineralization to be achieved, yielding nanostructured composites closely resembling the fundamental nanostructure of natural bone [2,9,20,24-26]. Without the presence of an anionic polymer, mineralization of collagen involves the heterogeneous nucleation of hydroxyapatite clusters on the surface of the collagen fibrils, which is drastically different than the structure of natural bone. Characterization of collagen fibrils mineralized with and without poly-L-aspartic acid clearly illustrates these structural differences [2,20,27]. Although our results with the PILP process are promising, we have found that thick collagen scaffolds are not homogenously mineralized, with the outer regions being fully mineralized while the inner regions have limited depth of mineral penetration. We believe this occurs due to solidification of the metastable precursor phase once it enters the collagen fibrils, thereby blocking further infiltration of more precursor phase.
The overall objective of this study was to determine if the use of other anionic polymers with the PILP model system could increase the homogeneity, kinetics, and quantity of mineralization of collagen scaffolds. For this study, poly-L-glutamic acid (PGLU), polyvinylphosphonic acid (PVPA), and polyacrylic acid) (PAA) were selected as the polymers to be used individually, and in combination with PASP, for the mineralization of collagen scaffolds through the PILP process. The proposed hypothesis was that the use of different anionic polymers could mimic more efficiently the functions of the NPCs in biomineralization, resulting in an improved in-vitro mineralization of collagen scaffolds compared to mineralization with PASP alone.
PGLU was investigated due to its presence as one of the primary residues of the non-collagenous proteins, as well as its structural similarity to PASP. PGLU differs structurally from polyaspartic acid only in that it has an additional methylene group separating the carboxylate group from the amino acid backbone. Studies of the PGLU homopolymer by Garcia-Ramos and Carmona demonstrated its effect on the nucleation and growth of hydroxyapatite crystals, and the correlation between the polymer structure and crystal lattice [28]. Our view is that this anionic polymer could behave similarly to PASP in sequestering mineral ions to form a precursor phase. Other polyelectrolytes that have been used as biomimetic models of the carboxylated and phosphorylated residues of the NPCs are PAA and PVPA, respectively. Tay and Pashley utilized these polymers in combination to achieve intrafibrillar and interfibrillar mineralization of demineralized human dentin. They theorized that PAA serves as a stabilizer of amorphous nanoprecursors of the calcium phosphate phase, while PVPA facilitated mineralization by templating the binding to collagen [29,30]. This hypothesis is similar to ours with respect to nanoprecursors, except they have found that both polymer analogues are required to cause intrafibrillar mineralization in their system, while we find that PASP alone is effective under our reaction conditions. Nevertheless, this prompted us to consider using a combination of polymers to see if there might be a synergistic effect in our system as well.
The objective of the present study was to investigate the use of various anionic polymers as process-directing agents in the mineralization of collagen scaffolds via the polymer-induced liquid-precursor process. In our prior studies, it was observed that the highest degree of mineralization was obtained with PASP at a concentration of 50 μg/mL, such that the solution remained clear, where the PILP phase was stabilized as nanodroplets roughly 16 nm [20] in size (too small to scatter light). If the precursor droplets aggregate/grow into larger precursors, they tend to densify and precipitate out of solution, and are less effective at mineralizing the collagen. Therefore, to determine which polymer(s) and concentrations would stabilize and control the dimensions of the calcium-phosphate precursor droplets, turbidity measurements of the polymer-containing mineralization solutions were obtained. The polymers forming a stable amorphous precursor phase (i.e., with no solution precipitates) were subsequently used to mineralize collagen scaffolds. Electron microscopy, wide-angle X-ray diffraction, and thermal analysis were used to characterize the resulting hydroxyapatite/collagen structure, mineral content, and mineral penetration depth.
2. Results and Discussion
2.1. Turbidity Assessment of Calcium Phosphate Polymer-Containing Solutions
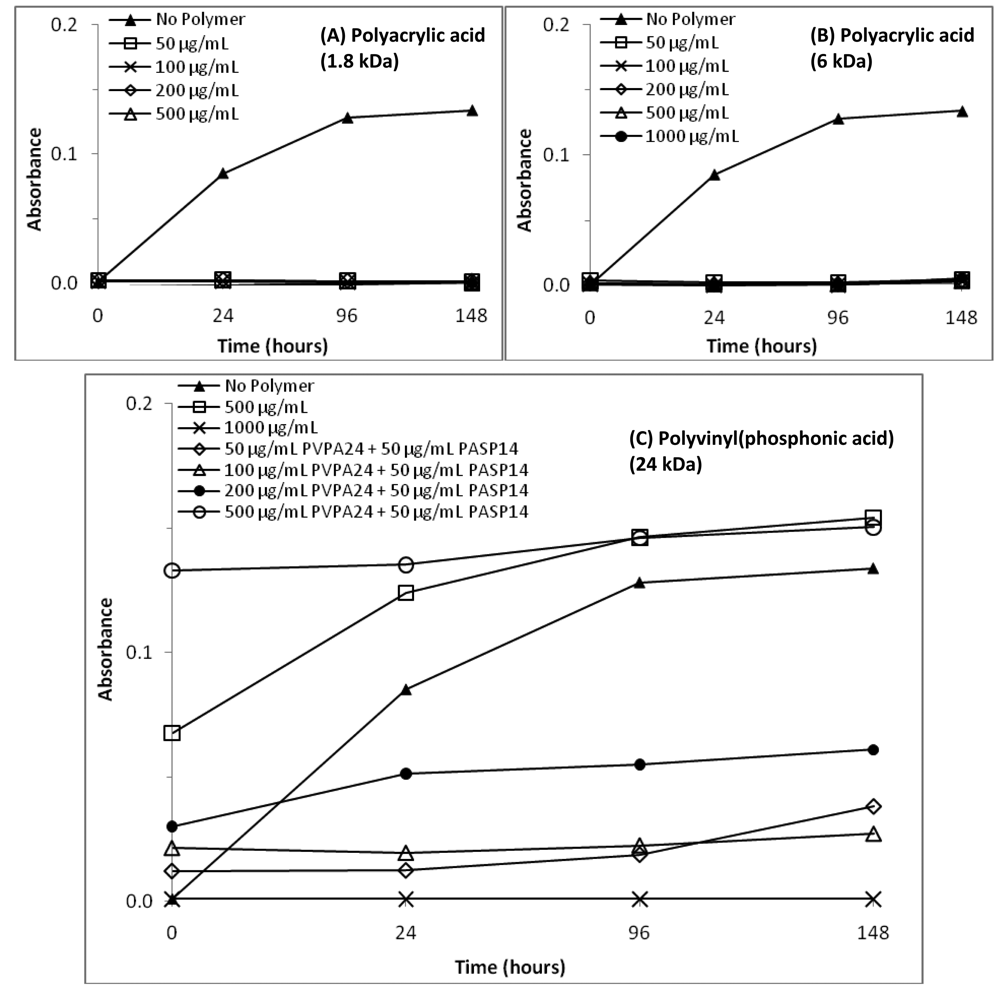
The time-dependent changes in optical density of the polymer-containing calcium phosphate (CaP) solutions are shown in Figures 1 and 2, comparing the poly(amino acids) and vinyl polymers, respectively. Optical density profiles of the solutions were characterized by the increase in turbidity as precipitation started to occur. CaP solution containing no polymer served as the negative control and deionized water was used as the baseline. The overall turbidity of the CaP containing 50 μg/mL polyaspartic acid was lower than that of the control that contained no polymer (Figure 1(A)). The optical density profiles of PASP-containing solutions showed that both the 14- and 27-kDa molecular weight polymers yielded stable (non-precipitated) precursor-containing solutions over a seven-day period. These results were expected based on our prior work with PASP [2,20], and demonstrate the role of PASP as an inhibitor of apatite nucleation and/or growth. The important point though, is that while it is inhibiting nucleation/growth of apatite, it is stabilizing the precursor nanodroplets.
