1. Introduction
In the present work, we investigate by computer experiments new ways of separating charged polymers in electric fields. We aim to distinguish between chains of different length by influencing their direction of movement, rather than their electrophoretic velocities. The latter topic has been investigated by us in previous papers [
1,
2]. We focus on very dilute solutions of polyelectrolytes in the presence of multivalent counterions. Here one can neglect other complex phenomena such as induced attraction between charged molecules mediated by multivalent ions [
3].
The motivation of our work comes from the observation that in an electric field, in free solution and without a sieving media charged polymers move with the same velocity, irrespective of their size. An excellent review of this topic is presented in Niedringhaus
et al. [
4]. If a sieving media is present, the disentanglement time of the charged polymers from the topologically static obstacles depends on the polyelectrolyte length [
5]. The Ogston model envisions the charged polymer as a spherical random coil that diffuses through the pores of the polymer network. At high applied fields the charged molecules are pinned against the gel fibers, and the capillary electrophoresis method becomes ineffective in separating polyelectrolytes. Further progress was possible by using pulsed electric fields [
6]. The pinned molecules retraced part of the path they had been traveling before and therefore the rate of advance in the forward direction changed with molecular size. The assumption was that longer molecules backtracked further than shorter molecules. In this non-equilibrium process, the duration of the pulse defines how much the molecules are allowed to approach their steady state (which is fully stretched), at either direction of the applied field.
Alternatively, ordered sieving media, in the form of small ordered confinements achieved by a colloidal self-assembly (CSA) approach was used by Nazemifard
et al. [
7]. The authors showed that stronger confinement (
i.e., pore sizes smaller than the persistence length of the charged molecule) lead to full stretching and more efficient separation as compared to larger pores, where fluctuations in size and loops, or hernia formation affect the reorientation time. Recent experiments in arrays of nano-grooves, separated by very thin slits, are reported in [
8]. The authors showed that there were two distinct ways in which the molecules preferred to migrate: (1) a folded state where the molecule advanced its monomers from one groove to the next, just as water is poured from one glass into the other; and (2) a side movement similar to sand flowing in an hour-glass from the top side to the bottom side. It was shown that the dynamic interplay between these two modes of transport created a size-dependent velocity that could be utilized for separation. The authors remarked that pure motion in either state exhibited a size-independent velocity at fixed buffer velocity, or external pressure gradient.
Other novel separation methods are stretching of fluorescently labeled charged molecules in channels, which are then imaged [
9], threading of the charged chains through small openings in membranes—either protein [
10] or solid state pores [
11,
12,
13,
14,
15,
16], or translocation though conical-shaped pores contained within glass membranes [
17]. Here the amount of time that the ionic current signal is altered during translocation can be measured, and this gives an indication of the total length of the molecule. For larger pores, the translocation process is controlled by overcoming a free energy barrier, which for not too long chains may depend on the chain length [
18].
It is apparent that the trend in sequencing technologies is to use smaller and smaller devices with exquisite control over the motion of each structural unit of the charged polymer, without perturbations due to the counterion cloud or thermal fluctuations [
19]. Here we study charged polymers separation in straight cylinders of uniform diameter under the action of a constant electric field that has two components: one component is parallel to the cylinder axis and the second component is orthogonal to the cylinder axis. We call the latter component the transversal field. The separation principle is based on two effects: (1) collapse of the chains in the presence of multivalent counterions [
20,
21] and (2) partial removal of condensed counterions from the polyelectrolyte cloud by transversal electric field
. In short, the chains are overcharged in the presence of multivalent counterions and move along the cylinder axis in one direction or another, depending on how many counterions are still bound. This happens because the role of the transversal field is to remove counterions from the chains and therefore switch the sign of their effective charge. The direction of the motion depends on chain length, the valency of the counterions, the strength of Coulomb interaction [
22] between polymer monomers and bound counterions, and
. The relative electrophoretic velocity depends on the diameter of the cylinder and concentration of free ions. In this work, we focus on scaling laws for velocities, effective charge, radii of gyration and chain extensions in the direction of motion, without quantitative calculations of electrolyte friction or hydrodynamic friction effects. The collapse of the chains may be triggered by attractive dipole-dipole and correlation-induced electrostatic interactions [
23,
24].
2. Method
By molecular dynamics simulations we investigate the electrophoretic motion of charged polymers in solution, in cylindrical confinement, in the presence of longitudinal
(along the cylinder axis) and transversal electric
fields. The charged polymers are coarse-grained models where each monomer carries a unit negative electric charge, −
e. The polymer monomers are connected by finitely extensible nonlinear elastic (FENE) and Lennard–Jonnes potentials. The Lennard–Jones potential
ULJ acts between all particle pairs in the system and is defined by unit energy
ϵ and unit length
σ:
The FENE potential
UFENE is given by the following relation:
where
k = 30 and maximum bond extension is
R0 = 3
σ. The system contains added salt, and all coions, counterions and fluid monomers are explicitly modeled. The walls of the cylinder are made of uncharged monomers with fixed positions in space. We investigate cylinders with the following radii
R = 7.74
σ, 11.62
σ, 15.5
σ and 23.24
σ.
All monomers interact through Lennard-Jones potential, Equation (1), and Coulomb potential
UC =
zizjkBTlB/
r, if they are charged. The variables
zi and
zj are valencies of interacting charges. The temperature of the system is set at
kBT = 1.2
ϵ. The Bjerrum length [
25] is set to
lB =
e2/
kBT ≈ 12
σ. (The bond length of the polymer is approximately
σ). When mapping the present simulations to real systems, one may consider that in water the Bjerrum length is 0.7 nm, where the dielectric constant of bulk water is about 80 at room temperature. This would give a very small value of parameter
σ. However, the dielectric constant of water in confinement is not clearly defined. For example, in [
26], the authors showed that the dielectric constant of water can be as low as 37 in micro-sized confinements. Notice further that additional binding forces such as hydrogen bonds between more complex counter ions and the charged groups have a similar effect as to increase the local electric attraction. The present polymer physics study extends therefore the parameter space to explore new phenomena.
The multivalent counterions may carry charges from +1e up to +4e. Apart from coions of charge −e, the MD (molecular dynamics) simulation box may contain therefore monovalent counterions, or mixture of counterions with +e and +2e charges, or +e and +3e charges, etc. In all cases, the multivalent monomer concentration is set to Cz = 3.56 × 10−3σ−3. The monovalent coions concentration C−1 is 0.031σ−3, while monovalent counterions of concentration C+1 are added to the system to fulfill the electric neutrality condition. For the range of chain lengths investigated here, C+1 varies between 0.017σ−3 and 0.024σ−3. The number density of fluid monomers is set to 0.82σ−3.
We consider a very low concentration of charged polymers, which means that inter-chain interactions are negligible. The Debye length
λD = ((
z2Cz +
C+1 +
C−1)
e2/
kBT )
−1/2 for each counterion valency
z = 1, 2, 3, 4 is
λD ∼ 2
σ, 1.6
σ, 1.4
σ and 1.25
σ, respectively. Given that the Debye lengths are so small, one may alternatively consider a calculation of the Coulomb potential using Debye-Hückel approximation. For computational reasons we simply use a bare Coulomb potential and use a cutoff radius of about half the simulation box length. This is of the order of 300
σ. We assume that all monomers have the same mass and LJ parameters,
ϵ and
σ. A few snapshots from MD simulations are presented in
Figure 1. We remark here that monovalent counterions are not able to fully collapse the chains, while higher valent counterions lead to the collapse of the polyelectrolytes (e.g.,
Figure 1d).
Figure 1.
Sideview snapshots of equilibrium configurations for chains of length (a) N = 70; (b) N = 400 with monovalent counterions; and (c) N = 70 and (d) N = 400 with tetravalent counterions. The polymer monomers and coions are represented in white, the gray is for solvent and wall particles, the red color represents monovalent counterions, and the bluecolor is used for monovalent or tetravalent counterions. The radius of the straight cylinder is R = 7.74. A schematic of the applied electric field components is shown under the graphs. The longitudinal field is orientated along the cylinder axis, while the transversal field is perpendicular to the cylinder axis.
