Impact of the Enhanced Permeability and Retention (EPR) Effect and Cathepsins Levels on the Activity of Polymer-Drug Conjugates



Abstract
:1. Introduction


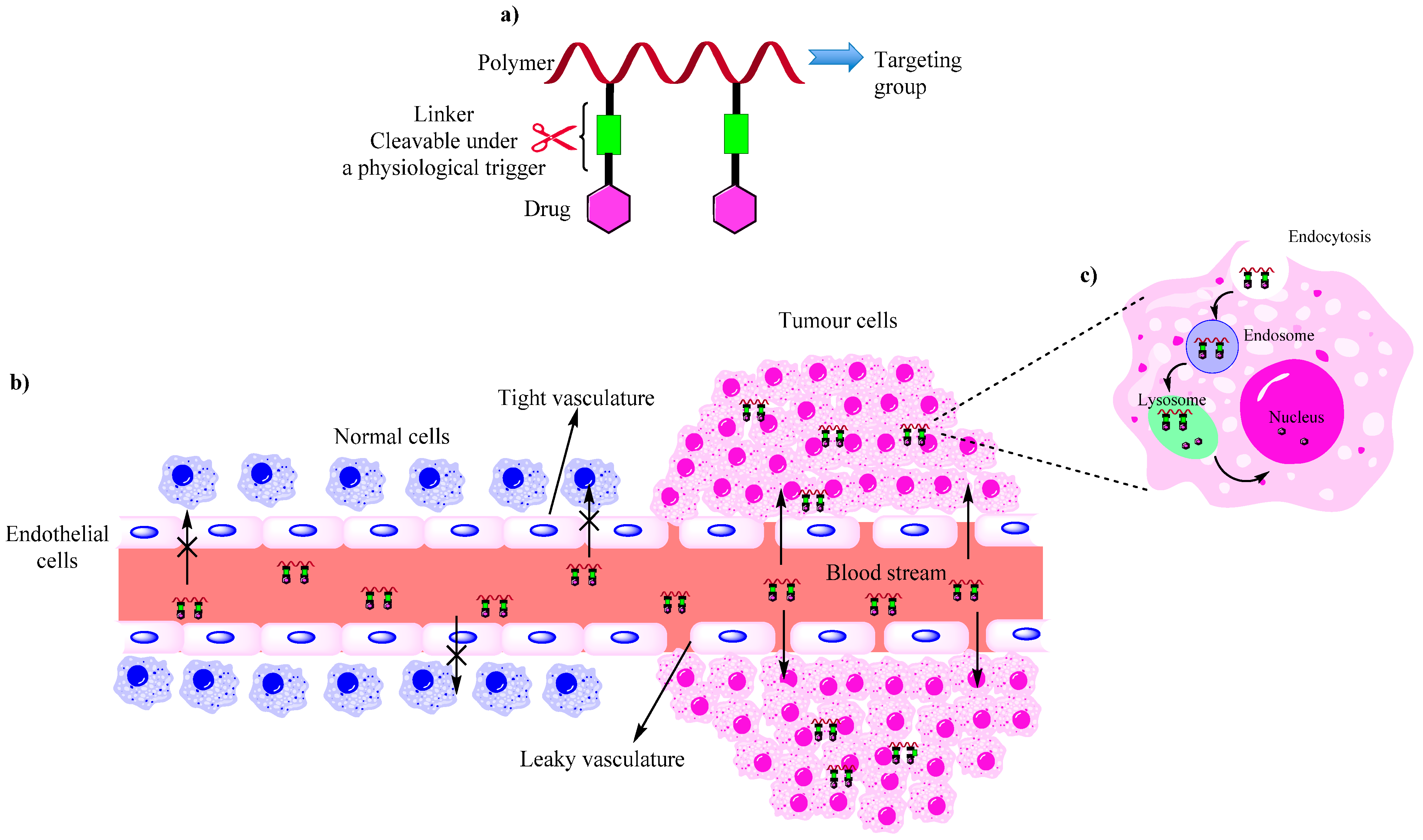{kind=link}
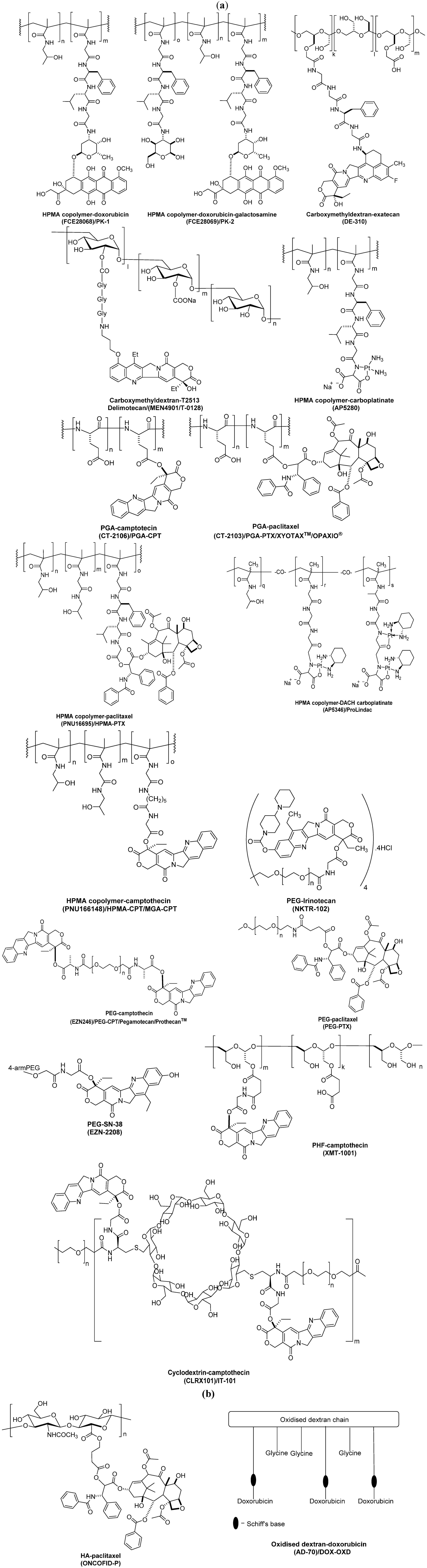{kind=link}
{kind=link}
{kind=link}
| Release Trigger | Code/Product Name | Composition | Linker/Spacer | Status | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Enzymatic | |||||||||
| Cathepsins | FCE28068/PK1 | HPMA copolymer-doxorubicin | Amide/Peptide d | Phase II | [4,20] | ||||
| Cathepsins | FCE28069/PK2 | HPMA copolymer-doxorubicin-galctosamine | Amide/Peptide d | Phase I | [21] | ||||
| Cathepsins | DE-310 | Carboxymethyldextran-exatecan | Amide/Peptide d | Phase I | [22] | ||||
| Cathepsins | Delimotecan (MEN 4901/T-0128) | Carboxymethyldextran-T2513 | Triglycine | Phase I | [23] | ||||
| Esterases/Acid hydrolysis a Cathepins b | CT-2106/PGA-CPT | PGA-camptothecin | Ester | Phase I | [24] | ||||
| Esterases/Acid hydrolysis a Cathepins b | CT-2103/PGA-PTX XYOTAX™/OPAXIO® | PGA-paclitaxel | Ester | Phase III | [5,6,19,25,26,27,28,29,30,31,32,33,34] | ||||
| Acid hydrolysis/Cathepsins c | AP5280 | HPMA copolymer-carboplatinate | Aminomalonate/Peptide d | Phase I/II | [35] | ||||
| Acid hydrolysis/Cathepsins c | AP5346/ProLindac® | HPMA copolymer-DACH oxiplatinate | Aminomalonate/Peptide e | Phase I | [36] | ||||
| Hydrolysis/Esterases | PNU166945/HPMA-PTX | HPMA copolymer-paclitaxel | Ester | Phase I discontinued | [37] | ||||
| Hydrolysis/Esterases | PNU166148//HPMA-CPT/MAG-CPT | HPMA copolymer-camptothecin | Ester | Phase I discontinued | [38,39,40] | ||||
| Hydrolysis/Esterases | EZN246/PEG-CPT/Pegamotecan/Prothecan™ | PEG-camptothecin | Ester | Phase II discontinued | [41,42,43] | ||||
| Hydrolysis/Esterases | PEG-PTX | PEG-paclitaxel | Ester | Phase I discontinued | [44] | ||||
| Hydrolysis/Esterases | EZN-2208/PEG-SN-38 | PEG-SN-38 | Glycinamidoester | Phase II | [45,46] | ||||
| Hydrolysis/Esterases | NKTR-102 | PEG-Irinotecan | Glycinamidoester | Phase II/III | [47,48,49,50] | ||||
| Hydrolysis/Esterases | NKTR-105 | PEG-Docetaxel | - | Phase I | [51] | ||||
| Hydrolysis/Esterases | XMT-1001 | PHF-camptothecin | Succinamidoester | Phase I | [52] | ||||
| Esterases | CRLX101/IT-101 | Cyclodextrin-camptothecin | Glycinamidoester | Phase II | [53] | ||||
| Non-enzymatic | |||||||||
| pH-sensitive | ONCOFID-PTM | HA-paclitaxel | Hydrazone | Phase I/II | [54] | ||||
| AD-70, DOX-OXD | Oxidised dextran-doxorubicin | Schiff’s base | Phase I discontinued | [55] | |||||

2. Experimental Section
2.1. Terminology
| Tumour Type (Terminology Used in This Article) | Tumour Type (Terminology Used in the Original Reference) |
|---|---|
| Breast | Breast, mammary gland, MCF-7, MDA-MB-231, BT 20 and DU4476-cell lines, Walker 256-murine model, MX-1, MAXF 449-human xenograft. |
| Colon and/or rectum | Colon, rectum, colorectal, anus, C26 NL-17, HT-29, LS174T-human xenograft. |
| Head/Neck and brain | Brain, glioblastoma, head/neck, thyroid, salivary gland, follicular, papillary, tongue, maxillary sinus, parotid gland, IMR-32, SK-N-SH, SK-N-DZ- human xenograft. |
| Lung | Non-small cell lung cancer, small cell lung cancer, bronchial, Meta-7-murine model, H522, COR L23-human xenograft. |
| Oesophagus, stomach and intestine | Oesophageal, cardioesophageal, stomach, gastroesophageal, gastrointestinal, intestine, small bowel, peritoneal carcinosis, OCUM-2MLN-human xenograft. |
| Ovary | Ovarian, Oca-1-murine model, A2780 cell line/human xenograft, SK-OV-3, OVCAR-3-human xenograft. |
| Pancreas | Pancreatic, PAXF 546-human xenograft. |
