Functionalized Polymers from Lignocellulosic Biomass: State of the Art

Department of Microbiology & Cell Science and UF Genetics Institute, University of Florida, Gainesville, FL 32610, USA
*
Author to whom correspondence should be addressed.
Polymers 2013, 5(2), 600-642; https://doi.org/10.3390/polym5020600
Submission received: 25 February 2013
/
Revised: 12 April 2013
/
Accepted: 14 May 2013
/
Published: 28 May 2013
(This article belongs to the Collection Polysaccharides)
Abstract
:Since the realization that global sustainability depends on renewable sources of materials and energy, there has been an ever-increasing need to develop bio-based polymers that are able to replace petroleum-based polymers. Research in this field has shown strong potential in generating high-performance functionalized polymers from plant biomass. With the anticipated large-scale production of lignocellulosic biomass, lignin, cellulose and hemicellulosic polysaccharides will be abundantly available renewable feedstocks for biopolymers and biocomposites with physico-chemical properties that match or exceed those of petroleum-based compounds. This review examines the state of the art regarding advances and challenges in synthesis and applications of specialty polymers and composites derived from cellulose, hemicellulose and lignin, ending with a brief assessment of genetic modification as a route to tailor crop plants for specific applications.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Bio-Based Polymers Can Lead the Way to a Future with Reduced Reliance on Fossil Fuels
Renewable polymers refer to polymers produced from recently harvested biological materials. This is in contrast to the production of polymers from the finite supply of fossil resources, especially petroleum that cannot be replaced once used. The interest in renewable polymers is driven by several factors, outlined below, which make the production of these polymers a necessity, a challenge and an opportunity.
The interest in renewable polymers is driven primarily by the same factors that have stimulated the interest in renewable energy: the realization that petroleum and other fossil resources are finite in supply, that access to much of the fossil resources is controlled by a relatively small number of countries, many of which do not have stable governments, and that the continued burning of fossil resources is expected to have a major impact on the climate as a result of the increase in atmospheric greenhouse gases [1]. According to the US Energy Information Administration (EIA), approximately 5% of the petroleum in the US is used in plastics; an additional 1.5% of the petroleum is used to generate energy for manufacturing [2]. A quick look around the living room or office makes our dependence of petroleum-derived products abundantly clear.
While renewable energy (solar, wind, biomass, hydroelectric) is perceived as a sustainable, long-term strategy to ensure continued supply of electricity and transportation fuels, investments in renewable energy have been limited for several reasons: (1) several major world economies rely heavily on the export of fossil fuels; (2) the use of fossil fuels generates large revenues for the oil and gas industry and tax revenues to governments, whereas implementation of renewables reduces the profits of these industries and, for the time being, requires government incentives and/or subsidies; (3) companies switching to alternative sources of energy run the risk of undermining their market positions in the short term, as competitors continue their business as usual, often at lower cost, unless new business models are implemented concomitantly [3,4].
In contrast, bio-based products other than fuels do not face these challenges in the same way, as they are not a major source of tax revenues, and represent less of a threat to the revenues of oil companies due to their smaller market share relative to fuels. Their smaller scale of production also presents less of a perceived threat to food security compared to biofuel production, although the “food vs. fuel” debate often ignores concurrent demographic and economic developments [1,5,6]. Furthermore, bio-based products have the ability to outcompete petroleum due to the richness in chemical building blocks, especially optically pure compounds. The production of bio-based chemicals from the waste stream of biofuel production facilities (biorefineries) has the potential to generate enough extra revenue that the cost of biofuels can be reduced, thereby rendering them more competitive with fossil fuels. As a consequence, the investment in bio-based products has the potential to lead the way to a future in which fossil fuels play a less prominent role.
Bio-based polymers can be based on lipids, oils and fatty acids produced from oil crops (e.g., oil palm, canola, soybean) and algae, chitin from insects and marine invertebrates, and polysaccharides and lignin from biomass crops. This review will focus on the latter, as this source of polymers is expected to become highly abundant in the years to come as a result of the U.S. Energy Independence and Security Act of 2007 and similar initiatives in other countries.
1.2. Lignocellulosic Biomass Consists of Different Cell Wall Polymers
Biomass crops, either woody species (e.g., pine, poplar, spruce, eucalyptus, willow), or grasses (e.g., sugarcane, sorghum, miscanthus, switchgrass, corn stover) consist primarily of cell walls, which is a complex matrix in which cellulose microfibrils are embedded in a network of cross-linking hemicellulosic polysaccharides [7]. The phenolic polymer lignin acts to harden the wall, providing structural rigidity and a physical barrier to fungi and insects, and facilitates the transport of water through the vascular tissue due to its hydrophobic nature. Cellulose is a crystalline polymer made of β-1,4-linked d-glucopyranose residues (Figure 1, structure 1). As neighboring glucose residues are rotated 180° relative to each other, the repeating unit of cellulose is cellobiose. The glucan chains in cellulose are generally present in bundles of 36 that form a cellulose microfibril. Cellulose is synthesized from the monomer UDP-d-glucose (a nucleotide sugar consisting of uridine diphosphate linked to d-glucose) through the action of cellulose synthases, membrane-bound enzyme complexes present in rosette structures containing six catalytic CesA subunits that are distinct between primary and secondary cell wall. Recent reviews on cellulose biosynthesis can be found in [8,9,10].
The term hemicellulose is technically a misnomer, as there are several different classes of hemicellulosic polysaccharides that vary substantially between species. In fact, the composition of hemicellulosic polysaccharides is sometimes used to classify different orders in the plant kingdom [11]. In dicot angiosperms and non-commelinoid monocots (e.g., lilies), the main hemicellulosic polysaccharide is xyloglucan (Figure 1, structure 2), which, as the name implies, contains a linear backbone of β-1,4-linked d-glucopyranose residues, with substitutions of α-d-xylose on the O-6 position of the glucose, typically the first three out of four adjacent glucose residues. Several other sugars, including d-galactose and l-fucose can be present on some of the xylose residues. The cell walls of softwood species contain galactoglucomannans as the primary hemicellulosic polysaccharide (Figure 1, structure 3). This polymer consists of a backbone containing both β-1,4-linked d-mannosyl and d-glucosyl residues, substituted with α-(1,6)-linked d-galactosyl residues. Approximately 50% of the mannosyl residues are acetylated on C2 or C3.
The main cross-linking polysaccharide in grasses is glucuronoarabinoxylan (Figure 1, structure 4), which consists of a backbone of β-1,4-linked d-xylopyranosyl residues with arabinose substitutions on the O-3 position of the xylose. Some of the xylose residues contain α-d-glucuronic acid substitutions on the O-2position, while some of the arabinose residues contain ferulate esterified at the O-5 position. A related hemicellulosic polysaccharide is present in woody species: 4-O-methylglucuronoxylan. This polymer contains a β-1,4-linked d-xylopyranosyl backbone which is substituted with 4-O-methyl-α-d-glucuronic acid residues. In hardwood species the polymer is acetylated, whereas in softwood species substitutions with arabinosyl residues occur. The primary wall of grasses also contains mixed-linkage β-glucans, which, as a result of alternating β-1,3 and β-1,4 linkages in the glucan backbone, form a corkscrew-like structure. Due to their transient nature, this class of hemicellulosic polysaccharides is typically no longer present in the biomass harvested at the end of the season, and therefore not a major source of bio-based products. The enzymes involved in the synthesis of the different classes of hemicellulosic polysaccharides are still under active investigation. At this time the genes encoding the enzymes responsible for synthesizing the backbones of xyloglucan and mixed-linkage β-glucans have been identified [12,13], as have several genes involved in decorating the backbones (reviewed in [14,15]).
Figure 1.
Structures of the cell wall polymers cellulose (1), (fucogalacto)xyloglucan (2), galactoglucomannan (3), glucuronoarabinoxylan (4). Sugar residues are color coded. Also shown are the phenolic compounds p-coumaryl alcohol (5), coniferyl alcohol (6), sinapyl alcohol (7), p-coumaric acid (8), and ferulic acid (9) and the breakdown products furfural (10) and hydroxymethylfurfural (11) derived from pentose and hexose sugars, respectively.
Figure 1.
Structures of the cell wall polymers cellulose (1), (fucogalacto)xyloglucan (2), galactoglucomannan (3), glucuronoarabinoxylan (4). Sugar residues are color coded. Also shown are the phenolic compounds p-coumaryl alcohol (5), coniferyl alcohol (6), sinapyl alcohol (7), p-coumaric acid (8), and ferulic acid (9) and the breakdown products furfural (10) and hydroxymethylfurfural (11) derived from pentose and hexose sugars, respectively.
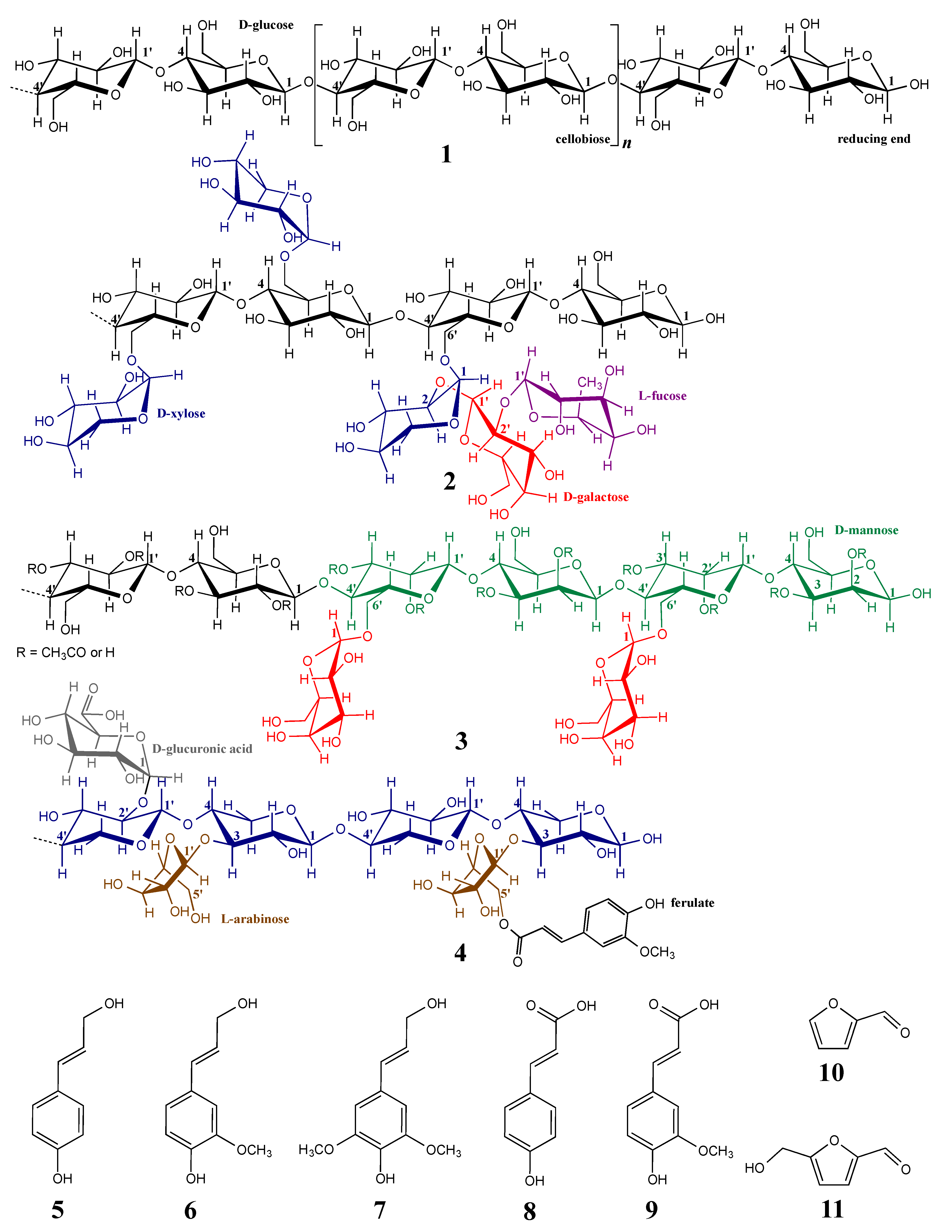
Lignin is formed from the oxidative coupling of hydroxycinnamyl alcohols and related compounds such as hydroxycinnamaldehydes. These precursors are synthesized via the concerted action of the shikimate and general phenylpropanoid pathways. The polymerization occurs in the cell wall, especially in the secondary cell walls in the water-conducting xylem vessels and in sclerenchyma fibers, and is mediated by peroxidases and/or laccases [16]. Lignin is not optically active due to its polymerization via combinatorial chemistry [17,18]. Variation in lignin composition is a function of plant species, age, and tissue type. Lignin from grasses contains a small proportion of p-hydroxyphenyl (H) residues, formed from p-coumaryl alcohol (Figure 1, structure 5), in addition to guaiacyl (G) residues derived from coniferyl alcohol (structure 6) and syringyl (S) residues derived from sinapyl alcohol (structure 7). Grasses also contain substantial amounts of cell wall-bound p-coumarate (structure 8) and ferulate (structure 9). Lignin from hardwood species consists of a mixture of G- and S-residues, whereas softwood species contain lignin consisting primarily of G-residues, although compression wood formed in response to gravitational stress contains H-residues. Detailed reviews on lignin biosynthesis can be found in [19,20].
1.3. The Biorefinery
The concept of the biorefinery is similar to that of an oil refinery, in that a crude product is converted into individual compounds of high purity. In the case of the biorefinery, biomass is converted to a variety of products, which can include fuels and chemicals [21], most commonly via microbial conversion of fermentable sugars derived from cellulose and, ideally, hemicellulose. The biomass needs to first be subjected to a pretreatment that enables separation of the individual cell wall polymers. Production of value-added co-products alongside biofuels during integrated biorefinery processes requires selectivity during pretreatment. Choosing the appropriate pretreatment is frequently a compromise between minimizing the degradation of the hemicellulose and cellulose to acetic acid, furfural (structure 10), and 5-hydoxymethylfurfural (HMF; structure 11), while maximizing the ease of subsequent hydrolysis of the cellulosic substrate by a cocktail of cellulolytic enzymes. Recent reviews summarize biomass pretreatment technologies with emphasis on concepts, mechanism and practicability of pretreatments [22,23,24,25,26,27].
1.4. Bio-Based Polymers from Plant Cell Walls
The three main sources of bio-based products derived from lignocellulosic biomass are:
- (1)
- The cell wall polymers themselves. In this case cellulose, hemicellulosic polysaccharides or lignin are isolated, processed, and converted to end products.
- (2)
- Fermentable sugars generated in a biorefinery. The production of second-generation cellulosic biofuels relies on the deconstruction of the cell wall polysaccharides into monomeric hexose and pentose sugars that are then converted to fuels such as ethanol or butanol via microbial fermentation [21]. The use of different microbial strains enables the production of chemical feedstocks that can be used for the production of bio-based products, including polymers, with poly-lactic acid (PLA) used for bio-degradable plastics currently representing the main application [28,29].
- (3)
- The waste stream of the biorefinery or paper mill. In the former, these residues include most of the lignin, and a portion (~5%) of the cellulose that is resistant to deconstruction, along with monomeric sugars that cannot be converted microbially, and compounds formed from the monomeric sugars during processing (e.g., furfural, HMF), as well as various extractives. In the case of paper mills, the waste stream consists primarily of lignin and extractives. Both the lignin and cellulose can be used for the production of biopolymers and composites. The remaining compounds are often not present in high enough concentrations to enable cost-effective recovery.
This review will focus on the use of cellulose, hemicellulosic polysaccharides and lignin for the production of bio-polymers (sources 1 and 3 from above). Specifically, we will introduce recent developments in the production of functionalized polymers from cellulose, hemicellulose and lignin for specific applications. The ease of functionalization, biocompatibility and biodegradability enable these polymers to be used in agricultural, biomedical, pharmaceutical, and electro-mechanical industries. The synthesis and characterization of some of these materials are still in their initial stage and their commercial large-scale applications are anticipated to reach the market in the coming years.
2. Cellulose Fibers and Composites
Being the most abundant renewable polymer on Earth, cellulose as a feedstock for functionalized materials has been of keen interest to the chemistry audience for over a century. As a result, a well-established knowledge base of isolation processes, modifications and applications has been developed [30]. However, with the anticipated increase in demand for renewable energy from biomass, the development of cost-efficient synthesis and production of value-added materials has become a crucial requirement for the continued growth of the chemical industry. We will outline new developments in the area of cellulose acetate, and modification of microfibrillated and nanocrystalline cellulose.
2.1. Cellulose Acetate
Cellulose acetate (CA) was first synthesized in 1865 by Schutzenberger [31]. This author reported that cellulose acetate could be prepared by heating cellulose to 180 °C in a sealed glass tube with acetic anhydride. Due to their good permeability, solubility in various organic solvents and biocompatible nature, cellulose acetates are among the most common commercial cellulose esters. CAs have been used for many industrial products such as textiles, filters and membranes, moldings, lacquers and protective coatings, liquid crystalline displays (LCD) and controlled drug delivery systems. Several industries relied solely on cellulose acetate products. Photographic and cinematographic films, as well as X-ray films, graphic and microfilms were made from CA. Recently, with the shift from traditional film-based photography and cinematography to digital production, new directions of cellulose acetate modification and application have been explored. Given the dynamic development and implementation of nanomaterials, CA has been successfully employed as a precursor for matrices, nanoparticles and nanofibers. In the year 2012 alone, CA was used to prepare membranes for nanofiltration [31,32,33,34,35,36,37,38,39,40,41,42], organoclay nanocomposites [43,44,45], antimicrobial wound dressing [46], bone regeneration material [47], biosensors [48], bioreactors [49], polymer electrolyte [50,51], energetic materials (composite explosives and propellants) [52], insulating [53] and fireproofing materials [54], and novel drug delivery systems [49,55].
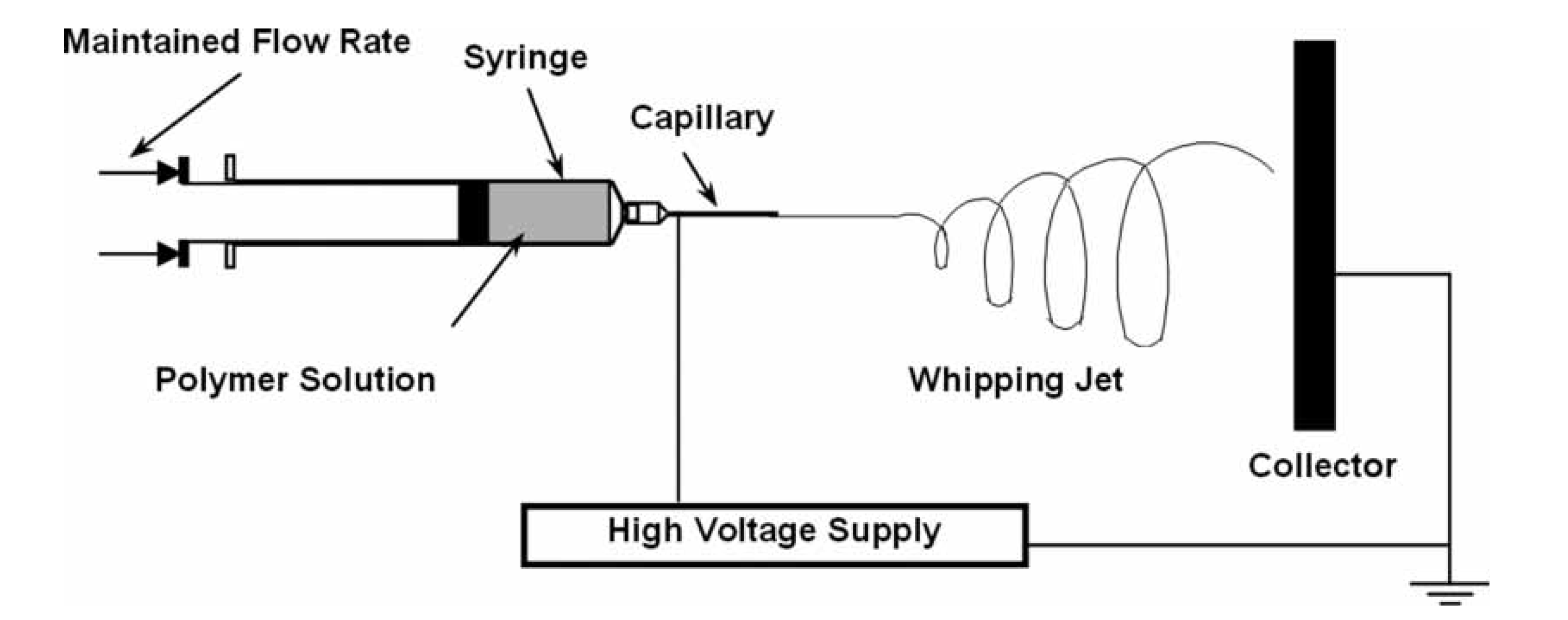
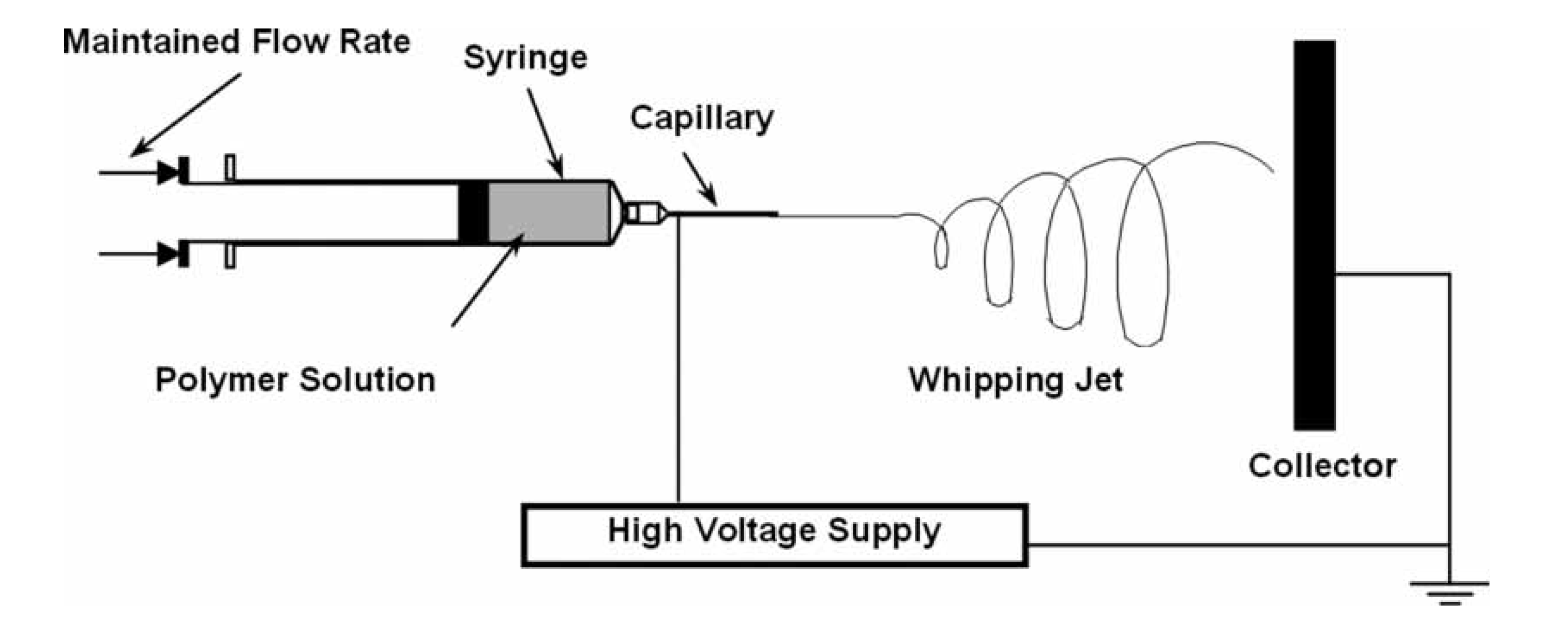
Promising results have been obtained using nanoprecipitation or interfacial deposition for the fabrication of nanoparticles and electrospinning for the production of nanofibers. One of the most significant developments in the latter technique is regarded to be coaxial electrospinning, whereby a concentric spinneret can accommodate two different liquids [56]. A spinneret is a metal nozzle (typically a syringe needle) connected to a high-voltage supply. The electrospun fibers are formed by applying an electric field that facilitates polymer solution or by a melt jet going through the spinneret (Figure 2). A concentric spinneret enables encapsulation of functional liquids, drugs or other bio-active agents into the polymer nanofibers and offers precise control over secondary structures of nanofibers [57]. Although it is common that the sheath fluids are polymer-solvent systems having high viscosity in order to overcome the interfacial tension between the two solutions [58], a recent study of cellulose acetate nanofibers showed that only organic solvents (acetone, N,N-dimethylacetamide (DMAc) and their mixture) could be used as sheath fluids [59]. By using sheath solvents, the core jet is subjected to electrical drawing for a longer period, facilitating homogeneous core jet solidification and resulting in CA nanofibers with high quality in terms of the nanofibers’ diameter and distribution, structural uniformity and surface smoothness [59]. This technique was used to employ CA as a filament-forming matrix with ketoprofen (KET) as an active pharmaceutical ingredient. With optimized sheath-to-core flow rate ratio, nanofibers prepared from the coaxial process had a smaller average diameter, narrower size distribution (240 ± 30 nm), more uniform structures and smoother surface morphologies than those generated via single fluid electrospinning (which had diameters of 680 ± 150 nm). In vitro dissolution tests were carried out in physiological saline and the amount of KET released from the fibers was determined by UV-Vis spectroscopy. While the single-fluid process did result in KET dispersed in an amorphous state in CA-based fibers, with the fibers providing a sustained drug release profile over 144 h via a Fickian diffusion mechanism, the fibers from the modified electrospinning process offered a better zero-order drug release profile with a smaller tailing residue [60].