Figure 1.
Sideview snapshots of equilibrium configurations for chains of length (a) N = 70; (b) N = 400 with monovalent counterions; and (c) N = 70 and (d) N = 400 with tetravalent counterions. The polymer monomers and coions are represented in white, the gray is for solvent and wall particles, the red color represents monovalent counterions, and the bluecolor is used for monovalent or tetravalent counterions. The radius of the straight cylinder is R = 7.74. A schematic of the applied electric field components is shown under the graphs. The longitudinal field is orientated along the cylinder axis, while the transversal field is perpendicular to the cylinder axis.
The time step in the simulations is 0.008σ. The applied electric field has two components: a longitudinal component along the cylinder axis and a transversal component perpendicular to the cylinder axis. In the present work, the longitudinal field is set to E‖ = 0.81kBT/(eσ). The transversal component E⊥ may have one of the values 0, 0.81, 1.62, 2.43, 3.24, or 4.86 kBT/(eσ). The system is periodic along the cylinder axis. For convenience, we drop the units in the rest of the paper and keep in mind that they are given in LJ units.
3. Results and Discussion
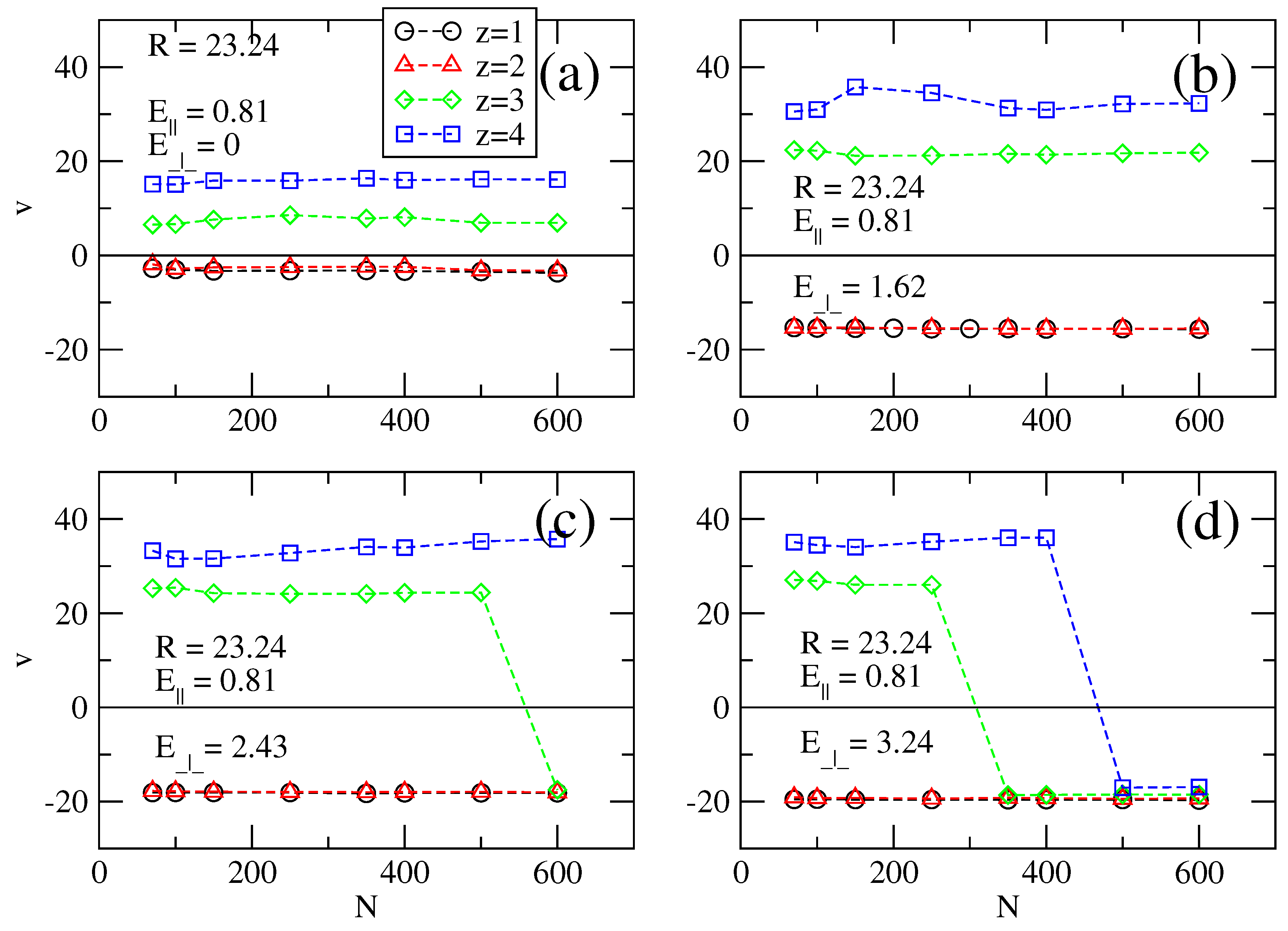
The electrophoretic velocities
v of polyelectrolytes as a function of chain length
N for various transversal field values
E⊥ (at constant
E‖) and valency
z of the counterions are presented in
Figure 2,
Figure 3,
Figure 4 and
Figure 5. The velocities are obtained from measurements of the center of mass motion in systems with periodic boundary conditions along the longitudinal axis of the straight cylinder. The graphs are grouped by the same cylinder radius
R. The electrophoretic velocities may be positive or negative, depending on the effective charge of the chains. They depend strongly on chain length for the smallest cylinder of radius
R = 7.74 (
Figure 2), at
z > 2 and small
E⊥, and are independent on
N for
z ≤ 2 for any value of the applied field or cylinder radius. What is important for us, and what we focus on in the present work is the critical chain length
N* of the polyelectrolyte when electrophoretic velocities change sign. Particularly, in the case of trivalent or tetravalent counterions. The separation principle works by splitting a group of charged chains with lengths up to
N in two subgroups: one with lengths
N <
N* and another one formed by chains with lengths
N >
N*. Chains with length
N <
N* move in, say, positive direction along the cylinder axis, while chains with
N >
N* move in the opposite direction. The critical length is controlled by
E⊥ and is a complex function of
R and
z. For convenience, the critical chain lengths
N* for trivalent and tetravalent counterions, at different radii
R of the cylinder are read from
Figure 2,
Figure 3,
Figure 4 and
Figure 5 and summarized in
Table 1.
Table 1.
Table 1. Minimum critical chain length N* of the polyelectrolyte required for charge inversion, for various radii R of the cylinder, valency z of the multivalent counterions and transversal field values E⊥. In all cases, E‖ = 0.81.
Table 1.
Table 1. Minimum critical chain length N* of the polyelectrolyte required for charge inversion, for various radii R of the cylinder, valency z of the multivalent counterions and transversal field values E⊥. In all cases, E‖ = 0.81.
| R | z | E⊥ |
|---|
| 0 | 1.62 | 2.43 | 3.24 |
|---|
| 7.74 | 3 | 100 | 100 | 100 | 100 |
| 4 | 600 | 450 | 100 | 50 |
| 11.62 | β | 600 | 250 | 150 | 50 |
| 4 | 600 | 600 | 600 | 400 |
| 15.5 | 3 | 600 | 450 | 300 | 100 |
| 4 | 600 | 600 | 600 | 400 |
| 23.24 | 3 | 600 | 600 | 500 | 250 |
| 4 | 600 | 600 | 600 | 400 |
Figure 2.
Electrophoretic velocities v as a function of chain length N for (a–d) transversal fields E⊥ = 0, 1.62, 2.43 and 3.24, respectively. The radius of the cylinder is R = 7.74 and the longitudinal field is E‖ = 0.81. For clarity of definition, in Figure (b,c), we show the minimum critical chain length N*.