| Skin | Melanoma, basal cell, histosarcoma, B16F10, A431-murine model, MEXF 276-human xenograft. |
| Urinary | Bladder, urinary tract, urethal, urothelial, urachus. |
| Others: The term indicates the tumour types which were either studied in a very low sample size (n < 3) and/or those for which low responses were observed. | Adrenal, adenoid cystic, adenocarcinoma (unknown primary), bone (ewing sarcoma, osteosarcoma), cervix (uterine, leiomysarcoma uteri, ME180-human xenograft), fibrosarcoma (S-180, Meth A-murine model), gall bladder, kidney, leiomysarcoma, liver (cholangiocarcinoma, ampullary, bile duct, L1210-murine model, VX-2 carcinoma), lymphoma, mesothelioma, neuroendocrine, prostrate, sarcoma (unknown), soft tissue sarcoma, squamous cell sarcoma (unknown origin), solid tumours (unknown), unknown primary tumours, MAC 15A-murine model, MAC 26-murine model, RXF 486, RXF 1220, ME 180-human xenograft. |
2.2. Clinical Status of the PDCs
2.3. Cathepsin Level
2.4. EPR Effect
3. Results and Discussion
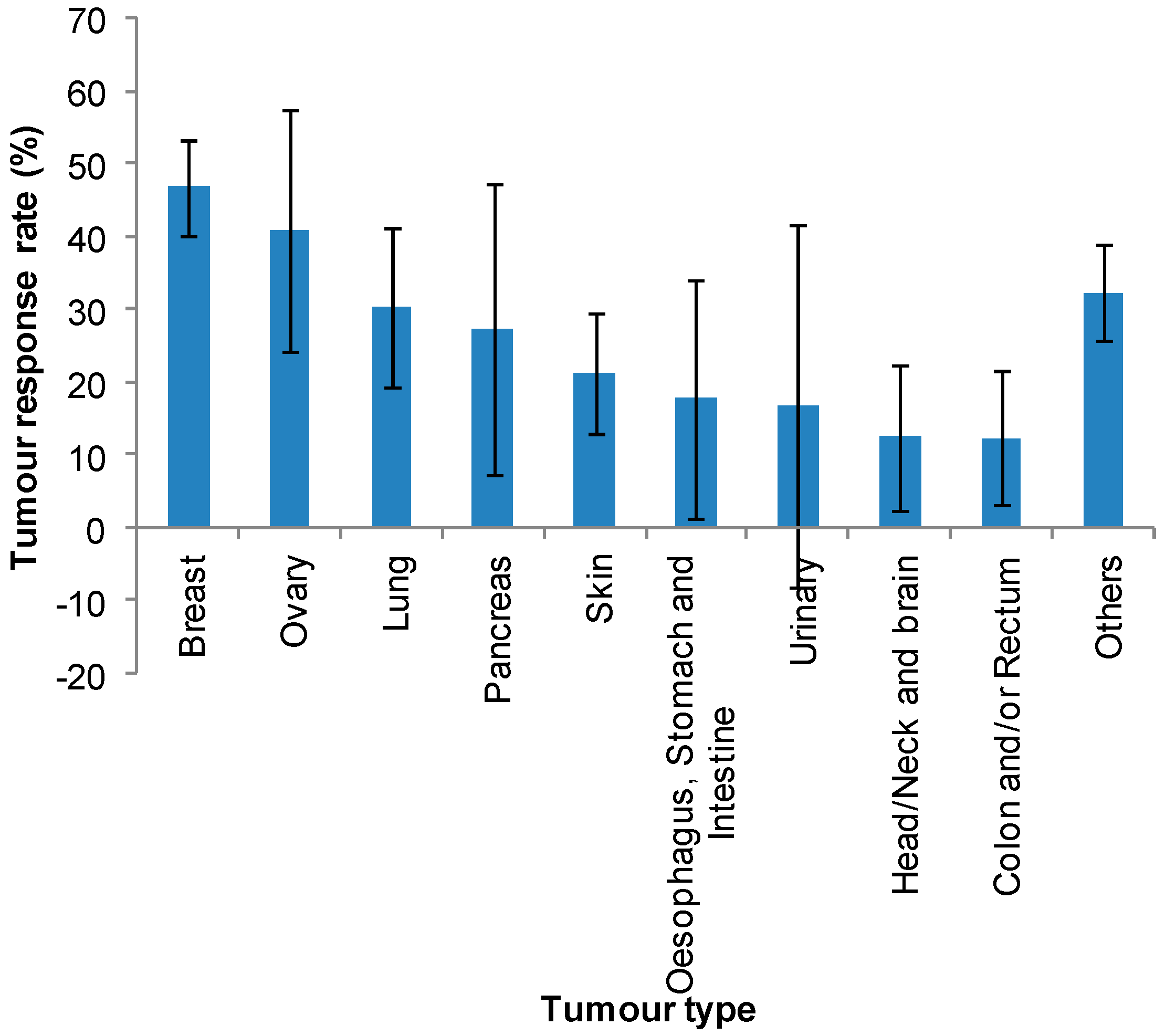
3.1. Effect of Tumour Type on Clinical Response
| Tumour Type | No. of Patients per Tumour Total a [Ph I/Ph II/Ph III] | Clinical Responses b Total (Ph I/Ph II/Ph III) | Tumour Response Rate c (%) Total [Ph I/Ph II/Ph III] | |||||
|---|---|---|---|---|---|---|---|---|
| No. of SD | No. of PR | No. of MR | No. of CR | No. of OS | No. of NR | |||
| HPMA copolymer-doxorubicin (PK1; FCE28068) [4,20] | ||||||||
| Lung | 31[(2/29(21) */-] | 8[-/8/-] | 5[2/3/-] | - | - | - | 10[0/10/-] | 57[100/52/-] |
| Breast | 20[3/17(14) */-] | 5[-/5/-] | 3[-/3/-] | 1[1/-/-] | - | - | 8[2/6] | 53[33/57/-] |
| Colon and/or rectum | 24[8/16/-] | - | - | 1[1/-/-] | - | - | 23[7/16/-] | 4[4/-/-] |
| HPMA copolymer-doxorubicin-galactosamine (PK2; FCE28069) [21] | ||||||||
| Liver | 25[25/-/-] | - | 2[2/-/-] | 1[1/-/-] | - | - | 22[22/-/-] | 12[12/-/-] |
| Colon and/or rectum | 6[6/-/-] | - | - | - | - | - | 6[6/-/-] | 0 |
| Carboxymethyldextran-exatecan; DE-310 [22] | ||||||||
| Adenocarcinoma (Unknown primary) | 2[2/-/-] | 1[1/-/-] | - | - | 1[1/-/-] | - | 0 | 100[100/-/-] |
| Pancreas | 3[3/-/-] | 2[2/-/-] | 1[1/-/-] | - | - | - | 0 | 100[100/-/-] |
| Urinary | 1[1/-/-] | 1[1/-/-] | - | - | - | - | 0 | 100[100/-/-] |
| Delimotecan; MEN 4901/T-0128 [23] | ||||||||
| Head/Neck and brain | 2[2/-/-] | - | 1[1/-/-] | - | - | - | 1[1/-/-] | 50[50/-/-] |
| Colon and/or rectum | 7[7/-/-] | - | 1[1/-/-] | - | - | - | 6[6/-/-] | 14[14/-/-] |
| Mesothelioma | 3[3/-/-] | - | - | - | - | - | 3[3/-/-] | 0 |
| Poly-l-glutamic acid-camptothecin; PGA-CPT; CT-2106 [24] | ||||||||
| Breast | 4[4/-/-] | 1[1/-/-] | - | - | - | - | 3[3/-/-] | 25[25/-/-] |
| Skin | 14[14/-/-] | 2[2/-/-] | - | - | - | - | 12[12/-/-] | 14[14/-/-] |
| Bone | 1[1/-/-] | - | - | - | - | - | 1[1/-/-] | 0 |
| Poly-l-glutamic acid-paclitaxel; PGA-PTX; CT2103; XYOTAX; OPAXIO® [5,6,19,24,25,26,27,28,29,30,31,32,33] | ||||||||
| Breast | 18[-/18/-] | 2[-/2/-] | 4[-/4/-] | 4[-/4/-] | - | - | 8[-/8/-] | 56[-/56/-] |
| Ovary | 99[-/99/-] | 32[-/32/-] | 10[-/10/-] | - | - | - | 57[-/57/-] | 42[-/42/-] |
| Mesothelioma | 3[3/-/-] | - | - | 1[1/-/-] | - | - | 2[2/-/-] | 33[33/-/-] |
| HPMA copolymer-carboplatin; HPMA-carboplatin; AP5280 [35] | ||||||||
| Lung | 4[4/-/-] | 2[2/-/-] | - | - | - | - | 2[2/-/-] | 50[50/-/-] |
| Ovary | 2[2/-/-] | 1[1/-/-] | - | - | - | - | 1[1/-/-] | 50[50/-/-] |
| Colon and/or rectum | 12[12/-/-] | 2[2/-/-] | - | - | - | - | 10[10/-/-] | 17[17/-/-] |
| HPMA copolymer-platinate; HPMA-Pt; AP5346 [36] | ||||||||
| Cervix | 1[1/-/-] | 1[1/-/-] | - | - | - | - | 0 | 100[100/-/-] |
