Polymers for Protein Conjugation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction, the Success of Polyethylene Glycol (PEG) in Protein Conjugation
- (1)
- As it is not biodegradable, high molecular weight PEGs should be used carefully and always at molecular weights below the kidney clearance threshold;
- (2)
- Specific anti-PEG antibodies have been detected in the serum of patients treated with PEG-asparaginase [30] and PEG-uricase [31], resulting in a neutralizing effect with a loss of therapeutic efficacy. Some investigators have detected the presence of pre-existing IgM and IgG anti-PEG antibodies in about 25% of patients who have never received prior treatment with PEG drugs [32,33];
- (3)
- While controversial kidney cell vacuolization has been observed in animals following multiple dose administration of PEG-conjugated hemoglobin [34] and TNF-R binding proteins [35], it has never been found when the polymer was administered alone or with other conjugates, and this has confirmed its safety. The finding, nonetheless, highlights another potential problem linked to the use of non-biodegradable, high molecular weight PEGs;
- (4)
- The large number of patents connected to the use of PEGs in drug delivery compositions might hamper or prevent the development of new conjugates.
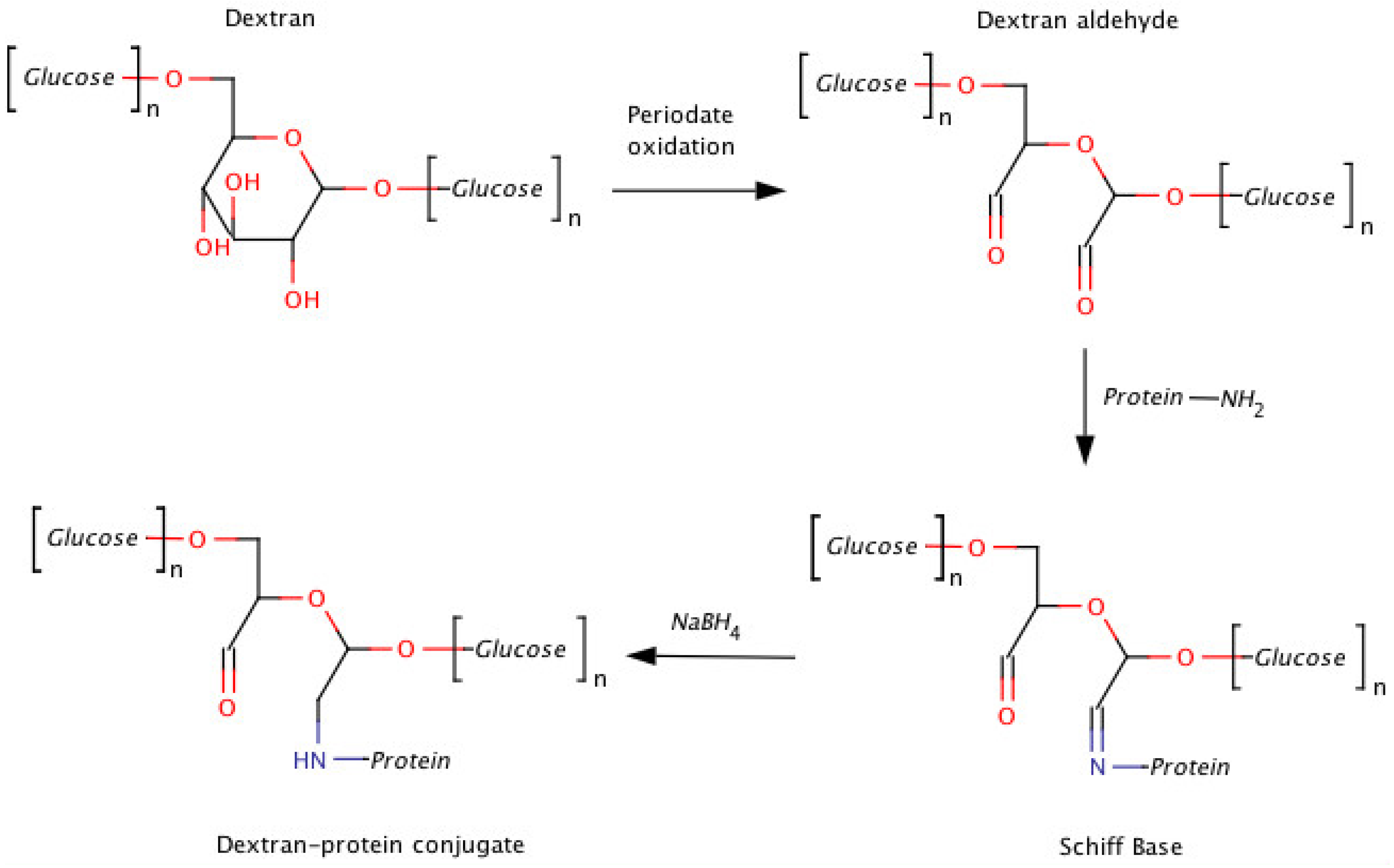
2. Dextran

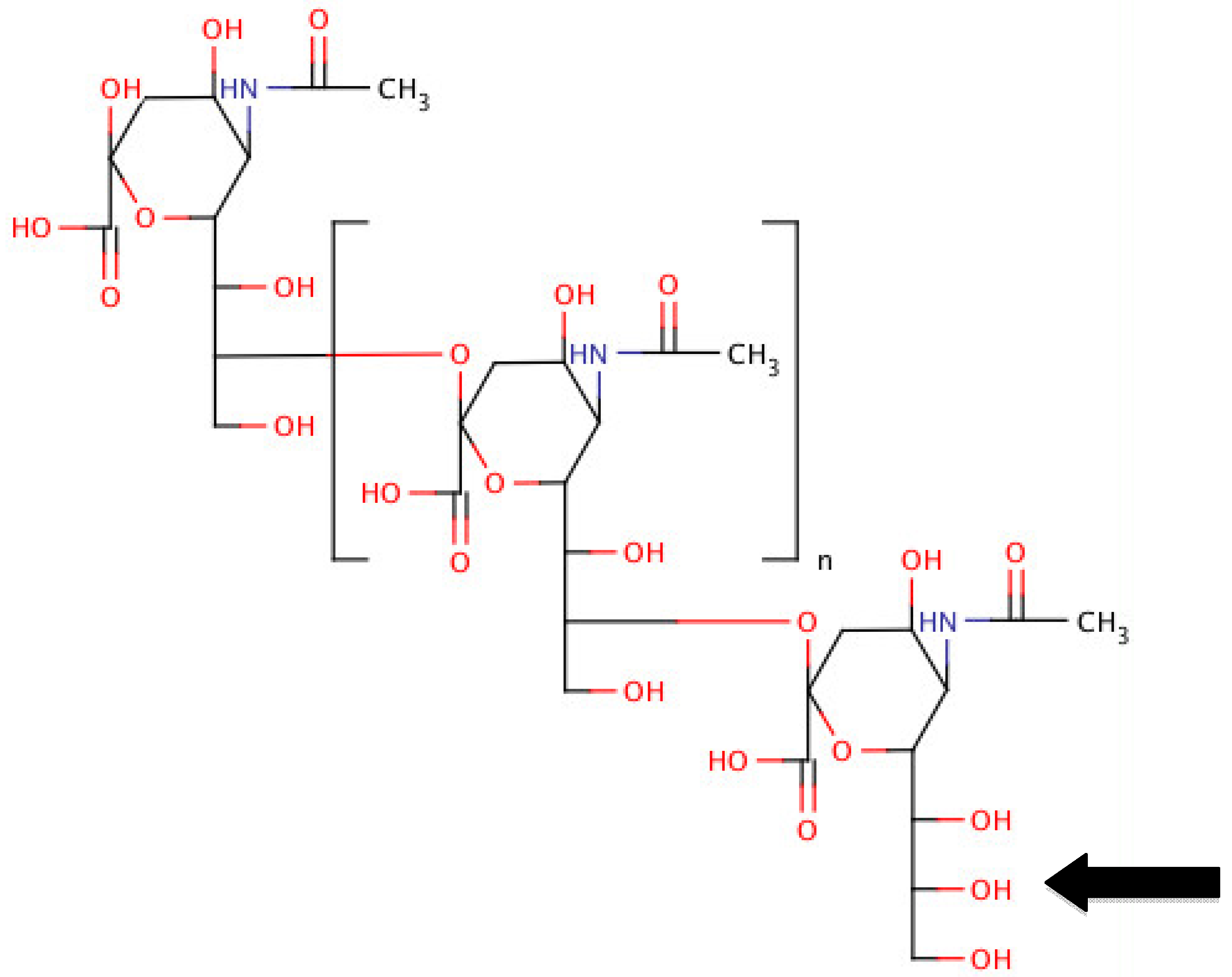
3. Polysialic Acids (PSAs)

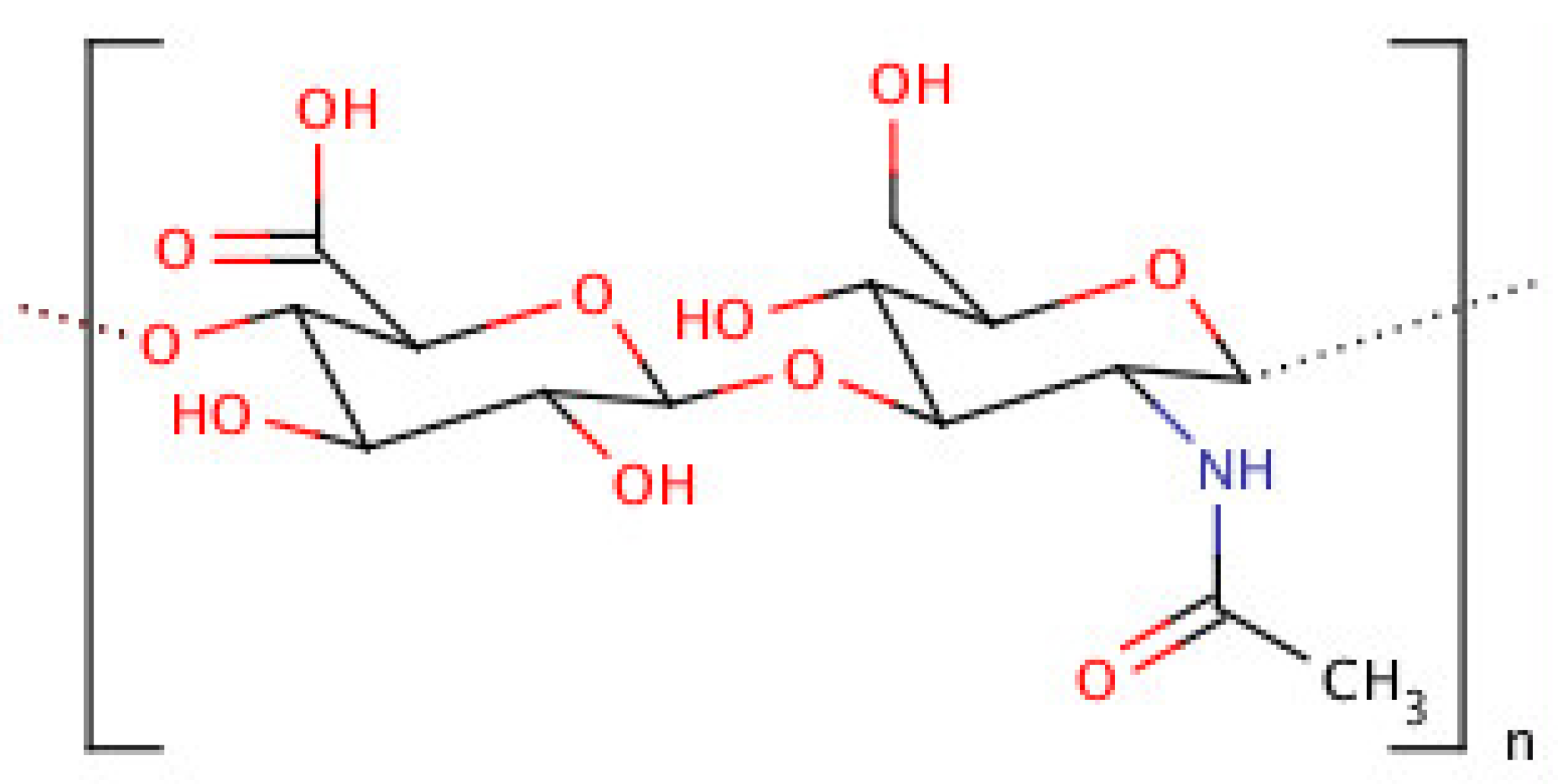
4. Hyaluronic Acid (HA)

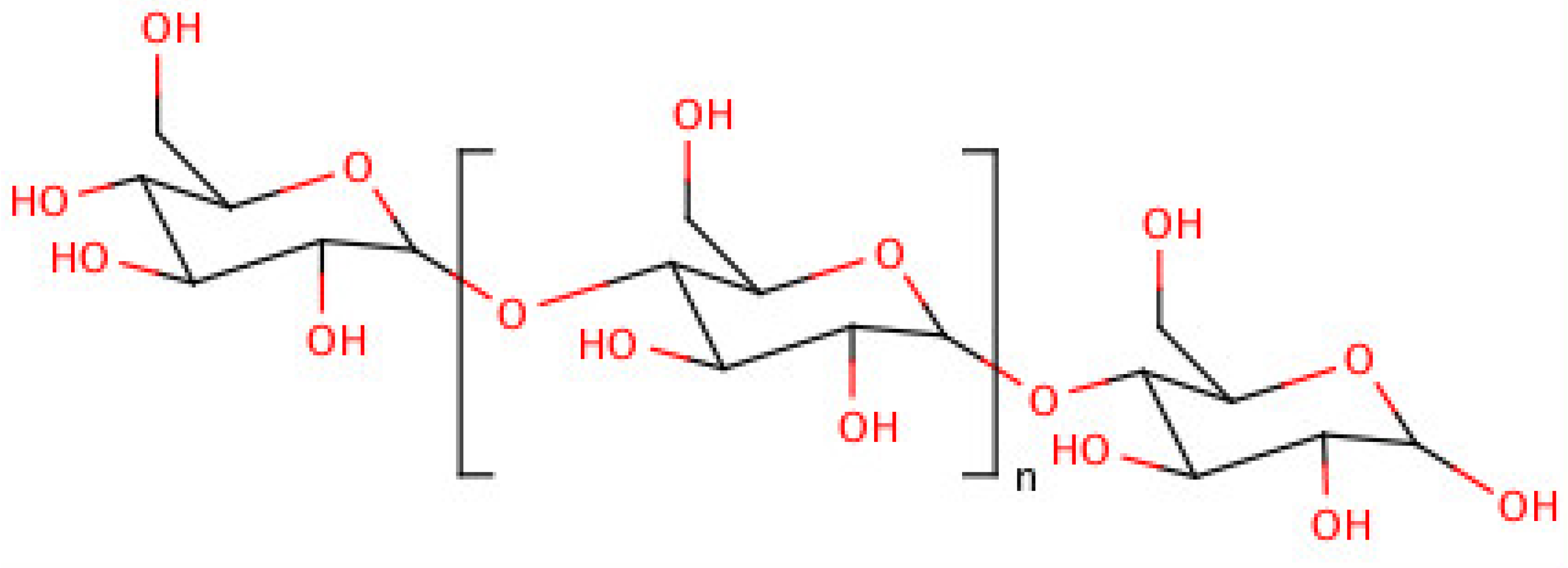
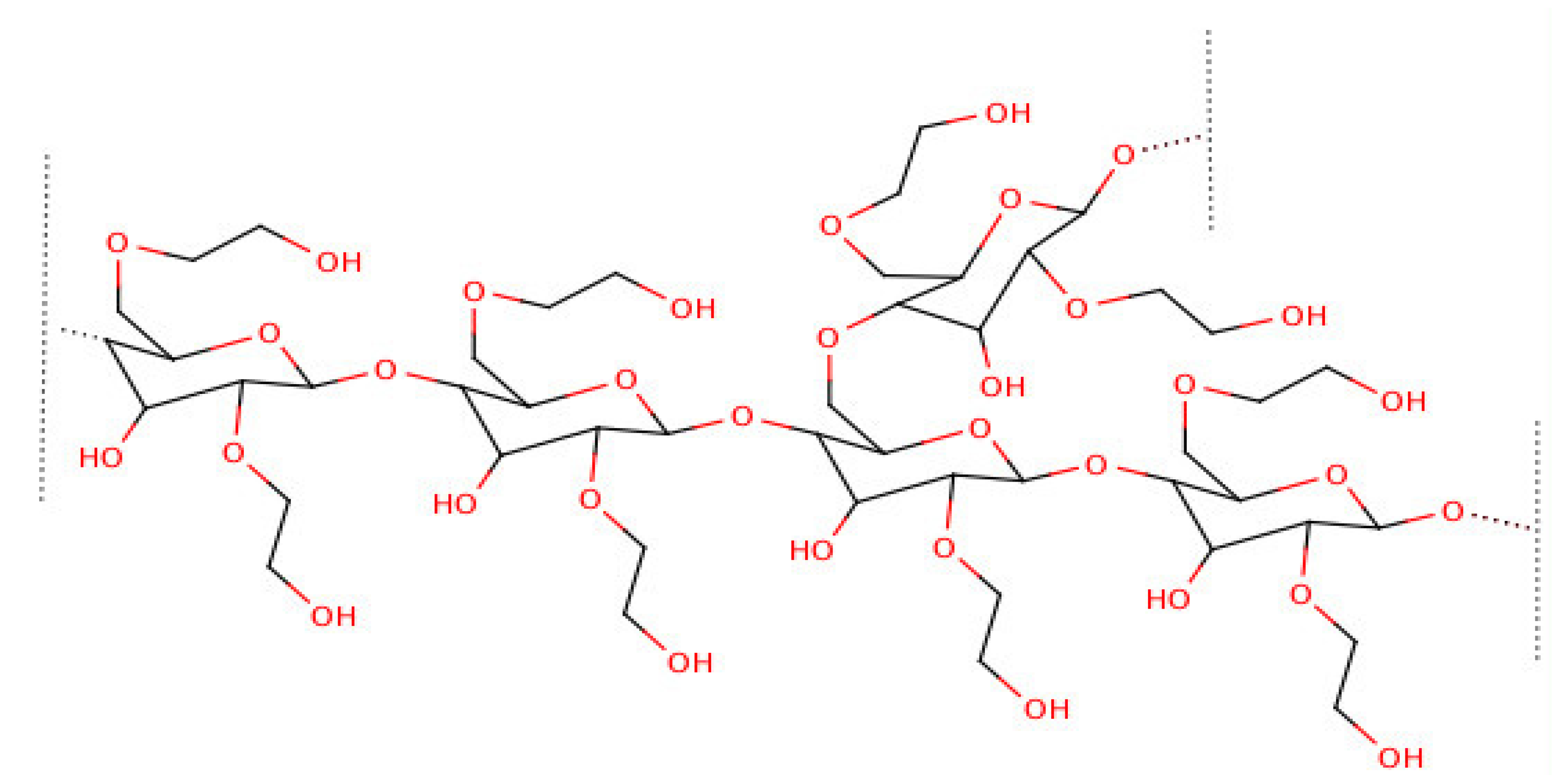
5. Dextrin

6. Hydroxyethyl-Starch (HES)

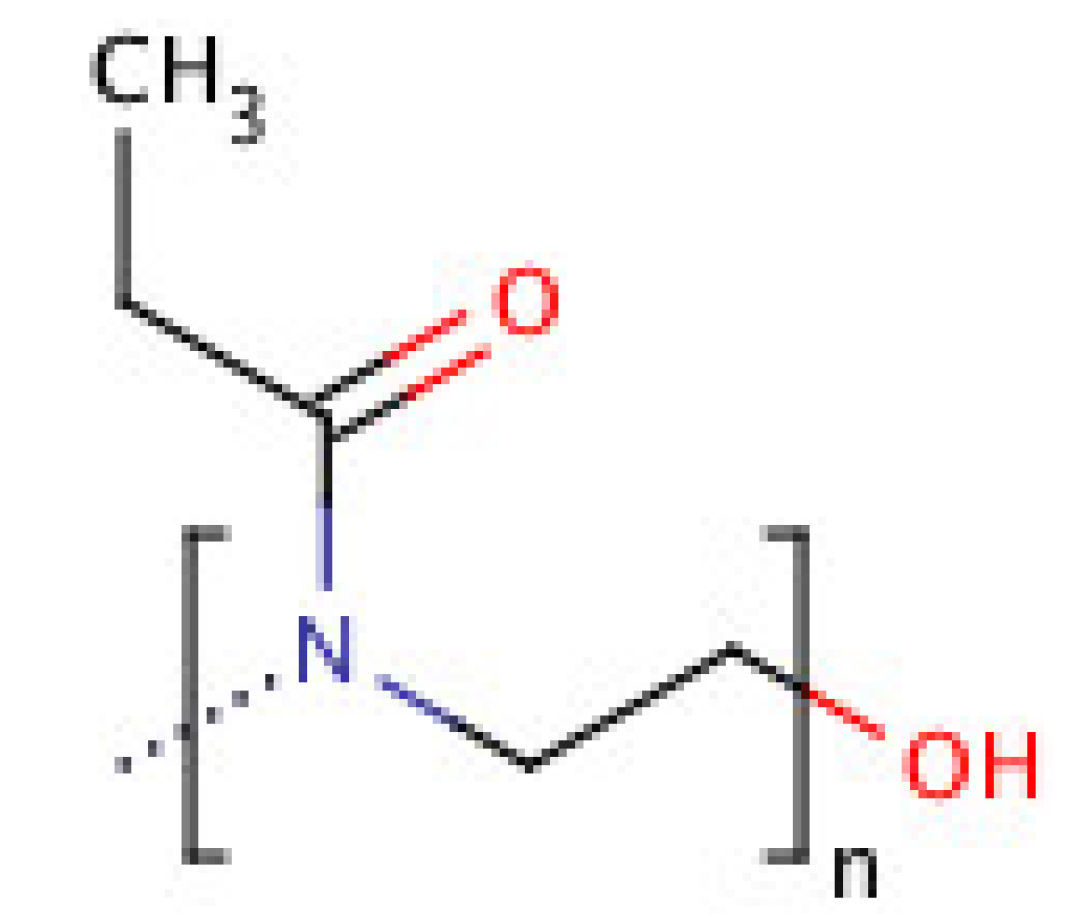
7. Poly(2-ethyl 2-oxazoline) (PEOZ)

8. Protein Conjugates Exploiting Polypeptides as Carrying Polymers
8.1. XTEN Technology
8.2. PASylation
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Biosimilars and Follow-on Biologics Report: The Global Outlook 2010–2025. Visiongain Ltd.: London, UK, 2010. Available online: http://www.visiongain.com/Report/474/Biosimilars-and-Follow-On-Biologics-Global-Market-Outlook-2010-2025 (accessed on 9 January 2014).
- Abuchowski, A.; van Es, T.; Palczuk, N.; Davis, F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar]
- Abuchowski, A.; McCoy, J.R.; Palczuk, N.C.; van Es, T.; Davis, F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977, 252, 3582–3586. [Google Scholar]