Figure 2.
Electrophoretic velocities v as a function of chain length N for (a–d) transversal fields E⊥ = 0, 1.62, 2.43 and 3.24, respectively. The radius of the cylinder is R = 7.74 and the longitudinal field is E‖ = 0.81. For clarity of definition, in Figure (b,c), we show the minimum critical chain length N*.
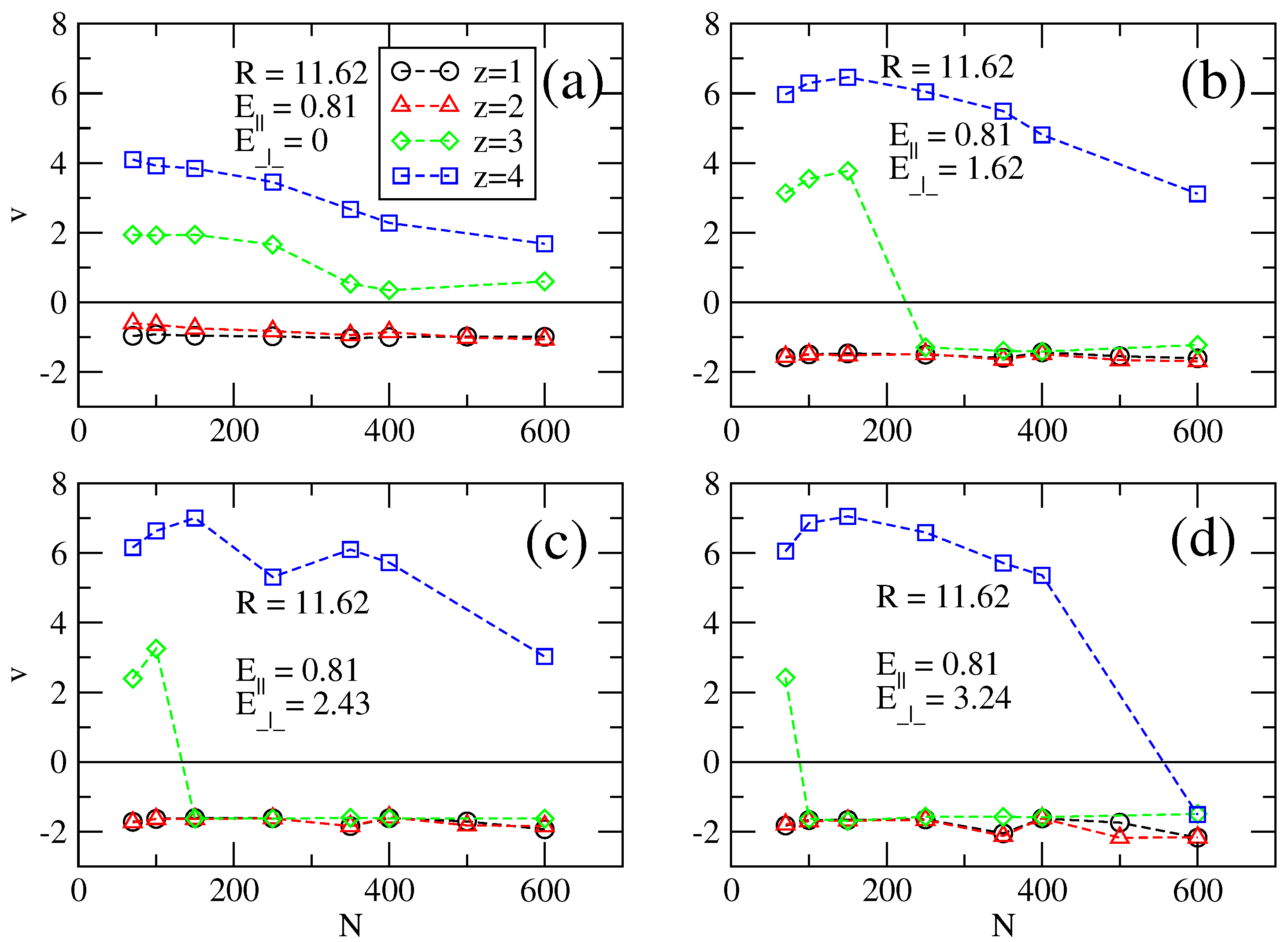
Figure 3.
Electrophoretic velocities v = v(N) for cylinder of radius R = 11.62 for (a–d) transversal fields of E⊥ = 0, 1.62, 2.43 and 3.24, respectively. The longitudinal field is constant, E‖ = 0.81.
Figure 3.
Electrophoretic velocities v = v(N) for cylinder of radius R = 11.62 for (a–d) transversal fields of E⊥ = 0, 1.62, 2.43 and 3.24, respectively. The longitudinal field is constant, E‖ = 0.81.
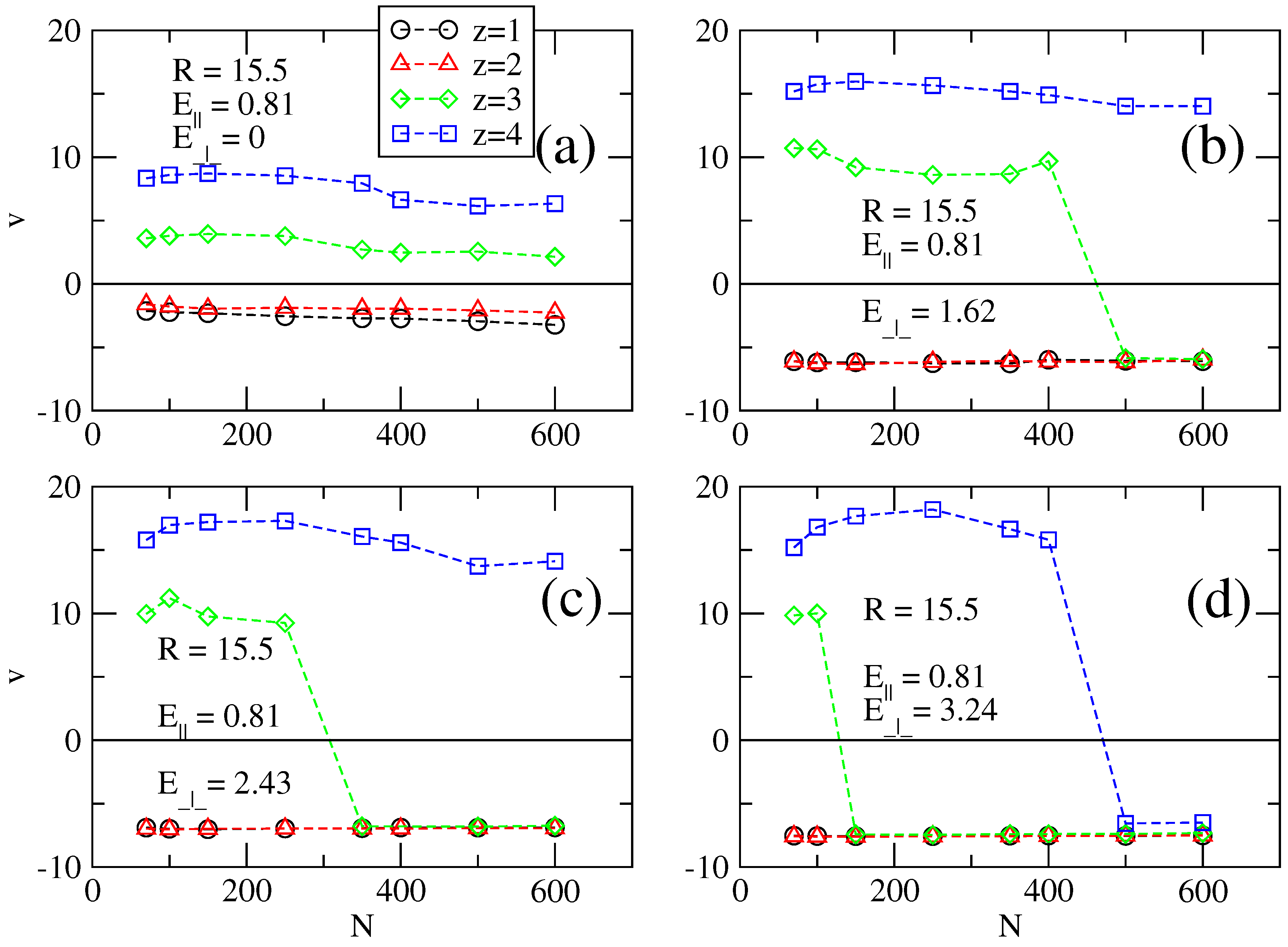
Figure 4.