| Oesophagus, stomach and intestine | 1[1/-/-] | 1[1/-/-] | - | - | - | - | 0 | 100[100/-/-] |
| Skin | 5[5/-/-] | 1[1/-/-] | 1[1/-/-] | - | - | - | 3[3/-/-] | 40[40/-/-] |
| Polyethylene-camptothecin; PEG-CPT; EZN246; Pegmaotecan; Prothecan™ [41,42,43] | ||||||||
| Bone | 1[1/-/-] | - | - | 1[1/-/-] | - | - | 0[0/-/-] | 100[100/-/-] |
| Oesophagus, stomach and intestine | 42[7/35/-] | 14[-/14/-] | 5[-/5/-] | 3[2/1/-] | - | - | 20[5/15/-] | 52[29/57/-] |
| Unknown primary | 5[5/-/-] | - | - | 1[1/-/-] | - | - | 4[4/-/-] | 20[20/-/-] |
| Multiarm-polyethylene-SN38; EZN-2208 [45,46] | ||||||||
| Urinary | 1[1/-/-] | 1[1/-/-] | - | - | - | - | 0[0/-/-] | 100[100/-/-] |
| Oesophagus, stomach and intestine | 3[3/-/-] | 2[2/-/-] | - | - | - | - | 1[1/-/-] | 67[67/-/-] |
| Breast | 3[3/-/-] | 2[2/-/-] | - | - | - | - | 1[1/-/-] | 67[67/-/-] |
| PHF-CPT; MER-1001; XMT-1001 [52] | ||||||||
| Skin | 2[2/-/-] | 1[1/-/-] | - | - | - | - | 1[1/-/-] | 50[50/-/-] |
| Lung | 7[7/-/-] | 3[3/-/-] | - | - | - | - | 4[4/-/-] | 43[43/-/-] |
| Solid tumours (Unspecified) | 8[8/-/-] | 3[3/-/-] | - | - | - | - | 5[5/-/-] | 38[38/-/-] |
| Cyclodextrin-camptothecin; CRLX101; IT-101 [53] | ||||||||
| Lung | 27[27/-/-] | 16[16/-/-] | - | - | - | - | 11[11/-/-] | 59[59/-/-] |
| Solid tumours * | 35[35/-/-] | 28[28/-/-] | - | - | - | - | 7[7/-/-] | 80[80/-/-] |
| HA-paclitaxel (ONCOFID-P™) [54] | ||||||||
| Urinary | 15[15/-/-] | - | - | - | 9[9/-/-] | - | 6[6/-/-] | 60[60/-/-] |
| Oxidized dextran-Dox; OXD-DOX (AD-70) [55] | ||||||||
| Colon and/or rectum | 6[6/-/-] | 1[1/-/-] | - | - | - | - | 5[5/-/-] | 17[17/-/-] |
| Oesophagus, stomach and intestine | 2[2/-/-] | - | - | - | - | - | 2[2/-/-] | 0 |
| Lung | 2[2/-/-] | - | - | - | - | - | 2[2/-/-] | 0 |
| HPMA copolymer-paclitaxel; HPMA-PTX; PNU166945 [37] | ||||||||
| Solid tumours 1 | 12[12/-/-] | 2[2/-/-] | 1[1/-/-] | - | - | - | 9[9/-/-] | 25[25/-/-] |
| HPMA copolymer-camptothecin; HPMA-CPT; PNU166148 [38,39,40] | ||||||||
| Solid tumours 2 | 40[40/-/-] | 5[5/-/-] | - | 1[1/-/-] | - | - | 34[34/-/-] | 15[15/-/-] |
| Polyethylene glycol-paclitaxel; PEG-PTX [44] | ||||||||
| Solid tumours 3 | 13 | NA | NC | NC | ||||
| Multi-arm-polyethylene glycol-pacliaxtel; PEG-PTX; NKTR-102 [47,48,49] | ||||||||
| Solid tumours 4 | 125[32/68/-] | 28[-/28/-] | 24[7/17/-] | 6[6/-/-] | 2[2/-/-] | - | 65[8/23/-] | 48[47/66/-] |
| Multi-arm-polyethylene glycol-docetaxel; NKTR-105 [51] | ||||||||
| NA | 17 | NA | NA | NA | NA | NA | NC | NC |

| Tumour Type | Tumour response rate (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PDC | Breast | Ovary | Lung | Pancreas | Skin | Oesophagus, Stomach and Intestine | Urinary | Head/Neck and Brain | Colon and/or Rectum | |
| PK1 | 53 | 0 | 56 | 0 | - | 0 | 0 | 0 | 4 | |
| PK2 | - | - | - | - | - | - | - | - | 0 | |
| DE-310 | - | 100 | 67 | 100 | 33 | 0 | 100 | - | 67 | |
| MEN4901 | - | - | 0 | - | 0 | 0 | 0 | 50 | 14 | |
| CT-2106 | 25 | - | 0 | 0 | 14 | - | - | - | 0 | |
| CT-2103 | 56 | 42 | 30 | - | - | 21 | - | 0 | 0 | |
| AP5280 | - | 50 | 50 | 0 | 0 | 0 | - | 0 | 16 | |
| AP5346 | 0 | 25 | 0 | 0 | 40 | 100 | 0 | 0 | - | |
| Normalised Average * | 47 | 41 | 30 | 27 | 21 | 18 | 17 | 13 | 12 | |
| SD | 26 | 37 | 29 | 45 | 19 | 40 | 50 | 22 | 24 | |
| SE | 13 | 17 | 11 | 20 | 8 | 16 | 25 | 10 | 9 | |

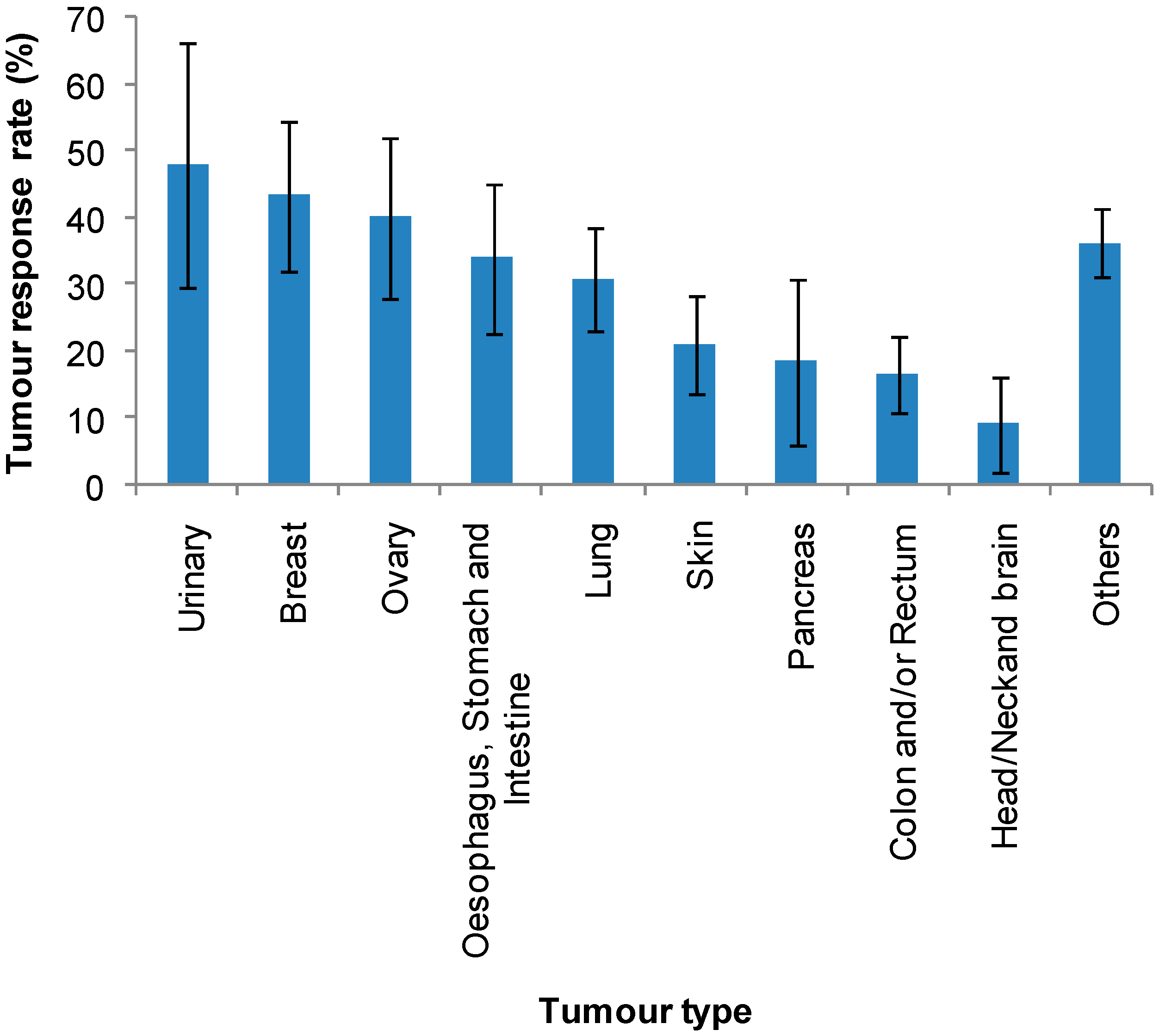
3.2. Cathepsin Levels in Different Tumour Types
| Tumour type | Tumour response rate (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PDC | Urinary | Breast | Ovary | Oesophagus, Stomach and Intestine | Lung | Skin | Pancreas | Colon and/or Rectum | Head/Neck and Brain | |
| PK1 | 0 | 53 | 0 | 0 | 56 | - | 0 | 4 | 0 | |