- Torchilin, V.; Voronkov, Y.I.; Mazaev, A. Use of immobilized streptokinase (Streptodekaza) for thrombosis treatment. Ter. Arkhiv 1982, 54, 21–26. [Google Scholar]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N.; Stadler, J.; Tilbury, L.; Smith, D. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos. 2007, 35, 9–16. [Google Scholar]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Manjula, B.; Tsai, A.; Upadhya, R.; Perumalsamy, K.; Smith, P.; Malavalli, A.; Vandegriff, K.; Winslow, R.; Intaglietta, M.; Prabhakaran, M. Site-specific PEGylation of hemoglobin at Cys-93(β): Correlation between the colligative properties of the PEGylated protein and the length of the conjugated PEG chain. Bioconjug. Chem. 2003, 14, 464–472. [Google Scholar] [CrossRef]
- Kinstler, O.; Moulinex, G.; Treheit, M.; Ladd, D.; Gegg, C. Mono-N-terminal poly(ethylene glycol)-protein conjugates. Adv. Drug Deliv. Rev. 2002, 54, 477–485. [Google Scholar] [CrossRef]
- Basu, A.; Yang, K.; Wang, M.; Liu, S.; Chintala, R.; Palm, T.; Zhao, H.; Peng, P.; Wu, D.; Zhang, Z. Structure-function engineering of interferon-β-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug. Chem. 2006, 17, 618–630. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A.; Caboi, F.; Sergi, M.; Marongiu, C.; Pasut, G. Site-specific pegylation of G-CSF by reversible denaturation. Bioconjug. Chem. 2007, 18, 1824–1830. [Google Scholar] [CrossRef]
- Weir, N.; Athwal, D.; Brown, D.; Foulkes, R.; Kollias, G.; Nesbitt, A.; Popplewell, A.; Spitali, M.; Stephens, S. A new generation of high-affinity humanized PEGylated fab fragment anti-tumor necrosis factor-α monoclonal antibodies. Therapy 2006, 3, 535–545. [Google Scholar]
- Shaunak, S.; Godwin, A.; Choi, J.; Balan, S.; Pedone, E.; Vijayarangam, D.; Heidelberger, S.; Teo, I.; Zloh, M.; Brocchini, S. Site-specific PEGylation of native disulfide bonds in therapeutic proteins. Nat. Chem. Biol. 2006, 2, 312–313. [Google Scholar] [CrossRef]
- Mero, A.; Schiavon, M.; Veronese, F.M.; Pasut, G. A new method to increase selectivity of transglutaminase mediated PEGylation of salmon calcitonin and human growth hormone. J. Control. Release 2011, 154, 27–34. [Google Scholar] [CrossRef]