Figure 2.
Schematic of the general set up for electrospinning. Reproduced with permission from [58]. Copyright 2008 Taylor & Francis.
Figure 2.
Schematic of the general set up for electrospinning. Reproduced with permission from [58]. Copyright 2008 Taylor & Francis.
While electrospinning is a simple, straightforward and inexpensive method to prepare CA nanofibers, nanoprecipitation is considered to be an efficient method for the preparation of CA nanoparticles. The potential applications of CA nanoparticles include their use in drug delivery systems, pharmaceuticals, and the bio- and food industries. Precipitation is achieved by mixing a low-concentration polymer solution (solvent) with a dispersive medium (non-solvent) in which the polymer is not soluble. The successive addition of a non-solvent to a dilute solution of the polymer leads to precipitation of nanoparticles. Solvent and non-solvent have to be miscible. CA nanoprecipitation was first reported by Hornig and Heinze in 2008 [61]. Mixing of the solvents was achieved through dialysis of the polymer solution against the non-solvent, or by dropping techniques [61]. The nanoparticle size was in the range from 86 to 368 nm and the nanoparticles were stable in aqueous suspensions over time. Nanoparticles were successfully produced applying both routes of nanoprecipitation. Dialysis of polymer dissolved in DMAc yielded regular nanospheres, whereas the preparation in acetone by drop-wise addition of water to the polymer solution or vice versa led to bean-shaped nanoparticles [61].
More recently, Kulterer et al. [62] developed a synthesis protocol for CA nanoparticles with a tunable particle diameter, ranging from 60 to 140 nm, and a recovery of 87%. Addition of tetrahydrofuran (THF) to the non-solvent led to smaller particles than obtained with pure water or standard stabilizing agents like surfactants and block copolymers. Changing the THF content and stirrer velocity allowed an easy and sensitive adjustment of the mean nanoparticle diameter. These CA nanoparticles can be used to produce ultrathin layers deposited onto positively charged surfaces. Potential applications include surface modifications and surface functionalization of CA foils and membranes.
The same group recently reported an in-situ technique for the preparation of composite nanoparticles using nanoprecipitation from hydrophobic cellulose acetate with a number of hydrophilic polysaccharides (hydroxyethyl cellulose (HEC), carboxymethyl cellulose (CMC), low-molecular-weight chitosan (L-CHI), and amino cellulose (AC)) [63]. Kulterer et al. [63] stated that the integration of different polysaccharides enabled the formation of composite nanoparticles with different sizes, charges, and effective Zeta-potentials, indicating that these hydrophilic polysaccharides bound to the CA nanoparticle matrix during the precipitation process and determined their surface characteristics. The successful entrapment of pyrene (a hydrophobic fluorescent dye) into CA/CMC nanoparticles demonstrates great potential to be used in the pharmaceutical industry and bio- or food technology, as delivery of hydrophobic substances in aqueous media.
CA can also be used as a template for nanoparticle preparation. Specifically, polyaniline (PANI) nanospheres were prepared by adding aniline in a mixture of CA, acetone and DMAc. During immobilization aniline could polymerize in the CA micelle and formed a spherical core–shell structure. After the core–shell spheres were immersed in acetone, pure PANI nanospheres were obtained. The authors speculated that CA not only played an important role in the formation of PANI nanoparticles, but also affected the size and uniformity of the PANI nanospheres [64]. Meanwhile, studies on NH3 gas sensitivity and reversibility properties of the PANI nanospheres indicated that the PANI nanospheres synthesized in the non-aqueous system had superior gas-response behavior.
2.2. Microfibrillated Cellulose and Cellulose Nanocrystals for Use in Composites
In the plant cell wall the β-1,4-d-glucan chains aggregate into cellulose microfibrils, which in turn aggregate into macrofibrils. Cellulose contains, in decreasing degrees of order, crystalline, subcrystalline and non-crystalline chains. Due to this hierarchical structure of native cellulose, different types of cellulose fibrils can be easily obtained using a combination of mechanical and chemical treatments. Repeated mechanical shearing processes can be used to obtain individual microfibrils usually called “microfibrillated cellulose” (MFC) or “nanofibrillated cellulose” (NFC). The network structure resulting from the high number of entanglements and high aspect ratio of MFC restrict the mobility of microfibrillated cellulose in polymer matrices, which in turn significantly influence mechanical properties of MFC-reinforced composites. The non-crystalline domains are weak spots along MFC, which can be hydrolyzed by sulfuric or hydrochloric acid. The remaining highly crystalline, rod-like nanostructures commonly referred to as “cellulose nanocrystals” (CNC) or “cellulose nanowhiskers” (CNW), are of particular value in composites.
Microfibrillated cellulose has a diameter between 10 and 100 nm and a length in the micron scale. It is usually difficult to measure the exact length of MFC due to high number of entanglements (Figure 3). Reducing the processing energy consumption is a crucial requirement to produce MFCs on an industrial scale. MFCs are typically produced by one of four mechanical methods: homogenization, microfluidization, microgrinding, or cryo-crushing, which consume different amounts of energy. Cooling during grinding was deemed necessary to prevent the adverse effect of heat generation on the mechanical properties of MFC for the production of paper sheets [65]. The addition of a cooling system enables the stabilization of temperature preventing thermal degradation of fibers. Moreover, the disintegration of fibers from cell walls was easier than in high-temperature conditions. Consequently, fewer passes through the grinder were required, leading to reduced energy consumption. Chemical and enzymatic pretreatments were also reported to reduce energy consumption, because pretreatments promote fiber swelling, making defibrillation and production of fine MFC easier. Mostly individualized cellulose nanofibers dispersed in water can be obtained by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical-mediated oxidation of native cellulose, followed by mechanical treatment of the oxidized cellulose in water [66]. Other chemical pretreatments involve dissolution in lithium chloride/DMAc) [67] and 1-butyl-3-methylimidazolium chloride ([C4mim]Cl) [68,69], peroxide alkaline (NaOH/H2O2), or peroxide alkaline with hydrochloric acid and potassium hydroxide [70]. On the other hand, besides lowering processing cost by facilitating disintegration of cellulose fibers, the use of enzymatic pretreatment is beneficial from an environmental point of view, compared with chemical methods. Different enzymes, such as cellulase prepared from Aspergillus spp. [65], xylanases [71], and endoglucanases [72] have been used in pretreatments. Moreover, while HCl-hydrolyzed MFC from wood showed an uneven distribution of fiber geometry and a large extent of thick cell wall fragments of low aspect ratio, the MFC produced from endoglucanase-pretreated cellulosic fibers showed higher average molar mass and larger aspect ratio [72].
Hardwood pulp samples processed by homogenization, microfluidization, or microgrinding were shown to exhibit different specific surface areas of the MFCs, and the MFC films varied in opacity, roughness, density, water interaction properties, and tensile properties [73]. Relative to the energy consumption, microfluidization with a refining pretreatment and the micro-grinding of wood fibers are more energy-efficient production methods than homogenization, and produce MFC films with better mechanical properties. Production of MFCs with a homogenizer, however, resulted in microfibrils with the highest specific surface area and films with the lowest water vapor transmission rate. Overall, it was concluded that films produced with a microfluidizer and micro-grinder had superior physical, optical, and water interaction properties compared to those formed via homogenization, suggesting that these materials could be produced in a more economical way for packaging applications [73,74]. Indeed, MFC coated with cooked starch, beeswax or paraffin resulted in water vapor transmission rates lower than that observed for low-density polyethylene. The improvement was associated with surface pore closure and filling of the pore network [74]. The effect of freezing was also investigated as an alternative to the most conventional final step of cellulose preparation, which consists of freeze-drying the suspensions. Freezing was shown to preserve the rheological properties of the suspensions, contrary to the freeze-drying process [75]. The use of recycled paper as starting material for MFC can potentially reduce the processing costs as well. The hornification (irreversible or partially reversible H-bonding upon drying or water removal) of fibers through a drying and rewetting cycle prior to refining and homogenization did not result in apparent improvements compared to films from never-dried fibers, indicating that MFC films can potentially be made from low-cost, recycled cellulosic materials [74,76].
Figure 3.
TEM image of dispersions of the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-oxidized celluloses with carboxylate contents of 0.9 and 1.5 mmol/g. Images were taken after stirring in water for 10 days [66]. Reproduced with permission from [37]. Copyright 2007 American Chemical Society.

Besides TEMPO oxidation [66], other chemical pretreatments have been used to reduce energy consumption and improve processability of MFC and compatibility of MFC with polymeric matrices. Carboxymethylation pretreatment increases the anionic charges in the formation of carboxyl groups on the surface of the MFC and make them easier to liberate. Moreover, the net specific energy consumption can be reduced from 5.5 MWh/t for un-treated fibers to 2.2 MWh/t for carboxymethylated MFC per pass through a microfluidizer [77]. However, carboxymethylation might produce shorter fibers [77]. An acetylation process involves the grafting of acetyl moieties in order to decrease the hydrophilicity of MFC and enhance the chemical affinity between MFC and non-polar polymers. For example, Tingaut et al. [78] reported that when added to poly(lactic acid), thermal stability, filler dispersion and hygroscopicity can be tailored by adjusting both acetyl content and MFC concentration. Acetyl content above 4.5% promotes significant changes in the crystalline structure of MFC and prevents hornification upon drying. This ability of acetylated MFC to be stored in a dry form opens the door to industrial-scale production.
Although the vast majority of reports are focused on MFC’s ability to reinforce [79,80,81,82,83,84,85,86,87] and toughen [85,86,87,88,89,90,91] polymer matrices, there is an increasing number of publications dedicated to the superior barrier properties of MFC for packaging materials [76,84,92,93,94,95,96,97,98,99]. The recent reviews of Siró et al. [97], Lavoine et al. [98] and Johansson et al. [99] give an overview of these competitive nanomaterials. More recent applications also include materials for polyelectrolyte systems in batteries [100,101,102,103,104,105,106], ionic diodes [107], superoleophobic (oil-repellant) aerogels [108], superabsorbents [109], emulsion stabilizers [110,111], foaming agents [112], coatings [83], membranes [113], tissue engineering [114], and electrostatically actuated mechanical switch devices [115]. A number of these applications are described in more detail in the following sections.
2.2.1. Polyelectrolyte Multilayers
As renewable sources of energy are being explored on a larger scale, there will likely be an increase in the use of devices that operate on electricity rather than on fossil fuels. Especially if the electricity is generated from wind and solar energy, both intermittent sources, there will be a need to store electrical charge, ideally at high density for the sake of cost-efficiency and space utilization. Polyelectrolyte multilayers (PEMs) are used to form batteries, semiconductors and sensors in organic electronics and can be prepared by combining different types of polyelectrolytes and MFC [100]. A polyelectrolyte with a three-dimensional structure leads to the build-up of thick layers of MFC, whereas the use of a highly charged linear polyelectrolyte leads to the formation of thinner layers of MFC. The image shown in Figure 4 illustrates how films of polyelectrolytes and MFC can be so smooth and well-defined that they show different interference colors as a function of film thickness [100].
Figure 4.
Interference colors of films of microfibrillated cellulose (MFC) and the polyelectrolyte polyethyleneimine (PEI) as a function of the number of layers. For example, 12 in this figure means a combination of six layers of PEI and six layers of MFC. No additional electrolyte was added. Reproduced with permission from [100]. Copyright 2008 American Chemical Society.
Figure 4.
Interference colors of films of microfibrillated cellulose (MFC) and the polyelectrolyte polyethyleneimine (PEI) as a function of the number of layers. For example, 12 in this figure means a combination of six layers of PEI and six layers of MFC. No additional electrolyte was added. Reproduced with permission from [100]. Copyright 2008 American Chemical Society.

2.2.2. Superoleophobic Surfaces
Superoleophobic surfaces—those that display contact angles greater than 150° with organic liquids having appreciably lower surface tensions than that of water—are extremely rare. Calculations suggest that creating such a surface would require a surface energy lower than that of any known material [116]. It is highly desirable for superhydrophobic surfaces also to be oil-repellent in order to maintain their superhydrophobicity. MFC aerogels with suitable surface textures were developed allowing the systematical adjustment of their surface-wettability characteristics. Aerogels of microfibrillated cellulose with a high porosity and a very low density (<0.03 g·cm−3) were prepared by freeze-drying [108]. The density and surface texture of the aerogels can be tuned by selecting the concentration of the MFC dispersions before freeze-drying. Chemical vapor deposition (CVD) of 1H, 1H, 2H, 2H-perfluorodecyltrichlorosilane (PFOTS) was used to uniformly coat the aerogel to tune their wetting properties towards non-polar liquids. The most oleophobic aerogel exhibited contact angles of approximately 166° and 144° for castor oil and hexadecane, respectively, which are among the highest contact angles reported in the literature for any substrate against liquids with similar surface tensions. By combining this understanding with a CVD process that provides a conformal fluorinated coating, it is possible to switch the wettability behavior of the cellulose surfaces between super-wetting and super-repellent, using different scales of roughness and porosity created by the freeze-drying technique and change of concentration of the NFC dispersion [108].
2.2.3. MFC as a Matrix for Hepatocyte Cell Cultures
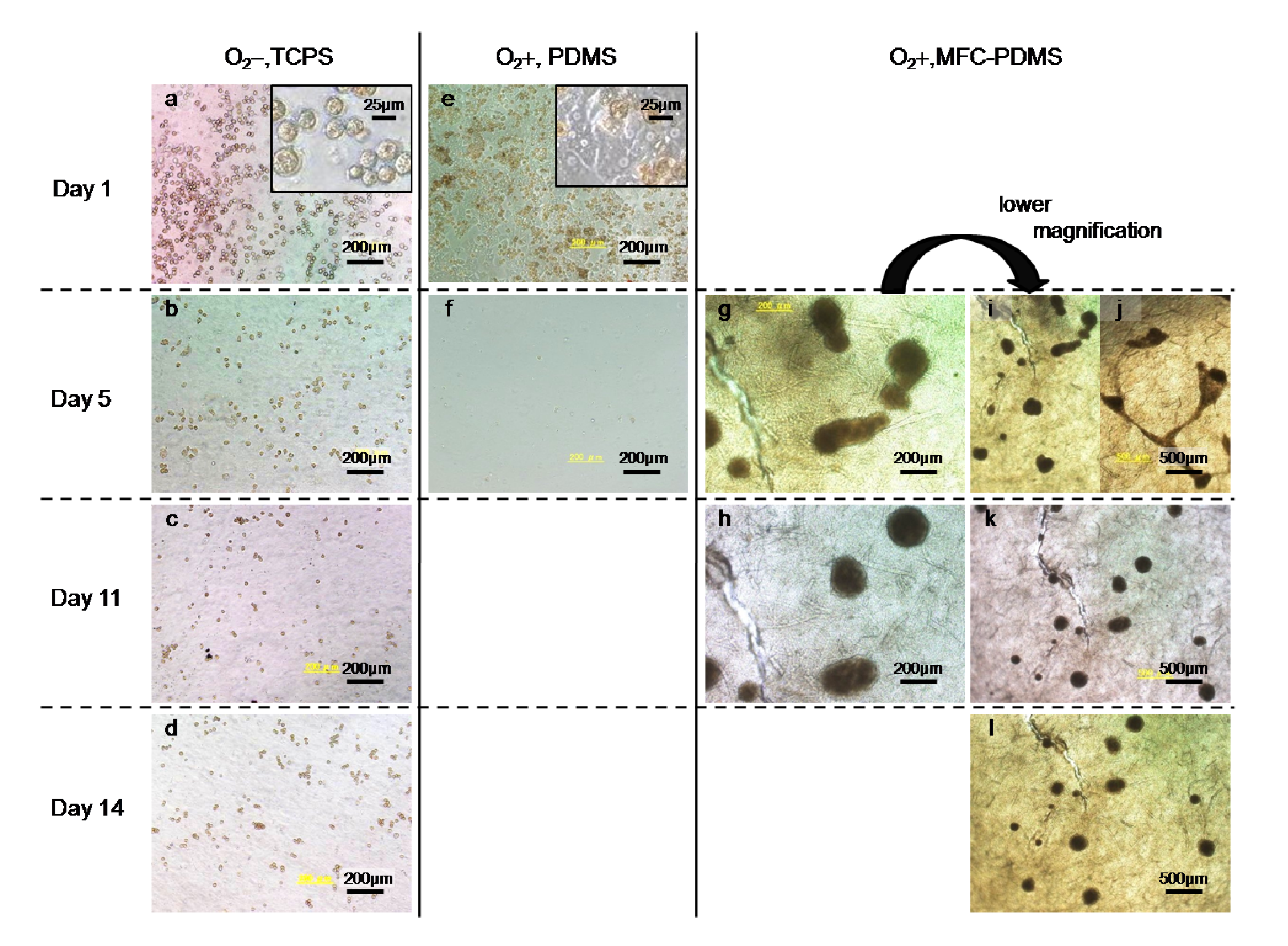
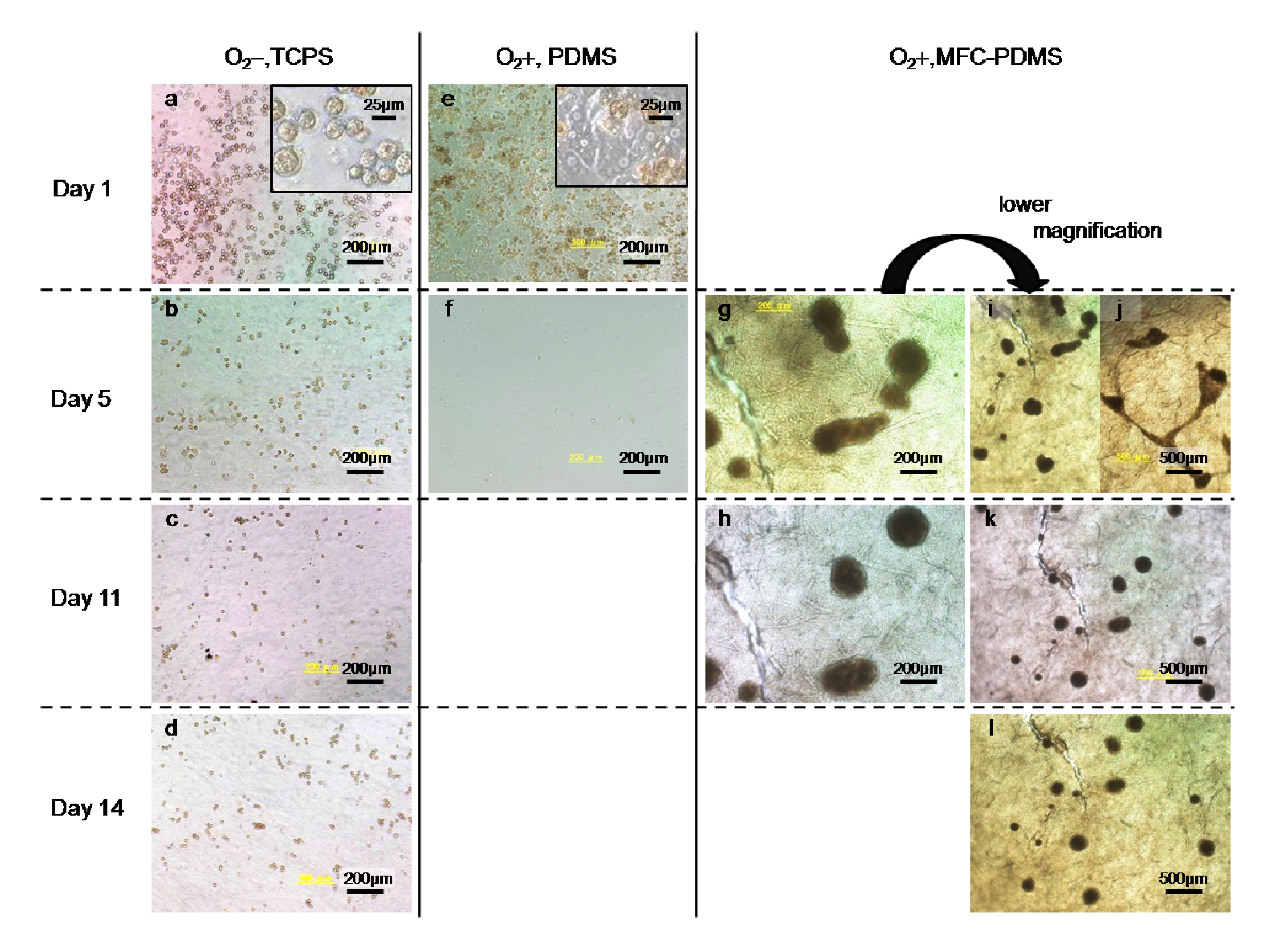
Evenou et al. [114] used MFC as a matrix for liver cell (hepatocyte) culture for evaluation of the potential toxicity of drugs and chemicals. The study focused on the development of alternative methods to animal experimentations. Inside the liver, hepatocytes are organized within a 3-D structure that promotes extensive cell–cell contacts and cell differentiation. In traditional tissue culture hepatocytes form 2-D monolayers, which do not have the same level of cell-to-cell contact. In contrast, 3-D multicellular aggregates called spheroids exhibit greater differentiated functions and better cell maintenance [114]. Cellulose sheets prepared from MFC pulp can be used as matrix materials for hepatocyte culture. The combination of such thin fibrous sheets and direct oxygenation through O2-permeable MFC-coated polydimethylsiloxane (PDMS) membranes promotes cell aggregation into stably-attached 3-D hemispheroids which exhibit enhanced liver-specific functions (Figure 5). Such a simple design appears suitable for engineering miniature 3-D liver-tissue models for microplate-based drug/chemical screenings, but also appears transposable to microfluidic device applications [114].