Figures 1(B) and 1(C) show the optical density of solutions containing 15- and 30-kDa polyglutamic acid, respectively. When a concentration range of 50 to 1,000 μg/mL of polyglutamic acid was analyzed, turbidity measurements for the 15-kDa PGLU were lower than the negative control at concentrations below 200 μg/mL. Conversely, turbidity results for the other four 15-kDa GLU concentrations (400, 600, 800 and 1,000 μg/mL) were all higher than that of the control, implying that the role of polyglutamic acid (Mw 15 kDa) as a promoter or inhibitor of mineral precipitation depends on its concentration. In the case of the higher molecular weight polyglutamic acid (30 kDa), turbidity measurements for all concentrations were lower than that of the negative control, suggesting that 30-kDa PGLU is effective at inhibiting mineral precipitation. Optical density profiles of solutions containing combinations of PASP and PGLU at 1:1 ratio were also analyzed, and exhibited turbidity measurements below the negative control (data not shown).
Figures 2(A) and 2(B) show the optical density profiles for the polyacrylic-acid-containing solutions. As can be seen from the data, all the turbidity measurements for the 1.8- and 6-kDa PAA were lower than that of the negative control. All PAA-containing solutions remained stable, non-precipitated for the seven-day period study. The optical density measurements for the PVPA solutions are displayed in Figure 2(C), showing that only solutions containing PVPA at 1,000 μg/mL resulted in no precipitation over seven days. The combination of PVPA and PASP acid did not seem to decrease turbidity of the solutions, suggesting that the stabilizing role of PASP is hindered by the presence of PVPA.
2.2. Mineralization of Collagen Sponges
The polymer-containing CaP solutions with a turbidity profile below that of the negative control (no polymer additive) were used to proceed with collagen scaffold mineralization. Table 1 summarizes the polymer combinations, molecular weights, concentrations and pH values of the solutions utilized in mineralizing collagen scaffolds. Commercially available collagen sponges (Ace Surgical Supply, Inc.) composed of reconstituted bovine type-I collagen were used as substrates for the mineralization experiments. Collagen sponges were mineralized either by the conventional nucleation and growth method, without polymer additive, or by the polymer-induced liquid-precursor (PILP) process developed by Gower and coworkers [2,25] using the polymer(s) from Table 1 as the process-directing agents.
2.2.1. Polyaspartic Acid as Process-Directing Agent
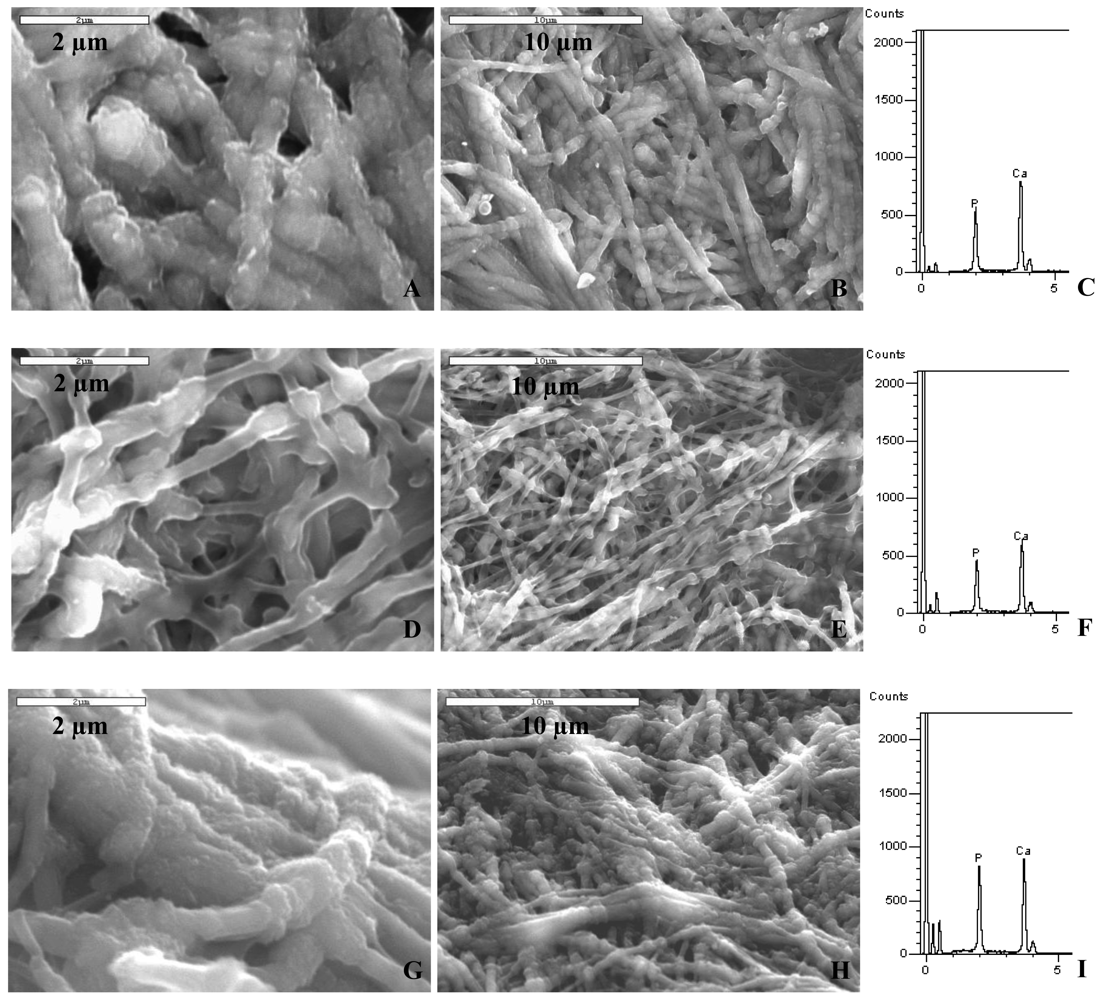
The morphology and elemental composition of each of the mineralized collagen scaffolds was characterized using scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS). Figure 3 shows scanning electron micrographs (SEM) of the sponge mineralized with polyaspartate (14 kDa) to induce the PILP process. The mineralized collagen took on a distinctly different appearance, with physical changes evident even at the macroscopic level. Differences could be seen in the color and rigidity of the sponges, which became white and stiff (while still in solution) as they mineralized. Microscopically, mineralized collagen sponges showed a fibrillar morphology which was very different than the amorphous, gel-like topography of untreated specimens (Figure 4(A)). Mineralized collagen samples developed a pronounced fibrous and rough texture (Figures 3(A) and 3(B)) with no evidence of surface deposits of mineral clusters, as seen in the control reaction (Figure 4(B)). It is interesting to note that something about the mineralization reaction brings out the fibrous texture of the collagen. One possible explanation is that the amorphous gelatinous constituents might dissolve during the time for mineralization. Alternatively, something about the PILP reaction media may cause further fibrillogenesis. The second explanation is supported by the observation that the fibrils have quite large diameters, ranging from a couple hundred nanometers to micron size. This seems to suggest that the PILP media has caused further assembly of multiple collagen fibrils. Figures 3(D) and 3(E) show the inner region of the collagen sponge after three days of reaction. The interior of the sponge (which is 3 mm thick) was obtained by the freeze-fracture technique. The interior of the sponge exhibited a fibrillar and nodular morphology looking slightly different than the topology of the surface.