- DeFrees, S.; Wang, Z.; Xing, R.; Scott, A.E.; Wang, J.; Zopf, D.; Gouty, D.L.; Sjoberg, E.R.; Panneerselvam, K.; Brinkman-Van, der Linden; Els, C.M. GlycoPEGylation of recombinant therapeutic proteins produced in Escherichia coli. Glycobiology 2006, 16, 833–843. [Google Scholar] [CrossRef]
- Popp, M.W.; Dougan, S.K.; Chuang, T.; Spooner, E.; Ploegh, H.L. Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. USA 2011, 108, 3169–3174. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, K.; Martinez, A.; Basu, A.; Chintala, R.; Liu, H.; Janjua, A.; Wang, M.; Filpula, D. Linear and branched bicin linkers for releasable PEGylation of macromolecules: Controlled release in vivo and in vitro from mono-and multi-PEGylated proteins. Bioconjug. Chem. 2006, 17, 341–351. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Yang, K.; Zhao, H.; Conover, C.D.; Lee, S.; Filpula, D. Controlled release of proteins from their poly(ethylene glycol) conjugates: Drug delivery systems employing 1, 6-elimination. Bioconjug. Chem. 2003, 14, 395–403. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Choe, Y.H.; Conover, C.D.; Shum, K.; Wu, D.; Royzen, M. Drug delivery systems based on trimethyl lock lactonization: Poly(ethylene glycol) prodrugs of amino-containing compounds. J. Med. Chem. 2000, 43, 475–487. [Google Scholar] [CrossRef]
- Tsubery, H.; Mironchik, M.; Fridkin, M.; Shechter, Y. Prolonging the action of protein and peptide drugs by a novel approach of reversible polyethylene glycol modification. J. Biol. Chem. 2004, 279, 38118–38124. [Google Scholar] [CrossRef]
- Wylie, D.C.; Voloch, M.; Lee, S.; Liu, Y.; Cannon-Carlson, S.; Cutler, C.; Pramanik, B. Carboxyalkylated histidine is a pH-dependent product of pegylation with SC-PEG. Pharm. Res. 2001, 18, 1354–1360. [Google Scholar] [CrossRef]
- Pasut, G.; Caboi, F.; Schrepfer, R.; Tonon, G.; Schiavon, O.; Veronese, F. New active poly(ethylene glycol) derivative for amino coupling. React. Funct. Polym. 2007, 67, 529–539. [Google Scholar] [CrossRef]
- Pasut, G.; Mero, A.; Caboi, F.; Scaramuzza, S.; Sollai, L.; Veronese, F.M. A new PEG-β-alanine active derivative for releasable protein conjugation. Bioconjug. Chem. 2008, 19, 2427–2431. [Google Scholar] [CrossRef]