2.2.4. Micro-Electro-Mechanical Systems
Micro-electro-mechanical systems (MEMS) are small electronic devices that can sense their environment and respond to changes in conditions. MEMS are widely used for accelerometers (to measure seismic activity, machine motion/vibration, navigation), biochips and biosensors. Couderc et al. [115] reported on an electrostatically actuated micromechanical switch device made of an MFC sheet coated with a thin polyimide layer. The MFC sheet was first patterned to enable its use as a substrate. Spin coating with a thin polyimide layer greatly improved the dielectric properties, moisture sensitivity and sheet surface roughness (by factor of 5). In addition, the leakage currents were lower by three orders of magnitude. Gold electrodes were added onto the sheet for electrostatic actuation and switch detection. The pull-down voltage of this switch, defined as the actuation voltage needed to establish a contact between the free end of the cantilever beam and the substrate, was measured to be about 55 V for a gap height of ~30 μm. With mechanical resonance of 700 Hz, the device reaction time is 1.4 ms. Although device response time is high compared to silica-based MEMS, the value is comparable to polymer-based mechanical switches. A theoretical model based on a free-clamped silicon MEMS switch fits these measurements, demonstrating standard behavior [115]. This device is inexpensive and, with a biodegradability ratio of 83%, eco-friendly. These devices open the way to promising applications such as flexible sensors and e-paper.
Figure 5.
Morphological changes over time of primary rat hepatocyte cultures in the different conditions: (a–d) under standard tissue-culture plates as control condition (O2−, TCPS); (e–f) undersmooth polydimethylsiloxane (PDMS) membranes (O2+, PDMS) and (g–l) under improved oxygenation on MFC-coated PDMS membranes (O2+, MFC-PDMS). The insets in images (a) and (e) show the respective cell morphologies at higher magnification. Reproduced with permission from [114]. Copyright 2011 Taylor & Francis.
Figure 5.
Morphological changes over time of primary rat hepatocyte cultures in the different conditions: (a–d) under standard tissue-culture plates as control condition (O2−, TCPS); (e–f) undersmooth polydimethylsiloxane (PDMS) membranes (O2+, PDMS) and (g–l) under improved oxygenation on MFC-coated PDMS membranes (O2+, MFC-PDMS). The insets in images (a) and (e) show the respective cell morphologies at higher magnification. Reproduced with permission from [114]. Copyright 2011 Taylor & Francis.
2.3. Cellulose Nanocrystals/Nanowhiskers
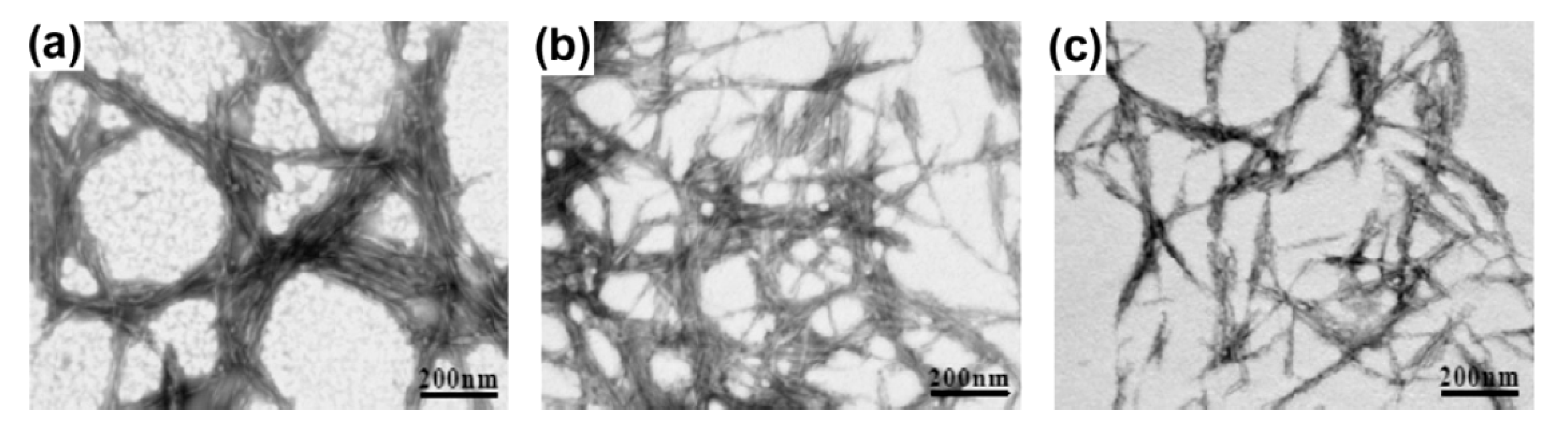
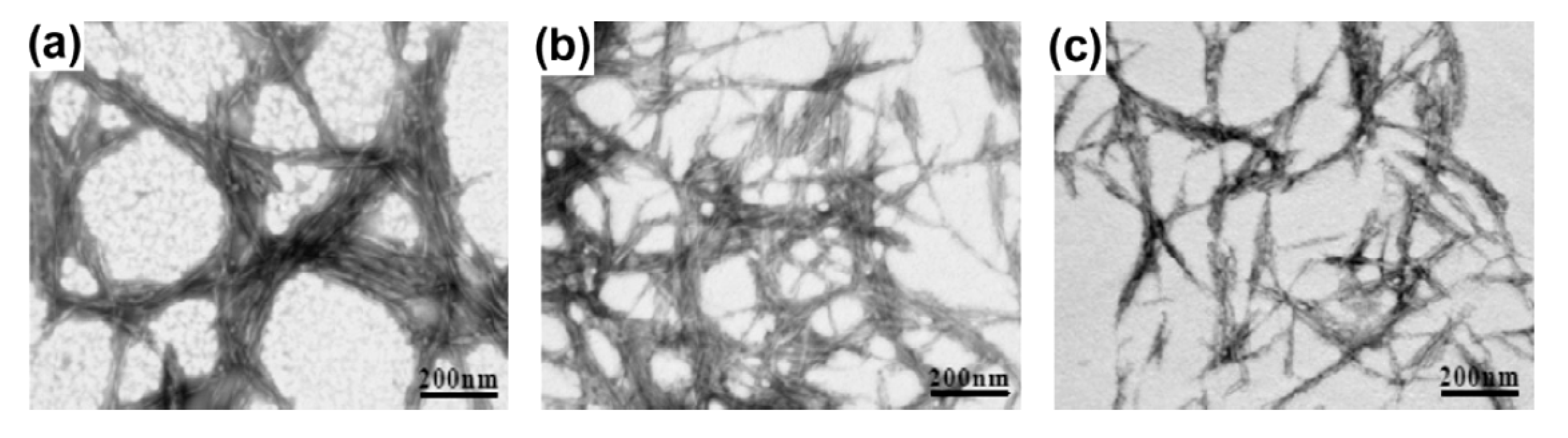
Cellulose nanowhiskers (CNW) or nanocrystals (CNC) can be prepared from any plant source containing cellulose via strong-acid hydrolysis. The dimensions and stiffness of CNW depend on cellulose source and conditions of hydrolysis such as time and temperature [117,118,119,120,121,122,123]. Several recent studies optimized the preparation conditions for pea hull fibers [117], coconut husk [118], kenaf bast fibers [119], grass (Zoysa japonica and Z. tenuifolia) [120], curauá (pineapple) fibers [121], rice straw [122] and microcrystalline cellulose [123]. A novel approach of CNW isolation was reported by Li et al. [124]. The authors used a purely physical method of high-intensity ultrasonication to prepare CNW from microcrystalline cellulose. The CNW had diameters between 10 and 20 nm and lengths between 50 and 250 nm. The length and crystallinity of NCC decreased with increasing time of ultrasonication. Therefore, it was concluded that the ultrasonication could remove both the amorphous and crystalline cellulose. The effect of ultrasonication time on morphology of CNW is evident in Figure 6.
Figure 6.
TEM images of cellulose nanowhiskers (CNW) treated using different ultrasonication times: (a) NCC-5 min; (b) NCC-10 min and (c) NCC-15 min. Reproduced with permission from [124]. Copyright 2012 Elsevier.
Figure 6.
TEM images of cellulose nanowhiskers (CNW) treated using different ultrasonication times: (a) NCC-5 min; (b) NCC-10 min and (c) NCC-15 min. Reproduced with permission from [124]. Copyright 2012 Elsevier.
Owing to their high elastic modulus of 143 GPa [125], CNW are recognized to be highly suitable reinforcing agents for various water-soluble [124,126,127,128] and water-insoluble polymers [129,130,131,132,133,134,135]. Several research groups compared the reinforcing effect of MFC and CNW in composites [136,137,138,139]. Chemically modified sisal MFC-reinforced polycaprolactone composites exhibited higher modulus than CNW-reinforced composites [136]. On the other hand, the elongation at break of MFC-reinforced composites was lower for a given loading level. The difference was ascribed to the possibility of entanglements of MFC contrarily to rod-like nanoparticles. Similar results were published for acrylic-based composites with cellulosic nanoparticles from alfa (also named Esparto grass; Stipa tenacissima) [137], and natural rubber latex composites with nanocelluloses from palm rachis [138]. Moreover, for the latter system it was concluded that higher filler-matrix adhesion dominated the behavior of MFC-based composites. This resulted in lower water uptake and higher stiffness. However, the ductility (high strain at break under tensile stresses) of MFC-reinforced materials was much lower compared to CNW-filled nanocomposite films resulting in a lower shrinkage for the latter [138]. The effect of cellulosic nanoparticles on molecular transport was evaluated for bamboo MFC and CNW incorporated in natural rubber composites [139]. Though the two types of nanocelluloses effectively reduced the molecular transport, the entanglements of MFC were responsible for significant reduction of solvent uptake, which makes MFC more efficient than CNW for selective separation membranes [139].
The high reinforcing potential of CNW is still far below theoretically predicted values because of the limited compatibility between the hydrophilic cellulose nanocrystals and the more hydrophobic polymer matrices. Chemical modifications of CNW were performed prior to [140,141] and after acid hydrolysis [136,142,143] to improve compatibility. Long-chain polymer grafting on cellulose is another way to provide covalent linkage between CNW and the matrix. It can be accomplished by two strategies, namely, the “grafting-onto” and “grafting from” [144]. The “grafting-onto” method relies on the attachment of pre-synthesized polymer chains onto cellulosic hydroxyl groups with the use of a coupling agent. However, steric hindrance can restrict optimal attachment because the polymer chains must diffuse through the grafted “brushes” to reach the reactive sites. Therefore, the “grafting-onto” method is limited to low surface density grafts. To increase the grafting density, the “grafting from” approach can be employed. In this method, polymer chains are formed by in situ polymerization of monomers onto immobilized initiators [145]. Molecular growth can be achieved by conventional radical, ionic, and ring-opening polymerizations. The main advantage of this method is that the reaction is simple, fast and easy, because there is no steric hindrance and the viscosity of the reaction medium remains low. The main drawback is that the grafted polymer is not fully characterized. Considering simplicity, higher grafting density and better control over chain length, the “grafting-from” approach is generally preferred. “Grafting from” via ring-opening polymerization has been developed for polylactide (PLA)- [146], polycaprolactone (PCL)- [147,148] and PCL/PLA blends-based nanocomposites [149]. Surface-initiated radical polymerization has been successfully applied to graft CNW with poly(N-isopropylacrylamide) brushes [145].
Unidirectional fiber-reinforced composites are ideally suited for applications requiring high mechanical performance. Attempts have been made to align CNW in all-cellulose or polymer composites. Several methods were successfully developed recently by applying mechanical forces (shearing and spin coating) [150,151,152,153,154,155], magnetic [156,157,158,159,160,161] and electric fields [130,162,163]. Electrospinning has been used to orient CNW along the fiber axis [164,165,166,167]. The addition of the CNWs had a positive impact on the fiber stiffness and reduced the fiber diameter. For example, for cellulose acetate composites, the dynamic mechanical analysis showed a ten-fold increase in storage modulus with only a 1 wt % addition of CNW [166]. Electrospinning can also induce anisotropy on the molecular scale, as polymer chains show preference for uniaxial alignment along the fiber axis. Besides CNW orientation within electrospun fibers, aligned and isotropic mats (webs) can be prepared from electrospun nanocomposites. Cellulose nanowhiskers have been shown to have a more significant reinforcement effect on the aligned mats than the isotropic mats [167].
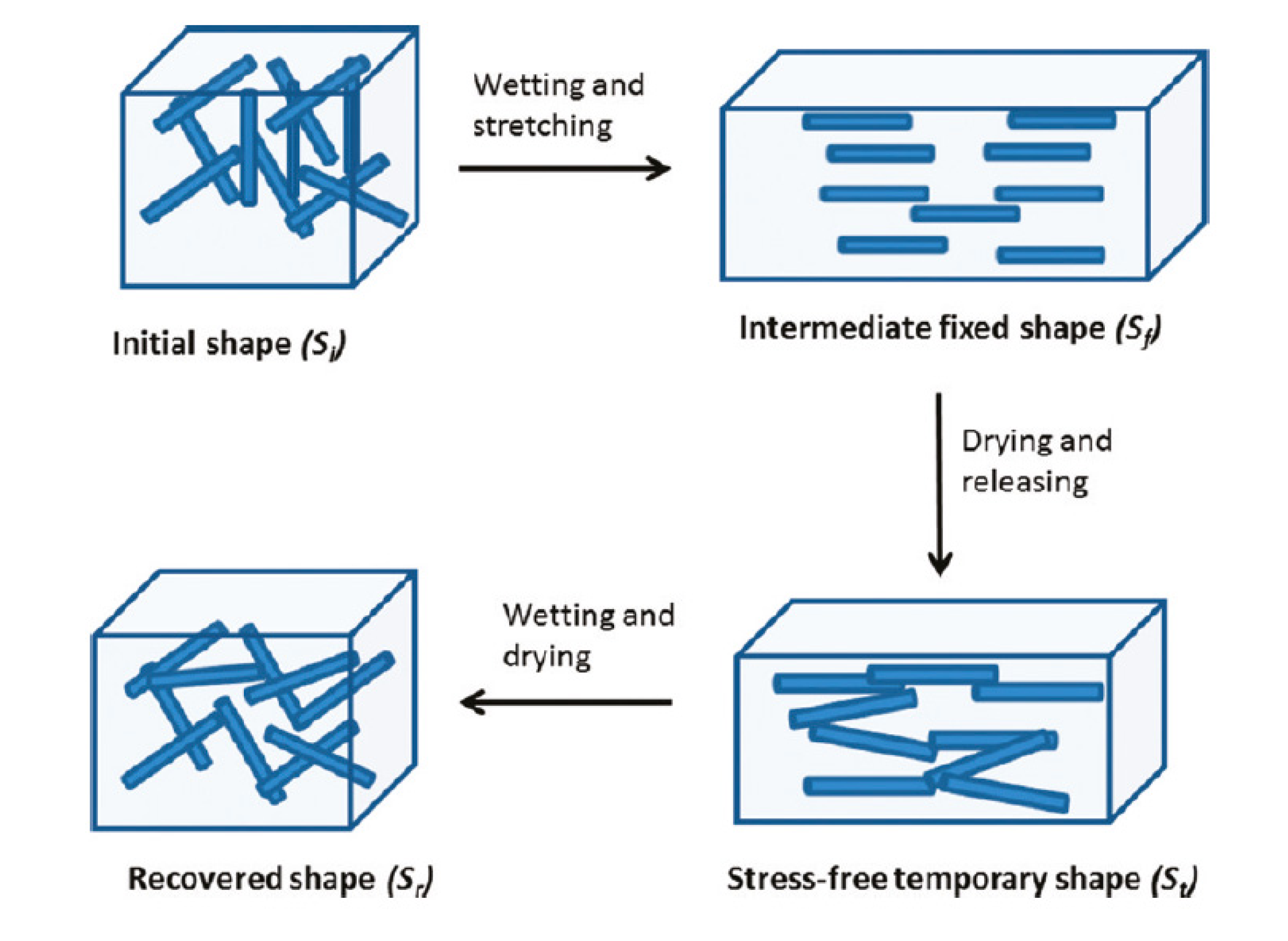
Aside from improving mechanical properties of composites, CNW can serve as effective water and gas barriers [168,169,170,171]. Because of the highly versatile functionality of cellulose, additional applications of CNW include three-dimensional conducting structures [172] for application in electronics, computing, sensors, and bioelectrochemical assemblies; catalysts and ion exchange systems [173] for filtration and water recycling; polyelectrolyte multilayer (PEM) thin films [174] for bioactive coatings, drug delivery, sensors or optically active surfaces. Using nano-sized CNW provides better control over thickness, roughness and surface properties compared to MFC. Novel biomimetic water-activated shape-memory materials were prepared by introducing rigid cotton cellulose nanowhiskers into a rubbery polyurethane (PU) matrix [175]. The nanocomposite changes its mechanical properties upon exposure to water and display a reversible water-activated shape-memory effect (Figure 7). One of the proposed applications is an artificial, self-propelled fishing lure that twists when wetted and may attract fish in different manner than static artificial lures.
Figure 7.
Schematic representation of the shape-memory materials explored here, which rely on switching the interactions between cellulose nanowhiskers within a polymer matrix upon addition and removal of water. Reproduced with permission from [175]. Copyright 2011 American Chemical Society.
Figure 7.
Schematic representation of the shape-memory materials explored here, which rely on switching the interactions between cellulose nanowhiskers within a polymer matrix upon addition and removal of water. Reproduced with permission from [175]. Copyright 2011 American Chemical Society.