| PK2 | - | - | - | - | - | - | - | 0 | - | |
| DE-310 | 100 | - | 100 | 0 | 67 | 33 | 100 | 67 | - | |
| MEN4901 | 0 | - | - | 0 | 0 | 0 | - | 14 | 50 | |
| CT-2106 | - | 25 | - | - | 0 | 14 | 0 | 0 | - | |
| CT-2103 | - | 56 | 42 | 21 | 30 | - | - | 0 | 0 | |
| AP5280 | - | - | 50 | 0 | 50 | 0 | 0 | 16 | 0 | |
| AP5346 | 0 | 0 | 25 | 100 | 0 | 40 | 0 | - | 0 | |
| PNU166945 a | ||||||||||
| PNU166148 a | ||||||||||
| EZN246 | - | 0 | 0 | 47 | 14 | 0 | 0 | 0 | 0 | |
| PEG-PTX | NC | |||||||||
| EZN2208 | 100 | 67 | 0 | - | 50 | - | 33 | 20 | - | |
| NKTR-102 | - | - | - | 1 | - | - | - | - | - | |
| NKTR-105 | NC | |||||||||
| XMT-1001 | - | 0 | 33 | 33 | 43 | 50 | 11 | 18 | - | |
| IT-101 | - | - | - | - | 59 | - | - | - | - | |
| ONCOFID-P | 60 | - | - | - | - | - | - | - | - | |
| AD-70 | 0 | - | - | 0 | 0 | 0 | - | 17 | 0 | |
| Normalised average * | 48 | 43 | 40 | 34 | 31 | 21 | 18 | 16 | 9 | |
| SD | 48 | 30 | 34 | 36 | 26 | 21 | 35 | 20 | 19 | |
| SE | 18 | 11 | 12 | 13 | 8 | 7 | 12 | 6 | 7 | |
| Tumour Type | Type of Cathepsin (CAT) | Clinical (C)/Pre-Clinical (PC)/In vitro (IV) | Sample Size (n) | Cathepsin Content | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Lung | CAT B | C | 105 | 10.65 ng/mL | [66] | ||||
| CAT B | C | 17 | 448 ng/mg of protein | [67] | |||||
| CAT B | PC | 159 | High * | [68] | |||||
| CAT D | C | 17 | 1304 ng/mg of protein | [67] | |||||
| CAT S | C | 60 | 4.2 ± 0.22 ng/mg of protein | [69] | |||||
| CAT H | C | 123 | 172 ± 86 ng/mg of protein | [70] | |||||
| CAT L | C | 105 | 26.16 ng/mL | [66] | |||||
| CAT L | C | 17 | 3835 ng/mg of protein | [68] | |||||
| Head/Neck and brain | CAT B | C | 84 | High * | [71] | ||||
| CAT B | C | 47 | High * | [72] | |||||
| CAT B | C | 32 | High * | [73] | |||||
| CAT B | PC | NA | High * | [74] | |||||
| CAT B | PC | NA | High * | [75] | |||||
| CAT B | PC | 11 | High * | [76] | |||||
| CAT B | PC | NA | High * | [77] | |||||
| CAT D | PC | 7 | 1300 ng/mg of protein | [78] | |||||
| CAT S | PC | 11 | Low * | [76] | |||||
| CAT H | PC | 7 | 1500 ng/mg of protein | [79] | |||||
| CAT L | PC | 11 | Low * | [76] | |||||
| Oesophagus, stomach and intestine | CAT B | C | 25 | 325.9 ng/mg of protein | [80] | ||||
| CAT B | C | 175 | 10.83 ± 1.8 ng/mL | [81] | |||||
| CAT B | PC | NA | Low * | [67] | |||||
| CAT L | C | 25 | 43.6 ng/mg of protein | [80] | |||||
| Colon and/or rectum | CAT B | C | 72 | 13.38 ng/mL | [81] | ||||
| CAT B | C | 108 | 168 ± 86 ng/mg of protein | [82] | |||||
| CAT B | C | 60 | 253.5 ng/mg of protein | [82] | |||||
| Colon and/or rectum | CAT B | C | 74 | 55 ± 5 hg/mg of protein | [83] | ||||
| CAT B | PC | 40 | High * | [84] | |||||
| CAT X | C | 77 | 17.4 ng/mL | [85] | |||||
| CAT H | C | 74 | 7 ± 1 ng/mg of protein | [83] | |||||
| CAT L | C | 74 | 50 ± 10 ng/mg of protein | [83] | |||||
| CAT L | C | 60 | 274 ng/mg of protein | [82] | |||||
| Breast | CAT B | C | 30 | 74 ng/mg of protein | [86] | ||||
| CAT B | PC (DU4475) | 4 | High * | [86] | |||||
| CAT B | IV (BT20) | NA | Low * | [87] | |||||
| CAT D | C | 57 | High * | [88] | |||||
| CAT X | IV (MCF-7) | NA | 2.5 ng/mL | [89] | |||||
| CAT X | IV (MDA-MB-231) | NA | 37 ng/mL | [89] | |||||
| Ovary | CAT L | C | 318 | 16.1 ± 5.1 ng/mL | [90] | ||||
| Pancreas | CAT B | PC | NA | High * | [91] | ||||
| Urinary | CAT B | PC | 7 | High * | [67] | ||||
| Others | |||||||||
| Liver | CAT B | C | 28 | 13.46 ng/mL | [81] | ||||
3.3. Magnitude of the EPR Effect in Different Tumour Types
| Tumour Type | Clinical (C)/Pre-Clinical (PC) | Sample Size (n) | Macromolecule System Used | Remarks | Reference | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Breast | C | 2 | PDC | 1.8%–5.9% dose of PDC uptake in tumour | [20] | ||||||||||||||