- Mueller, C.; Capelle, M.A.; Arvinte, T.; Seyrek, E.; Borchard, G. Noncovalent pegylation by dansyl-poly(ethylene glycol)s as a new means against aggregation of salmon calcitonin. J. Pharm. Sci. 2011, 100, 1648–1662. [Google Scholar] [CrossRef]
- Mueller, C.; Capelle, M.A.; Arvinte, T.; Seyrek, E.; Borchard, G. Tryptophan-mPEGs: Novel excipients that stabilize salmon calcitonin against aggregation by non-covalent PEGylation. Eur. J. Pharm. Biopharm. 2011, 79, 646–657. [Google Scholar] [CrossRef]
- Mero, A.; Ishino, T.; Chaiken, I.; Veronese, F.M.; Pasut, G. Multivalent and flexible PEG-nitrilotriacetic acid derivatives for non-covalent protein pegylation. Pharm. Res. 2011, 28, 2412–2421. [Google Scholar] [CrossRef]
- Liu, M.; Tirino, P.; Radivojevic, M.; Phillips, D.J.; Gibson, M.I.; Leroux, J.; Gauthier, M.A. Molecular sieving on the surface of a protein provides protection without loss of activity. Adv. Funct. Mater. 2012, 23, 2007–2015. [Google Scholar]
- Miyaji, Y.; Kasuya, Y.; Furuta, Y.; Kurihara, A.; Takahashi, M.; Ogawara, K.; Izumi, T.; Okazaki, O.; Higaki, K. Novel comb-shaped PEG modification enhances the osteoclastic inhibitory effect and bone delivery of osteoprotegerin after intravenous administration in ovariectomized rats. Pharm. Res. 2012, 29, 3143–3155. [Google Scholar] [CrossRef]
- Ryan, S.M.; Frías, J.M.; Wang, X.; Sayers, C.T.; Haddleton, D.M.; Brayden, D.J. PK/PD modelling of comb-shaped PEGylated salmon calcitonin conjugates of differing molecular weights. J. Control. Release 2011, 149, 126–132. [Google Scholar] [CrossRef]
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 2007, 110, 103–111. [Google Scholar] [CrossRef]
- Sherman, M.R.; Saifer, M.G.; Perez-Ruiz, F. PEG-Uricase in the management of treatment-resistant gout and hyperuricemia. Adv. Drug Deliv. Rev. 2008, 60, 59–68. [Google Scholar] [CrossRef]
- Leger, R.M.; Arndt, P.; Garratty, G.; Armstrong, J.K.; Meiselman, H.J.; Fisher, T.C. Normal donor sera can contain antibodies to polyethylene glycol (PEG). Transfusion 2001, 41, 29S. [Google Scholar]
- Fisher, T.C.; Armstrong, J.K.; Wenby, R.B.; Meiselman, H.J.; Leger, R.; Garratty, G. Isolation and identification of a human antibody to polyethylene glycol (abstract). Blood 2003, 102, 559A. [Google Scholar]
- Bendele, A.; Seely, J.; Richey, C.; Sennello, G.; Shopp, G. Short communication: Renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicol. Sci. 1998, 42, 152–157. [Google Scholar] [CrossRef]