Graphs similar to
Figure 2 and
Figure 3 for cylinder radius of
R = 15.5.
Figure 4.
Graphs similar to
Figure 2 and
Figure 3 for cylinder radius of
R = 15.5.
The sign change of the electrophoretic velocities is due to overcharging. In this case, without a transversal field, a chain attracts and retains counterions around it, such that the overall electric charge of the ensemble is positive. When the transversal field is switched on, some of these counterions are unbound if the transversal field is strong, which leads to a reduced effective charge. Naturally, the loss of counterions may also happen indirectly because of the action of a longitudinal field alone, without a transversal field.
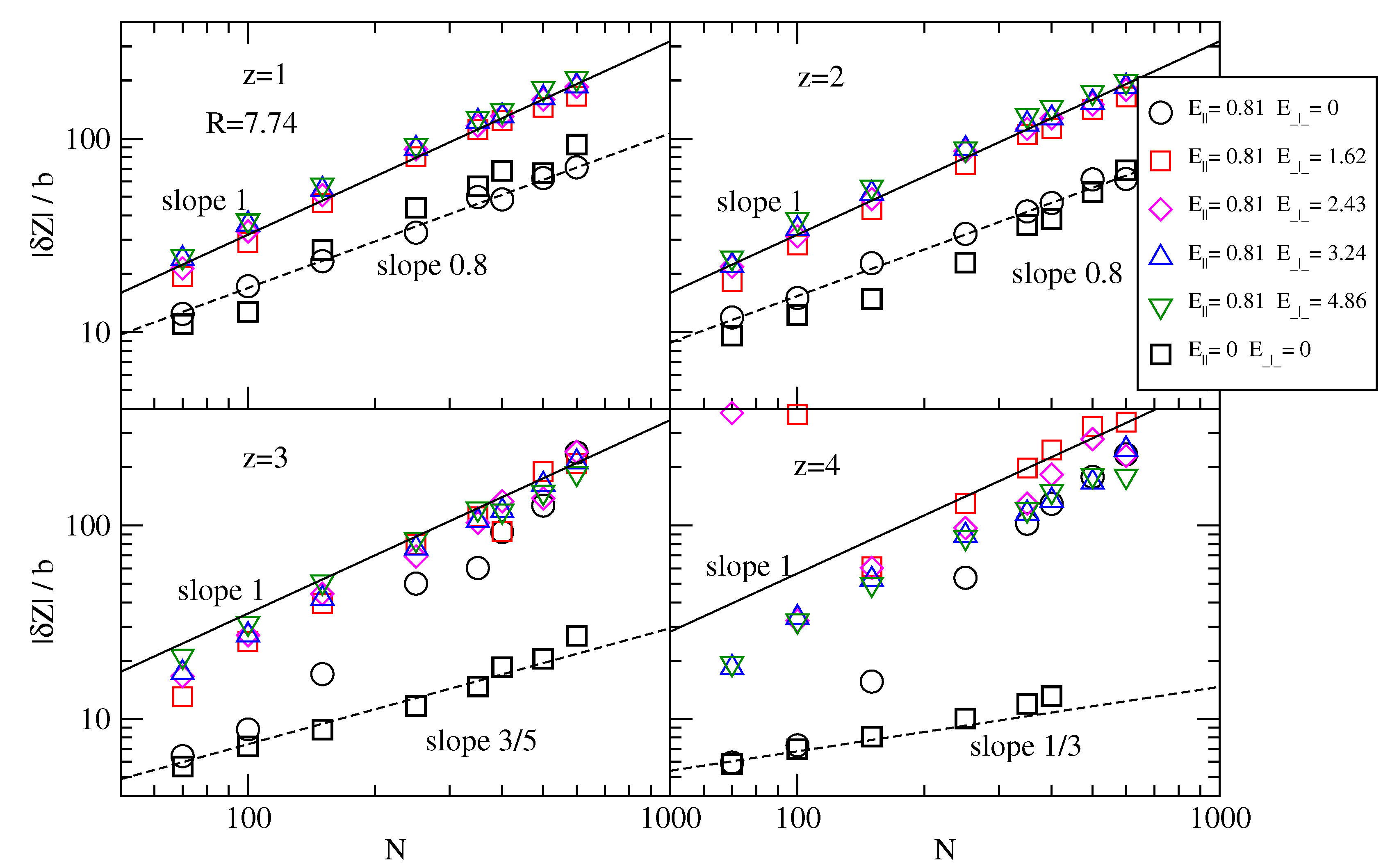
For the smallest cylinder of radius
R = 7.74, we show in
Figure 6 the chain extension |
δZ| = |
min(
zi) −
max(
zj)| along the cylinder axis, where
zi and
zj are any of the
z-coordinates of polymer monomers. Clearly, the static conformations of the chains (at
E = 0), in the presence of monovalent or divalent counterions indicates |
δZ|/
b ∼
N0.8, which is close to the scaling of chain size in non-confinement. On the other hand, chains are fully collapsed by tetravalent counterions, which is indicated by the scaling |
δZ|/
b ∼
N1/3 . If a driving field is applied,
E‖ = 0.81, these collapsed chains unfold, even if no transversal field is applied yet, which may be a hydrodynamic effect specific to strong confinements. We note that longer chains are easier to unfold than shorter chains.
Figure 6.
Axial extension |δZ| = |min(zi) − max(zi)| (i = 1, N) along the cylinder axis normalized by average bond length b as a function of chain length N, in a cylinder of radius R = 7.74, for multivalent counterions of valency z = 1, 2, 3, 4, transversal field values E⊥ = 0, 1.62, 2.43, 3.24, 4.86 and longitudinal field E‖ = 0.81. The static case of zeroapplied field, E⊥ = 0 and E‖ = 0, is also shown.
Figure 6.
Axial extension |δZ| = |min(zi) − max(zi)| (i = 1, N) along the cylinder axis normalized by average bond length b as a function of chain length N, in a cylinder of radius R = 7.74, for multivalent counterions of valency z = 1, 2, 3, 4, transversal field values E⊥ = 0, 1.62, 2.43, 3.24, 4.86 and longitudinal field E‖ = 0.81. The static case of zeroapplied field, E⊥ = 0 and E‖ = 0, is also shown.
Before we comment on the conformations of the chains at
E⊥ ≠ 0, we remark here on chain extensions in the largest cylinder,
R = 23.24. These data are shown in
Figure 7. At zero field (
E = 0) we have the known excluded volume result |
δZ|/
b ∼
N3/5 for chains in non-confinement at
z = 1, and full chain collapse for
z ≥ 2, which means that |
δZ|/
b ∼
N1/3. A small driving field (
E‖ = 0.81) leads to chain stretching for mono- and divalent counterions, but the availability of space leads to internal segmental reorientations such that previously collapsed chains by tri- and tetravalent counterions cannot be unfolded solely by
E‖ (
Figure 7, lower panels).
Figure 7.
Graph similar to
Figure 6 of axial extension normalized by the bond length, |
δZ|/
b, as a function of chain length
N, for the largest cylinder of radius
R = 23.24.