2.3.1. CNW Biodegradability
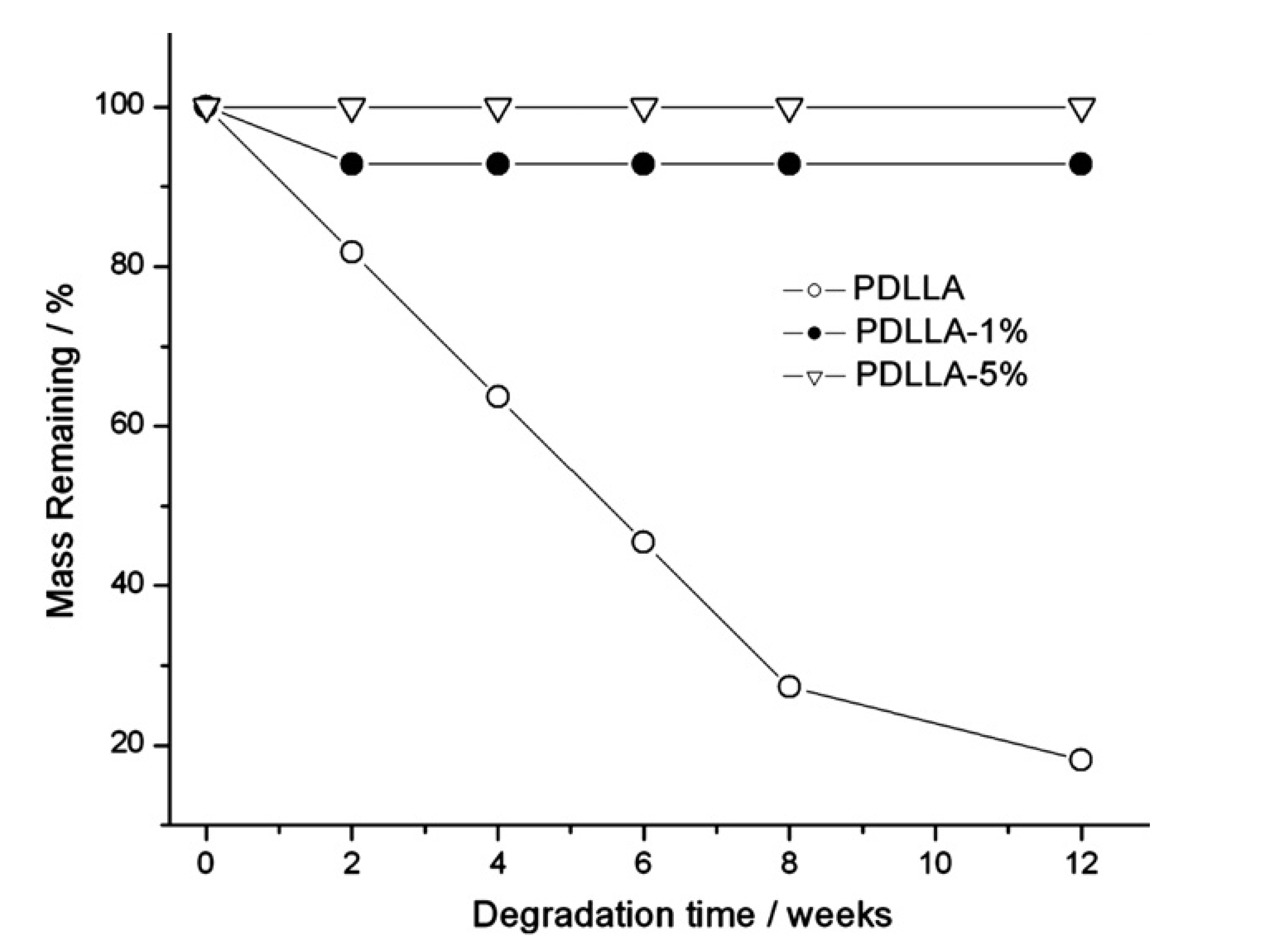
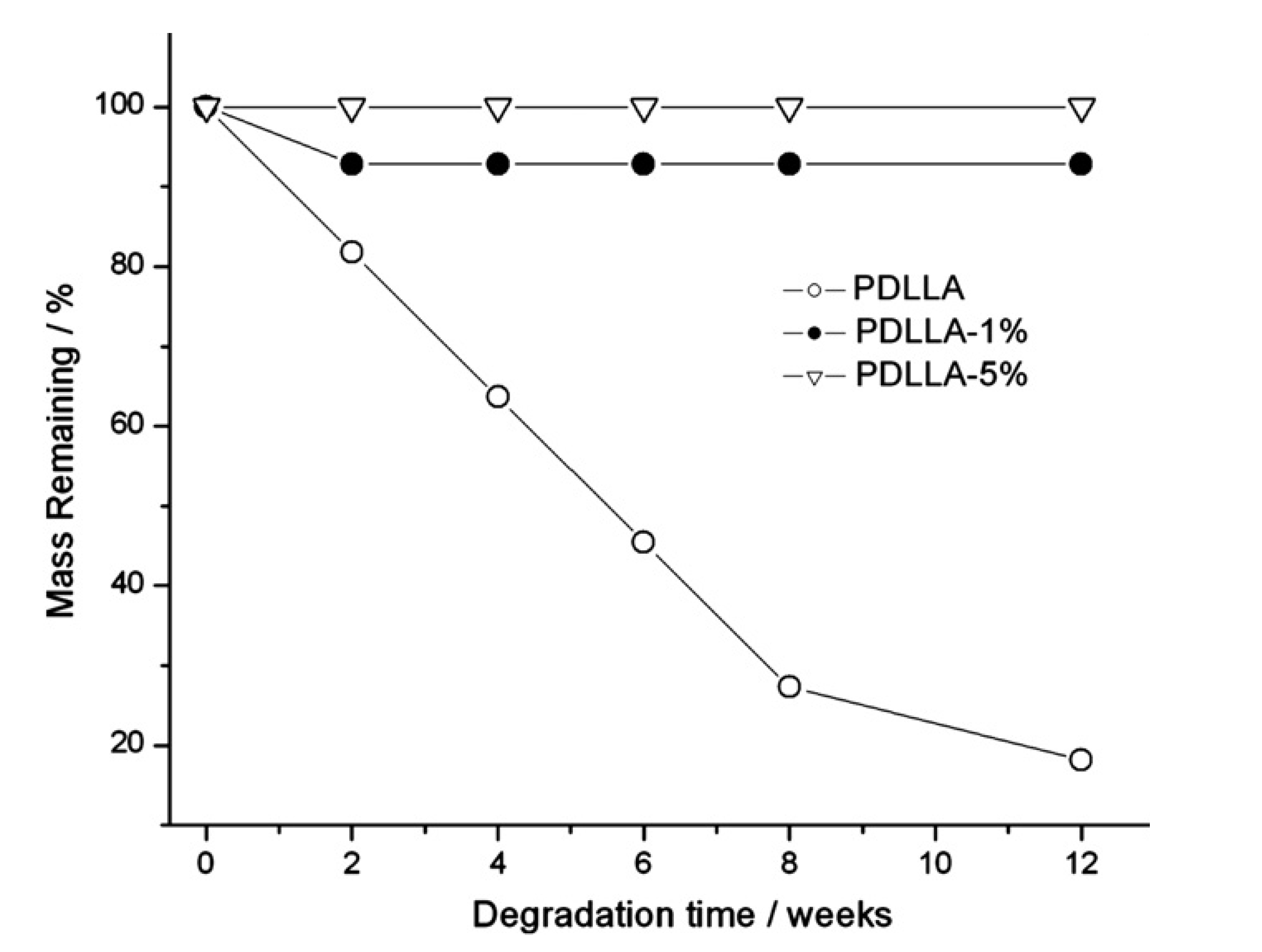
An attractive feature associated with the use of bio-based polymers is the potential of biological degradation by microorganisms. This is of particular relevance for applications where it is undesirable for the biopolymers to linger around beyond their useful lifespan, such as the use of biopolymers for drug delivery, or as gene vectors. Fullerenes, single-, double-, and multi-walled as well as COOH-functionalized carbon nanotubes (CNTs) have been used for these applications, but they tend to not be as biodegradable as delivery vehicles based on CNWs [176]. In fact, fullerenes and CNTs were reported to not be biodegradable at all. Toxicity can be assessed by comparing oxygen consumption, with high levels of oxygen consumption indicating degradation, and low levels of oxygen consumption indicating cell death. A compound is considered toxic if the difference between the predicted and measured amount of oxygen consumption exceeds 25% [176]. None of the particles (CNW and CNT) were found to be toxic. Another study showed that CNW with a concentration in the cell culture medium from 0.01% to 0.2% had low cytotoxicity [177]. CNW can also be used to tailor the biodegradability of composites. CNW addition induced a strong delay in the hydrolytic degradation of poly(d,l-lactide) (PDLLA) in phosphate buffer, even when the concentration of the nanofillers was only 1% (Figure 8). The cellulose nanocrystals delay the degradation because they act as a physical barrier, hindering the absorption and/or the diffusion of water among the polymer chains and consequently modifying the kinetics of the hydrolytic process [178].
Figure 8.
Residual mass of neat poly(d,l-lactide) (PDLLA), PDLLA with 1%, and PDLLA 5% of CNWs as a function of degradation time. Reproduced with permission from [178]. Copyright 2011 Elsevier.
Figure 8.
Residual mass of neat poly(d,l-lactide) (PDLLA), PDLLA with 1%, and PDLLA 5% of CNWs as a function of degradation time. Reproduced with permission from [178]. Copyright 2011 Elsevier.
A novel cellulose nanowhisker-based drug delivery system was obtained for amine-containing biologically active molecules and drugs. The nanowhiskers were grafted with a spacer molecule, γ-aminobutyric acid, using a periodate oxidation and Schiff’s base condensation reaction sequence. To achieve controlled and rapid delivery of the targeting moiety, syringyl alcohol, a releasable linker, was then attached to it. The reported method could be widely adapted for the controlled delivery of enzymes, proteins and amine-containing drugs with the selection of desired linker molecules [179].
Due to the number of attractive properties discussed above, industrial production of CNW has gained increased attention. Large-scale production of CNW can be adapted to extend the platform of common pulp/paper mills [180]. Recently, a viability of the residue from a bioethanol process as a low-cost source of cellulose nanowhiskers was investigated to add value to the production of bioethanol. The results showed that the lignin-rich residue from the bioethanol process is an excellent feedstock for large-scale production of cellulose nanowhiskers. The residue contained 49.4 wt % cellulose, 42.1 wt % lignin and 8.4 wt % extractives. The total yield of CNW separated through ultrasonication and homogenization was calculated and found to be as high as 48% [181].
3. Hemicellulosic Polysaccharides
Isolated and purified hemicellulosic polysaccharides can also be used for production of value-added renewable polymers. Current isolation and purification strategies including alkali peroxide extraction, organic solvent extraction, steam explosion, ultrasound-assisted extraction, microwave-assisted extraction, column chromatography, and membrane separation, which have been reviewed and summarized by Peng et al. [182]. Among the isolation and purification methods, alkali treatment has been proven to be an efficient method for extracting available hemicelluloses from the cell walls of biomass, but the alkali extractions have the disadvantage of de-acetylating the hemicelluloses. The hemicelluloses extracted with a higher alkaline concentration have the features of more linear structure and a higher MW [183]. Alkali peroxide treatment is an effective way for both delignification and solubilization of hemicelluloses from lignocellulosic materials. Dimethylsulfoxide (DMSO) is the most common neutral solvent for extracting hemicelluloses, which has minimal impact on their structural integrity. Anti-solvent precipitation of xylans and mannans from DMSO or DMSO/water mixtures, and subsequent drying with supercritical carbon dioxide (scCO2) were developed into a useful technique for preparing spherical hemicellulose microparticles. Potential applications of these microparticles include use as chromatographic separation materials or the encapsulation of active compounds to ensure slow release [184,185].
Physical methods, such as steam explosion, microwave, and ultrasound-assisted extraction, accelerate hemicellulose extraction and reduce the consumption of chemicals. Membrane technologies such as ultrafiltration, microfiltration, nanofiltration and reverse osmosis have drawn great attention in the biorefinery for separation and purification of lignocellulosic products. They provide relatively cost-effective separation steps, and offer a commercial alternative to chromatographic methods for purification of hemicelluloses. The combination of twin-screw extrusion, ultrafiltration, and anion exchange chromatography efficiently produced hemicellulosic polymers with high purity rates [182].
The effect of hemicellulose extraction from cellulosic fibers on multiple properties of cellulose fibers and cellulose fiber-reinforced composites has been extensively investigated. Hemicellulose removal can alter the properties as well as the morphology of natural fibers. Since hemicellulose degrades more rapidly than cellulose, thermal analysis of hemicellulose-extracted hemp fibers confirmed improved thermal stability [186]. The decrease in the content of hemicellulose resulted in increased Young’s modulus and elongation at break for alkali-treated Borassus fruit fibers [187]. Moreover, hot-water-pretreated wood flakes showed a decrease in wettability consistent with a decrease in the hydrophilic character of wood strands. The amount of water taken up by strands subjected to hot-water extraction decreased as the percentage of mass loss increased [188]. When blended with 5% liquid phenol-formaldehyde resin and 1% wax emulsion and hot pressed, the hemicellulose-extracted flakeboards showed remarkable decrease in water absorption and thickness swelling without a decrease in mechanical properties. Resistance of the panels to mold growth also increased as less hemicellulosic polysaccharides remained [189]. In another study, FT-IR measurements indicated that there was an increase in the number of hydroxyl groups on the cellulose surface after alkali treatment. Mechanical testing revealed that alkali treatment of wood particulates improved both composite strength and modulus when polypropylene (PP) grafted with maleic acid used as a coupling agent. The strength increase was ascribed to improved adhesion between the fiber and matrix due to improved bonding resulting from the increase in the number of hydroxyl groups, while the improved modulus was due to the removal of lignin and hemicellulose that are not as stiff as cellulose [190]. Similar results were reported for sisal fibers. The alkaline treatment of sisal increased the number of active sites and therefore, significantly increased the surface area (from 1.633 to 2.392 m2/g) and decreased the diffusion coefficient [191].
Purified hemicelluloses have fewer commercial applications than cellulose and lignin, but in recent years, interest in production of value-added products, such as xylitol [192,193,194,195,196], packaging films [197,198,199] and hydrogels [200,201,202,203,204,205,206,207,208,209] based on hemicelluloses has increased. Xylitol is used for sweetening owing to its dietetic and anticariogenic properties. Several types of naturally xylose-utilizing yeasts from the genus Candida are capable of producing xylitol effectively from xylose by fermentation of hydrolyzed agricultural biomass [192,193,194,195]. Polymers derived from hemicellulose applied in packaging films and hydrogels are discussed further below.
3.1. Packaging Films
Aside from offering an opportunity for marketing, food packaging provides physical protection of the food inside, and chemical protection against spoiling. Spoiling can be the result of oxygen-induced damage, especially to fatty acids and vitamins, which affects taste and nutritional benefits. For the use in packaging films and as matrices for composites, thermal and mechanical properties of acetylated arabinoxylans films were investigated [197]. Prior to film casting of arabinoxylans/chloroform solutions, arabinoxylans from rye were partly debranched by oxalic acid hydrolysis, and arabinoxylans differing in arabinosyl substitution were acetylated using acetic acid anhydride. Nuclear magnetic resonance (NMR) showed that all free hydroxyl groups (of both xylose and arabinose) were acetylated. Thermo-gravimetric analysis confirmed that the arabinose content contributes slightly to the thermal stability of the acetylated arabinoxylans. Tensile tests showed that the strain at break, which reflects the flexibility of the films, was significantly higher at higher arabinosyl substitution levels. The elastic Young’s modulus was not significantly affected, although a tendency was seen toward a less stiff material at higher arabinosyl substitution [197].
3.2. Hydrogels
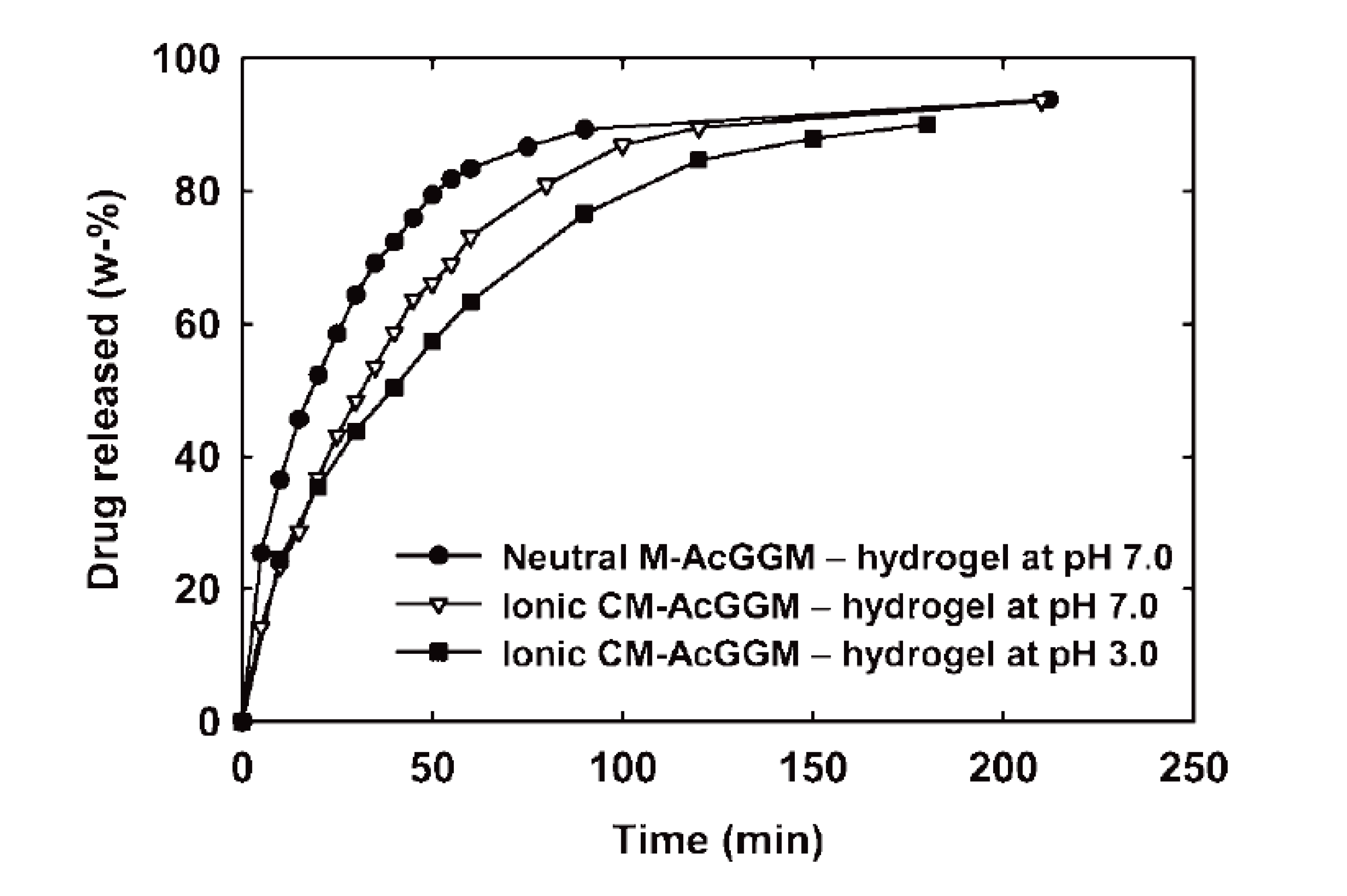
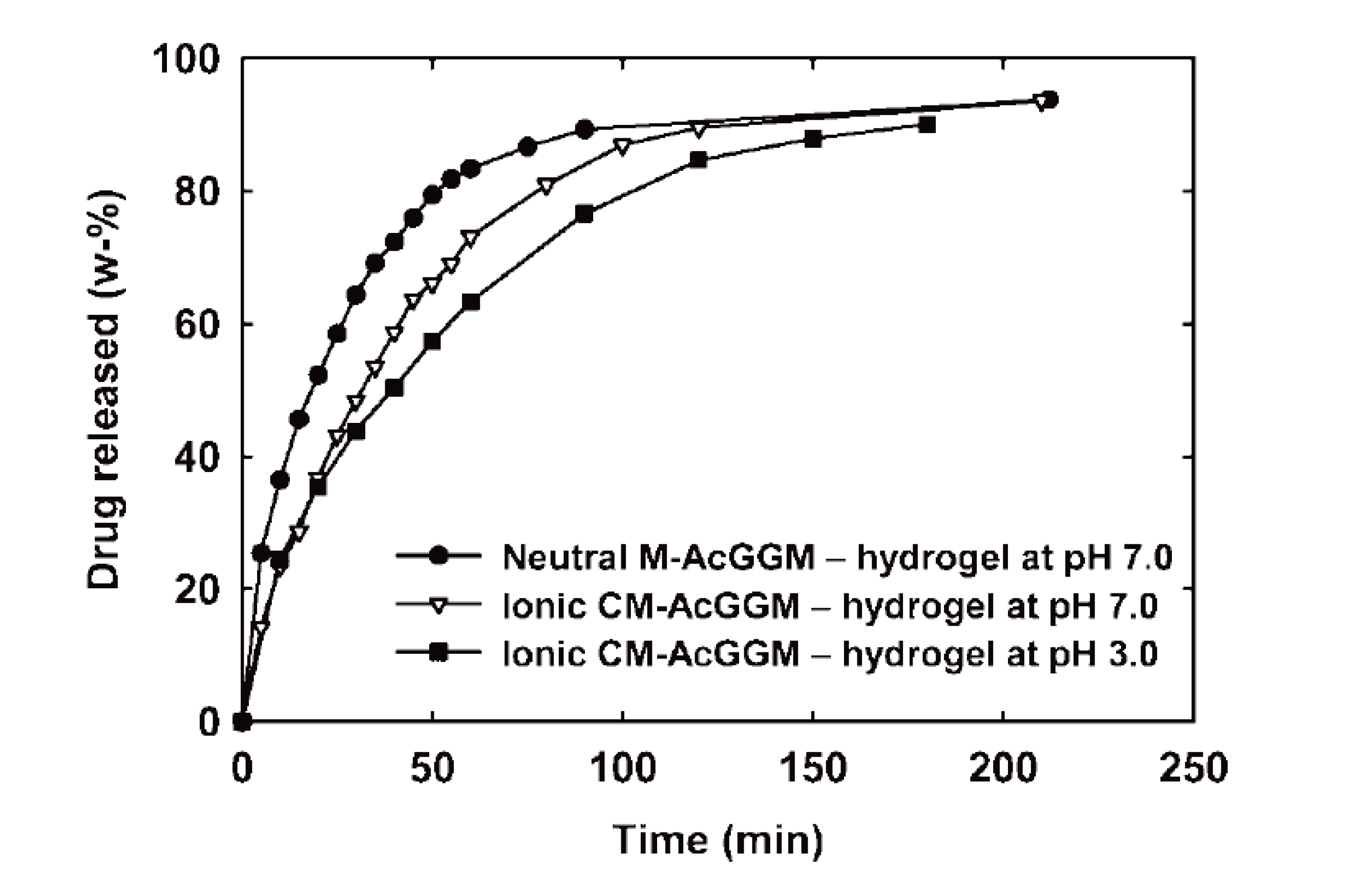
A series of hydrogels based on acetylated galactoglucomannan (AcGGM) was successfully synthesized and evaluated for drug delivery systems applications [200,201]. The modified AcGGM was radically crosslinked to form hydrogels, using a redox initiation and photo initiation in water and DMSO, respectively. Varying the linked alkenyl species and its degree of substitution (DS) on the AcGGM backbone and the crosslinking method were found to be important tools for adjusting the resulting gels properties. Controlling all these parameters gave rise to a variety of properties, ranging from dense gels with very low swelling to soft gels with considerably higher swelling as well as gels of varying modulus [200]. Furthermore, neutral hydrogels were produced from functionalized AcGGM using 2-hydroxyethylmethacrylate (HEMA) coupled via carbonyldiimidazole (CDI) and a co-monomer in a radical-initiated polymerization. Through a second modification reaction between the HEMA-modified AcGGM (M-AcGGM-methacrylated AcGGM) and maleic anhydride, a “double-modified” AcGGM (CM-AcGGM-carboxylated M-AcGGM) was successfully formed that could be cross-linked to form ionic hydrogels by the very same polymerization method [201]. In neutral hydrogels the drug (caffeine and vitasyn blue) release rate and the Fickian swelling decreased with an increase in the relative amount of the methacrylated AcGGM and its corresponding degree of methacrylation. In Figure 9, it is shown that the release was faster at pH 7.0 in comparison to at pH 3.0. Under acidic conditions, the release speed was lowered as expected because of protonation of carboxylic functionalities which leads to a swelling of the matrix due to electrostatic repulsion. This effect explains the release results obtained from the buffered neutral conditions which gave a slower release in comparison to the corresponding nonbuffered conditions. These novel hemicellulose-containing hydrogels have future prospects in oral drug administration technology [201].