- Conover, C.; Lejeune, L.; Linberg, R.; Shum, K.; Shorr, R.G. Transitional vacuole formation following a bolus infusion of PEG-hemoglobin in the rat. Artif. Cell. Blood Sub. 1996, 24, 599–611. [Google Scholar] [CrossRef]
- Garay, R.P.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Del. 2012, 9, 1319–1323. [Google Scholar] [CrossRef]
- Ganson, N.; Kelly, S.; Scarlett, E.; Sundy, J.; Hershfield, M. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res. Ther. 2005. [Google Scholar] [CrossRef]
- Zhang, C.; Fan, K.; Ma, X.; Wei, D. Impact of large aggregated uricases and PEG diol on accelerated blood clearance of PEGylated canine uricase. PLoS One 2012, 7, e39659. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control. Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef]
- Jevševar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar] [CrossRef]
- Pasut, G.; Veronese, F.M. PEG conjugates in clinical development or use as anticancer agents: An overview. Adv. Drug Deliv. Rev. 2009, 61, 1177–1188. [Google Scholar] [CrossRef]
- Mehvar, R. Recent trends in the use of polysaccharides for improved delivery of therapeutic agents: Pharmacokinetic and pharmacodynamic perspectives. Curr. Pharm. Biotechnol. 2003, 4, 283–302. [Google Scholar] [CrossRef]
- Mehvar, R. Dextrans for targeted and sustained delivery of therapeutic and imaging agents. J. Control. Release 2000, 69, 1–25. [Google Scholar]
- Larsen, C. Dextran prodrugs—Structure and stability in relation to therapeutic activity. Adv. Drug Deliv. Rev. 1989, 3, 103–154. [Google Scholar] [CrossRef]
- Walker, G.J. Dextrans. In Biochemistry of Carbohydrates; Manners, D.J., Ed.; University Park Press: Baltimore, MD, USA, 1978; Volume 16, pp. 75–126. [Google Scholar]
- Mehvar, R.; Shepard, T.L. Molecular-weight-dependent pharmacokinetics of fluorescein-labeled dextrans in rats. J. Pharm. Sci. 1992, 81, 908–912. [Google Scholar] [CrossRef]
- Vercauteren, R.; Bruneel, D.; Schacht, E.; Duncan, R. Effect of the chemical modification of dextran on the degradation by dextranase. J. Bioact. Compat. Pol. 1990, 5, 4–15. [Google Scholar] [CrossRef]
- Schacht, E.; Vercauteren, R.; Vansteenkiste, S. Some aspects of the application of dextran in prodrug design. J. Bioact. Compat. Pol. 1988, 3, 72–80. [Google Scholar] [CrossRef]