From previous studies [20], we correlate the fibrillar structure to a more advanced degree mineralization, and the nodular structure to a lower mineral content. At the initial stages of mineralization, collagen fibrils are not fully mineralized and the observed nodules represent the areas within the fibril where mineral is present. Mineral infiltrating the collagen fibril displaces water within the fibril. When samples are dehydrated for SEM examination, the fibril areas infiltrated by the mineral maintain their shape while areas with no mineral collapse, giving the sample a nodular appearance. Once the collagen substrate reaches a high mineral content, mineral penetration throughout the fibril is more homogeneous. When fibrils are fully mineralized, they maintain their shape after dehydration and the nodular structure is no longer observed. In addition, samples from the exterior of the scaffold appeared to have a rough texture, indicating that mineralization might be occurring on the outside of the fibrils as well. These differences in morphology illustrate the progressive intra- and inter-fibrillar mineralization of the samples, where inter-fibrillar mineralization often occurs after the fibrils have become well infiltrated with mineral [27]. EDS data, shown in Figures 3(C) and 3(F), corroborate the observations made from the morphology analysis. Calcium and phosphorous signals were present on the surface of the scaffolds as well as on the inside, but a lower signal intensity of the elements was observed on the inside of the sponge. When more time was allowed for the collagen sponge to become mineralized (6 days), no differences were observed between the surface and the inner regions of the mineralized scaffold (Figures 3(G–I) show the inner region).
When collagen samples were mineralized using the higher molecular weight PASP27 to induce PILP mineralization, the fibrillar structure was observed on the surface as well as on the inner regions of the sponges after three days of reaction (Figures 5(A) and 5(B)). The exterior looked similar to that produced with the PASP14 (Figure 3(A,B)), while the interior seemed to be more uniformly mineralized throughout the fibrils than the interior of the PASP14 (Figures 3(D,E)). Also, EDS analysis determined that the collagen fibrils contained a high amount of calcium and phosphorous when compared to the carbon peak at around 0 keV (Figure 5(C), right). These results were expected since previous studies have shown that higher molecular weight polyaspartic acid appears to stabilize the precursor in the form of nanodroplets that more effectively mineralize the collagen compared to polyaspartates with lower molecular weights [20].
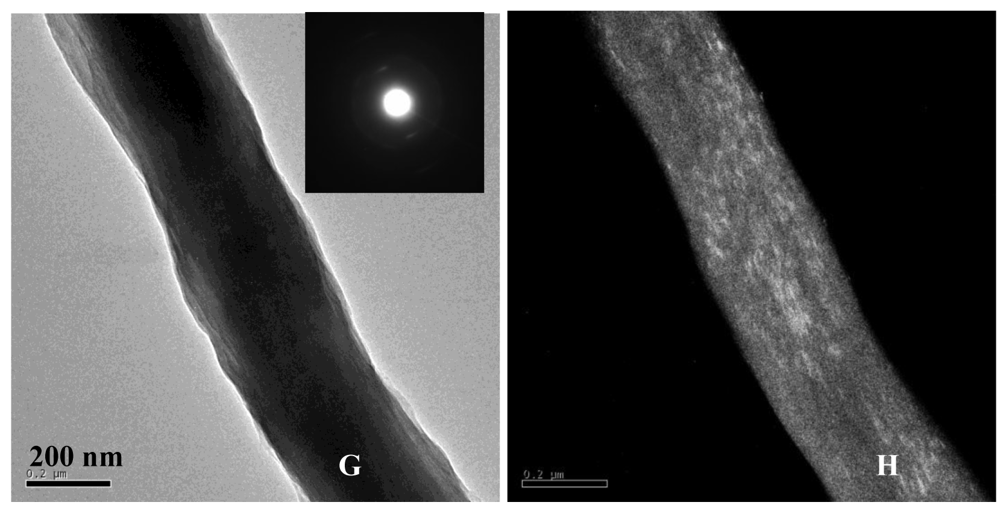
Transmission electron microscopy (TEM) analysis was also used to verify that the mineralized collagen fibrils contained a high degree of intrafibrillar mineral. Bright-field, dark-field and selected area electron diffraction (SAED) modes were used to determine the orientation and phase of the calcium phosphate crystals present in the PILP-mineralized specimens. TEM sample preparation consisted of crushing mineralized collagen sponges in liquid nitrogen to isolate individual fibrils. Figures 5(D) and 5(E) show TEM bright-field and dark-field images of an isolated fibril, respectively. It should be noted that none of the TEM samples were stained with phosphotungstic acid or any other electron-dense stain. Therefore the dark contrast observed in the bright-field image is due to the presence of mineral embedded within the fibril. The fibril in Figure 5(D) looks highly mineralized, which is further corroborated by the dark-field TEM mode of the same fibril, which shows numerous crystals aligned within the fibril (Figure 5(E)). Although a banding pattern can sometimes be seen in lightly mineralized fibrils, it becomes less distinct when there is a high degree of mineralization, as is the case here. SAED was performed on individual fibrils to assess the crystallographic orientation of the intrafibrillar hydroxyapatite crystals (Figure 5(F)). The SAED pattern of the collagen fibril shown in Figure 5(D) demonstrates that the orientation of the hydroxyapatite platelets is in the [001] direction parallel to the long axis of the collagen fibril. In addition, the SAED pattern looks similar to that of native bone [4,24], where tilting and rotational disorder of the crystals create arcs for most of the planes [20,24].
2.2.2. Polyglutamic Acid or Combinations with Polyaspartic Acid as Process-Directing Agent
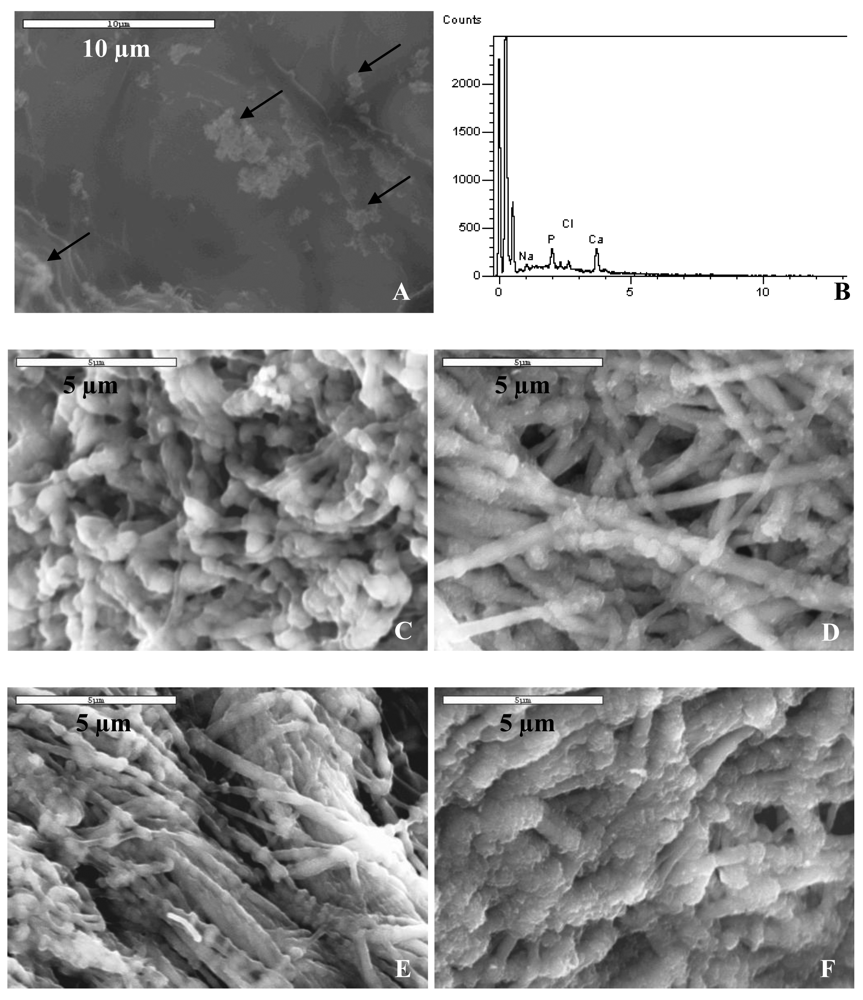
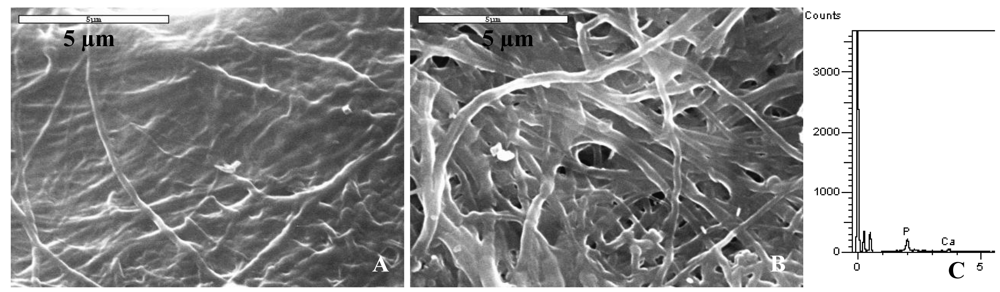
A comparison of the differences observed macroscopically among the collagen sponges mineralized using different polymers as process-directing agents is shown in Table 2. Sponges that were rigid and white upon collection indicated a high content of mineral, while sponges that did not hold their shape and were translucent upon collection indicated that there was little to no mineral present in the sample. When collagen sponges were mineralized with different molecular weights of polyglutamic acid at concentrations of 50, 100 and 200 μg/mL, we noted that precipitation of the mineralization solutions was observed after two days of reaction. These results contradicted the optical density measurements since turbidity profiles for PGLU concentrations below 200 μg/mL (without the collagen sponge) did not differ from that of the baseline signal of deionized water. The morphology of collagen scaffolds mineralized using PGLU was very similar to the morphology of collagen mineralized via the conventional solution crystallization reaction with no polymer additive. Spherulitic clusters of hydroxyapatite crystals were found randomly distributed along the surface of the collagen substrate treated with PGLU (Figure 6(A)). In addition, the EDS spectrum for PGLU treated samples did not show strong calcium and phosphorous peaks (Figure 6(B)).