| C | 5 | Liposome | 5.3% ± 2.6% ID/Kg of drug uptake in tumour # | [97] | |||||||||||||||
| C | 6 | Liposome | 4–16 fold higher drug accumulation in tumour than the free drug | [98] | |||||||||||||||
| PC (MX-1, Human xenograft) | 10 | Protein-conjugate | 33% higher drug accumulation in tumour than the free drug | [99] | |||||||||||||||
| PC (MAXF 449, Human xenograft) | NA | PDC | 1.0%–0.1% dose/g/tumour drug accumulation | [13] | |||||||||||||||
| PC (Mouse) | NA | PDC | 5.29% dose/g for HMW 3.18% dose/g for LMW | [100] | |||||||||||||||
| PC (MX-1) | NA | PDC | 207 fold higher tumour exposure than the free drug (SN-38) | [101] | |||||||||||||||
| PC (Walker 256, Rat) | NA | Protein-conjugate | 7 fold higher drug accumulation in tumour than the free drug | [102] | |||||||||||||||
| PC (Mouse) | NA | Nanoparticle | Drug accumulation observed in tumour * | [103] | |||||||||||||||
| Pancreas | C | NA | Liposomes | Low drug accumulation observed in tumour * | [95] | ||||||||||||||
| PC (Mouse) | 40 | Micelle | 3 fold higher drug accumulation in tumour than the free drug | [104] | |||||||||||||||
| Lung | C | 6 | PDC | No drug accumulation observed in tumour * | [20] | ||||||||||||||
| C | 4 | Liposome | 18.3% ± 5.7% ID/Kg of drug uptake in tumour # | [97] | |||||||||||||||
| C | 3 | Liposome | 4–16 fold higher drug accumulation in tumour than the free drug | [98] | |||||||||||||||
| C | 15 | Liposome | Higher drug accumulation observed in tumour * | [105] | |||||||||||||||
| PC (Meta-7, Mouse) | NA | PDC | 3.5%–4.7% dose/g/tumour drug accumulation | [13] | |||||||||||||||
| PC (COR L23, Human xenograft) | NA | PDC | 4.7%–12.2% dose/g/tumour drug accumulation | [13] | |||||||||||||||
| PC (B16, Mouse) | NA | Liposomes | 10.6% ± 0.2% ID/g of tumour | [106] | |||||||||||||||
| PC (B16F10, Mouse) | NA | PDC | 8.82% dose/g for HMW 3.23% dose/g for LMW | [100] | |||||||||||||||
| PC (B16, Mouse) | NA | Micelle | 2–3 fold higher drug accumulation in tumour than the free drug | [107] | |||||||||||||||
| PC (B16, Mouse) | NA | PDC | 6–12 fold higher drug accumulation in tumour than the free drug | [28] | |||||||||||||||
| PC (B16, Mouse) | 3 | PDC | Higher drug accumulation observed in tumour * | [108] | |||||||||||||||
| Lung | PC (A431, Mouse) | NA | Protein-conjugate | 24 fold higher drug accumulation in tumour than the free drug | [109] | ||||||||||||||
| PC (B16, Mouse) | NA | PDC | 30–63 fold higher drug accumulation in tumour than the free drug | [110] | |||||||||||||||
| PC (B16, Mouse) | NA | PDC | 16.3 fold higher drug accumulation in tumour than the free drug | [111] | |||||||||||||||
| Ovary | C | 3 | Liposome | 4–16 fold higher drug accumulation in tumour than the free drug | [98] | ||||||||||||||
| PC (Mouse) | NA | PDC | Drug accumulation observed in tumour * | [28] | |||||||||||||||
| PC (Oca-1, Mouse) | NA | PDC | 5 fold higher drug accumulation in tumour than the free drug | [112] | |||||||||||||||
| PC (Oca-1, Mouse) | NA | PDC | 28–38 times higher drug accumulation in tumour than the free drug | [113] | |||||||||||||||
| PC (A2780, Human xenograft) | NA | PDC | Drug accumulation observed in tumour * | [114] | |||||||||||||||
| PC (OVCAR-3, Human xenograft) | NA | PDC | 45 fold higher drug accumulation in tumour than the free drug | [115] | |||||||||||||||
| Oesophageal, stomach and intestine | PC (Mouse) | NA | PDC | Drug accumulation observed in tumour * | [116] | ||||||||||||||
| PC (OCUM-2MLN, Human xenograft) | NA | Micelle | Drug accumulation observed in tumour * | [117] | |||||||||||||||