Xylan can also be used to prepare poly(2-hydroxyethyl methacrylate)-based hydrogels via facile crosslinking induced by methacrylic monomers using standard radical polymerization. Effect of xylan-based acetyl substituents on the morphology and physical properties of a novel polysaccharide-based hydrogel biomaterial was thoroughly investigated [202]. The presence of acetyl groups introduced high stiffness to the hydrogels, which ultimately reduced their water swelling capacity, and, moreover, significantly enhanced their drug release properties as evidenced by the time release profile obtained for a representative drug, i.e., doxorubicin.
pHEMA hydrogels with modified hemicellulose and cellulose nanowhiskers were also synthesized and characterized for a potential replacement material of articular cartilage [204]. HEMA-modified hemicellulose, isolated from aspen wood was adsorbed onto the cellulose nanowhiskers in aqueous medium and hydrogels were synthesized by in situ radical polymerization of the methacrylic groups of the adsorbed coating to form a network of pHEMA matrix reinforced with cellulose nanowhiskers. The properties of the produced pHEMA hydrogels, such as water holding capacity, mechanical properties and viscoelasticity, appeared to be similar to load-bearing natural tissue having hydrogel-like characteristics [204].
Figure 9.
Caffeine release (in 900 mL H2O, 37 °C, 50 rpm stirring) from initially dry neutral (degree of 2-hydroxyethylmethacrylate (HEMA)-Im–substitution is 0.10) and ionic hydrogels (degree of HEMA-Im–substitution is 0.10, degree of carboxylation is 0.31) loaded during polymerization. The hydrogels were all produced with 50 wt % HEMA as co-monomer. Reproduced with permission from [201]. Copyright 2009 Wiley Periodicals, Inc.
Figure 9.
Caffeine release (in 900 mL H2O, 37 °C, 50 rpm stirring) from initially dry neutral (degree of 2-hydroxyethylmethacrylate (HEMA)-Im–substitution is 0.10) and ionic hydrogels (degree of HEMA-Im–substitution is 0.10, degree of carboxylation is 0.31) loaded during polymerization. The hydrogels were all produced with 50 wt % HEMA as co-monomer. Reproduced with permission from [201]. Copyright 2009 Wiley Periodicals, Inc.
4. Lignin
Due to the fact that lignin is an amorphous aromatic polymer, the chemical and physical properties of lignin are fundamentally different from those of cellulose and hemicellulose. Depending on the origin and isolation method, lignin has a glass transition temperature (Tg) between 110 and 160 °C [210], which is substantially lower than the Tg of cellulose (approximately 220 °C) [211]. Furthermore, lignin has a redox potential and contains more diverse reactive chemical groups (aromatic and aliphatic hydroxyl, carboxyl, and carbonyl moieties). Lignin has historically been viewed as a waste product with little intrinsic value. Lignin generated by pulp/paper mills relying on the Kraft process, also known as the sulfate process, is present as a low-MW fraction in so-called “black liquor” that can be concentrated and burned to generate heat or power at the paper mill, or, alternatively, gasified or carbonized [212,213]. An example of the latter is to convert the lignin via carbonization to a solid fuel similar in properties to coal. Lignin isolated in pulp/paper mills using the sulfite process is present as lignosulfonate with a MW between 5100 and 61,000 Da depending on the source of the lignin and the exact processing conditions [214]. Lignosulfonates have been used in low-value, high-volume applications, including as a dispersive agent in cement to enhance rheological properties [215], binder in particle boards and plywood [216], additive to enhance the lubrication qualities of grease [217], and as a thinning agent of the drilling fluid (“mud”) used to facilitate the drilling of oil wells [218]. A detailed review on these types of applications is provided by Windeisen and Wegener [219]. With the desire to maximize the use of all components of the biomass, efforts are being made to identify additional uses of lignin that capitalize on the intrinsic features of lignin, and that ideally have a higher market value than the above-mentioned uses. Furthermore, biorefineries aimed at producing biofuels from biomass will likely utilize different processes to decompose the biomass into its component polymers than pulp/paper mills, and will use additional biomass feedstocks (especially grasses). Consequently, additional sources of lignin displaying different physico-chemical properties will become available.
4.1. Lignin as an Additive to Enhance Composites
Unlike natural lignin, lignosulfonates are water soluble due to the presence of sulfonate groups. The presence of both hydrophilic and hydrophobic domains in lignosulfonates enables them to be mixed with different kinds of polymers, including poly(alpha-hydroxy acid) (e.g., polylactate, polyglycolic acid) and poly(beta-hydroxy alkanoate) (e.g., poly(beta-hydroxybutyrate), poly (beta-hydroxybutyrate-co-valerate). Blending lignin with these polymers often results in enhanced thermochemical and mechanical characteristics. Several excellent recent reviews on the different kinds of lignin-polymer composites exist [220,221], and the reader is referred to these for further detail. A number of novel applications are discussed further below.
4.2. Lignosulfonates in Active Packaging
Active packaging refers to packaging that protects against oxidative damage with the use of oxygen scavengers, such as glucose oxidase. Johansson et al. [222] investigated the use of laccase (EC 1.10.3.2) from the white-rot fungus Trametes versicolor as a scavenging agent. In the presence of oxygen this copper-containing enzyme generates phenoxy radicals from phenolic hydroxyl groups. The radicals formed as a result of the enzymatic activity can form polymeric structures. Laccase and its lignosulfonate substrate were applied to three-ply packaging barrier board to test oxygen-scavenging ability in airtight containers. At high relative humidity (RH) the oxygen concentration was reduced from the initial 3% to 0.3%, although this did require 3 days. At lower RH and with enzyme-free coatings, there was no reduction in oxygen content. These data suggest that the coating of packaging board with lignosulfonate-laccase will improve the packaging properties of items that are stored under high RH, especially if the packaging can be modified so that the effect is noticeable before 3 days. The water stability of latex-based coatings on aluminum foil and of cast starch-based films also improved noticeably after addition of laccase and lignosulfonate. The E′ modulus of the latter film had increased, reflecting a stiffer material, likely due to the higher degree of polymerization resulting from the laccase activity [222].
4.3. Lignin-Coated Electrodes in Sensors and Batteries
The redox potential of lignin is based on the formation of quinone structures involving the phenolic hydroxyl moiety on C4 of the aromatic ring and a neighboring hydroxyl group on C3 or C5, formed via electrochemical oxidation from the methoxyl group that was originally present. The quinone structure can be reversibly cycled between an oxidized and reduced form. This feature of lignin makes it suitable for applications in electrodes, sensors and batteries. For example, Milczarek [223] prepared a suspension of multi-walled carbon nanotubes (MWCNTs) and Kraft lignin in DMSO and applied this on a glassy carbon electrode. Kraft lignin in the resulting film functioned as a redox system, while the MWCNTs facilitated charge transfer. One of the benefits of using a lignin-MWCNT-modified electrode is a faster electron transfer rate and a reduction of the reaction overpotential. The latter feature was demonstrated by oxidizing dihydronicotinamide adenine dinucleotide (NADH), a compound involved in providing reducing capacity to living cells, comparing a standard versus lignin-MWCNT-modified electrode. These modified electrodes may be employed as electrochemical sensors to track biochemical reactions.
One of the risks associated with the application of lignin-based films containing MWCNTs is the potential instability over time, as the film disintegrates. Faria et al. [224] therefore chemically crosslinked Kraft lignin doped with MWCNTs in a polymer formed from poly(propylene glycol) and deposited the resulting composite on a glassy carbon electrode. At a concentration of just 0.18% (w/w), the MWCNTs enhanced the conductivity of the resulting film without impacting the thermal or viscoelastic properties. The presence of lignin in the composite enabled an even distribution of the MWCNTs along the lignin clusters. The composite did not leach and was thermally stable. The resulting potentiometric sensor was tested against a number of ions and shown to be very sensitive to Cr(VI). A follow-up study by Rudnitskaya et al. [225] indicated that different sources of lignin—Kraft lignin, lignosulfonate and organosolv lignin—affected the performance of sensors. Sensors made from lignosulfonate or organosolv lignin were more sensitive than sensors made from Kraft lignin. This was attributed to different degrees of dispersion of MWCNTs in the composite.
In addition to sensors, which respond to changes in conductivity, the redox potential of lignin can also be exploited in batteries. Milczarek and Inganäs [226] co-polymerized lignosulfonate derived from the waste stream of a sulfite-based paper mill with pyrrole onto gold electrodes. The formation of quinones from free phenolic end-groups in the lignin allowed the storage of electrons and protons, whereas the polypyrrole matrix provided conductivity. The charge density of this system was higher than comparable standard electrochemical systems.
4.4. Lignin Nanotubes
Carbon nanotubes with the fullerene structure have many uses, including the smart delivery of therapeutic agents to target cells in humans and animals. This approach relies on nanotubes functionalized with the therapeutic agent and a ligand that interacts with receptors on the target cells [227,228]. Carbon nanotubes can be produced from graphite using several different methods (reviewed by [229]), from carbonization of biomass with loss of the original structure [230,231], or while maintaining the original structural features [232]. One of the challenges associated with these types of nanotubes, however, is their chemical inertness and sharp, needle-like shape that can mimic asbestos [233,234]. The production of nanotubes derived from lignin, as described by Caicedo et al. [235], may overcome some of these challenges (Figure 10). A template-mediated synthesis protocol was developed in which a lignin base layer was cross-linked to an alumina membrane, followed by peroxidase-mediated addition of dehydrogenation polymer (“synthetic lignin”). Subsequent dissolution of the membrane in phosphoric acid resulted in flexible nanotubes or nanowires that could be easily functionalized due to the presence of many reactive groups, and whose optical and physical properties could be tailored depending on the monomers employed in the polymerization reaction.
Figure 10.
Scanning electron micrographs of lignin nanotubes prepared according to the protocol in reference 235 (a) Overview image showing a bundle of lignin nanotubes from a partially dissolved alumina membrane; bar = 2 μm. (b) An individual lignin nanotube with an incomplete wall, revealing the lumen of the tube; bar = 1 μm. Images were acquired on a JEOL JSM 6335F field-emission scanning electron microscope at the Major Analytical Instrumentation Center, University of Florida, Gainesville, FL, USA.
Figure 10.
Scanning electron micrographs of lignin nanotubes prepared according to the protocol in reference 235 (a) Overview image showing a bundle of lignin nanotubes from a partially dissolved alumina membrane; bar = 2 μm. (b) An individual lignin nanotube with an incomplete wall, revealing the lumen of the tube; bar = 1 μm. Images were acquired on a JEOL JSM 6335F field-emission scanning electron microscope at the Major Analytical Instrumentation Center, University of Florida, Gainesville, FL, USA.

5. Genetic Modification of Biomass Composition: Tailoring Plants for Specific Applications
Expanding bioenergy and bio-polymer production to commercial scales requires a significant increase in the production of the feedstock. Since metabolic pathways in plants are under genetic control, it is possible to modify the chemical composition of plant biomass through genetic approaches, leading to crops that are more amenable to down-stream applications, either because the cell walls can be broken down more easily, or because of physico-chemical properties optimized for certain applications. Plant breeding and plant biotechnology have primarily focused on improving food and feed quality (grain yield, starch and protein composition, grain digestibility) and overall crop performance (disease and pest resistance, drought tolerance), whereas the quality of the vegetative parts of the plant have typically only been considered in forage crops (biomass fed to ruminant animals; e.g., forage maize and sorghum, alfalfa) and fiber crops (e.g., flax, jute, kenaf) [236]. The optimal biomass composition for the production of fuels and chemicals is, however, likely to be very different, due to the different biomass conversion process. Moreover, there is no need to consider palatability in the selection and breeding process of bioenergy crops.
Most of the efforts aimed at modifying plant cell wall composition have focused on lignin [237,238]. The enzymatic steps involved in lignin biosynthesis are relatively well characterized and have been the subject of numerous manipulation experiments [reviewed in 26]. This was initially motivated by the desire to improve the efficiency of paper making from wood pulp [239], with transgenic approaches offering changes in cell wall chemistry that could not be obtained through traditional tree breeding approaches. Despite the obvious environmental benefits of woody biomass that can be pulped with reduced chemical inputs, this approach met serious opposition from environmentalists [240]. The interest in cellulosic biofuels has led to evaluations of transgenic plants [241] and naturally occurring mutants [242] with modified cell wall compositions. Total lignin concentration in the cell wall can be reduced, lignin subunit composition (higher or lower S/G ratio) can be changed, and plants that incorporate intermediates from the monolignol biosynthetic pathway at levels higher than normally observed (e.g., coniferaldehyde, sinapaldehyde) can be obtained (reviewed by [19,243]). High S/G ratios favor biomass conversion in angiosperm dicot species (poplar, alfalfa) [241,244], whereas in grasses low S/G ratios are more favorable [242,245,246,247]. This difference most likely reflects spatio-temporal differences in lignification among these two classes of plant species.
In contrast, the more limited understanding of the genetic control of the biosynthetic pathways leading to cellulose and hemicellulosic polysaccharides have hampered efforts to tailor polysaccharide composition to specific down-stream applications.
Most investigations on the biosynthesis of cell wall polysaccharides have been performed on the model species Arabidopsis thaliana, which has as benefits short generation times, a sequenced genome, and the availability of mutant collections. Translation to species with relevance for large-scale agricultural production, however, is not always easy. Among grass species, maize and sorghum are currently the best studied.
Maize genetics has a long history and many mutants exist, including cell wall mutants. The brown midrib mutants, named after the brown vascular tissue in the main leaf vein (midrib), display differences in lignin subunit composition, and/or the spatio-temporal deposition of lignin in the plant (reviewed by [248]). Furthermore, the brittle stalk2 mutant is a mutant with aerial parts that are easily broken as a result of altered interactions among cell wall polymers [249]. A collection of maize mutants identified based on altered near infrared reflectance spectra of their leaves is available [250], and similar resources exist for sorghum [251,252]. It is likely that more detailed characterization of these collections will reveal mutants with modified cell wall polymers. In addition, natural genetic variation affecting biomass conversion to fermentable sugars has been shown to exist in sorghum [253]. Some of the observed variation is likely due to different proportions of cell wall polymers. Natural genetic variation in cell wall composition has also been identified in poplar [254].
Care must be taken that the agronomic performance of plants with altered cell wall composition is not compromised. Plant cell wall composition has evolved to withstand attack by fungi and insects, to enable the efficient transport of water through the lignified xylem cells, and to provide mechanical strength to cells and tissues. Hence, the targeted modification of cell wall composition has the intrinsic risk that the resulting plants display reduced fitness. For example, the maize brown midrib double mutant bm2-bm4 is severely compromised in its ability to grow and reproduce as a result of changes to the vascular tissue, and the addition of a third bm mutation, bm1, rendered the plants unable to develop beyond the seedling stage [255] (Figure 11). In contrast, some of the sorghum brown midrib mutants were shown to be more resistant to the pathogenic fungus Fusarium as a result of the accumulation of phenolic precursors for monolignols that were toxic to the fungus [256]. Modification of lignin subunit composition has also been shown to impact plant performance in alfalfa [257].
Figure 11.
(a) The bm2-bm4 double mutant (blue arrow) is significantly shorter and thinner than the single mutants bm2 (shown to the right) and bm4; (b) The vascular tissue of the bm2-bm4 mutant is dark-brown; (c) Six-week old, field-grown seedlings of inbred A619 (1) and near-isogenic mutants bm1-bm2 (2), bm2-bm4 (3), and bm1-bm2-bm4 (4). Inset: Gel image showing the PCR [polymerase chain reaction] product obtained with SSR [simple sequence repeat] marker dupSSR10. Numbers correspond to image 1C. M is a 100 bp [basepair]ladder. Reproduced with permission from [255]. Copyright 2010 Oxford University Press.
Figure 11.
(a) The bm2-bm4 double mutant (blue arrow) is significantly shorter and thinner than the single mutants bm2 (shown to the right) and bm4; (b) The vascular tissue of the bm2-bm4 mutant is dark-brown; (c) Six-week old, field-grown seedlings of inbred A619 (1) and near-isogenic mutants bm1-bm2 (2), bm2-bm4 (3), and bm1-bm2-bm4 (4). Inset: Gel image showing the PCR [polymerase chain reaction] product obtained with SSR [simple sequence repeat] marker dupSSR10. Numbers correspond to image 1C. M is a 100 bp [basepair]ladder. Reproduced with permission from [255]. Copyright 2010 Oxford University Press.

Computational methods for analyzing monolignol biosynthesis that account for lignin biosynthesis in various transgenic lines and different developmental stages has been proposed by Lee et al. [258] for alfalfa and by Vanholme et al. [259] for Arabidopsis. Critical evaluation of the transgenic data against the metabolic pathway structure can provide mechanistic insights that will aid the design of combined genetic modification strategies toward the generation of crops with cell wall compositions that are ideally suited for the production of specific biopolymers.
In summary, a better understanding of the biosynthesis of secondary wall components will help us to identify genes for genetically modifying pathways that determine the quantity and property of the biomass, thus further enhancing the conversion technologies leading to biofuel and biopolymers, while reducing the cost of production.
6. Conclusion and Prospects
From the review of the publications discussed in this article it is clear that bio-polymers can provide an alternative to a number of petroleum-derived polymers and enable the development of novel applications, especially as bio-compatible compounds, sometimes with properties that exceed those of synthetic polymers made from petroleum. Economic benefits, however, are currently the primary driving force behind wide-scale adoption of bio-polymers. In some cases the lower cost of the feedstock and/or lower cost of production provide obvious advantages to bio-based products, but the relatively long history of refining petroleum has made this process very efficient and cost-effective and environmental costs other than disposal or waste management are generally not yet included in economic analyses. Hence, the development of efficient and cost-effective processes for the synthesis of renewable polymers is a key requirement for the gradual replacement of petroleum-based chemicals and materials. Despite significant recent advances, some major aspects remain to be addressed, including infrastructure, process integration, scale up, market development, and political and public support, briefly discussed below.
Biomass, especially from grasses, has a low bulk density. This impacts transportation and storage, with a risk of relatively high feedstock costs and loss of quality. Biomass processing in rural areas where the biomass is produced would help reduce transportation costs and stimulate local economies, but also reduce the financial benefits associated with economy of scale. Pre-processing of biomass leading to higher density and better storage properties may be a viable way to address this challenge [260].
The development of integrated biorefineries that produce biofuels, chemical feedstocks, bio-based polymers and a waste stream compatible with generation of heat and power will maximize efficiency and enhance economic sustainability. This will require flexible plant designs that can adapt to varying needs and demands. In addition, the pretreatment method is a crucial factor governing the quality of the cellulose, hemicellulose and lignin fractions from the biorefinery processes, and therefore the potential value-added products that can be produced. When selecting a particular pretreatment method, it may be worth considering the combined value of biofuels and bio-products rather than just the cost of production of biofuels.
Continuing investments in academic and private research efforts are necessary for the development of a mature bio-products industry. This research will benefit from interdisciplinary education at the interface of biology, chemistry, engineering and marketing, whereby both energy generation and bio-products are considered. Widespread societal adoption of bioenergy and bio-based products will require education of the general public, as well as environmental and economic policies that encourage the transition from fossil to renewable resources. Partnerships between public and private entities and between different businesses will create new economic opportunities that will further this transition. A number of such partnerships already exist. Examples include large chemical companies that have partnered with plant breeding companies and manufacturers of industrial enzymes, and pulp/paper mills that have partnered with tree breeders in industry and academia. The diversity in bio-polymers and their applications offers many opportunities for new partnerships, especially in the areas of specialty chemicals, therapeutics and energy storage.
Acknowledgements
The authors are grateful for financial support from the Biomass Research and Development Initiative grant No. 2011-10006-30358 from the USDA National Institute of Food and Agriculture for research on renewable polymers. We thank Amelia Dempere from the Major Analytical Instrumentation Center at the University of Florida for her support.
References
- Vermerris, W. Why Bioenergy Makes Sense. In Genetic Improvement of Bioenergy Crops; Vermerris, W., Ed.; Springer: New York, NY, USA, 2008; pp. 3–42. [Google Scholar]
- Independent Statistics & Analysis U.S. Energy Information Administration Web Page. Frequently Asked Questions. Available online: http://www.eia.gov/tools/faqs/faq.cfm?id=34&t=6 (accessed on 22 February 2013).