Figures 6(C–F) show SEM images of the inner-sections of PILP-mineralized collagen samples using combinations of poly-L-aspartic and poly-L-glutamic acid as the process-directing agents. The images are all taken from the inside of the samples to demonstrate that the mineralization was not only near the surface of the scaffold, but penetrated the entire thickness of the sample. The mineralized collagen sponges treated with PGLU-PASP presented a morphology similar to the samples treated with the high molecular weight PASP, where a fibrillar structure was observed with no difference between the inside and outside (not shown) of the sample. In addition, several of the samples appeared to have a rough surface indicating that mineralization might be occurring on the outside of the fibrils as well. The EDS spectrums for PGLU-PASP treated samples showed strong calcium and phosphorous peaks, which are consistent with the high degree of mineralization (data not shown). Figures 6(G) and 6(H) show TEM bright- and dark-field images of a mineralized collagen fibril treated with the PGLU-PASP combination. As in the case of fibrils mineralized with PASP alone, the high z-contrast observed in the TEM image could be attributed to the presence of hydroxyapatite crystals embedded within the collagen fibril. Also, the SAED pattern of the mineralized fibril (Figure 6(G), inset) shows an orientation of the HA crystals very similar to bone′s, which are illuminated in the dark-field image.
2.2.3. Polyvinylphosphonic or Polyacrylic Acid as Process-Directing Agent
Regarding polyvinylphosphonic and polyacrylic acid as process-directing agents, the PVPA and PAA treated samples showed little to no mineral content, illustrated by a mixture of fibrous and amorphous appearing collagen (Figure 7(A)), which had a drastically different appearance than that of the mineralized fibrils of the PASP and PASP-PGLU samples. The EDS spectrums for these samples showed insignificant calcium and phosphate peaks, indicating a low mineral content. To determine if the use of PASP could enhance the mineralization of collagen when using PVPA and PAA, a combination of the polymers was used as the process directing agent. Figure 7(B) shows SEM of the surface of a collagen sponge mineralized using a combination of PVPA and PASP. The mixture of polymers did not improve the mineralization of collagen, as it did for the case of the polyaspartic and polyglutamic acid mixture. The topology of the PVPA-PASP treated sample was similar to the sample treated with just PVPA, although there appears to be less amorphous collagen. The EDS spectra for PVPA-PASP treated samples showed very low levels of calcium and phosphorous, with an insignificant Ca peak and a small P peak, which is probably from the PVPA (Figure 7(C)).
2.3. Wide Angle X-ray Diffraction (WAXD) Analysis
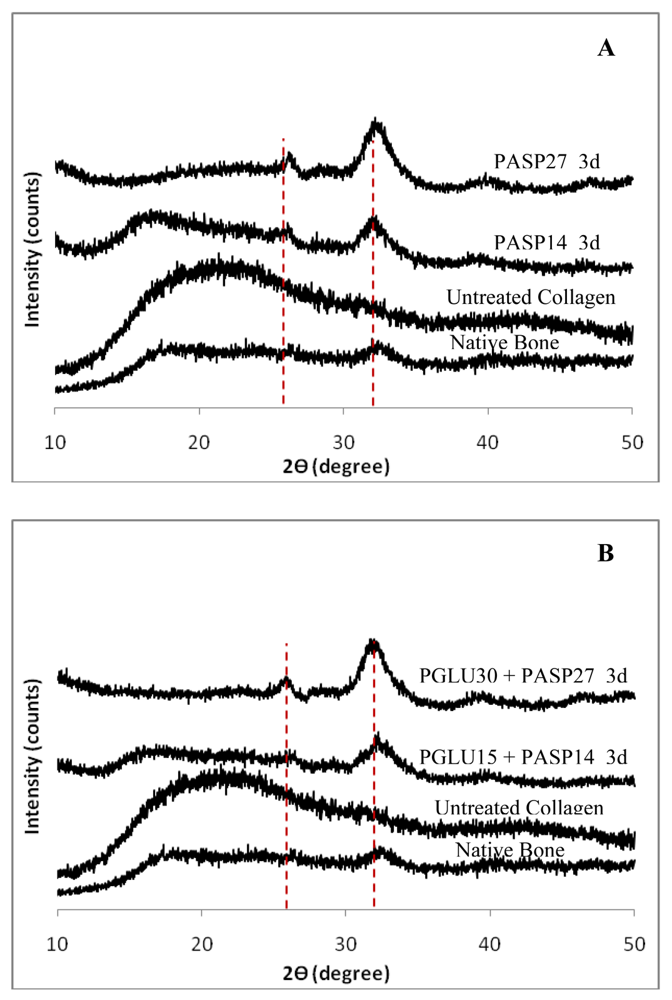
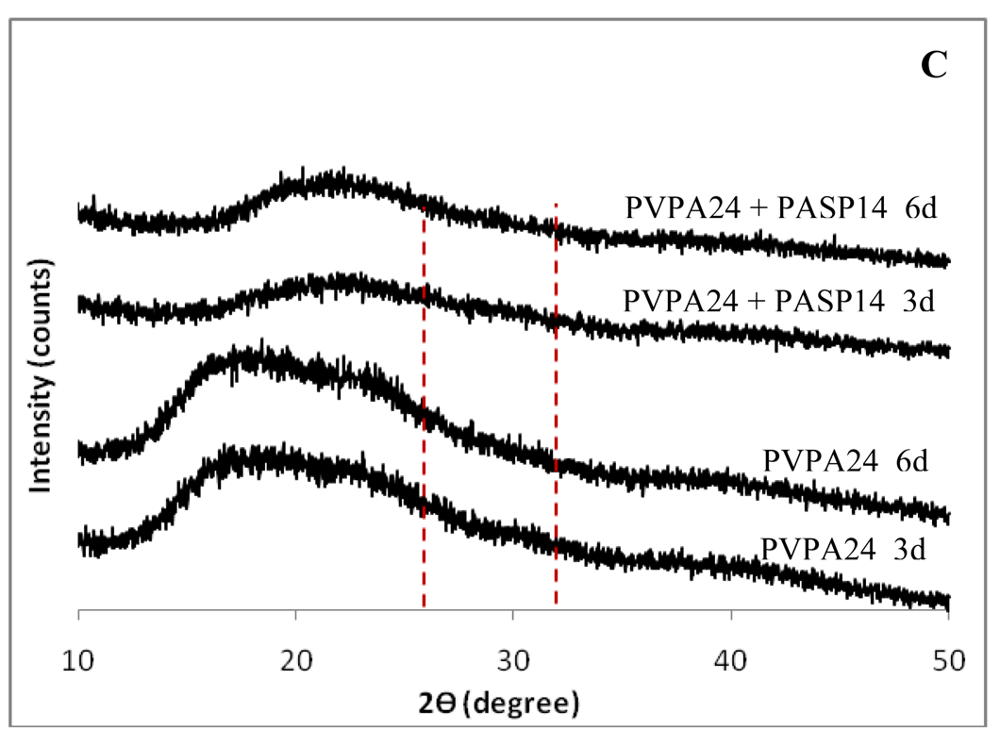
The wide angle X-ray diffraction (WAXD) results for each of the samples are shown in Figure 8. The sponges mineralized with 50 μg/mL PASP at both molecular weights, as well as the higher and lower molecular weight combinations of 50 μg/mL PGLU with 50 μg/mL PASP, show peaks characteristic of hydroxyapatite. In past studies, undesirable calcium phosphate mineral phases, such as brushite, have occasionally been observed in mineralized collagen scaffolds, but the WAXD data showed that none of the samples from this investigation exhibited those phases. After 3 days of reaction, broad peaks of hydroxyapatite were observed at around 26° and 32°, corresponding to the (002) and the combination of overlapping (211), (112), (300) peaks, respectively (Figures 8(A) and 8(B)). The phase can be further verified correlating the spacings and angles in the SAED patterns, as described in our prior report [2]. This WAXD pattern of PILP mineralized collagen using PASP or the combination of PASP and PGLU exhibits similar peak widths to those seen in the WAXD patterns of bone. The (211), (112), (300) peaks of carbonated hydroxyapatite in bone are normally overlapped into one broad peak due to the extremely small crystal size and defects such as carbonate, impurities and calcium vacancies [31-36].