| Colon and/or rectum | C | 5 | PDC | No drug accumulation observed in tumour * | [20] | ||||||||||||||
| C | 10 | PDC | 64 fold higher drug accumulation in tumour than the free drug | [118] | |||||||||||||||
| PC (HT29, Human xenograft) | 4 | Liposome | 1.7 fold higher drug accumulation for 0.6 mol % PEG-conjugate in tumour than the free drug | [119] | |||||||||||||||
| PC (C26 NL-17, Mouse) | NA | Liposome | Higher drug accumulation observed in tumour * | [120] | |||||||||||||||
| PC (Mouse) | NA | Micelle | Drug accumulation observed in tumour * | [121] | |||||||||||||||
| Colon and/or rectum | PC (LS174T, Human xenograft) | NA | PDC | 160 fold higher drug accumulation in tumour than the free drug | [122] | ||||||||||||||
| PC (LS174T, Human xenograft) | NA | Liposomes | 6.3% ± 2.9% ID/g of tumour | [106] | |||||||||||||||
| PC (Mouse) | NA | PDC | Drug accumulation observed in tumour * | [123] | |||||||||||||||
| PC (LS174T, Human xenograft) | NA | PDC | Drug accumulation observed in tumour * | [124] | |||||||||||||||
| PC (HT29, Human xenograft) | NA | PDC | Drug accumulation observed in tumour * | [125] | |||||||||||||||
| PC (HT29, Human xenograft) | 111 | Nano crystal (3H-PTX) | Low drug accumulation observed in tumour * | [126] | |||||||||||||||
| Head/Neck and brain | C | 6 | PDC | 2.2% ± 2.1% dose at 2–3 h 1.3% ± 0.4% dose at 24 h 0.5% ± 0.3% dose at 8 days uptake in tumour | [4] | ||||||||||||||
| C | 10 | Liposome | 13–19 times higher accumulation in tumour as compared to the normal brain tissue | [127] | |||||||||||||||
| C | 5 | Liposome | 7–13 times higher accumulation in tumour as compared to the normal brain tissue | [127] | |||||||||||||||
| C | 7 | Liposome | 33.0% ± 15.8% ID/Kg of drug uptake in tumour # | [97] | |||||||||||||||
| Others | |||||||||||||||||||
| Adenocarcinoma (Unknown) | PC (MAC 26, Mouse) | NA | PDC | 6.9%–10.8% dose/g/tumour drug accumulation | [13] | ||||||||||||||
| Adenocarcinoma (Unknown) | PC (MAC 15A, Mouse) | NA | PDC | 8.2%–12.6% dose/g/tumour drug accumulation | [13] | ||||||||||||||
| Cervix | PC (ME180, Human xenograft) | NA | Liposome | Drug accumulation observed in tumour * | [128] | ||||||||||||||
| Fibrosarcoma | PC (S-180, Mouse) | NA | Polymer conjugates | Drug accumulation observed in tumour * | [129] | ||||||||||||||
| PC (S-180, Mouse) | NA | Micelle | 13 fold higher drug accumulation in tumour than the free drug | [130] | |||||||||||||||
| PC (S-180, Mouse) | NA | PDC | Drug accumulation observed in tumour * | [131] | |||||||||||||||
| PC (S-180, Mouse) | NA | Protein-conjugate | Drug accumulation observed in tumour* | [10] | |||||||||||||||
| Fibrosarcoma | PC (Meth A, Mouse) | NA | PDC | Drug accumulation observed in tumour * | [132] | ||||||||||||||
| PC (S-180, Mouse) | NA | Protein-conjugate | 4 fold higher drug accumulation in tumour than the free drug | [8] | |||||||||||||||
| PC (S-180, Mouse) | NA | PDC | Drug accumulation observed in tumour * | [133] | |||||||||||||||
| Liver | C | 31 | Liposomes | Low drug accumulation observed in tumour * | [95] | ||||||||||||||
| C | 3 | Liposomes | Low drug accumulation observed in tumour * | [96] | |||||||||||||||
| PC (Mouse) | NA | Micelle | 4 fold higher drug accumulation in tumour than the free drug | [134] | |||||||||||||||
| PC (VX-2, Rabbit) | NA | Polymer-protein conjugate (SMANCS-Lipidol) | Drug accumulation observed in tumour * | [135] | |||||||||||||||
| PC (VX-2, Rabbit) | NA | Polymer-protein conjugate (Lipidol) | Drug accumulation observed in tumour * | [136] | |||||||||||||||
| PC | NA | PDC | Drug accumulation observed in tumour * | [137] | |||||||||||||||