- Loock, M. Going beyond best technology and lowest price: On renewable energy investors’ preference for service-driven business models. Energy Policy 2012, 40, 21–27. [Google Scholar] [CrossRef]
- Masini, A.; Menichetti, E. The impact of behavioral factors in the renewable energy investment decision making process: Conceptual framework and empirical findings. Energy Policy 2012, 40, 28–38. [Google Scholar]
- Chakravorty, U.; Hubert, M.-H.; Nøstbakken, L. Fuel versus food. Annu. Rev. Resour. Econ. 2009, 1, 645–663. [Google Scholar] [CrossRef]
- Thompson, P.B. The agricultural ethics of biofuels: Climate ethics and mitigation arguments. Agriculture 2012, 8, 169–189. [Google Scholar]
- Carpita, N.C.; Gibeaut, D.M. Structural models of primary cell walls in flowering plants: Consistency of molecular structure with the physical properties of the walls during growth. Plant J. 1993, 3, 1–30. [Google Scholar] [CrossRef]
- Crowell, E.F.; Gonneau, M.; Stierhof, Y.-D.; Höfte, H.; Vernhettes, S. Regulated trafficking of cellulose synthases. Curr. Opin. Plant Biol. 2010, 13, 700–705. [Google Scholar]
- Guerriero, G.; Fugelstad, J.; Bulone, V. What do we really know about cellulose biosynthesis in higher plants? J. Integr. Plant Biol. 2010, 52, 161–175. [Google Scholar]
- Mizrachi, E.; Mansfield, S.; Myburg, A. Cellulose factories: Advancing bioenergy production from forest trees. New Phytol. 2012, 194, 54–62. [Google Scholar] [CrossRef]
- Dahlgren, G. An updated angiosperm classification. Bot. J. Linn. Soc. 1989, 100, 197–203. [Google Scholar]
- Burton, R.A.; Wilson, S.M.; Hrmova, M.; Harvey, A.J.; Shirley, N.J.; Medhurst, A.; Stone, B.A.; Newbigin, E.J.; Bacic, A.; Fincher, G.B. Cellulose Synthase-like CSLF genes mediate the synthesis of cell wall (1,3;1,4)-β-d-glucans. Science 2006, 311, 1940–1942. [Google Scholar] [CrossRef]
- Cocuron, J.; Lerouxel, O.; Drakakaki, G.; Alonso, A.; Liepman, A.; Keegstra, K.; Raikhel, N.; Wilkerson, C. A gene from the cellulose synthase-like C family encodes a beta-1,4 glucan synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 8550–8555. [Google Scholar] [CrossRef]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef]
- Zabotina, O.A. Xyloglucan and its biosynthesis. Front. Plant Sci. 2012, 3, 134. [Google Scholar] [CrossRef]
- Vermerris, W.; Nicholson, R. Phenolic Compound Biochemistry; Springer: Dordrecht, The Netherlands, 2006; p. 276. [Google Scholar]
- Ralph, J.; Peng, J.; Lu, F.; Hatfield, R.; Helm, R. Are lignins optically active? J. Agric. Food Chem. 1999, 47, 2991–2996. [Google Scholar] [CrossRef]
- Hatfield, R.; Vermerris, W. Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol. 2001, 126, 1351–1357. [Google Scholar] [CrossRef]
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; Boerjan, W. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 2004, 3, 29–60. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Ragauskas, A.; Williams, C.; Davison, B.; Britovsek, G.; Cairney, J.; Eckert, C.; Frederick, W.; Hallett, J.; Leak, D.; Liotta, C.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J. Hydrolysis of lignocellulosic materials for ethanol production: A review. Bioresour. Technol. 2002, 83, 1–11. [Google Scholar] [CrossRef]
- Saritha, M.; Arora, A.; Lata. Biological pretreatment of lignocellulosic substrates for enhanced delignification and enzymatic digestibility. Indian J. Microbiol. 2012, 52, 122–130. [Google Scholar] [CrossRef]
- Singh, P.; Sulaiman, O.; Hashim, R.; Rupani, P.F.; Peng, L.C. Biopulping of lignocellulosic material using different fungal species: A review. Rev. Environ. Sci. Biotechnol. 2010, 9, 141–151. [Google Scholar] [CrossRef]
- Pu, Y.; Kosa, M.; Kalluri, U.C.; Tuskan, G.A.; Ragauskas, A.J. Challenges of the utilization of wood polymers: How can they be overcome? Appl. Microbiol. Biotechnol. 2011, 91, 1525–1536. [Google Scholar] [CrossRef]
- Gullón, P.; Romaní, A.; Vila, C.; Garrote, G.; Parajó, J.C. Potential of hydrothermal treatments in lignocellulose biorefineries. Biofuels Bioprod. Bioref. 2012, 6, 219–232. [Google Scholar] [CrossRef]
- Nampoothiri, K.; Nair, N.; John, R. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar]
- Inkinen, S.; Hakkarainen, M.; Albertsson, A.; Sodergard, A. From lactic acid to poly(lactic acid) (PLA): Characterization and analysis of PLA and its precursors. Biomacromolecules 2011, 12, 523–532. [Google Scholar] [CrossRef]
- Klemm, D.; Heinze, T.; Wagenknecht, W. Comprehensive Cellulose Chemistry; Wiley-VCG: Weinheim, Germany, 1998; pp. 115–133. [Google Scholar]
- Schutzenberger, P. Action de l'acide acétique anhydre sur la cellulose, l'amidon, les sucres, la mannite et ses congénères, les glucosides et certaines matières colorantes végétales. [in French]. Compt. Rend. Hebd. Séances Acad. Sci. 1865, 61, 484–487. [Google Scholar]
- Ghaemi, N.; Madaeni, S.S.; Alizadeh, A.; Daraei, P.; Zinatizadeh, A.A.; Rahimpour, F. Separation of nitrophenols using cellulose acetate nanofiltration membrane: Influence of surfactant additives. Sep. Purif. Technol. 2012, 85, 147–156. [Google Scholar] [CrossRef]
- Zaky, A.; Escobar, I.; Motlagh, A.M.; Gruden, C. Determining the influence of active cells and conditioning layer on early stage biofilm formation using cellulose acetate ultrafiltration membranes. Desalination 2012, 286, 296–303. [Google Scholar]
- Koseoglu-Imer, D.Y.; Nadir Dizge, N.; Koyuncu, I. Enzymatic activation of cellulose acetate membrane for reducing of protein fouling. Colloids Surf. B 2012, 92, 334–339. [Google Scholar] [CrossRef]
- Jayalakshmi, A.; Rajesh, S.; Senthilkumar, S.; Mohan, D. Epoxy functionalized poly(ether-sulfone) incorporated cellulose acetate ultrafiltration membrane for the removal of Chromium ions. Sep. Purif. Technol. 2012, 90, 120–132. [Google Scholar]
- Kumari, A.; Sarkhel, G.; Choudhury, A. Preparation and characterization of polyvinylpyrollidone incorporated cellulose acetate membranes for ultrafiltration of metal ion. J. Appl. Polym. Sci. 2012, 124, E300–E308. [Google Scholar] [CrossRef]
- Harper, M.; Ashley, K. Preliminary studies on the use of acid-soluble cellulose acetate internal capsules for workplace metals sampling and analysis. J. Occup. Environ. Hyg. 2012, 9, D125–D129. [Google Scholar] [CrossRef]
- Magosso, H.A.; Fattori, N.; Arenas, L.T.; Gushikem, Y. New promising composite materials useful in the adsorption of Cu(II) in ethanol based on cellulose and cellulose acetate. Cellulose 2012, 19, 913–923. [Google Scholar] [CrossRef]
- Ghaemi, N.; Madaeni, S.S.; Alizadeh, A.; Rajabi, H.; Daraei, P.; Falsafi, M. Effect of fatty acids on the structure and performance of cellulose acetate nanofiltration membranes in retention of nitroaromatic pesticides. Desalination 2012, 301, 26–41. [Google Scholar]
- Rana, D.; Scheier, B.; Narbaitz, R.M.; Matsuura, T.; Tabe, S.; Jasimd, S.Y.; Khulbe, K.C. Comparison of cellulose acetate (CA) membrane and novel CA membranes containing surface modifying macromolecules to remove pharmaceutical and personal care product micropollutants from drinking water. J. Membr. Sci. 2012, 409–410, 346–354. [Google Scholar] [CrossRef]
- Kiran; Rana, D.S.; Balokhra, R.L.; Umar, A.; Chauhan, S. A thermodynamic study of 1,4-dioxane across cellulose acetate membrane under different conditions. Fluid Phase Equilib. 2012, 322–323, 148–158. [Google Scholar] [CrossRef]
- Nista, S.V.G.; Peres, L.; D’Ávila, M.A.; Schmidt, F.L.; Mei, L.H.I. Nanostructured membranes based on cellulose acetate obtained by electrospinning, part 1: Study of the best solvents and conditions by designof experiments. J. Appl. Polym. Sci. 2012, 126, E70–E78. [Google Scholar]
- De Lima, J.A.; Pinotti, C.A.; Felisberti, M.I.; do Carmo Gonçalves, M. Blends and clay nanocomposites of cellulose acetate and poly(epichlorohydrin). Compos. B 2012, 43, 2375–2381. [Google Scholar] [CrossRef]
- De Lima, J.A.; Pinotti, C.A.; Felisberti, M.I.; do Carmo Gonçalves, M. Morphology and mechanical properties of nanocomposites of cellulose acetate and organic montmorillonite prepared with different plasticizers. J. Appl. Polym. Sci. 2012, 124, 4628–4635. [Google Scholar]
- Rodriguez, F.J.; Galotto, M.J.; Guarda, A.; Bruna, J.E. Modification of cellulose acetate films using nanofillers based on organoclays. J. Food Eng. 2012, 110, 262–268. [Google Scholar] [CrossRef]
- Liu, X.; Lin, T.; Gao, Y.; Xu, Z.; Huang, C.; Yao, G.; Jiang, L.; Tang, Y.; Wang, X. Antimicrobial electrospun nanofibers of cellulose acetate and polyester urethane composite for wound dressing. J. Biomed. Mater. Res. B 2012, 100, 1556–1565. [Google Scholar]
- Gouma, P.; Xue, R.; Goldbeck, C.P.; Perrotta, P.; Balázsi, C. Nano-hydroxyapatite-cellulose acetate composites for growing of bone cells. Mater. Sci. Eng. C 2012, 32, 607–612. [Google Scholar] [CrossRef]
- Kanyong, P.; Pemberton, R.M.; Jackson, S.K.; Hart, J.P. Development of a sandwich format, amperometric screen-printed uric acid biosensor for urine analysis. Anal. Biochem. 2012, 428, 39–43. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, T.; Chen, L.; Ren, M.; Zhang, B.; Wang, Z.; Wang, Y. Bifunctional polyethersulfone hollow fiber with a porous, single-layer skin for use as a bioartificial liver bioreactor. J. Mater. Sci. 2012, 23, 2001–2011. [Google Scholar]
- Harun, N.I.; Ali, R.M.; Ali, A.M.M.; Yahya, M.Z.A. Dielectric behaviour of cellulose acetate-based polymer electrolytes. Ionics 2012, 18, 599–606. [Google Scholar] [CrossRef]
- Ramesh, S.; Shanti, R.; Morris, E. Plasticizing effect of 1-allyl-3-methylimidazolium chloride in cellulose acetate based polymer electrolytes. Carbohydr. Polym. 2012, 87, 2624–2629. [Google Scholar] [CrossRef]
- Shamsipur, M.; Pourmortazavi, S.M.; Hajimirsadeghi, S.S.; Atifeh, S.M. Effect of functional group on thermal stability of cellulose derivative energetic polymers. Fuel 2012, 95, 394–399. [Google Scholar] [CrossRef]
- Gutiérrez, M.C.; de Paoli, M.-A.; Felisberti, M.I. Biocomposites based on cellulose acetate and short curauá fibers: Effect of plasticizers and chemical treatments of the fibers. Compos. A 2012, 43, 1338–1346. [Google Scholar] [CrossRef]
- Stiubianu, G.; Nicolescu, A.; Nistor, A.; Cazacu, M.; Varganici, C.; Simionescu, B.C. Chemical modification of cellulose acetate by allylation and crosslinking with siloxane derivatives. Polym. Int. 2012, 61, 1115–1126. [Google Scholar]
- Wan, S.; Sun, Y.; Qi, X.; Tan, F. Improved bioavailability of poorly water-soluble drug curcumin in cellulose acetate solid dispersion. AAPS PharmSciTech 2012, 13, 159–166. [Google Scholar] [CrossRef]
- Díaz, J.E.; Barrero, A.; Márquez, M.; Loscertales, I.G. Controlled encapsulation of hydrophobic liquids in hydrophilic polymer nanofibers by co-electrospinning. Adv. Funct. Mater. 2006, 16, 2110–2116. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Wang, X.; Feng, Y.; Li, J.; Lim, C.T.; Ramakrishna, S. Coaxial electrospinning of (fluorescein isothiocyanate-conjugated bovine serum albumin)-encapsulated poly(ε-caprolactone) nanofibers for sustained release. Biomacromolecules 2006, 7, 1049–1057. [Google Scholar] [CrossRef]
- Moghe, A.K.; Gupta, B.S. Co-axial electrospinning for nanofiber structures: Preparation and applications. Polym. Rev. 2008, 48, 353–377. [Google Scholar] [CrossRef]
- Yan, J.; Yu, D.-G. Smoothening electrospinning and obtaining high-quality cellulose acetate nanofibers using a modified coaxial process. J. Mater. Sci. 2012, 47, 7138–7147. [Google Scholar] [CrossRef]
- Yu, D.-G.; Yu, J.-H.; Chen, L.; Williams, G.R.; Wang, X. Modified coaxial electrospinning for the preparation of high-quality ketoprofen-loaded cellulose acetate nanofibers. Carbohydr. Polym. 2012, 90, 1016–1023. [Google Scholar] [CrossRef]
- Hornig, S.; Heinze, T. Efficient approach to design stable water-dispersible nanoparticles of hydrophobic cellulose esters. Biomacromolecules 2008, 9, 1487–1492. [Google Scholar] [CrossRef]
- Kulterer, M.R.; Reichel, V.E.; Hribernik, S.; Wu, M.; Kostlerd, S.; Kargl, R.; Ribitsch, V. Nanoprecipitation of cellulose acetate using solvent/nonsolvent mixtures as dispersive media. Colloids Surf. A Eng. Asp. 2011, 375, 23–29. [Google Scholar] [CrossRef]
- Kulterer, M.R.; Reichel, V.E.; Kargl, R.; Köstler, S.; Sarbova, V.; Heinze, T.; Stana-Kleinschek, K.; Ribitsch, V. Functional polysaccharide composite nanoparticles from cellulose acetate and potential applications. Adv. Funct. Mater. 2012, 22, 1749–1758. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, H.; Cao, F.; Gong, J.; Su, Z. Nonaqueous synthesis of uniform polyaniline nanospheres via cellulose acetate template. J. Polym. Sci. A Polym. Chem. 2012, 50, 912–917. [Google Scholar] [CrossRef]
- Yoo, S.; Hsieh, J.S. Enzyme-assisted preparation of fibrillated cellulose fibers and its effect on physical and mechanical properties of paper sheet composites. Ind. Eng. Chem. Res. 2010, 49, 2161–2168. [Google Scholar] [CrossRef]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Soykeabkaew, N.; Nishino, T.; Peijs, T. All-cellulose composites of regenerated cellulose fibres by surface selective dissolution. Compos. A Appl. Sci. Manuf. 2009, 40, 321–328. [Google Scholar] [CrossRef]
- Duchemin, B.J.C.; Mathew, A.P.; Oksman, K. All-cellulose composites by partial dissolution in the ionic liquid 1-butyl-3-methylimidazolium chloride. Compos. A 2009, 40, 2031–2037. [Google Scholar] [CrossRef]
- Shakeri, A.; Mathew, A.P.; Oksman, K. Self-reinforced nanocomposite by partial dissolution of cellulose microfibrils in ionic liquid. J. Compos. Mater. 2012, 46, 1305–1311. [Google Scholar] [CrossRef]
- Zuluaga, R.; Putaux, J.L.; Cruz, J.; Vélez, J.; Mondragon, I.; Gañán, P. Cellulose microfibrils from banana rachis: Effect of alkaline treatments on structural and morphological features. Carbohydr. Polym. 2009, 76, 51–59. [Google Scholar] [CrossRef]
- Hassan, E.A.; Hassan, M.L.; Oksman, K. Improving bagasse pulp paper sheet properties with microfibrillated cellulose isolated from xylanase-treated bagasse. Wood Fiber Sci. 2011, 43, 76–82. [Google Scholar]
- Henriksson, M.; Henriksson, G.; Berglund, L.A.; Lindstrom, T. An environmentally friendly method for enzyme-assisted preparation of microfibrillated cellulose (MFC) nanofibers. Eur. Polym. J. 2007, 43, 3434–3441. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Habibi, Y.; Pawlak, J.J. A comparative study of energy consumption and physical properties of microfibrillated cellulose produced by different processing methods. Cellulose 2011, 18, 1097–1111. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Habibi, Y.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films from wood pulps: Water interactions and physical properties for packaging applications. Cellulose 2010, 17, 835–848. [Google Scholar] [CrossRef]
- Agoda-Tandjawa, G.; Durand, S.; Berot, S.; Blassel, C.; Gaillard, C.; Garnier, C.; Doublier, J.-L. Rheological characterization of microfibrillated cellulose suspensions after freezing. Carbohydr. Polym. 2010, 80, 677–686. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Habibi, Y.; Rojas, O.J.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films fromwood pulps: Mechanical processing and physical properties. Bioresour. Technol. 2010, 101, 5961–5968. [Google Scholar] [CrossRef]
- Taipale, T.; Österberg, M.; Nykänen, A.; Ruokolainen, J.; Laine, J. Effect of microfibrillated cellulose and fines on the drainage of kraft pulp suspension and paper strength. Cellulose 2010, 17, 1005–1020. [Google Scholar]
- Tingaut, P.; Zimmermann, T.; Lopez-Suevos, F. Synthesis and characterization of bionanocomposites with tunable properties from poly(lactic acid) and acetylated microfibrillated cellulose. Biomacromolecules 2010, 11, 454–464. [Google Scholar] [CrossRef]
- Mikkonen, K.S.; Pitkänen, L.; Liljeström, V.; Bergström, E.M.; Serimaa, R.; Salmén, L.; Tenkanen, M. Arabinoxylan structure affects the reinforcement of films by microfibrillated cellulose. Cellulose 2012, 19, 467–480. [Google Scholar] [CrossRef]
- Qiu, K.; Netravali, A.N. Fabrication and characterization of biodegradable composites based on microfibrillated cellulose and polyvinyl alcohol. Compos. Sci. Technol. 2012, 72, 1588–1594. [Google Scholar] [CrossRef]
- Fortunato, G.; Zimmermann, T.; Lübben, J.; Bordeanu, N.; Hufenus, R. Reinforcement of polymeric submicrometer-sized fibers by microfibrillated cellulose. Macromol. Mater. Eng. 2012, 297, 576–584. [Google Scholar] [CrossRef]
- Tanpichai, S.; Sampson, W.W.; Eichhorn, S.J. Stress-transfer in microfibrillated cellulose reinforced poly(lactic acid) composites using Raman spectroscopy. Compos. A 2012, 43, 1145–1152. [Google Scholar] [CrossRef]
- Ridgway, C.J.; Gane, P.A.C. Constructing NFC-pigment composite surface treatment for enhanced paper stiffness and surface properties. Cellulose 2012, 19, 547–560. [Google Scholar] [CrossRef]
- Syverud, K.; Stenius, P. Strength and barrier properties of MFC films. Cellulose 2009, 16, 75–85. [Google Scholar] [CrossRef]
- Bulota, M.; Jääskeläinen, A.S.; Paltakari, J.; Hughes, M. Properties of biocomposites: Influence of preparation method, testing environment and a comparison with theoretical models. J. Mater. Sci. 2011, 46, 3387–3398. [Google Scholar]
- Okubo, K.; Fujii, T.; Thostenson, E.T. Multi-scale hybrid biocomposite: Processing and mechanical characterization of bamboo fiber reinforced PLA with microfibrillated cellulose. Compos. A 2009, 40, 469–475. [Google Scholar] [CrossRef]
- Nakagaito, A.N.; Fujimura, A.; Sakai, T.; Hama, Y.; Yano, H. Production of microfibrillated cellulose (MFC)-reinforced polylactic acid (PLA) nanocomposites from sheets obtained by a papermaking-like process. Compos. Sci. Technol. 2009, 69, 1293–1297. [Google Scholar] [CrossRef]
- Bulota, M.; Kreitsmann, K.; Hughes, M.; Paltakari, J. Acetylated microfibrillated cellulose as a toughening agent in poly(lactic acid). J. Appl. Polym. Sci. 2012, 126, E448–E457. [Google Scholar]
- Bulota, M.; Hughes, M. Toughening mechanisms in poly(lactic) acid reinforced with TEMPO-oxidized cellulose. J. Mater. Sci. 2012, 47, 5517–5523. [Google Scholar] [CrossRef]
- Gabr, M.H.; Elrahman, M.A.; Okubo, K.; Fujii, T. Effect of microfibrillated cellulose on mechanical properties of plain-woven CFRP reinforced epoxy. Compos. Struct. 2010, 92, 1999–2006. [Google Scholar] [CrossRef]
- Gabr, M.H.; Elrahman, M.A.; Okubo, K.; Fujii, T. Interfacial adhesion improvement of plain woven carbon fiber reinforced epoxy filled with micro-fibrillated cellulose by addition liquid rubber. J. Mater. Sci. 2010, 45, 3841–3850. [Google Scholar]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Pawlak, J.J.; Hubbe, M.A. Water vapor barrier properties of coated and filled microfibrillated cellulose composite films. Bioresources 2011, 6, 4370–4388. [Google Scholar]