The remainder of the collagen scaffolds showed no mineral presence even after six days of mineralization. The nodular and fibrillar appearance, combined with the EDS results and WAXD analyses, correlate to the presence of hydroxyapatite in the samples after three days of mineralization with PASP and the PASP-PGLU combination. Furthermore, given that hydroxyapatite clusters were not observed on the surface of collagen sponges, as seen in samples treated without the polymer additive, the large calcium and phosphorous peaks of the EDS spectra indicate that the mineral must be within the scaffold. The broad WAXD peaks, along with the TEM analysis, suggest that the majority of the hydroxyapatite crystals are embedded within the collagen fibrils (intrafibrillar), which is further supported by the TG/DTA data discussed below.
2.5. Thermogravimetric and Differential Thermal Analysis (TG/DTA)
Following examination of the hydroxyapatite mineral using WAXD, the amount of mineral was determined using thermogravimetric analysis (TGA). Figure 9 shows a drastic difference in mineral content between the samples mineralized using PASP alone or the PASP-PGLU combination, both of which had approximately 50% (w/w) mineral after 3 days, compared to the PVPA and PAA samples with 5 and 0% mineral (not shown), respectively. After six days of mineralization, the PASP and PASP-PGLU samples exhibited very high mineral content at around 60 to 65% (w/w) mineral, but there was not a distinguishable difference between the mineralization using polyaspartic acid alone compared to polyaspartic in combination with polyglutamic acid. The mineral content increase could be correlated to the mineralization progression through to the inner regions of the scaffold, as observed in the SEM images described earlier. We presume the increased mineralization relies on the time it takes for the amorphous precursor nano-droplets to diffuse through solution before they can reach the collagen scaffold and then absorb into the collagen fibrils.
In addition to TGA, differential thermal analysis (DTA) of the mineralized collagen sponges was performed. Thermal stability of collagen is affected by the presence of intrafibrillar mineral, and it can be observed by changes of the DTA peaks. Figure 10 displays the DTA curves for untreated and PILP-mineralized collagen sponges. According to the literature, exothermic peaks in the range of 260–360 °C correspond to collagen decomposition, while exothermic peaks in the range of 450–550 °C correspond to collagen combustion [38]. In the case of the untreated collagen sponge, the high temperature peak observed at around 500 °C is typical of non-mineralized collagen and is found in both biological and reconstituted collagen [38,39]. On the other hand, DTA measurements for the PILP-mineralized specimens showed a shift of the high temperature combustion peak to lower temperatures of approx. 320 °C. The altered thermal behavior of the mineralized specimens is comparable to the thermal behavior of collagen in bone and dentin, showing very similar DTA peaks [38]. This change in thermal stability is thought to be attributed to the intimate structural relationship between hydroxyapatite and the collagen fibrils, as seen in native bone and dentin. Therefore, thermal analysis of PILP mineralized specimens is another tool (in addition to TEM analysis) for discriminating intrafibrillar mineralization, where hydroxyapatite crystals embedded within the fibrils are intimately associated and alter its thermal stability.
The discrepancies in mineral content and degree of mineralization observed in collagen sponges mineralized with different polymers could possibly be explained by full inhibition of the formation of the amorphous precursor [23], or by variations of the physicochemical characteristics of the precursor. The use of polyvinylphosphonic acid and polyacrylic acid in stable solution concentrations, as observed by the optical density profiles, resulted in little to no mineralization of the collagen scaffolds and inhibited mineralization when used in combination with polyaspartate. This may suggest that the inhibitory activity of these polymers was too large, which even inhibited the formation of the amorphous precursor, particularly for the case of the PVPA, which was added at very high concentration (where we were striving to form a stable solution). Unfortunately, PVPA did not behave as a functional biomimetic analogue of phosphorylated proteins under the conditions studied. Other possible explanations include: (1) PVPA containing a phosphonate group rather than phosphate, (2) no linker between the phosphonate and the polymer backbone compared to the CH2–O– linker in phosphorylated serine, (3) hydrophilicity difference between the backbone of vinyl polymers and the amide backbone in proteins, and (4) changes in the isoelectric point of collagen due to electrolyte condition differences in the presence of the polymer [40]. In contrast, Kim et al. found that a combination of PVPA and PAA, the two polymers that didn't work here, worked well in their system, which utilized a different approach to generating the reactant ions (calcium release from a Portland cement-containing resin composite combined with a phosphate-containing fluid) [30]. Thus, it seems that a synergistic effect can only be obtained with a certain set of reaction conditions.
Although polyacrylic acid has been used as an analog of the carboxylated residues of NCPs [29,37], it did not yield mineralization of the collagen substrates under the conditions tested in this study. Differences in molecular weight and in ion concentrations might explain these results, where the molecular weights used in these studies were on the low side (1.8 and 6-KDa). The polymer net surface charge could change as the molecular weight changes, affecting the amorphous precursor formation. Furthermore, alterations in the formation of amorphous precursor, including size and surface charge, could influence its infiltration within the collagen matrix. Polyglutamic acid alone did not produce mineralization of collagen sponges, while the addition of poly-L-aspartic acid with poly-L-glutamic acid yielded intra- and inter-fibrillar mineralization of collagen sponges. An advantage of using a mixture of the polyelectrolytes over polyaspartate alone could not be determined from these results.
2.6. Mineralization of Rat Tail Tendon and Demineralized Manatee Bone
Previous studies have demonstrated that it is difficult to reach large depths of penetration into denser scaffolds that are mineralized through the PILP process. Therefore, dense collagen scaffolds, such as rat tail tendon and demineralized manatee bone, might be expected to show a distinguishable difference between mineralization using PASP versus PASP in combination with PGLU. The manatee bone was pre-treated in an EDTA solution to remove the native mineral that was present in the samples. SEM and TGA experiments were performed to analyze the surface morphology and mineral content, respectively. The results shown in Figure 11 are fairly narrowly distributed with a mineral content of 65% (w/w) and 35% (w/w) for the rat tail tendon (Figure 11(A)) and the demineralized manatee bone substrates (Figure 11(B)), respectively. The polyaspartic acid alone appears to have been a little more effective in both systems.