| Prostate | PC (Human xenograft) | NA | 89Zr-DFO-mAlb (Polymer-protein conjugate) | Drug accumulation observed in tumour * | [138] | ||||||||||||||
| PC (Human xenograft) | NA | PDC | Drug accumulation observed in tumour * | [139] | |||||||||||||||
| PC (Rat) | NA | PDC | Drug accumulation observed in tumour * | [140] | |||||||||||||||
| PC (Rat) | NA | PDC | Drug accumulation observed in tumour * | [141] | |||||||||||||||
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, T.E.; Murray, T.; et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl) methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents—Drug-polymer conjugates. Clin. Cancer. Res. 1999, 5, 83–94. [Google Scholar]
- Albain, K.S.; Belani, C.P.; Bonomi, P.; O’Byrne, K.J.; Schiller, J.H.; Socinski, M. PIONEER: A phase III randomized trial of paclitaxel poliglumex versus paclitaxel in chemotherapy-naive women with advanced-stage non-small-cell lung cancer and performance status of 2. Clin. Lung Cancer 2006, 7, 417–419. [Google Scholar]
- Langer, C.J.; O’Byrne, K.J.; Socinski, M.A.; Mikhailov, S.M.; Leśniewski-Kmak, K.; Smakal, M.; Ciuleanu, T.E.; Orlov, S.V.; Dediu, M.; Heigener, D.; et al. Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin vs. standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naïve advanced non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 623–630. [Google Scholar] [CrossRef]
- Li, C.; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev. 2008, 60, 886–898. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Maeda, H.; Bharate, G.Y.; Daruwalla, J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur. J. Pharm. Biopharm. 2009, 71, 409–419. [Google Scholar]
- Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjug. Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef]
- Maeda, H.; Matsumura, Y. EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv. Drug Deliv. Rev. 2011, 63, 129–130. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Duncan, R.; Sat-Klopsch, Y.N.; Burger, A.M.; Bibby, M.C.; Fiebig, H.H.; Sausville, E.A. Validation of tumour models for use in anticancer nanomedicine evaluation: The EPR effect and cathepsin B-mediated drug release rate. Cancer Chemother. Pharmacol. 2013, 72, 417–427. [Google Scholar] [CrossRef]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Satchi, R.; Connors, T.A.; Duncan, R. PDEPT: Polymer directed enzyme prodrug therapy. I. HPMA copolymer-cathepsin B and PK1 as a model combination. Br. J. Cancer 2001, 85, 1070–1076. [Google Scholar] [CrossRef]
- Zhong, Y.J.; Shao, L.H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy. Int. J. Oncol. 2013, 42, 373–383. [Google Scholar]
- Chipman, S.D.; Oldham, F.B.; Pezzoni, G.; Singer, J.W. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer-drug conjugate. Int. J. Nanomed. 2006, 1, 375–383. [Google Scholar]
- Ross, H.; Bonomi, P.; Langer, C.; O’Brien, M.; O’Byrne, K.; Paz-Ares, L.; Sandler, A.; Socinski, M.; Oldham, F.; Singer, J. Effect of gender on outcome in two randomized phase III trials of paclitaxel poliglumex (PPX) in chemo-naïvepts with advanced NSCLC and poor performance status (PS2). J. Clin. Oncol. 2006, 24. Abstract 7039. [Google Scholar]
- Seymour, L.W.; Ferry, D.R.; Kerr, D.J.; Rea, D.; Whitlock, M.; Poyner, R.; Boivin, C.; Hesslewood, S.; Twelves, C.; Blackie, R.; et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int. J. Oncol. 2009, 6, 1629–1636. [Google Scholar]
- Seymour, L.W.; Ferry, D.R.; Anderson, D.; Hesslewood, S.; Julyan, P.J.; Poyner, R.; Doran, J.; Young, A.M.; Burtles, S.; Kerr, D.J. Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J. Clin. Oncol. 2002, 20, 1668–1676. [Google Scholar] [CrossRef]