- Wileman, T.E. Properties of asparaginase-dextran conjugates. Adv. Drug Deliv. Rev. 1991, 6, 167–180. [Google Scholar] [CrossRef]
- Wileman, T.E.; Foster, R.L.; Elliott, P.N. Soluble asparaginase-dextran conjugates show Increased circulatory persistence and lowered antigen reactivity. J. Pharm. Pharmacol. 1986, 38, 264–271. [Google Scholar] [CrossRef]
- Melton, R.G.; Wiblin, C.N.; Foster, R.L.; Sherwood, R.F. Covalent linkage of carboxypeptidase G2 to soluble dextrans: I. Properties of conjugates and effects on plasma persistence in mice. Biochem. Pharmacol. 1987, 36, 105–112. [Google Scholar] [CrossRef]
- Molteni, L. Dextrans as Drug Carriers. In Drug Carriers in Biology and Medicine; Gregoriadis, G., Ed.; Academic Press: New York, NY, USA, 1979; pp. 107–125. [Google Scholar]
- Yasuda, Y.; Fujita, T.; Takakura, Y.; Hashida, M.; Sezaki, H. Biochemical and biopharmaceutical properties of macromolecular conjugates of uricase with dextran and polyethylene glycol. Chem. Pharm. Bull. (Tokyo) 1990, 38, 2053–2056. [Google Scholar] [CrossRef]
- Fujita, T.; Nishikawa, M.; Tamaki, C.; Takakura, Y.; Hashida, M.; Sezaki, H. Targeted delivery of human recombinant superoxide dismutase by chemical modification with mono-and polysaccharide derivatives. J. Pharmacol. Exp. Ther. 1992, 263, 971–978. [Google Scholar]
- Baudyš, M.; Letourneur, D.; Liu, F.; Mix, D.; Jozefonvicz, J.; Kim, S.W. Extending insulin action in vivo by conjugation to carboxymethyl dextran. Bioconjug. Chem. 1998, 9, 176–183. [Google Scholar] [CrossRef]
- Caron, A.; Menu, P.; Faivre-Fiorina, B.; Labrude, P.; Vigneron, C. The effects of stroma-free and dextran-conjugated hemoglobin on hemodynamics and carotid blood flow in hemorrhaged guinea pigs. Art. Cell. Blood Sub. 1999, 27, 49–64. [Google Scholar] [CrossRef]
- Faivre, B.; Labaeye, V.; Menu, P.; Labrude, P.; Vigneron, C. Assessment of dextran 10-benzene-tetracarboxylate-hemoglobin, an oxygen carrier, using guinea pig isolated bowel model. Art. Cell. Blood Sub. 1995, 23, 495–504. [Google Scholar] [CrossRef]
- Hreczuk-Hirst, D.; Jain, S.; Genkin, D.; Laing, P.; Gregoriadis, G. Preparation and Properties of Polysialylated Interferon-α-2b. In Proceedings of the AAPS Annual Meeting, Toronto, ON, Canada, 10–14 November, 2002. M1056.