The amount of mineral present in the rat tail tendon samples represented the maximum amount of mineral available in the PILP solution. Conversely, the mineral content for the remineralized manatee bone samples was small and below the theoretical value for cortical bone [1,41,42], even though enough mineral was available in the PILP solution to restore the mineral value of bone. It is possible that the collagen is crosslinked in the bone specimen, which may make it more difficult to mineralize. In any case, no significant improvement in the mineral content of the dense scaffolds was observed by the addition of polyglutamic acid. Although polyaspartic and polyglutamic acid constitute the two primary residues of non-collagenous proteins, using both polyelectrolytes to induce the PILP process did not enhance the mineralization of collagen in vitro, at least not under the conditions employed.
3. Conclusions
In this study, different polyanionic polymers, namely poly-L-glutamic acid, polyvinylphosphonic acid, polyacrylic acid, and combinations of these with poly-L-aspartic acid were analyzed concerning their mineralization-mediating abilities on collagen sponges. The use of PVPA and PAA in stable (non-precipitating) concentrations in the reactant solutions resulted in little to no mineralization of the collagen scaffolds, and inhibited mineralization when used in combination with PASP.
The study also revealed that mineralization of collagen sponges was only obtained when using poly-L-aspartic acid, or combinations of it with different ratios of poly-L-glutamic acid. PGLU alone did not yield mineralization of the collagen sponges, but combinations of PGLU and PASP yielded scaffolds with high mineral content (65% w/w). A distinguishable improvement in collagen mineralization from using PASP and PGLU combinations versus using PASP alone could not be determined, even when dense collagen substrates such as rat tail tendon and demineralized manatee bone were used. These results demonstrate that 27-KDa poly-L-aspartic acid alone, under this set of in vitro mineralization conditions, was the most successful at mimicking the role of the acidic proteins present during bone formation.
4. Experimental Section
4.1. Turbidity Assessment of Calcium Phosphate Polymer-Containing Solutions
The effects of different concentrations and molecular weight of four polymer additives in the stability of the calcium phosphate amorphous precursor was examined. Poly-L-aspartic acid sodium salt (Mw: 14,000 and 27,000 Da; Alamanda Polymers, Huntsville, AL) at a 50 μg/mL concentration; poly-L-glutamic acid sodium salt (Mw: 15,000 and 30,000 Da; Alamanda Polymers, Huntsville, AL) at 50, 100, 200, 400, 600, 800 and 1,000 μg/mL concentrations; polyacrylic acid (1,800 and 6,000 Da; Sigma, St. Louis, MO) at 50, 100, 200, 500 and 1,000 μg/mL concentrations; polyvinyl phosphonic acid (24,000 Da; Polysciences, Warrington, PA) at 50, 100, 200, 500 and 1,000 μg/mL concentrations; and combinations of polyaspartic acid with all the other polymers were examined. Solutions of 4.5 mM CaCl2•2H2O (Sigma, St. Louis, MO), 2.1 mM K2HPO4 (Sigma, St. Louis, MO), 0.02% (w/v) sodium azide (Sigma, St. Louis, MO) and polymer in Tris buffer saline (TBS) were prepared in 20 mL amounts. The polymer combinations, concentrations, and molecular weights were varied for each of the solutions while all other conditions were held constant. The solutions were placed in an incubator at 32 °C for a seven day period, and optical density measurements at a fixed wavelength of 650 nm were taken at 2 hours, 24 hours, 4 days, and 7 days. The solution conditions which remained stable (did not precipitate) over the seven day period were used to mineralize collagen scaffolds.
4.2. Mineralization of Collagen Sponges
Synthetic collagen sponges were purchased from Ace Surgical Supply, Inc. (Brockton, MA). The collagen sponges had a rectangular shape of 40 × 20 mm and 3-mm thickness. According to the company specifications, collagen sponges were composed of reconstituted type-I collagen from bovine tendon and had 90% porosity. Prior to mineralization, samples were prepared by cutting the collagen sponges into a 10 × 10 mm square shape and pre-washing them with de-ionized (dH2O) water to remove any salts or chemical agents introduced during fabrication. Sponge scaffolds were mineralized with calcium phosphate (CaP) via the polymer-induced liquid-precursor (PILP) process. In this study, the mineralization solution was prepared by mixing equal volumes of 9 mM CaCl2•2H2O and 4.2 mM K2HPO4 solutions. To maintain the pH of the mineralization solution at 7.4, calcium and potassium solutions were made in Tris-buffered saline (TBS).
Poly-aspartates with different molecular weights were used as the PILP process-directing agent at a 50-μg/mL concentration. Polyglutamic (50, 100, 200 μg/mL), polyacrylic (50 μg/mL), and polyvinyl phosphonic acid (1,000 μg/mL), as well as combinations of these polymers with polyaspartic acid (50 μg/mL), were also used to mineralized collagen sponges. The polyelectrolyte or combination of polyelectrolytes was added to 250 mL of calcium solution before mixing an equal volume of the phosphate counterion solution. Collagen sponges were incubated in the mineralization solution under vacuum conditions for 30 minutes to remove any air bubbles trapped in the pores of the sponge. After degassing the samples, the mineralization reaction was kept in a 37 °C oven to emulate physiological conditions. A control reaction, with no polymer additive, was run for each set of experiments. At predetermined reaction times, mineralized samples were removed from the solution, copiously washed with de-ionized water, lyophilized and stored at −20 °C until use.
4.3. Wide Angle X-ray Diffraction (WAXD) Analysis
WAXD analysis was used to determine the crystallographic phase of the samples (untreated and mineralized). To observe the major hydroxyapatite peaks, which develop at around 26° (002) and 32° (combination of (211), (112) and (300)), the samples were scanned with Cu-Kα X-ray radiation from a Philips XRD ADP 3720 Diffractometer at 40 KV and 20 mA, using a step size of 0.01θ mrad/s with a time of 1.25 s/step, over a 2θ range of 24–40θ.
4.4. Scanning Electron Microscopy (SEM) Analysis
Lyophilized samples were mounted on an aluminum stub covered in double-sided copper tape, and then sputter coated with amorphous carbon. The surface morphology of mineralized samples was then analyzed using a 6400 JEOL SEM instrument equipped with an energy dispersive spectrometer, at an accelerating voltage of 15 kV. For elemental analysis of mineralized samples, energy dispersive x-ray spectroscopy (EDS) analysis was done during SEM examination.
4.5. Transmission Electron Microscopy (TEM) Analysis
Nano-structural analysis of mineralized samples was performed on a 200CX JEOL TEM instrument with an accelerating voltage of 200 kV. To determine the crystallographic orientation of the embedded hydroxyapatite nanocrystals, bright field (BF) and selected area electron diffraction (SAED) modes were used. For TEM analysis, samples were pulverized in liquid nitrogen, dispersed in methanol and added dropwise onto a copper TEM grid.
4.6. Thermogravimetric and Differential Thermal Analysis (TG/DTA)
To determine the degree of mineralization of the samples, TG/DTA analyses were conducted using a TG/DTA 320 (Seiko, Thermo Haake, Germany) instrument. A heating rate of 5 °C/min was applied in the temperature range of 30–800 °C under air at a flow rate of 100 mL/min. For TG/DTA analysis, samples were pulverized with a mortar and pestle while frozen in liquid nitrogen, and then air-dried for 24 hours. The amount of sample examined was between 10–20 mg and alumina powder was used as the standard. To compare the degree of mineralization, the material remaining at 600 °C was interpreted as the mineral content since the organic portion of the samples should be totally combusted by 600 °C in an oxygen-containing environment.