Figure 7.
Graph similar to
Figure 6 of axial extension normalized by the bond length, |
δZ|/
b, as a function of chain length
N, for the largest cylinder of radius
R = 23.24.
We discuss now the effect of the transversal field
E⊥ ≠ 0 on chain extension. In the smallest channel,
R = 7.74, at
z = 1 and
z = 2, the chain extension follows the scaling |
δZ|/
b ∼
N, independent of the exact value of
E⊥ (
Figure 6). This means it is a strong confinement. As we shall see below, the effective charge also follows the scaling
Qeff ∼
N, and it not surprising that the electrophoretic velocities in buffers containing monovalent or divalent counterions are independent of chain length. On the other hand, in the largest channel at
R = 23.24 (
Figure 7), the chain extension varies as |
δZ|/
b ∼
N0.7. The effective charge still remains proportional to
N, and it is not immediately apparent why the electrophoretic velocities are independent on
N (at
z = 1,
z = 2). If we consider that chain extension is an indicator of hydrodynamic friction, it follows that hydrodynamic friction in these cases is a small perturbation and the effective charge of the chain dictates in fact the electrophoretic velocity.
In the largest channel, for
z > 2,
δZ decreases with increasing
E⊥. It can be argued that stronger transversal fields lead to higher axial extension of chains because transversal fields remove condensed counterions, which in turn causes chain stretching due to Coulomb repulsive self interactions among the charges on the polymer backbone. Although appealing, this argument is however not supported in large channels (compare for example |
δZ| for
N = 600 and
z = 4, at
E⊥ = 1.62 with same chain at
E⊥ = 4.86 in
Figure 7). The reason is that the Coulomb self repulsive interactions are not strong enough to prevent hydrodynamic effects, such as chain folding. This works by noting that fluid resistance is highest at the tip of the polymer chain and weaker towards the tail. The redistribution of tension along the polymer bonds is simply realized by chain thickening in the direction of motion which translates in a reduced size of the chain. The discussion must be however extended with care to cases where the transversal field succeeds in removing condensed counterions and have the direction of motion of the chains reversed.
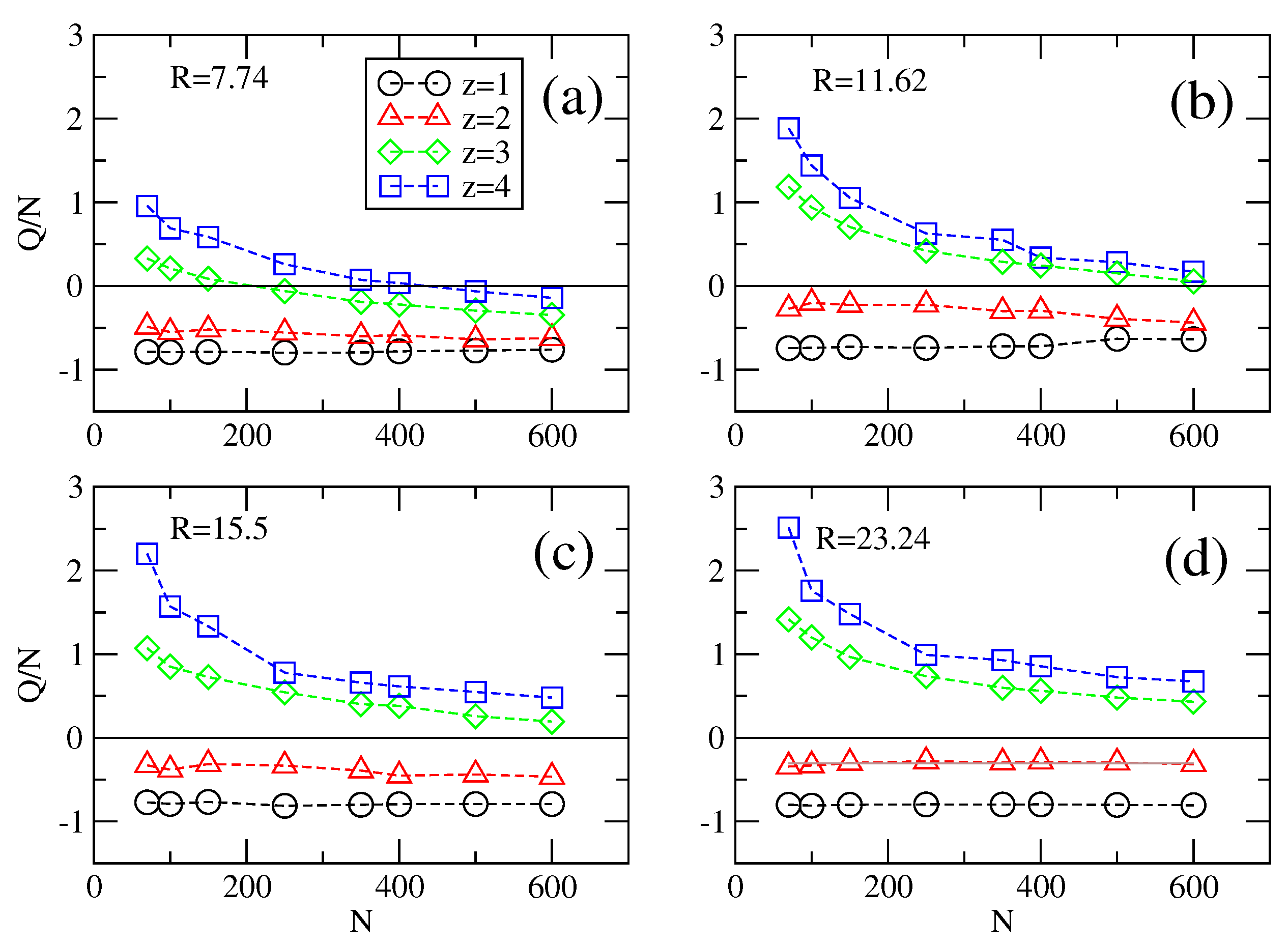
We define next the effective charge of the chain Qeff as the number of condensed counterions on the chain which are found within a set distance from individual polymer monomers. In practice, the effective charge is simply obtained by summing up all charges present within a set distance of about two bond lengths around individual polymer monomers. These numbers are time averages over series of conformations adopted by polyelectrolytes.
The effective charge is shown in three representative cases: (1) the static case when longitudinal and transversal fields are zero,
E‖ = 0 and
E⊥ = 0, respectively; (2) the longitudinal field case,
E‖ ≠ 0 and
E⊥ = 0; and (3) a normal operating case when both fields are not zero,
E‖ ≠ 0 and
E⊥ ≠ 0. In the static case (
Figure 8), both for monovalent and for divalent counterions the effective charge of the polymer scales linearly with
N, irrespective of cylinder radius
R. Here we note that the effective charge at
z = 1 is smaller than the effective charge at
z = 2, for any
R. It means that divalent counterions are better at neutralizing the charge of the chains than monovalent counterions. The effective charge
Q switches over to the positive domain for
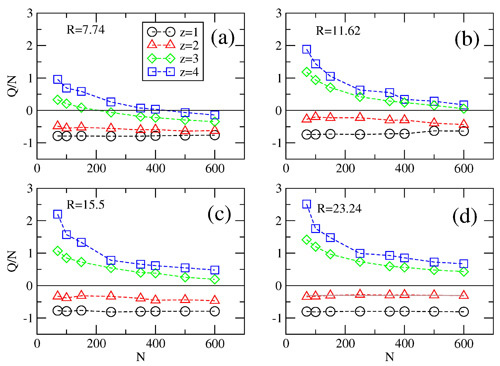
z > 2, first for short chains in strong confinement and then all chain lengths at
R > 7.74. Considering that short chains are overcharged in strong confinements (
Figure 8a, it can be inferred from the simulation data that multivalent counterions of
z > 2 may approach and condense easier on shorter chains than on longer chains. This explains the decrease of
Q with
N seen also in channels of larger radii,
R > 7.74. These observations on the variation of
Q with
z are supported by our theoretical model. In particular, numerical calculation based on Equation (5) show that
Q is independent of
N and
R for mono and divalent counterions.