- Fernández, A.; Sánchez, M.D.; Ankerfors, M.; Lagaron, J.M. Effects of ionizing radiation in ethylene-vinyl alcohol copolymers and in composites containing microfibrillated cellulose. J. Appl. Polym. Sci. 2008, 109, 126–134. [Google Scholar] [CrossRef]
- Minelli, M.; Baschetti, M.G.; Doghieri, F.; Ankerfors, M.; Lindström, T.; Siró, I.; Plackett, D. Investigation of mass transport properties of microfibrillated cellulose (mfc) films. J. Membr. Sci. 2010, 358, 67–75. [Google Scholar] [CrossRef]
- Aulin, C.; Gällstedt, M.; Lindström, T. Oxygen and oil barrier properties of microfibrillated cellulose films and coatings. Cellulose 2010, 17, 559–574. [Google Scholar] [CrossRef]
- Rodionova, G.; Lenes, M.; Eriksen, Ø.; Gregersen, Ø. Surface chemical modification of microfibrillated cellulose: Improvement of barrier properties for packaging applications. Cellulose 2011, 18, 127–134. [Google Scholar] [CrossRef]
- Siró, I.; Plackett, D. Microfibrillated cellulose and new nanocomposite materials: A review. Cellulose 2010, 17, 459–494. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Johansson, C.; Bras, J.; Mondragon, I.; Nechita, P.; Plackett, D.; Šimon, P.; Svetec, D.G.; Virtanen, S.; Baschetti, M.G.; Breen, C.; et al. Renewable fibers and bio-based materials for packaging applications—A review of recent developments. Bioresources 2012, 7, 2506–2552. [Google Scholar]
- Wågberg, L.; Decher, G.; Norgren, M.; Lindström, T.; Ankerfors, M.; Axnäs, K. The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 2008, 24, 784–795. [Google Scholar] [CrossRef]
- Larsson, P.A.; Wågberg, L. Diffusion-Induced dimensional changes in papers and fibrillar films: Influence of hydrophobicity and fibre-wall cross-linking. Cellulose 2010, 17, 891–901. [Google Scholar] [CrossRef]
- Aulin, C.; Varga, I.; Claesson, P.M.; Wågberg, L.; Lindström, T. Buildup of polyelectrolyte multilayers of polyethyleneimine and microfibrillated cellulose studied by in situ dual-polarization interferometry and quartz crystal microbalance with dissipation. Langmuir 2008, 24, 2509–2518. [Google Scholar]
- Enarsson, L.-E.; Wågberg, L. Polyelectrolyte adsorption on thin cellulose films studied with reflectometry and quartz crystal microgravimetry with dissipation. Biomacromolecules 2009, 10, 134–141. [Google Scholar] [CrossRef]
- Nyström, G.; Mihranyan, A.; Razaq, A.; Lindstro¨m, T.; Nyholm, L.; Strømme, M. A nanocellulose polypyrrole composite based on microfibrillated cellulose from wood. J. Phys. Chem. B 2010, 114, 4178–4182. [Google Scholar] [CrossRef]
- Jabbour, L.; Gerbaldi, C.; Chaussy, D.; Zeno, E.; Bodoardo, S.; Beneventi, D. Microfibrillated cellulose-graphite nanocomposites for highly flexible paper-like Li-ion battery electrodes. J. Mater. Chem. 2010, 20, 7344–7347. [Google Scholar]
- Chiappone, A.; Nair, J.R.; Gerbaldi, C.; Jabbour, L.; Bongiovanni, R.; Zeno, E.; Beneventi, D.; Penazzi, N. Microfibrillated cellulose as reinforcement for li-ion battery polymer electrolytes with excellent mechanical stability. J. Power Sources 2011, 196, 10280–10288. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Lu, C.; Wang, Y.; Deng, Y. Flexible and transparent paper-based ionic diode fabricated from oppositely charged microfibrillated cellulose. J. Phys. Chem. C 2012, 116, 9227–9234. [Google Scholar]
- Aulin, C.; Netrval, J.; Wågberg, L.; Lindström, T. Aerogels from nanofibrillated cellulose with tunable oleophobicity. Soft Matter 2010, 6, 3298–3305. [Google Scholar]
- Larsson, M.; Zhou, Q.; Larsson, A. Different types of microfibrillated cellulose as filler materials in polysodium acrylate suprasorbents. Chin. J. Polym. Sci. 2011, 29, 407–413. [Google Scholar] [CrossRef]
- Xhanari, K.; Syverud, K.; Stenius, P. Emulsions stabilized by microfibrillated cellulose: The effect of hydrophobization, concentration and O/W ratio. J. Disper. Sci. Technol. 2011, 32, 447–452. [Google Scholar] [CrossRef]
- Lif, A.; Stenstad, P.; Syverud, K.; Nydén, M.; Holmberg, K. Fischer–Tropsch diesel emulsions stabilised by microfibrillated cellulose and nonionic surfactants. J. Colloid Interface Sci. 2010, 352, 585–592. [Google Scholar] [CrossRef]
- Boissard, C.I.R.; Bourban, P.-E.; Tingaut, P.; Zimmermann, T.; Månson, J.-A.E. Water of functionalized microfibrillated cellulose as foaming agent for the elaboration of poly(lactic acid) biocomposites. J. Reinf. Plast. Compos. 2011, 30, 709–719. [Google Scholar] [CrossRef]
- Qu, P.; Tang, H.; Gao, Y.; Zhang, L.; Wang, S. Polyethersulfone composite membrane blended with cellulose fibrils. Bioresources 2010, 5, 2323–2336. [Google Scholar]
- Evenou, F.; Couderc, S.; Kim, B.; Fujii, T.; Sakai, Y. Microfibrillated cellulose sheets coating oxygen-permeable PDMS membranes induce rat hepatocytes 3D aggregation into stably-attached 3D hemispheroids. J. Biomater. Sci. 2011, 22, 1509–1522. [Google Scholar] [CrossRef]
- Couderc, S.; Ducloux, O.; Kim, B.J.; Someya, T. A mechanical switch device made of a polyimide-coated microfibrillated cellulose sheet. J. Micromech. Microeng. 2009, 19, 055006:1–055006:11. [Google Scholar]
- Tuteja, A.; Choi, W.; Ma, M.; Mabry, J.M.; Mazzella, S.A.; Rutledge, G.C.; McKinley, G.H.; Cohen, R.E. Designing superoleophobic surfaces. Science 2007, 318, 1618–1622. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Chang, P.R.; Cao, X.; Anderson, D.P. Bionanocomposites based on pea starch and cellulose nanowhiskers hydrolyzed from pea hull fibre: Effect of hydrolysis time. Carbohydr. Polym. 2009, 76, 607–615. [Google Scholar] [CrossRef]
- Rosa, M.F.; Medeiros, E.S.; Malmonge, J.A.; Gregorski, K.S.; Wood, D.F.; Mattoso, L.H.C.; Glenn, G.; Orts, W.J.; Imam, S.H. Cellulose nanowhiskers from coconut husk fibers: Effect of preparation conditions on their thermal and morphological behavior. Carbohydr. Polym. 2010, 81, 83–92. [Google Scholar] [CrossRef]
- Kargarzadeh, H.; Ahmad, I.; Abdullah, I.; Dufresne, A.; Zainudin, S.Y.; Sheltami, R.M. Effects of hydrolysis conditions on the morphology, crystallinity, and thermal stability of cellulose nanocrystals extracted from kenaf bast fibers. Cellulose 2012, 19, 855–866. [Google Scholar] [CrossRef]
- Pandey, J.K.; Kim, C.-S.; Chu, W.-S.; Lee, C.S.; Jang, D.-Y.; Ahn, S.-H. Evaluation of morphological architecture of cellulose chains in grass during conversion from macro to nano dimensions. E-Polymers 2009, 102, 1–15. [Google Scholar]
- De Oliveira Taipina, M.; Ferrarezi, M.M.F.; do Carmo Goncalves, M. Morphological evolution of curauá fibers under acid hydrolysis. Cellulose 2012, 19, 1199–1207. [Google Scholar] [CrossRef]
- Lu, P.; Hsieh, Y.-L. Preparation and characterization of cellulose nanocrystals from rice straw. Carbohydr. Polym. 2012, 87, 564–573. [Google Scholar] [CrossRef]
- Sèbe, G.; Ham-Pichavant, F.; Ibarboure, E.; Koffi, A.L.C.; Tingaut, P. Supramolecular structure characterization of cellulose II nanowhiskers produced by acid hydrolysis of cellulose I substrates. Biomacromolecules 2012, 13, 570–578. [Google Scholar] [CrossRef]
- Li, W.; Yue, J.; Liu, S. Preparation of nanocrystalline cellulose via ultrasound and its reinforcement capability for poly(vinyl alcohol) composites. Ultrason. Sonochem. 2012, 19, 479–485. [Google Scholar] [CrossRef]
- Sturcova, A.; Davies, G.R.; Eichhorn, S.J. The elastic modulus and stress-transfer properties of tunicate cellulose whiskers. Biomacromolecules 2005, 6, 1055–1061. [Google Scholar] [CrossRef]
- Ahola, S.; Österberg, M.; Laine, J. Cellulose nanofibrils-adsorption with poly(amideamine) epichlorohydrin studied by QCM-D and application as a paper strength additive. Cellulose 2008, 15, 303–314. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Chang, P.R.; Anderson, D.P.; Huneault, M.A. Pea starch-based composite films with pea hull fibers and pea hull fiber-derived nanowhiskers. Polym. Eng. Sci. 2009, 49, 369–378. [Google Scholar] [CrossRef]
- Da Silva, J.B.A.; Pereira, F.V.; Druzian, J.I. Cassava starch-based films plasticized with sucrose and inverted sugar and reinforced with cellulose nanocrystals. J. Food Sci. 2012, 77, N14–N19. [Google Scholar]
- Yang, H.-S.; Gardner, D.J.; Nader, J.W. Characteristic impact resistance model analysis of cellulose nanofibril-filled polypropylene composites. Compos. A 2011, 42, 2028–2035. [Google Scholar]
- Ten, E.; Jiang, L.; Wolcott, M.P. Preparation and properties of aligned poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Carbohydr. Polym. 2013, 92, 206–213. [Google Scholar] [CrossRef]
- Ten, E.; Jiang, L.; Bahr, D.F.; Li, B.; Wolcott, M.P. Effects of cellulose nanowhiskers on mechanical, dielectric and rheological properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)(PHBV)/cellulose nanowhiskers (CNW) composites. Ind. Eng. Chem. Res. 2012, 51, 2941–2951. [Google Scholar] [CrossRef]
- Ten, E.; Turtle, J.; Bahr, D.; Jiang, L.; Wolcott, M.P. Thermal and mechanical properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Polymer 2010, 51, 2652–2660. [Google Scholar] [CrossRef]
- Yu, H.-Y.; Qin, Z.-Y.; Liu, Y.-N.; Chen, L.; Liu, N.; Zhou, Z. Simultaneous improvement of mechanical properties and thermal stability of bacterial polyester by cellulose nanocrystals. Carbohydr. Polym. 2012, 89, 971–978. [Google Scholar]
- Petersson, L.; Mathew, A.P.; Oksman, K. Dispersion and properties of cellulose nanowhiskers and layered silicates in cellulose acetate butyrate nanocomposites. J. Appl. Polym. Sci. 2009, 112, 2001–2009. [Google Scholar] [CrossRef]
- Siqueira, G.; Mathew, A.P.; Oksman, K. Processing of cellulose nanowhiskers/cellulose acetate butyrate nanocomposites using sol-gel process to facilitate dispersion. Compos. Sci. Technol. 2011, 71, 1886–1892. [Google Scholar] [CrossRef]
- Siqueira, G.; Bras, J.; Dufresne, A. Cellulose whiskers versus microfibrils: Influence of the nature of the nanoparticle and its surface functionalization on the thermal and mechanical properties of nanocomposites. Biomacromolecules 2009, 10, 425–432. [Google Scholar] [CrossRef]
- Mabrouk, A.B.; Kaddami, H.; Boufi, S.; Erchiqui, F.; Dufresne, A. Cellulosic nanoparticles from alfa fibers (Stipa tenacissima): Extraction procedures and reinforcement potential in polymer nanocomposites. Cellulose 2012, 19, 843–853. [Google Scholar] [CrossRef]
- Bendahou, A.; Kaddami, H.; Dufresne, A. Investigation on the effect of cellulosic nanoparticles’ morphology on the properties of natural rubber based nanocomposites. Eur. Polym. J. 2010, 46, 609–620. [Google Scholar] [CrossRef]
- Visakh, P.M.; Thomas, S.; Oksman, K.; Mathew, A.P. Cellulose nanofibres and cellulose nanowhiskers based natural rubber composites: Diffusion, sorption, and permeation of aromatic organic solvents. J. Appl. Polym. Sci. 2012, 124, 1614–1623. [Google Scholar] [CrossRef]
- Fahma, F.; Iwamoto, S.; Hori, N.; Iwata, T.; Takemura, A. Effect of pre-acid-hydrolysis treatment on morphology and properties of cellulose nanowhiskers from coconut husk. Cellulose 2011, 18, 443–450. [Google Scholar] [CrossRef]
- Cao, X.; Ding, B.; Yu, J.; Al-Deyab, S.S. Cellulose nanowhiskers extracted from TEMPO-oxidized jute fibers. Carbohydr. Polym. 2012, 90, 1075–1080. [Google Scholar] [CrossRef]
- Çetin, N.M.; Tingaut, P.; Özmen, N.; Henry, N.; Harper, D.; Dadmun, M.; Sébe, G. Acetylation of cellulose nanowhiskers with vinyl acetate under moderate conditions. Macromol. Biosci. 2009, 9, 997–1003. [Google Scholar] [CrossRef]
- Siqueira, G.; Bras, J.; Dufresne, A. New process of chemical grafting of cellulose nanoparticles with a long chain isocyanate. Langmuir 2010, 26, 402–411. [Google Scholar] [CrossRef]
- Dufresne, A. Processing of polymer nanocomposites reinforced with polysaccharide nanocrystals. Molecules 2010, 15, 4111–4128. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Habibi, Y.; Rojas, O.J.; Venditti, R.A.; Johansson, L.-S.; Efimenko, K.; Österberg, M.; Laine, J. Poly(N-isopropylacrylamide) brushes grafted from cellulose nanocrystals via surface-initiated single-electron transfer living radical polymerization. Biomacromolecules 2010, 11, 2683–2691. [Google Scholar] [CrossRef]
- Goffin, A.-L.; Raquez, J.-M.; Duquesne, E.; Siqueira, G.; Habibi, Y.; Dufresne, A.; Dubois, P. From interfacial ring-opening polymerization to melt processing of cellulose nanowhisker-filled polylactide-based nanocomposites. Biomacromolecules 2011, 12, 2456–2465. [Google Scholar] [CrossRef]
- Goffin, A.-L.; Raquez, J.-M.; Duquesne, E.; Siqueira, G.; Habibi, Y.; Dufresne, A.; Dubois, P. Poly(ε-caprolactone) based nanocomposites reinforced by surface-grafted cellulose nanowhiskers via extrusion processing: Morphology, rheology, and thermo-mechanical properties. Polymer 2011, 52, 1532–1538. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Citric acid as a benign alternative to metal catalysts for the production of cellulose-grafted-polycaprolactone copolymers. Polym. Chem. 2012, 3, 679–684. [Google Scholar] [CrossRef]
- Goffin, A.-L.; Habibi, Y.; Raquez, J.-M.; Dubois, P. Polyester-grafted cellulose nanowhiskers: A new approach for tuning the microstructure of immiscible polyester blends. ACS Appl. Mater. Interfaces 2012, 4, 3364–3371. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Kuga, S.; Wada, M.; Okano, T. Cellulose microcrystal film of high uniaxial orientation. Macromolecules 1997, 30, 6395–6397. [Google Scholar] [CrossRef]
- Edgar, C.D.; Gray, D.G. Smooth model cellulose I surfaces from nanocrystal suspensions. Cellulose 2003, 10, 299–306. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Morphological and optical characterization of polyelectrolyte multilayers incorporating nanocrystalline cellulose. Biomacromolecules 2006, 7, 2522–2530. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Birefringence in spin-coated films containing cellulose nanocrystals. Colloids Surf. A 2008, 325, 44–51. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G.; Rutland, M.W. Direct surface force measurements of polyelectrolyte multilayer films containing nanocrystalline cellulose. Langmuir 2010, 26, 17190–17197. [Google Scholar] [CrossRef]
- Hoeger, I.; Rojas, O.J.; Efimenko, K.; Velev, O.D.; Kelley, S.S. Ultrathin film coatings of aligned cellulosenanocrystalsfrom a convective-shearassemblysystem and their surface mechanical properties. Soft Matter 2011, 7, 1957–1967. [Google Scholar] [CrossRef]
- Sugiyama, J.; Chanzy, H.; Maretg, G. Orientation of cellulose microcrystals by strong magnetic fields. Macromolecules 1992, 25, 4232–4234. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Formation of cellulose-based electrostatic layer-by-layer films in a magnetic field. Sci. Technol. Adv. Mater. 2006, 7, 319–321. [Google Scholar] [CrossRef]
- Kimura, F.; Kimura, T.; Tamura, M.; Hirai, A.; Ikuno, M.; Horii, F. Three-dimensional crystal alignment using a time-dependent elliptic magnetic field. Langmuir 2005, 21, 2034–2037. [Google Scholar] [CrossRef]
- Kvien, I.; Oksman, K. Orientiation of cellulose nanowhiskers in polyvinyl alcohol. Appl. Phys. A 2007, 87, 641–643. [Google Scholar] [CrossRef]
- Li, D.; Liu, Z.; Al-Haik, M.; Tehrani, M.; Murray, F.; Tannenbaum, R.; Garmestani, H. Magnetic alignment of cellulose nanowhiskers in an all-cellulose composite. Polym. Bull. 2010, 65, 635–642. [Google Scholar] [CrossRef]
- Li, D.; Sun, X.; Khaleel, M.A. Materials design of all-cellulose composite using microstructure based finite element analysis. J. Eng. Mater. Technol. 2012, 134, 010911:1–010911:9. [Google Scholar]
- Bordel, D.; Putaux, J.-L.; Heux, L. Orientation of native cellulose in an electric field. Langmuir 2006, 22, 4899–4901. [Google Scholar] [CrossRef]
- Habibi, Y.; Heim, T.; Douillard, R. AC Electric field-assisted assembly and alignment of cellulose nanocrystals. J. Polym. Sci. B Polym. Phys. 2008, 46, 1430–1436. [Google Scholar] [CrossRef]
- Huang, J.; Liu, L.; Yao, J. Electrospinning of Bombyx mori silk fibroin nanofiber mats reinforced by cellulose nanowhiskers. Fiber Polym. 2011, 12, 1002–1006. [Google Scholar] [CrossRef]
- Changsarn, S.; Mendez, J.D.; Shanmuganathan, K.; Foster, E.J.; Weder, C.; Supaphol, P. Biologically inspired hierarchical design of nanocomposites based on poly(ethylene oxide) and cellulose nanofibers. Macromol. Rapid Commun. 2011, 32, 1367–1372. [Google Scholar] [CrossRef]
- Herrera, N.V.; Mathew, A.P.; Wang, L.Y.; Oksman, K. Randomly oriented and aligned cellulose fibres reinforced with cellulose nanowhiskers, prepared by electrospinning. Plast. Rubber Compos. 2011, 40, 57–64. [Google Scholar] [CrossRef]
- Lee, J.; Deng, Y. Increased Mechanical properties of aligned and isotropic electrospun PVA nanofiber webs by cellulose nanowhisker reinforcement. Macromol. Res. 2012, 20, 76–83. [Google Scholar] [CrossRef]
- Sanchez-García, M.D.; Hilliou, L.; Lagarón, J.M. Morphology and water barrier properties of nanobiocomposites of κ/ι-Hybrid carrageenan and cellulose nanowhiskers. J. Agric. Food Chem. 2010, 58, 12847–12857. [Google Scholar] [CrossRef]
- Belbekhouche, S.; Bras, J.; Siqueira, G.; Chappey, C.; Lebrun, L.; Khelifi, B.; Marais, S.; Dufresne, A. Water sorption behavior and gas barrier properties of cellulose whiskers and microfibrils films. Carbohydr. Polym. 2011, 83, 1740–1748. [Google Scholar] [CrossRef]
- Fortunati, E.; Peltzer, M.; Armentano, I.; Torre, L.; Jiménez, A.; Kenny, J.M. Effects of modified cellulose nanocrystals on the barrier and migration propertiesof PLA nano-biocomposites. Carbohydr. Polym. 2012, 90, 948–956. [Google Scholar] [CrossRef]
- Savadekar, N.R.; Mhaske, S.T. Synthesis of nano cellulose fibers and effect on thermoplastics starch based films. Carbohydr. Polym. 2012, 89, 146–151. [Google Scholar] [CrossRef]
- Eyley, S.; Shariki, S.; Dale, S.E.C.; Bending, S.; Marken, F.; Thielemans, W. Ferrocene-decorated nanocrystalline cellulose with charge carrier mobility. Langmuir 2012, 28, 6514–6519. [Google Scholar]
- Eyley, S.; Thielemans, W. Imidazolium grafted cellulose nanocrystals for ion exchange applications. Chem. Commun. 2011, 47, 4177–4179. [Google Scholar] [CrossRef]
- Moreau, C.; Beury, N.; Delorme, N.; Cathala, B. Tuning the architecture of cellulose nanocrystal−poly(allylamine hydrochloride) multilayered thin films: Influence of dipping parameters. Langmuir 2012, 28, 10425–10436. [Google Scholar] [CrossRef]
- Mendez, J.; Annamalai, P.K.; Eichhorn, S.J.; Rusli, R.; Rowan, S.J.; Foster, E.J.; Weder, C. Bioinspired mechanically adaptive polymer nanocomposites with water-activated shape-memory effect. Macromolecules 2011, 44, 6827–6835. [Google Scholar] [CrossRef]
- Kümmerer, K.; Menz, J.; Schubert, T.; Thielemans, W. Biodegradability of organic nanoparticles in the aqueous environment. Chemosphere 2011, 82, 1387–1392. [Google Scholar] [CrossRef]