- Gregoriadis, G.; McCormack, B.; Wang, Z.; Lifely, R. Polysialic acids: Potential in drug delivery. FEBS Lett. 1993, 315, 271–276. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Jain, S.; Papaioannou, I.; Laing, P. Improving the therapeutic efficacy of peptides and proteins: A role for polysialic acids. Int. J. Pharm. 2005, 300, 125–130. [Google Scholar] [CrossRef]
- Jain, S.; Hreczuk-Hirst, D.H.; McCormack, B.; Mital, M.; Epenetos, A.; Laing, P.; Gregoriadis, G. Polysialylated insulin: Synthesis, characterization and biological activity in vivo. Biochim. Biophys. Acta 2003, 1622, 42–49. [Google Scholar] [CrossRef]
- Epenetos, A.; Hreczuk-Hirst, D.; McCormack, B.; Gregoriadis, G. Polysialylated proteins: A potential role in cancer therapy. Clin. Pharm. 2002, 21, 2186. [Google Scholar]
- Gregoriadis, G.; Fernandes, A.; Mital, M.; McCormack, B. Polysialic acids: Potential in improving the stability and pharmacokinetics of proteins and other therapeutics. Cell. Mol. Life Sci. 2000, 57, 1964–1969. [Google Scholar] [CrossRef]
- Fernandes, A.I.; Gregoriadis, G. The effect of polysialylation on the immunogenicity and antigenicity of asparaginase: Implication in its pharmacokinetics. Int. J. Pharm. 2001, 217, 215–224. [Google Scholar] [CrossRef]
- Constantinou, A.; Epenetos, A.; Hreczuk-Hirst, D.; Jain, S.; Wright, M.; Chester, K.; Deonarain, M. Site-specific polysialylation of an antitumor single-chain Fv fragment. Bioconjug. Chem. 2009, 20, 924–931. [Google Scholar] [CrossRef]
- Lindhout, T.; Iqbal, U.; Willis, L.M.; Reid, A.N.; Li, J.; Liu, X.; Moreno, M.; Wakarchuk, W.W. Site-specific enzymatic polysialylation of therapeutic proteins using bacterial enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 7397–7402. [Google Scholar]
- Evered, D.; Whelan, J. The Biology of Hyaluronan; Wiley & Sons: Chichester, UK, 1989. [Google Scholar]
- Almond, A. Hyaluronan. Cell. Mol. Life Sci. 2007, 64, 1591–1596. [Google Scholar] [CrossRef]
- Laurent, T.C.; Fraser, J.R. Hyaluronan. FASEB J. 1992, 6, 2397–2404. [Google Scholar]
- Saravanakumar, G.; Choi, K.Y.; Yoon, H.Y.; Kim, K.; Park, J.H.; Kwon, I.C.; Park, K. Hydrotropic hyaluronic acid conjugates: Synthesis, characterization, and implications as a carrier of paclitaxel. Int. J. Pharm. 2010, 394, 154–161. [Google Scholar] [CrossRef]
- Homma, A.; Sato, H.; Okamachi, A.; Emura, T.; Ishizawa, T.; Kato, T.; Matsuura, T.; Sato, S.; Tamura, T.; Higuchi, Y. Novel hyaluronic acid-methotrexate conjugates for osteoarthritis treatment. Bioorg. Med. Chem. 2009, 17, 4647–4656. [Google Scholar] [CrossRef]
- Yang, J.; Park, K.; Jung, H.; Kim, H.; Hong, S.W.; Yoon, S.K.; Hahn, S.K. Target specific hyaluronic acid–interferon alpha conjugate for the treatment of hepatitis C virus infection. Biomaterials 2011, 32, 8722–8729. [Google Scholar] [CrossRef]
- Mero, A.; Pasqualin, M.; Campisi, M.; Renier, D.; Pasut, G. Conjugation of hyaluronan to proteins. Carbohydr. Polym. 2013, 92, 2163–2170. [Google Scholar] [CrossRef]
- D’Este, M.; Renier, D.; Pasut, G.; Rosato, A. Process for the Synthesis of Conjugates of Glycosaminoglycanes (GAG) with Biologically Active Molecules, Polymeric Conjugates and Relative Uses Thereof. WO2010145821 A1, 23 December 2010. [Google Scholar]
- Campisi, M.; Mero, A. Hyaluronic acid as polymeric carrier of drugs and proteins. Polymers 2014, in press. [Google Scholar]
- Ferguson, E.L.; Richardson, S.C.; Duncan, R. Studies on the mechanism of action of dextrin—Phospholipase A2 and its suitability for use in combination therapy. Mol. Pharm. 2010, 7, 510–521. [Google Scholar] [CrossRef]
- Duncan, R.; Gilbert, H.; Carbajo, R.; Vicent, M. Polymer masked-unmasked protein therapy (PUMPT) 1: Bioresponsive dextrin-trypsin and-MSH conjugates designed for α-amylase activation. Biomacromolecules 2008, 9, 1146–1154. [Google Scholar] [CrossRef]
- Hardwicke, J.T.; Hart, J.; Bell, A.; Duncan, R.; Thomas, D.W.; Moseley, R. The Effect of dextrin–rhEGF on the healing of full-thickness, excisional wounds in the (db/db) diabetic mouse. J. Control. Release 2011, 152, 411–417. [Google Scholar] [CrossRef]
- Hardwicke, J.; Moseley, R.; Stephens, P.; Harding, K.; Duncan, R.; Thomas, D.W. Bioresponsive dextrin−rhEGF conjugates: In vitro evaluation in models relevant to its proposed use as a treatment for chronic wounds. Mol. Pharm. 2010, 7, 699–707. [Google Scholar] [CrossRef]
- Besheer, A.; Hertel, T.C.; Kressler, J.; Mäder, K.; Pietzsch, M. Enzymatically catalyzed HES conjugation using microbial transglutaminase: Proof of feasibility. J. Pharm. Sci. 2009, 98, 4420–4428. [Google Scholar] [CrossRef]
- Besheer, A.; Hause, G.; Kressler, J.; Mäder, K. Hydrophobically modified hydroxyethyl starch: Synthesis, characterization, and aqueous self-assembly into nano-sized polymeric micelles and vesicles. Biomacromolecules 2007, 8, 359–367. [Google Scholar] [CrossRef]
- Schubert, S.; Autenrieth, I.B. Conjugation of hydroxyethyl starch to desferrioxamine (DFO) modulates the dual role of DFO in yersinia enterocolitica infection. Clin. Diagn. Lab. Immunol. 2000, 7, 457–462. [Google Scholar]
- Zander, N.; Conradt, H.; Eichner, W. Method of Producing Hydroxyalkyl Starch Derivatives. WO2004024776, 25 March 2004. [Google Scholar]
- Conradt, H.S.; Grabenhorst, E.; Nimtz, M.; Zander, N.; Frank, R.; Eichner, W. HASylated Polypeptides, especially HASylated Erythropoietin. WO2004024761 A1, 25 March 2004. [Google Scholar]
- Zarychanski, R.; Abou-Setta, A.M.; Turgeon, A.F.; Houston, B.L.; McIntyre, L.; Marshall, J.C.; Fergusson, D.A. Association of hydroxyethyl starch administration with mortality and acute kidney injury in critically Ill patients requiring volume resuscitationa systematic review and meta-analysis hydroxyethyl starch and outcomes in critically Ill. JAMA 2013, 309, 678–688. [Google Scholar] [CrossRef]
- Perel, P.; Roberts, I.; Pearson, M. Colloids versus crystalloids for fluid resuscitation in critically Ill patients. Cochrane Database Syst. Rev. 2007, 4, CD000567. [Google Scholar]
- PRAC Recommends Suspending Marketing Authorizations for Infusion Solutions Containing Hydroxyethyl Starch. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/ news_and_events/news/2013/06/news_detail_001814.jsp&mid=WC0b01ac058004d5c1 (accessed on 25 November 2013).