Figure 8.
Effective charge Q per chain length N at zero field (E‖ = 0 and E⊥ = 0) as a function of the length N of the polyelectrolyte for (a–d) different radii R = 7.74, 11.62, 15.5 and 23.24 of the cylinder and valency z of the multivalent counterions. In the presence of tri-, or tetra-valent counterions the effective charge charges sign at some critical chain length N*, which depends on cylinder radius and valency of the counterions.
Figure 8.
Effective charge Q per chain length N at zero field (E‖ = 0 and E⊥ = 0) as a function of the length N of the polyelectrolyte for (a–d) different radii R = 7.74, 11.62, 15.5 and 23.24 of the cylinder and valency z of the multivalent counterions. In the presence of tri-, or tetra-valent counterions the effective charge charges sign at some critical chain length N*, which depends on cylinder radius and valency of the counterions.
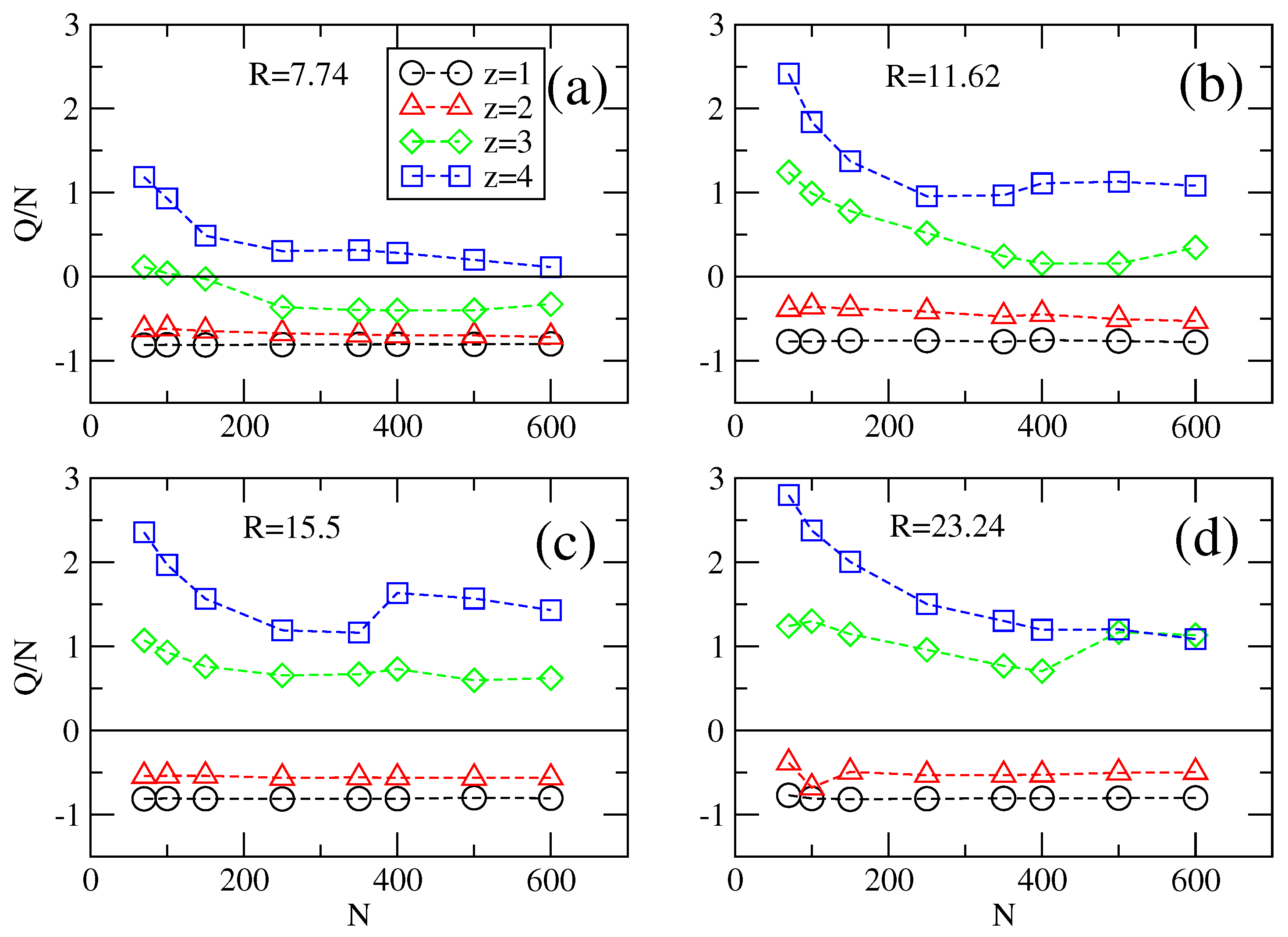
The effective charges in the longitudinal field case,
E‖ ≠ 0 and
E⊥ = 0, are shown in
Figure 9. For monovalent, divalent, trivalent or tetravalent counterions there are no remarkable effects to be noticed as compared with the static case in
Figure 8: monovalent and divalent counterions have effective charges proportional with
N and for
z > 2 we observe overcharging effects. It appears that the effective charge levels off for longer chains,
N > 400, which underlines once again the reason of why we need transversal fields. This case is shown in
Figure 10. Here, the chains remain hardly overcharged in strong confinements. The effective charge per chain length stays close to unity for about any value of
z. In weaker confinements, the chains are seen to loose bound counterions abruptly, depending on their length. This is one of the main results of our work and the critical chain length of the transition has been given in
Table 1. Another way to explain the transition is to recall from
Figure 8 that charged chains of length
N are packed with multivalent counterions in a ratio that depends on
N. The transversal field removes these counterions more easily from longer chains than from shorter chains. The transition is also abrupt, which we assume that it is a hydrodynamic effect. The physical separation boundary of the coions and counterions flows at high transversal fields is quite steep and in this region there are strong viscous shear forces. As soon as a charged polymer reaches this area, it is quickly unfolded by the shear forces, looses bound counterions and it is carried away by any of the two flows, irrespective of the initial direction of movement. This process is irreversible and it happens quite soon after the transversal field is switched on. Another noteworthy observation in
Figure 10 is that the ratio
Q/N appears to approach −2, below the limit of −1 which represents a chain with absolutely no bounded counterions. The surplus charge comes from the charge of nearby coions, which are, together with the polymer, pushed towards the walls of the confinement.
Figure 9.
Effective charge Q per chain length N for non-zero longitudinal field E‖ = 0.81 and zero transversal field E⊥ = 0, as a function of N for (a–d) different radii R = 7.74, 11.62, 15.5 and 23.24 of the cylinder, respectively.
Figure 9.
Effective charge Q per chain length N for non-zero longitudinal field E‖ = 0.81 and zero transversal field E⊥ = 0, as a function of N for (a–d) different radii R = 7.74, 11.62, 15.5 and 23.24 of the cylinder, respectively.
Figure 10.
Graphs similar to
Figure 8 and
Figure 9 of effective charge per chain length
Q/N as a function of
N for applied field components
E‖ = 0.81 and
E⊥ = 3.24 at (
a–
d) different radii
R = 7.74, 11.62, 15.5 and 23.24 of the cylinder and counterion valency
z. Strong
E⊥ leads to an almost step function behavior of
Q =
Q(
N).
Figure 10.
Graphs similar to
Figure 8 and
Figure 9 of effective charge per chain length
Q/N as a function of
N for applied field components
E‖ = 0.81 and
E⊥ = 3.24 at (
a–
d) different radii
R = 7.74, 11.62, 15.5 and 23.24 of the cylinder and counterion valency
z. Strong
E⊥ leads to an almost step function behavior of
Q =
Q(
N).