- Ni, H.; Zeng, S.; Wu, J.; Cheng, X.; Luo, T.; Wang, W.; Zeng, W.; Chen, Y. Cellulose nanowhiskers: Preparation, characterization and cytotoxicity evaluation. Biomed. Mater. Eng. 2012, 22, 121–127. [Google Scholar]
- De Paula, E.L.; Mano, V.; Pereira, F.V. Influence of cellulose nanowhiskers on the hydrolytic degradation behavior of poly(d,l-lactide). Polym. Degrad. Stabil. 2011, 96, 1631–1638. [Google Scholar] [CrossRef]
- Dash, R.; Ragauskas, A.J. Synthesis of a novel cellulose nanowhisker-based drug delivery system. RSC Adv. 2012, 2, 3403–3409. [Google Scholar] [CrossRef]
- Chirat, C.; Lachenal, D.; Dufresne, A. Biorefinery in a kraft pulp mill: From bioethanol to cellulose nanocrystals. Cellul. Chem. Technol. 2010, 44, 59–64. [Google Scholar]
- Oksman, K.; Etang, J.A.; Mathew, A.P.; Jonoobi, M. Cellulose nanowhiskers separated from a bio-residue from wood bioethanol production. Biomass Bioenergy 2011, 35, 146–152. [Google Scholar]
- Peng, F.; Peng, P.; Xu, F.; Sun, R.-C. Fractional purification and bioconversion of hemicelluloses. Biotechnol. Adv. 2012, 30, 879–903. [Google Scholar] [CrossRef]
- Peng, P.; Peng, F.; Bian, J.; Xu, F.; Sun, R.-C.; Kennedy, J.F. Isolation and structural characterization of hemicelluloses from the bamboo species Phyllostachys incarnata Wen. Carbohydr. Polym. 2011, 86, 883–890. [Google Scholar] [CrossRef]
- Haimer, E.; Wendland, M.; Potthast, A.; Rosenau, T.; Liebner, F. Precipitation of hemicelluloses from DMSO/water mixtures using carbon dioxide as an antisolvent. J. Nanomater. 2008, 2008, 1–5. [Google Scholar]
- Haimer, E.; Wendlana, M.; Potthast, A.; Henniges, U.; Rosenau, T.; Liebner, F. Controlled precipitation and purification of hemicellulose from DMSO and DMSO/water mixtures by carbon dioxide as anti-solvent. J. Supercrit. Fluids 2010, 53, 121–130. [Google Scholar] [CrossRef]
- Kaczmar, J.W.; Pach, J.; Burgstaller, C. The chemically treated hemp fibres to reinforce polymers. Polimery 2011, 56, 11–12. [Google Scholar]
- Reddy, K.O.; Guduri, B.R.; Rajulu, A.V. Structural characterization and tensile properties of Borassus fruit fibers. J. Appl. Polym. Sci. 2009, 114, 603–611. [Google Scholar] [CrossRef]
- Zhang, Y.; Hosseinaei, O.; Wang, S.; Zhou, Z. Influence of hemicellulose extraction on water uptake behavior of wood strands. Wood Fiber Sci. 2011, 43, 244–250. [Google Scholar]
- Hosseinaei, O.; Wang, S.; Rials, T.G.; Xing, C.; Taylor, A.M.; Kelley, S.S. Effect of hemicellulose extraction on physical and mechanical properties and mold susceptibility of flakeboard. For. Prod. J. 2011, 61, 31–37. [Google Scholar]
- Chang, W.-P.; Kim, K.-J.; Gupta, R.K. Ultrasound-assisted surface-modification of wood particulates for improved wood/plastic composites. Compos. Interfaces 2009, 16, 687–709. [Google Scholar] [CrossRef]
- Cordeiro, N.; Gouveia, C.; Moraes, A.G.O.; Amico, S.C. Natural fibers characterization by inverse gas chromatography. Carbohydr. Polym. 2011, 84, 110–117. [Google Scholar] [CrossRef]
- Rao, R.S.; Jyothi, C.P.; Prakasham, R.S.; Rao, C.S.; Sarma, P.N.; Rao, L.V. Xylitol production from corn fiber and sugarcane bagasse hydrolysates by Candida tropicalis. Bioresour. Technol. 2006, 97, 1974–1978. [Google Scholar] [CrossRef]
- Li, M.; Meng, X.; Diao, E.; Du, F. Xylitol production by Candida tropicalis from corn cob hemicellulose hydrolysate in a two-stage fed-batch fermentation process. J. Chem. Technol. Biotechnol. 2012, 87, 387–392. [Google Scholar] [CrossRef]
- Silva, C.J.S.M.; Mussatto, S.S.; Roberto, I.C. Study of xylitol production by Candida guilliermondii on a bench bioreactor. J Food Eng. 2006, 75, 115–119. [Google Scholar] [CrossRef]
- Mayerhoff, Z.D.V.L.; Roberto, I.C.; Silva, S.S. Xylitol production from rice straw hemicellulose using different yeast strains. Biotechnol. Lett. 1997, 19, 407–409. [Google Scholar] [CrossRef]
- Cruz, J.M.; Domínguez, J.M.; Domínguez, H.; Parajó, J.C. Solvent extraction of hemicellulosic wood hydrolysates: A procedure useful for obtaining both detoxified fermentation media and polyphenols with antioxidant activity. Food Chem. 1999, 67, 147–153. [Google Scholar] [CrossRef]
- Stepan, A.M.; Höije, A.; Schols, H.A.; de Waard, P.; Gatenholm, P. Arabinose content of arabinoxylans contributes to flexibility of acetylated arabinoxylan films. J. Appl. Polym. Sci. 2012, 125, 2348–2355. [Google Scholar] [CrossRef]
- Stevanic, J.S.; Bergström, E.M.; Gatenholm, P.; Berglund, L.; Salmén, L. Arabinoxylan/nanofibrillated cellulose composite films. J. Mater. Sci. 2012, 47, 6724–6732. [Google Scholar] [CrossRef]
- Edlund, U.; Yu, Y.; Zhu Ryberg, Y.; Krause-Rehberg, R.; Albertsson, A.-C. Positron lifetime reveals the nano level packing in complex polysaccharide-rich hydrolysate matrixes. Anal. Chem. 2012, 84, 3676–3681. [Google Scholar] [CrossRef]
- Voepel, J.; Edlund, U.; Albertsson, A.-C. Alkenyl-functionalized precursors for renewable hydrogels design. J. Polym. Sci. Polym. Chem. 2009, 47, 3595–3606. [Google Scholar] [CrossRef]
- Voepel, J.; Sjöberg, J.; Reif, M.; Albertsson, A.-C.; Hultin, U.-K.; Gasslander, U. Drug diffusion in neutral and ionic hydrogels assembled from acetylated galactoglucomannan. J. Appl. Polym. Sci. 2009, 112, 2401–2412. [Google Scholar] [CrossRef]
- Silva, T.C.F.; Habibi, Y.; Colodette, J.L.; Lucia, L.A. The influence of the chemical and structural features of xylan on the physical properties of its derived hydrogels. Soft Matter 2011, 7, 1090–1099. [Google Scholar] [CrossRef]
- Karaaslan, M.A.; Tshabalala, M.A.; Buschle-Diller, G. Wood Hemicellulose/chitosan-based semi-interpenetrating network hydrogels: Mechanical swelling and controlled drug release properties. Bioresources 2010, 5, 1036–1054. [Google Scholar]
- Karaaslan, M.A.; Tshabalala, M.A.; Yelle, D.J.; Buschle-Diller, G. Nanoreinforced biocompatible hydrogels from wood hemicelluloses and cellulose whiskers. Carbohydr. Polym. 2011, 86, 192–201. [Google Scholar] [CrossRef]
- Paës, G.; Chabbert, B. Characterization of arabinoxylan/cellulose nanocrystals gels to investigate fluorescent probes mobility in bioinspired models of plant secondary cell wall. Biomacromolecules 2012, 13, 206–214. [Google Scholar] [CrossRef]
- Peng, X.-W.; Ren, J.-L.; Zhong, L.-X.; Peng, F.; Sun, R.-C. Xylan-rich hemicelluloses-graft-acrylic acid ionic hydrogels with rapid responses to pH, salt, and organic solvents. J. Agric. Food Chem. 2011, 59, 8208–8215. [Google Scholar] [CrossRef]
- Peng, X.-W.; Zhong, L.-X.; Ren, J.-L.; Sun, R.-C. Highly effective adsorption of heavy metal ions from aqueous solutions by macroporous xylan-rich hemicelluloses-based hydrogel. J. Agric. Food Chem. 2012, 60, 3909–3916. [Google Scholar] [CrossRef]
- Pohjanlehto, H.; Setälä, H.; Kammiovirta, K.; Harlin, A. The use of N,N′-diallylaldardiamides as cross-linkers in xylan derivatives-based hydrogels. Carbohydr. Res. 2011, 346, 2736–2745. [Google Scholar]
- Oliveira, E.E.; Silva, A.E.; Nagashima, T.; Gomes, M.C.S.; Aguiar, L.M.; Marcelino, H.R.; Araújo, I.B.; Bayer, M.P.; Ricardo, N.M.P.S.; Oliveira, A.O.; et al. Xylan from corn cobs, a promising polymer for drug delivery: Production and characterization. Bioresour. Technol. 2010, 101, 5402–5406. [Google Scholar] [CrossRef]
- Olsson, A.; Salmen, L. Viscoelasticity of in situ lignin as affected by structure softwood vs. hardwood. ACS Symp. Ser. 1992, 489, 133–143. [Google Scholar] [CrossRef]
- Szczesniak, L.; Rachocki, A.; Tritt-Goc, J. Glass transition temperature and thermal decomposition of cellulose powder. Cellulose 2008, 15, 445–451. [Google Scholar] [CrossRef]
- Naqvi, M.; Yan, J.; Dahlquist, E. Black liquor gasification integrated in pulp and paper mills: A critical review. Bioresour. Technol. 2010, 101, 8001–8015. [Google Scholar] [CrossRef]
- Kang, S.; Li, X.; Fan, J.; Chang, J. Solid fuel production by hydrothermal carbonization of black liquor. Bioresour. Technol. 2012, 110, 715–718. [Google Scholar] [CrossRef]
- Braaten, S.; Christensen, B.; Fredheim, G. Comparison of molecular weight and molecular weight distributions of softwood and hardwood lignosulfonates. J. Wood Chem. Technol. 2003, 23, 197–215. [Google Scholar] [CrossRef]
- Yu, G.; Li, B.; Wang, H.; Liu, C.; Mu, X. Preparation of concrete superplasticizer by oxidation sulfomethylation of sodium lignosulfonate. Bioresources 2013, 8, 1055–1063. [Google Scholar]
- Mittal, M.; Sharma, C. Studies on lignin-based adhesives for plywood panels. Polym. Int. 2007, 29, 7–8. [Google Scholar] [CrossRef]
- Nadji, H.; Rodrigue, D.; Benaboura, A.; Bedard, Y.; Stevanovic, T.; Riedl, B. Value-added derivatives of soda lignin alfa grass (Stipa tenacissima). II. Uses as lubricants in plastic processing. J. Appl. Polym. Sci. 2009, 114, 3003–3007. [Google Scholar] [CrossRef]
- Ibrahim, M.; Azreena, M.; Ismail, M. Lignin graft copolymer as a drilling mud thinner for high temperature well. J. Appl. Sci. 2006, 6, 1808–1813. [Google Scholar] [CrossRef]
- Windeisen, E.; Wegener, G. Lignin as building unit for polymers. Polym. Sci. 2012, 10, 255–265. [Google Scholar]
- Kumar, M.; Mohanty, A.; Erickson, L.; Misra, M. Lignin and its applications with polymers. J. Biobased Mater. Bioenergy 2009, 3, 1–24. [Google Scholar] [CrossRef]
- Doherty, W.; Mousavioun, P.; Fellows, C. Value-adding to cellulosic ethanol: Lignin polymers. Ind. Crop Prod. 2011, 33, 259–276. [Google Scholar] [CrossRef] [Green Version]
- Johansson, K.; Winestrand, S.; Johansson, C.; Jarnstrom, L.; Jonsson, L. Oxygen-scavenging coatings and films based on lignosulfonates and laccase. J. Biotechnol. 2012, 161, 14–18. [Google Scholar]
- Milczarek, G. Kraft lignin as dispersing agent for carbon nanotubes. J. Electroanal. Chem. 2010, 638, 178–181. [Google Scholar] [CrossRef]
- Faria, F.; Evtuguin, D.; Rudnitskaya, A.; Gomes, M.; Oliveira, J.; Graça, M.; Costa, L. Lignin-basedpolyurethane doped with carbon nanotubes for sensor applications. Polym. Int. 2012, 61, 788–794. [Google Scholar] [CrossRef]
- Rudnitskaya, A.; Evtuguin, D.; Costa, L.; Graça, M.; Fernandes, A.; Correia, M.; Gomes, M.; Oliveira, J. Potentiometric chemical sensors from lignin-poly(propylene oxide) copolymers doped by carbon nanotubes. Analyst 2013, 138, 501–508. [Google Scholar] [CrossRef]
- Milczarek, G.; Inganäs, O. Renewable cathode materials from biopolymer/conjugated polymer interpenetrating networks. Science 2012, 335, 1468–1471. [Google Scholar] [CrossRef]
- Bhirde, A.; Sousa, A.A.; Patel, V.; Azari, A.; Gutkind, J.S.; Leapman, R.D.; Rusling, J.F. Imaging the distribution of individual platinum-based anticancer drug molecules attached to single-wall carbon nanotubes. Nanomedicine 2009, 4, 763–772. [Google Scholar] [CrossRef]
- Li, R.; Wu, R.; Zhao, L.; Wu, M.; Yang, L.; Zou, H. P-glycoprotein antibody functionalized carbon nanotube overcomes the multidrug resistance of human leukemia cells. ACS Nano 2010, 4, 1399–1408. [Google Scholar] [CrossRef]
- Thostenson, E.; Ren, Z.; Chou, T. Advances in the science and technology of carbon nanotubes and their composites: A review. Compos. Sci. Technol. 2001, 61, 1899–1912. [Google Scholar] [CrossRef]
- Kang, Z.; Wang, E.; Mao, B.; Su, Z.; Chen, L.; Xu, L. Obtaining carbon nanotubes from grass. Nanotechnology 2005, 16, 1192–1195. [Google Scholar] [CrossRef]
- Shin, Y.; Wang, C.; Li, X.; Exarhos, G. Synthesis of supportedcarbon nanotubes in mineralized silica-wood composites. Carbon 2005, 43, 1096–1098. [Google Scholar] [CrossRef]
- Xie, X.; Goodell, B.; Qian, Y.; Daniel, G.; Zhang, D.; Nagle, D.; Peterson, M.; Jellison, J. A Method of producing carbon nanotubes directly from plant materials. For. Prod. J. 2009, 59, 26–28. [Google Scholar]
- Magrez, A.; Kasas, S.; Salicio, V.; Pasquier, N.; Seo, J.; Celio, M.; Catsicas, S.; Schwaller, B.; Forro, L. Cellular toxicity of carbon-based nanomaterials. Nano Lett. 2006, 6, 1121–1125. [Google Scholar] [CrossRef]
- Poland, C.; Duffin, R.; Kinloch, I.; Maynard, A.; Wallace, W.; Seaton, A.; Stone, V.; Brown, S.; MacNee, W.; Donaldson, K. Carbon Nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat. Nanotechnol. 2008, 3, 423–428. [Google Scholar] [CrossRef]
- Caicedo, H.M.; Dempere, A.L.; Vermerris, W. Template-mediated synthesis and bio-functionalization of flexible lignin-based nanotubes and nanowires. Nanotechnology 2012, 23, 105605. [Google Scholar] [CrossRef]
- Vermerris, W. Composition and biosynthesis of lignocellulosic biomass. In Genetic Improvement of Bioenergy Crops; Vermerris, W., Ed.; Springer: New York, NY, USA, 2008; pp. 89–142. [Google Scholar]
- Vanholme, R.; Van Acker, R.; Boerjan, W. Potential of Arabidopsis systems biology to advance the biofuel field. Trends Biotechnol. 2010, 28, 543–547. [Google Scholar] [CrossRef]
- Vermerris, W. Survey of genomics approaches to improve bioenergy traits in maize, sorghum and sugarcane. J. Integr. Plant Biol. 2011, 53, 105–119. [Google Scholar] [CrossRef]
- Pilate, G.; Guiney, E.; Holt, K.; Petit-Conil, M.; Lapierre, C.; Leplé, J.; Pollet, B.; Mila, I.; Webster, E.; Marstorp, H.; et al. Field and pulping performances of transgenic trees with altered lignification. Nat. Biotechnol. 2002, 20, 607–612. [Google Scholar] [CrossRef]
- Herrera, S. Struggling to see the forest through the trees. Nat. Biotechnol. 2005, 23, 165–167. [Google Scholar] [CrossRef]
- Chen, F.; Dixon, R.A. Lignin modification improves fermentable sugar yields for biofuel production. Nat. Biotechnol. 2007, 25, 759–761. [Google Scholar] [CrossRef]
- Vermerris, W.; Saballos, A.; Ejeta, G.; Mosier, N.S.; Ladisch, M.R.; Carpita, N.C. Molecular breeding to enhance ethanol production from corn and sorghum stover. Crop Sci. 2007, 47, S142–S153. [Google Scholar]
- Vanholme, R.; Morreel, K.; Darrah, C.; Oyarce, P.; Grabber, J.; Ralph, J.; Boerjan, W. Metabolic engineering of novel lignin in biomass crops. New Phytol. 2012, 196, 978–1000. [Google Scholar] [CrossRef]
- Davison, B.H.; Drescher, S.R.; Tuskan, G.A.; Davis, M.F.; Nghiem, N.P. Variation of S/G ratio and lignin content in a Populus family influences the release of xylose by dilute acid hydrolysis. Appl. Biochem. Biotechnol. 2006, 129-132, 427–435. [Google Scholar]
- Fu, C.; Mielenz, J.; Xiao, X.; Ge, Y.; Hamilton, C.; Rodriguez, M.; Chen, F.; Foston, M.; Ragauskas, A.; Bouton, J.; Dixon, R.; Wang, Z. Genetic manipulation of lignin reduces recalcitrance and improves ethanol production from switchgrass. Proc. Natl. Acad. Sci. USA. 2011, 108, 3803–3808. [Google Scholar] [CrossRef]
- Jung, J.H.; Fouad, W.M.; Vermerris, W.; Gallo, M.; Altpeter, F. RNAi suppression of lignin biosynthesis in sugarcane reduces recalcitrance for biofuel production from lignocellulosic biomass. Plant Biotechnol. J. 2012, 10, 1067–1076. [Google Scholar] [CrossRef]
- Sattler, S.E.; Palmer, N.A.; Saballos, A.; Greene, A.M.; Xin, Z.; Sarath, G.; Vermerris, W.; Pedersen, J.F. Identification and characterization of 4 missense mutations in Brown midrib 12 (Bmr12); the caffeic O-methyltranferase (COMT) of sorghum. Bioenerg. Res. 2012, 5, 855–865. [Google Scholar] [CrossRef]
- Sattler, S.E.; Funnell-Harris, D.L.; Pedersen, J.F. Brown midrib mutations and their importance to the utilization of maize, sorghum, and pearl millet lignocellulosic tissues. Plant Sci. 2010, 178, 229–238. [Google Scholar] [CrossRef]
- Sindhu, A.; Langewisch, T.; Olek, A.; Multani, D.S.; McCann, M.C.; Vermerris, W.; Carpita, N.C.; Johal, G. Maize Brittle stalk2 encodes a COBRA-Like protein expressed in early organ development but required for tissue flexibility at maturity. Plant Physiol. 2007, 145, 1444–1459. [Google Scholar] [CrossRef]
- 250. Penning, B.W.; Hunter, C.T., III; Tayengwa, R.; Eveland, A.L.; Dugard, C.K.; Olek, A.T.; Vermerris, W.; Koch, K.E.; McCarty, D.R.; Davis, M.F.; Thomas, S.R.; McCann, M.C.; Carpita, N.C. Genetic resources for maize cell wall biology. Plant Physiol. 2009, 151, 1703–1728. [Google Scholar] [CrossRef]
- Xin, Z.; Wang, M.L.; Barkley, N.A.; Burow, G.; Franks, C.; Pederson, G.; Burke, J. Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol. 2008, 8, 103. [Google Scholar] [CrossRef]
- Vermerris, W.; Saballos, A. Genetic enhancement of sorghum for biomass utilization. In Genetics and Genomics of the Saccharinae; Paterson, A., Ed.; Springer: New York, NY, USA, 2012; pp. 391–428. [Google Scholar]
- Vandenbrink, J.; Hilten, R.; Das, K.; Paterson, A.; Feltus, F. Analysis of crystallinity index and hydrolysis rates in the bioenergy crop Sorghum bicolor. Bioenerg. Res. 2012, 5, 387–397. [Google Scholar] [CrossRef]
- Studer, M.; Brethauer, S.; DeMartini, J.; McKenzie, H.; Wyman, C. Co-hydrolysis of hydrothermal and dilute acid pretreated Populus slurries to support development of a high-throughput pretreatment system. Biotechnol. Biofuels 2011, 4, 19–29. [Google Scholar] [CrossRef]
- Vermerris, W.; Sherman, D.M.; McIntyre, L.M. Phenotypic plasticity in cell walls of maize brown midrib mutants is limited by lignin composition. J. Exp. Bot. 2010, 61, 2479–2490. [Google Scholar] [CrossRef]
- Funnell-Harris, D.; Pedersen, J.; Sattler, S. Alteration in lignin biosynthesis restricts growth of Fusarium spp. in brown midrib sorghum. Phytopathology 2010, 100, 671–681. [Google Scholar] [CrossRef]
- Jackson, L.A.; Shadle, G.L.; Zhou, R.; Nakashima, J.; Chen, F.; Dixon, R.A. Improving Saccharification Efficiency of alfalfa stems through modification of the terminal stages of monolignol biosynthesis. Bioenerg. Res. 2008, 1, 180–192. [Google Scholar] [CrossRef]
- Lee, Y.; Chen, F.; Gallego-Giraldo, L.; Dixon, R.A.; Voit, E.O. Integrative analysis of transgenic alfalfa (Medicago sativa L.) suggests new metabolic control mechanisms for monolignol biosynthesis. PLoS Comput. Biol. 2011, 7, 1–13. [Google Scholar]
- Vanholme, R.; Storme, V.; Vanholme, B.; Sundin, L.; Christensen, J.; Goeminne, G.; Halpin, C.; Rohde, A.; Morreel, K.; Boerjan, W. A Systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell. 2012, 24, 3506–3529. [Google Scholar] [CrossRef] [Green Version]
- Eranki, P.L.; Bals, B.D.; Dale, B.E. Advanced regional biomass processing depots: A key to the logistical challenges of the cellulosic biofuel industry. Biofuels Bioprod. Bioref. 2011, 5, 621–630. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Ten, E.; Vermerris, W. Functionalized Polymers from Lignocellulosic Biomass: State of the Art. Polymers 2013, 5, 600-642. https://doi.org/10.3390/polym5020600
AMA Style
Ten E, Vermerris W. Functionalized Polymers from Lignocellulosic Biomass: State of the Art. Polymers. 2013; 5(2):600-642. https://doi.org/10.3390/polym5020600
Chicago/Turabian StyleTen, Elena, and Wilfred Vermerris. 2013. "Functionalized Polymers from Lignocellulosic Biomass: State of the Art" Polymers 5, no. 2: 600-642. https://doi.org/10.3390/polym5020600