- Zalipsky, S.; Hansen, C.B.; Oaks, J.M.; Allen, T.M. Evaluation of blood clearance rates and biodistribution of poly(2-oxazoline)-grafted liposomes. J. Pharm. Sci. 1996, 85, 133–137. [Google Scholar] [CrossRef]
- Hoogenboom, R. Poly(2-oxazoline)s: A polymer class with numerous potential applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar] [CrossRef]
- Mero, A.; Pasut, G.; Via, L.D.; Fijten, M.W.; Schubert, U.S.; Hoogenboom, R.; Veronese, F.M. Synthesis and characterization of poly(2-ethyl 2-oxazoline)-conjugates with proteins and drugs: Suitable alternatives to PEG-conjugates? J. Control. Release 2008, 125, 87–95. [Google Scholar] [CrossRef]
- Goddard, P.; Hutchinson, L.E.; Brown, J.; Brookman, L.J. Soluble polymeric carriers for drug delivery: Part 2. Preparation and in vivo behaviour of N-acylethylenimine copolymers. J. Control. Release 1989, 10, 5–16. [Google Scholar] [CrossRef]
- Miyamoto, M.; Naka, K.; Tokumizu, M.; Saegusa, T. End capping of growing species of poly(2-oxazoline) with carboxylic acid: A novel and convenient route to prepare vinyl-and carboxy-terminated macromonomers. Macromolecules 1989, 22, 1604–1607. [Google Scholar] [CrossRef]
- Einzmann, M.; Binder, W.H. Novel functional initiators for oxazoline polymerization. J. Polym. Sci. A 2001, 39, 2821–2831. [Google Scholar] [CrossRef]
- Jordan, R.; Ulman, A. Surface initiated living cationic polymerization of 2-oxazolines. J. Am. Chem. Soc. 1998, 120, 243–247. [Google Scholar] [CrossRef]
- Kobayashi, S.; Masuda, E.; Shoda, S.; Shimano, Y. Synthesis of acryl-and methacryl-type macromonomers and telechelics by utilizing living polymerization of 2-oxazolines. Macromolecules 1989, 22, 2878–2884. [Google Scholar] [CrossRef]
- Kobayashi, S.; Iijima, S.; Igarashi, T.; Saegusa, T. Synthesis of a nonionic polymer surfactant from cyclic imino ethers by the initiator method. Macromolecules 1987, 20, 1729–1734. [Google Scholar] [CrossRef]
- Mero, A.; Fang, Z.; Pasut, G.; Veronese, F.M.; Viegas, T.X. Selective conjugation of poly(2-ethyl 2-oxazoline) to granulocyte colony stimulating factor. J. Control. Release 2012, 159, 353–361. [Google Scholar] [CrossRef]
- Gaertner, F.C.; Luxenhofer, R.; Blechert, B.; Jordan, R.; Essler, M. Synthesis, biodistribution and excretion of radiolabeled poly(2-alkyl-2-oxazoline)s. J. Control. Release 2007, 119, 291–300. [Google Scholar] [CrossRef]
- Viegas, T.X.; Bentley, M.D.; Harris, J.M.; Fang, Z.; Yoon, K.; Dizman, B.; Weimer, R.; Mero, A.; Pasut, G.; Veronese, F.M. Polyoxazoline: Chemistry, properties, and applications in drug delivery. Bioconjug. Chem. 2011, 22, 976–986. [Google Scholar] [CrossRef]
- Eskow Jaunarajs, K.L.; Standaert, D.G.; Viegas, T.X.; Bentley, M.D.; Fang, Z.; Dizman, B.; Yoon, K.; Weimer, R.; Ravenscroft, P.; Johnston, T.H. Rotigotine polyoxazoline conjugate SER-214 provides robust and sustained antiparkinsonian benefit. Mov. Disord. 2013, 28, 1675–1682. [Google Scholar] [CrossRef]
- Tong, J.; Luxenhofer, R.; Yi, X.; Jordan, R.; Kabanov, A.V. Protein modification with amphiphilic block copoly(2-oxazoline)s as a new platform for enhanced cellular delivery. Mol. Pharm. 2010, 7, 984–992. [Google Scholar] [CrossRef]
- Luxenhofer, R.; Han, Y.; Schulz, A.; Tong, J.; He, Z.; Kabanov, A.V.; Jordan, R. Poly(2-oxazoline)s as polymer therapeutics. Macromol. Rapid Commun. 2012, 33, 1613–1631. [Google Scholar] [CrossRef]
- Schmidt, S.R. Fusion-proteins as biopharmaceuticals—Applications and challenges. Curr. Opin. Drug Discov. Devel. 2009, 12, 284–295. [Google Scholar]
- Subramanian, G.M.; Fiscella, M.; Lamousé-Smith, A.; Zeuzem, S.; McHutchison, J.G. Albinterferon α-2b: A genetic fusion protein for the treatment of chronic hepatitis C. Nat. Biotechnol. 2007, 25, 1411–1419. [Google Scholar] [CrossRef]
- Schellenberger, V.; Wang, C.; Geething, N.C.; Spink, B.J.; Campbell, A.; To, W.; Scholle, M.D.; Yin, Y.; Yao, Y.; Bogin, O. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 2009, 27, 1186–1190. [Google Scholar] [CrossRef]
- Cleland, J.L.; Geething, N.C.; Moore, J.A.; Rogers, B.C.; Spink, B.J.; Wang, C.; Alters, S.E.; Stemmer, W.P.; Schellenberger, V. A novel long-acting human growth hormone fusion protein (vrs-317): Enhanced in vivo potency and half-life. J. Pharm. Sci. 2012, 101, 2744–2754. [Google Scholar] [CrossRef]
- Geething, N.C.; To, W.; Spink, B.J.; Scholle, M.D.; Wang, C.; Yin, Y.; Yao, Y.; Schellenberger, V.; Cleland, J.L.; Stemmer, W.P. Gcg-XTEN: An improved glucagon capable of preventing hypoglycemia without increasing baseline blood glucose. PLoS One 2010, 5, e10175. [Google Scholar] [CrossRef]
- Schlapschy, M.; Theobald, I.; Mack, H.; Schottelius, M.; Wester, H.; Skerra, A. Fusion of a recombinant antibody fragment with a homo-amino-acid polymer: Effects on biophysical properties and prolonged plasma half-life. Protein Eng. Des. Sel. 2007, 20, 273–284. [Google Scholar]
- Schlapschy, M.; Binder, U.; Börger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pasut, G. Polymers for Protein Conjugation. Polymers 2014, 6, 160-178. https://doi.org/10.3390/polym6010160
Pasut G. Polymers for Protein Conjugation. Polymers. 2014; 6(1):160-178. https://doi.org/10.3390/polym6010160
Chicago/Turabian StylePasut, Gianfranco. 2014. "Polymers for Protein Conjugation" Polymers 6, no. 1: 160-178. https://doi.org/10.3390/polym6010160