4. Theory
We consider in the following the effective charge of the chains and the minimum critical chain length for charge inversion. The statistical model of a polyelectrolyte in an electric field is considered first for a buffer solution containing monovalent counterions and coions. The chain model is a three states model where each polymer monomer can be (1) in a state where a counterion is bound to it and the dipole thus formed is orientated along the direction of the applied field; or (2) a state where the dipole formed with the bound counterion is orientated in the opposite direction to the applied field; or (3) a state where there is no counterion bound to it. Further, we note with the probability of a polymer monomer being in state (1), with the probability of state (2), and with the probability of state (3). The normalization condition requires that + + = 1. The electric dipoles are assumed to exist also in the absence of the externally applied electric fields.
The above probabilities depend on the interaction energies of the pairs of polymer monomers and condensed counterions or polymer charges without condensed counterions. These probabilities also depend on the concentration of the counterions in the buffer solution. We may assume that they are the product of a concentration dependent term and an interaction energy term.
The interaction energy of a dipole with an external field is − and we take the probability as a product of a concentration or counterion valency dependent term c1 and an exponential term of the form c1e+βpE⊥/C, where C is a normalization constant. The probability of the dipole being orientated in the direction opposite to the applied field is = c2e−βpE⊥/C. Similarly, the probability is taken as an exponential function that depends on the charge of a polymer monomer and the applied electric potential δV in the transversal direction, i.e., = c0eβeδV/C. We shall describe in the ulterior paragraphs how the constants c1, c2, and c0 are calculated, based on simulation data.
Generally, for a three states model without charge-charge, charge-dipole, dipole-dipole interactions, and without confinement effects (
i.e., all chain conformations are equally probable), the effective charge
is given by a classical result of statistical mechanics for
N non-interacting particles [
27]:
where
n′′ is the number of polymer monomers without bound counterions, and
N =
j +
n +
n′′. The number
N!/(
j!
n!
n′′!) represents the number of distinct states of
N polymer monomers where
j polymer monomers form dipoles aligned in the direction of the field,
n polymer monomers form dipoles orientated opposite to the field, and
n′′ polymer monomers are without condensed counterions. The Boltzmann distribution of the system, or
jnn′′ represents the probability of a single particular configuration of
j parallel dipoles,
n antiparallel dipoles, and
n′′ polymer monomers without condensed counterions to occur. The fundamental assumption is that
,
and
are statistically independent probabilities.
To account for charge-charge, charge-dipole and dipole-dipole interactions we remain in the framework of the three states model and consider that the total energy of the polymer chain is a separable sum of field-dipole, field-charge, dipole-dipole, and charge-dipole energies. The new dipole moment
p′ includes the induced polarization because of the other dipoles. Similarly, the new local potential
δV′ at the place of a single charge includes the potential
δVs of all the other charges. In the canonical ensemble, the Boltzmann distribution of the system of
j parallel with the field dipoles,
n antiparallel dipoles, and
n′′ polymer charges without condensed counterions is thus:
where
ϕuv is the full interaction potential between any two polymer monomers. The new probabilities are
′ =
c1e+βp'E⊥ /
C′,
′ =
c2e−βp'E⊥ /
C′, and
′ =
c0eqδV /
C′, where
p′ =
p +
α. The relation
q ≈
e[1 +
(
δVs/
δV)] is used to account for the change in the local potential. The
α,
γ and
q are parameters of the model. In Equation (4), the integration over the polymer coordinates signifies the fact that the Boltzmann distribution of the system is a sum of individual probabilities taken for any possible values of polymer monomers coordinates. The integration space of the
N-dimensional integral is a cylinder of set diameter and of infinite length. The double sum in Equation (4) can be reduced if we consider only pairwise interactions [
28]. In this case, there are
j(
j − 1)/2 pairwise interactions of dipoles aligned parallel with the applied field,
n(
n − 1)/2 pairwise interactions of dipoles antiparallel with the applied field, and
jn pairwise interactions of parallel-antiparallel dipoles. The dipole-dipole interaction terms vary with the distance
r between dipoles as 1/
r3. For the polymer charge- dipole interaction we note that there are
jn′′ pairwise interactions of charge-parallel dipoles and
nn′′ pairwise interactions of charge-antiparallel dipoles. The corresponding interaction energy shows a 1/
r2 distance dependency [
29]. The charge-charge interaction is given by Coulomb’s law. Altogether, in the pairwise approximation, the effective charge is given by the relation:
where
n′′ =
N −
j −
n, and
with
E1(
j,
n) given by the relation:
where
r12 is the distance between two dipoles
p,
dV1 =
dr1dΩ
1,
dV2 =
dr2dΩ
2, with
r1 and
r2 radial distances of the interacting dipoles,
dΩ
1 ,
dΩ
2 are solid angles, respectively,
β = 1/
kBT, and
is a constant taken unity. The integration volume
V of the confinement is
πR2L, where
R is the cylinder diameter and the length
L of the integration volume is taken arbitrarily large such that the integrals converge. We note here that the correction term
E1 does not depend explicitly on the transversal or longitudinal field, but through the variation of the dipole moment itself with the field. This term adjusts the effective charge as a function of
R and is responsible for confinement effects. The influence of transversal field on the number of dipoles that are fully aligned in the direction of the field or in the direction opposite to the field is contained in the probabilities
′ and
′, respectively.
Now, for the case of multivalent counterions of valency
z > 1, we start with two extreme cases. First, let us hypothetically consider that each single polymer monomer forms a pair with a condensed
z-valent counterion. Then, the maximum effective charge of the chain is (
z − 1)
Ne. For a buffer solution containing tetravalent counterions, for example, the maximum possible overcharge
Qeff /
N is 300%. This is more than what is observed in the simulations (
Figure 8). The second limiting case that we consider is when each multivalent counterion attracts around it exactly
z polymer monomers. Thus, a
N -mer chain can have at most
N/z multivalent condensed counterions. And thus the chain cannot become positively charged, irrespective of the value of
z. In this approximation, a number of
z polymer monomers and a
z-valent counterion may be virtually grouped together such that it forms an ensemble of dipole moment
z (
Figure 11a). The dipoles may be orientated in the field direction, or in the opposite direction, just as it was the case of monovalent counterions.
From the two limiting cases above we may assume that in fact some polymer monomers may have in their neighborhood an arbitrary number of multivalent counterions (
Figure 11b). In this sense, we refer to these monomers as shared monomers. The physical origin of monomer sharing may be thought of as a conformational effect, whereas the polymer is in a folded or collapsed state due to attractive dipole-dipole interactions. Liu
et al. [
30] showed that bridging between polymer segments leads to an enhanced shrinkage of polymer size. For two
z-valent counterions sharing at most
z polymer monomers, it follows that the maximum overcharge of a chain is at most 100% for any value of
z. In other words, we restrict ourselves to cases where any polymer monomer may belong to at most two multivalent counterions.
Figure 11.
Illustrations of (a) associated dipoles formed between condensed multivalent counterions and polymer monomers and (b) multivalent counterions sharing polymer monomers. The latter case allows for overcharged chains. The effective charge increases with the number of shared polymer monomers and is a nonlinear function of chain length.
Figure 11.
Illustrations of (a) associated dipoles formed between condensed multivalent counterions and polymer monomers and (b) multivalent counterions sharing polymer monomers. The latter case allows for overcharged chains. The effective charge increases with the number of shared polymer monomers and is a nonlinear function of chain length.
From the simulations we know the effective charge of the chains. At zero field
= 0 these are (
Figure 8):
Qeff /
N = −0.8 for monovalent counterions and
Qeff /
N = −0.3 for divalent counterions, both independent of cylinder radius
R. For trivalent and tetravalent counterions
Qeff /
N is not a constant, but depends on
R and slowly decreases to a plateau value as
N increases. In the limit of large
R and long chain lengths (
Figure 8d), we take
Qeff /
N = +0.43 for trivalent counterions, and
Qeff /
N = +0.67 for tetravalent counterions. We use these values from simulations as reference points in setting the theoretical zero-field probabilities
′,
′, and
′.