4.6. Mineralization of Rat Tail Tendon and Demineralized Manatee Bone
Rat tail tendons were harvested from 3-month-old Sprague Dawley rats. Manatee bone samples were kindly donated by J. Mecholsky from the Department of Materials Science and Engineering at the University of Florida. The samples used were rib bones composed of solid cortical bone [43,44]. Prior to demineralization, bone samples were cut into rectangular strips of 40 × 3 × 0.5 mm using a wet diamond saw (Exakt Technologies, Hamburg, Germany). Demineralization of bone pieces was carried out in a 0.5 M EDTA solution (Acros Organics, Morris Plains, NJ, pH adjusted to 8.0 with NaOH) containing 0.02% (w/v) sodium azide (Sigma, St. Louis, MO) to avoid bacterial contamination. Bone pieces as well as tendons were incubated in the demineralization medium while stirring at room temperature for 72 hours. After demineralization, samples were taken out from the demineralization solution, washed six times with large amounts of ultrapure water to remove all traces of EDTA, lyophilized and stored at −20 °C until use.
Rat tail tendon and demineralized bone samples were mineralized with calcium phosphate (CaP) via the polymer-induced liquid-precursor (PILP) process. The same recipe for the mineralization solution applied to collagen sponges was used for the natural scaffolds. To determine the influence of using a mixture of polyaspartate and polyglutamate in the mineralization of collagen, 27-KDa poly-aspartic acid and combinations of low and high molecular weights of polyaspartic and polyglutamic acid were used as the PILP process-directing agent at a 100-μg/mL concentration. Substrates were incubated in the mineralization solution under vacuum conditions for 30 minutes to remove any air bubbles potentially trapped in the pores of the specimens. After degassing the samples, the mineralization solution was kept in a 37 °C oven to emulate physiological conditions. A control reaction, with no polyaspartate additive, was run for each set of experiments. After seven days, mineralized samples were removed from the solution, copiously washed with de-ionized water, lyophilized and stored at −20 °C until further use.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer 1 | Concentration (μg/mL) | Polymer 2 | Concentration (μg/mL) | pH of Solution at day 0 | pH of Solution at day 6 |
|---|---|---|---|---|---|
| PASP14 | 50 | 7.4 | 7.2 | ||
| PASP14 | 50 | PGLU15 | 50 | 7.4 | 7.2 |
| PASP27 | 50 | 7.4 | 7.2 | ||
| PASP27 | 50 | PGLU30 | 50 | 7.4 | 7.2 |
| PGLU15 | 50, 100, 200 | - | - | ||
| PGLU30 | 50, 100, 200 | - | - | ||
| PAA1.8 | 50 | 7.3 | 7.2 | ||
| PASP14 | 50 | PAA1.8 | 50 | - | - |
| PAA6 | 50 | - | - | ||
| PVPA24 | 1,000 | 7.3 | 7.0 | ||
| PASP14 | 50 | PVPA24 | 1,000 | - | - |
| Polymer Combination | Sponge at Collection |
|---|---|
| PASP14 | Rigid, white |
| PASP27 | Rigid, white |
| PGLU15 | Didn't hold shape, translucent (solution precipitated) |
| PGLU30 | Didn't hold shape, translucent (solution precipitated) |
| PASP14 – PGLU15 | Rigid, white |
| PASP27 – PGLU30 | Rigid, white |
| PAA1.8 | Didn't hold shape, translucent |
| PAA1.8 – PASP14 | Didn't hold shape, translucent |
| PAA6 | Didn't hold shape, translucent |
| PVPA24 | Didn't hold shape, translucent |
| PVPA24 – PASP14 | Didn't hold shape, translucent |
Acknowledgments
We are grateful to John Mecholsky (Materials Science and Engineering Department, University of Florida) for the donation of the manatee bone specimens. We also would like to thank the Major Analytical Instrumentation Center, Department of Materials Science and Engineering, University of Florida for the assistance in sample preparation and analysis. This work was supported in part by NSF Grants BES-0404000 and DMR-0710605, and NIH grant RO1 DE16849.
References
- Weiner, S.; Wagner, H.D. The material bone: Structure mechanical function relations. Annu. Rev. Mater. Sci. 1998, 28, 271–298. [Google Scholar]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y-Y; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. R-Rep. 2007, 58, 77–116. [Google Scholar]
- Weiner, S.; Traub, W. Organization of crystals in bone. In Mechanisms and Phylogeny of Mineralization in Biological Systems; Suga, S., Nakahara, H., Eds.; Springer-Verlag: New York, NY, USA, 1991; pp. 247–253. [Google Scholar]
- Landis, W.J.; Song, M.J.; Leith, A.; McEwen, L.; McEwen, B.F. Mineral and organic matrix interaction in normally calcifying tendon visualized in three dimensions by high-voltage electron microscopic tomography and graphic image reconstruction. J. Struct. Biol. 1993, 110, 39–54. [Google Scholar]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar]
- Olszta, M.; Gajjeraman, S.; Kaufman, M.; Gower, L.B. Nanofibrous calcite synthesized via a solution-precursor-solid mechanism. Chem. Mater. 2004, 16, 2355–2362. [Google Scholar]
- Cheng, X.; Gower, L.B. Molding mineral within microporous hydrogels by a polymer-induced liquid-precursor (PILP) process. Biotechnol. Prog. 2006, 22, 141–149. [Google Scholar]
- Amos, F.F.; Dai, L.; Khan, S.R.; Gower, L.B. Mechanism of formation of concentrically laminated spherules: Implication to Randall's plaque and stone formation. Urol. Res. 2009, 37, 11–17. [Google Scholar]
- Amos, F.F.; Olszta, M.J.; Khan, S.R.; Gower, L.B. Biomineralization—Medical Aspects of Solubility; Königsberger, E., Königsberger, L., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 125–217. [Google Scholar]
- Amos, F.F.; Sharbaugh, D.M.; Talham, D.R.; Gower, L.B.; Fricke, M.; Volkmer, D. Formation of single-crystalline aragonite tablets/films via an amorphous precursor. Langmuir 2006, 23, 1988–1994. [Google Scholar]
- Kim, Y.-Y.; Douglas, E.P.; Gower, L.B. Patterning inorganic (CaCO3) thin films via a polymer-induced liquid-precursor process. Langmuir 2007, 23, 4862–4870. [Google Scholar]
- Fisher, L.W.; Torchia, D.A.; Fohr, B.; Young, M.F.; Fedarko, N.S. Flexible structures of SIBLING proteins, bone sialoprotein, andosteopontin. Biochem. Biophys. Res. Commun. 2001, 280, 460–465. [Google Scholar]
- Sreenath, T.; Thyagarajan, T.; Hall, B.; Longenecker, G.; D'Souza, R.; Hong, S.; Wright, J.T.; MacDougall, M.; Sauk, J.; Kulkarni, A.B. Dentin sialophosphoprotein knockout mouse teeth display widened predentin zone and develop defective dentin mineralization similar to human dentinogenesis imperfect-III. J. Biol. Chem. 2003, 278, 24874–24880. [Google Scholar]
- Xiao, S.; Yu, C.; Chou, X.; Yuan, W.; Wang, Y.; Bu, L.; Fu, G.; Qian, M.; Yang, J.; Shi, Y.; Hu, L.; Han, B.; Wang, Z.; Huang, W.; Liu, J.; Chen, Z.; Zhao, G.; Kong, X. Dentinogenesis imperfect 1 with or without progressive hearing loss associated with distinct mutations in DSPP. Nat. Genet. 2001, 27, 201–204. [Google Scholar]
- Xu, T.; Bianco, P.; Fisher, L.W.; Longenecker, G.; Smith, E.; Goldstein, S.; Bonadio, J.; Boskey, A.; Heegaard, A.M.; Sommer, B.; Satomura, K.; Dominguez, P.; Zhao, C.; Kulkarni, A.B.; Robey, P.G.; Young, M.F. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 1998, 20, 78–82. [Google Scholar]
- Zhang, X.; Zhao, J.; Li, C.; Gao, S.; Qiu, C.; Liu, P. DSPP mutation in dentinogenesis imperfecta Shields type II. Nat. Genet. 2001, 27, 151–152. [Google Scholar]
- Qin, C.; Baba, O.; Butler, W.T. Post-translational modifications of SIBLING proteins and their roles in osteogenesis and dentinogenesis. Crit. Rev. Oral Biol. Med. 2004, 15, 126–136. [Google Scholar]
- Gower, L.B.; Odom, D.J. Deposition of calcium carbonate films by a polymer-induced liquid-precursor (PILP) process. J. Crystal Growth 2000, 210, 719–734. [Google Scholar]
- Dai, L.; Douglas, E.P.; Gower, L.B. Compositional analysis of a polymer-induced liquid-precursor (PILP) amorphous CaCO3 phase. J. Non-Crystal. Solids 2008, 354, 1845–1854. [Google Scholar]
- Jee, S.-S.; Thula, T.T.; Gower, L.B. Development of bone-like composites via the polymer-induced liquid-precursor (PILP) process: 1) Influence of polymer molecular weight. Acta Biomater. 2010, 6, 3676–3686. [Google Scholar]
- Ben-Bassat, H.; Klein, B.Y.; Leichter, I.; Liebergall, M.; Segal, D.; Kahana, F.; Sarig, S. Novel bone graft substitute with composition mimicking bone mineral. In Biomaterials Engineering and Devices: Human Applications; Wise, D.L., Trantolo, D.J., Lewandrowski, K-U, Gresser, J.D., Cattaneo, M.V., Yaszemski, M.J., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 2, pp. 155–169. [Google Scholar]
- Sarig, S. Aspartic acid nucleates the apatite crystallites of bone: A hypothesis. Bone 2004, 35, 108–109. [Google Scholar]
- Chi, C.; Shi, Y.; Zheng, H.; Zhang, Y.; Chen, W.; Yang, W.; Tang, Y. Biomineralization process of calcium phosphate: Modulation of the poly-amino acid with different hydroxyl/carboxyl ratios. Mater. Chem. Phys. 2009, 115, 808–809. [Google Scholar]
- Olszta, M.J.; Douglas, E.P.; Gower, L.B. Materials Research Society Symposium: Materials Inspired by Biology; Thomas, J., Kiick, K., Gower, L., Eds.; MRS: San Francisco, CA, USA, 2003; pp. 127–134. [Google Scholar]
- Olszta, M.J.; Douglas, E.P.; Gower, L.B. Scanning electron microscopic analysis of the mineralization of type I Collagen via a polymer-induced liquid-precursor (PILP) process. Calcified Tissue Int. 2003, 72, 583–591. [Google Scholar]
- Olszta, M.J.; Odom, D.J.; Douglas, E.P.; Gower, L.B. A new paradigm for biomineral formation: Mineralization via an amorphous liquid-phase precursor. Connect. Tissue Res. 2003, 44, 326–334. [Google Scholar]
- Jee, S.S.; Culver, L.; Li, Y.; Douglas, E.P.; Gower, L.B. Biomimetic mineralization of collagen via an enzyme-aided PILP process. J. Crystal Growth 2010, 312, 1249–1256. [Google Scholar]
- Garcia-Ramos, J.V.; Carmona, P. The effect of some homopolymers on the crystallization of calcium phosphates. J. Crystal Growth 1982, 57, 336–342. [Google Scholar]
- Tay, F.R.; Pashley, D.H. Guided remineralisation of partially demineralised human dentine. Biomaterials 2007, 29, 1127–1129. [Google Scholar]
- Kim, Y.K.; Gu, L.S.; Bryan, T.E.; Kim, J.R.; Chen, L.A.; Liu, Y.; Yoon, J.C.; Breschi, L.; Pashley, D.H.; Tay, F.R. Mineralisation of reconstituted collagen using polyvinylphosphonic acid/polyacrylic acid templating matrix protein analogues in the presence of calcium, phosphate and hydroxyl ions. Biomaterials 2010, 31, 6618–6627. [Google Scholar]
- LeGeros, R.Z. Calcium Phosphates in Oral Biology and Medicine; S Karger Pub: Basel, Switzerland, 1991. [Google Scholar]
- Dorozhkin, S.V. Calcium orthophosphates in nature, biology and medicine. Materials 2009, 2, 399–498. [Google Scholar]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Glimcher, M.J. The nature of the mineral phase in bone: Biological and clinical implications. In Metabolic Bone Disease and Clinically Related Disorders; Avioli, L.V., Krane, S.M., Eds.; Academic Press: San Diego, CA, USA, 1998; pp. 23–50. [Google Scholar]
- Kikuchi, M.; Itoh, S.; Ichinose, S.; Shinomiya, K.; Tanaka, J. Self-organization mechanism in a bone-like hydroxyapatite/collagen nanocomposite synthesized in vitro and its biological reaction in vivo. Biomaterials 2001, 22, 1705–1711. [Google Scholar]
- Murugan, R.; Ramakrishna, S. Crystallographic study of hydroxyapatite bioceramics derived from various sources. Crystal Growth Des. 2005, 5, 111–112. [Google Scholar]
- He, G.; Gajjeraman, S.; Schultz, D.; Cookson, D.; Qin, C.; Butler, W.T.; Hao, J.; George, A. Spatially and temporally controlled biomineralization is facilitated by interaction between self-assembled dentin matrix protein 1 and calcium phosphate nuclei in solution. Biochemistry 2005, 44, 16140–16148. [Google Scholar]
- Lozano, L.F.; Pena-Rico, M.A.; Heredia, A.; N-Flores, J.O.; Velazquez, R.; Belio, I.A.; Bucio, L. Thermal analysis study of human bone. J. Mater. Sci. 2003, 38, 4777–4782. [Google Scholar]
- Bigi, A.; Ripamonti, A.; Cojazzi, G.; Pizzuto, G.; Roveri, N.; Koch, M.H.J. Structural analysis of turkey tendon collagen upon removal of the inorganic phase. Int. J. Biol. Macromol. 1991, 13, 110–114. [Google Scholar]
- Li, Y.; Asadi, A.; Monroe, M.; Douglas, E.P. pH effects on collagen fibrillogenesis in vitro: Electrostatic interactions and phosphate binding. Mater. Sci. Eng. C 2009, 29, 1643–1649. [Google Scholar]
- Baer, E.; Cassidy, J.J.; Hiltner, A. Hierarchical structure of collagen composite systems: Lessons from biology. In Biomimetics—Design and Processing of Materials; Sarikaya, M., Aksay, I.A., Eds.; AIP Press: Woodbury, NY, USA, 1995; pp. 13–34. [Google Scholar]
- Glimcher, M.J. Molecular biology of mineralized tissues with particular reference to bone. Rev. Mod. Phys. 1959, 31, 359–393. [Google Scholar]
- Clifton, K.B.; Reep, R.L.; Mecholsky, J.J., Jr. Quantitative fractography for estimating whole bone properties of manatee rib bones. J. Mater. Sci. 2008, 43, 2026–2034. [Google Scholar]
- Clifton, K.B.; Yan, J.; Mecholsky, J.J., Jr.; Reep, R.L. Material properties of manatee rib bone. J. Zool. 2008, 274, 150–159. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thula, T.T.; Svedlund, F.; Rodriguez, D.E.; Podschun, J.; Pendi, L.; Gower, L.B. Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents. Polymers 2011, 3, 10-35. https://doi.org/10.3390/polym3010010
Thula TT, Svedlund F, Rodriguez DE, Podschun J, Pendi L, Gower LB. Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents. Polymers. 2011; 3(1):10-35. https://doi.org/10.3390/polym3010010
Chicago/Turabian StyleThula, Taili T., Felicia Svedlund, Douglas E. Rodriguez, Jacob Podschun, Laura Pendi, and Laurie B. Gower. 2011. "Mimicking the Nanostructure of Bone: Comparison of Polymeric Process-Directing Agents" Polymers 3, no. 1: 10-35. https://doi.org/10.3390/polym3010010