This is done by a Monte Carlo algorithm as follows. We start with an empty array of arbitrary length N, which is the length of the polymer chain. We use then series of random values 0 or 1 or 2 to simulate probable states of the N -mer polymer chain. The construction of the random series obeys the rule that a polymer monomer may belong simultaneously to a maximum of two counterions. The values of 1 in the random series are assigned to polymer monomers with condensed counterions that form a dipole orientated in the field direction, the value of 2 is assigned to polymer monomers that form dipoles orientated in opposite direction to the field, and any value 0 in the series is assigned to polymer monomers without condensed counterions. The equilibrium effective charge per chain length is then obtained by the formula: −1 + z(C1 + C2), where C1 is the probability of a polymer monomer being part of a dipole aligned in the field direction and C2 is the probability of a polymer monomer being part of dipole aligned in the opposite direction to the field. The target effective charge at equilibrium is achieved by adjusting in the series, the weight given to polymer monomers without condensed counterions. The end result gives ′ = ′ = 0.1 and ′ = 0.8 for monovalent counterions, ′ = ′ = ′ = 0.3 for divalent counterions, ′ = ′ = 0.36 and ′ = 0.28 for trivalent counterions, and ′ = ′ = 0.41 and ′ = 0.18 for tetravalent counterions. We remark that at least for monovalent and divalent counterions these results were expected.
For buffers containing only monovalent counterions we have Qeff < 0 and we take the number of shared monomers zero. In the case of divalent counterions, we take again the number of shared monomers zero.
For multivalent counterions of valency
z > 2, the equivalent formulation of Equation (5) is obtained by working with a normalized chain length 2
N/
z, which comes from the fact that each multivalent counterion is allowed to bind to
z polymer monomers and each polymer monomer can be shared by two counterions:
where
N′ = 2
N/
z, and
n′′ =
N′ −
j −
n.
The effective charge, Equation (8), allows us to obtain the minimum critical chain length
N* for charge inversion. We recall that the removal of counterions, or the unbinding process is an essential step to charge inversion and we are interested in solutions of
N* =
N*(
E⊥). To do this, we start with a given
N =
N* and solve for
E⊥ the nonlinear implicit equation:
The theoretical plot of
N* =
N*(
E⊥) from Equation (9) together with data points from simulations is presented in
Figure 12. The values
N* =
N*(
E⊥) from Equation (9) are obtained by using
′ = 0.36
e+βp′E⊥/
C′,
′ = 0.36
e−βp′E⊥/
C′, and
′ = 0.28
e+βp′E⊥/
C′, where
p′ = 0.5 ×10
−3(1 + 16
) and
q = −9 × 10
−3 for trivalent counterions, and
′ = 0.41
e+βp′E⊥/
C′,
′ = 0.41
e−βp′E⊥/
C′,
′ = 0.18
e+qβδV /
C′, where
p′ = 0.5 × 10
−3(1 + 20
) and
q = −8 × 10
−3 for tetravalent counterions. The normalization constant
in Equation (7) is taken unity, and |
δV| =
E⊥.
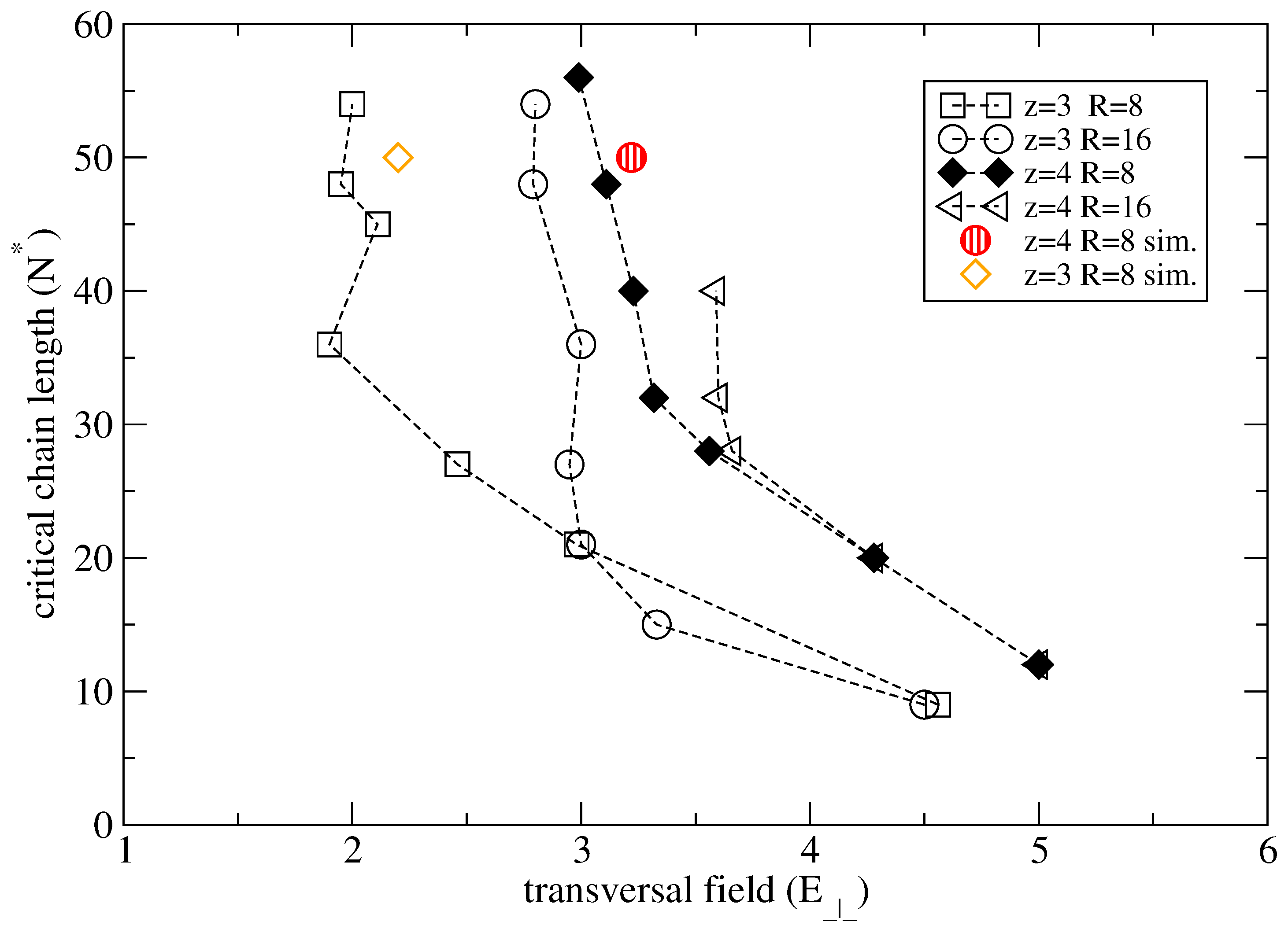
The range of comparison is restricted to short chain lengths and strong confinements. For clarity we note that some curves may extend vertically much more than shown. We note that the minimum critical chain length shows a decrease with increasing transversal field. This observation is in good agreement with data shown in
Table 1. In practical terms, this means that higher fields are required to remove multivalent counterions from short chains than from long chains. As shown in
Figure 12,
N* increases in weaker confinements (or larger cylinder radii). This further suggests that separation of chains of any length is best achieved under confinement conditions and using externally applied fields that gives the maximum variation of
N* with
E⊥.
Figure 12.
Theoretical and simulation data for the minimum critical chain length N* for charge inversion as a function of the transversal field E⊥ in strong confinements.
Figure 12.
Theoretical and simulation data for the minimum critical chain length N* for charge inversion as a function of the transversal field E⊥ in strong confinements.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