- Soepenberg, O.; de Jonge, M.J.; Sparreboom, A.; de Bruin, P.; Eskens, F.A.; de Heus, G.; Wanders, J.; Cheverton, P.; Ducharme, M.P.; Verweij, J. Phase I and pharmacokinetic study of DE-310 in patients with advanced solid tumors. Clin. Cancer Res. 2005, 11, 703–711. [Google Scholar]
- Veltkamp, S.A.; Witteveen, E.O.; Capriati, A.; Crea, A.; Animati, F.; Voogel-Fuchs, M.; van den Heuvel, I.J.; Beijnen, J.H.; Voest, E.E.; Schellens, J.H. Clinical and pharmacologic study of the novel prodrug delimotecan (MEN 4901/T-0128) in patients with solid tumors. Clin. Cancer Res. 2008, 14, 7535–7544. [Google Scholar] [CrossRef]
- Homsi, J.; Simon, G.R.; Garrett, C.R.; Springett, G.; de Conti, R.; Chiappori, A.A.; Munster, P.N.; Burton, M.K.; Stromatt, S.; Allievi, C.; et al. Phase I trial of poly-l-glutamate camptothecin (CT-2106) administered weekly in patients with advanced solid malignancies. Clin. Cancer Res. 2007, 13, 5855–5861. [Google Scholar] [CrossRef]
- Galic, V.L.; Wright, J.D.; Lewin, S.N.; Herzog, T.J. Paclitaxel poliglumex for ovarian cancer. Expert Opin. Invest. Drugs. 2011, 20, 813–821. [Google Scholar] [CrossRef]
- Veronese, M.L.; Flaherty, K.; Kramer, A.; Konkle, B.A.; Morgan, M.; Stevenson, J.P.; O’Dwyer, P.J. Phase I study of the novel taxane CT-2103 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2005, 55, 497–501. [Google Scholar] [CrossRef]
- Amato, R.J.; Khan, M.M.T. Phase II study of paclitaxel poliglumex (PPX) for androgen independent prostate cancer (AIPC). In Proceedings of the American Society of Clinial Oncology (ASCO), Prostate Cancer Symposium, Alexandria, VA, USA; 2007. Abstract 243. [Google Scholar]
- Singer, J.W.; Bhatt, R.; Tulinsky, J.; Buhler, K.R.; Heasley, E.; Klein, P.; de Vries, P. Water-soluble poly-(l-glutamic acid)-Gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J. Control. Release 2001, 74, 243–247. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Cunningham, C.; Senzer, N.; Gray, M.; Oldham, F.; Pippen, J.; Mennel, R.; Eisenfeld, A. Phase I study of CT-2103, a polymer-conjugated paclitaxel, and carboplatin in patients with advanced solid tumors. Cancer Invest. 2005, 23, 671–676. [Google Scholar] [CrossRef]
- Boddy, A.V.; Plummer, E.R.; Todd, R.; Sludden, J.; Griffin, M.; Robson, L.; Cassidy, J.; Bissett, D.; Bernareggi, A.; Verrill, M.W.; et al. A phase I and pharmacokinetic study of paclitaxel poliglumex (XYOTAX), investigating both 3-weekly and 2-weekly schedules. Clin. Cancer Res. 2005, 11, 7834–7840. [Google Scholar] [CrossRef]
- Dipetrillo, T.; Milas, L.; Evans, D.; Akerman, P.N.T.; Miner, T.; Cruff, D.; Chauhan, B.; Iannitti, D.; Harrington, D.; Safran, H. Paclitaxel poliglumex (PPX-Xyotax) and concurrent radiation for esophageal and gastric cancer: A phase I study. Am. J. Clin. Oncol. 2006, 29, 376–379. [Google Scholar] [CrossRef]
- Lin, N.U.; Parker, L.M.; Come, S.E.; Burstein, H.J.; Haldoupis, M.; Ryabin, N.; Gelman, R.; Winer, E.P.; Shulman, L.N. Phase II study of CT-2103 as first- or second-line chemotherapy in patients with metastatic breast cancer: Unexpected incidence of hypersensitivity reactions. Invest. New Drugs 2007, 25, 369–375. [Google Scholar] [CrossRef]
- Sabbatini, P.; Sill, M.W.; O’Malley, D.; Adler, L.; Secord, A.A. A phase II trial of paclitaxel poliglumex in recurrent or persistent ovarian or primary peritoneal cancer (EOC): A Gynecologic Oncology Group Study. Gynecol. Oncol. 2008, 111, 455–460. [Google Scholar] [CrossRef]
- Bonomi, P. Paclitaxel poliglumex (PPX, CT-2103): Macromolecular medicine for advanced non-small-cell lung cancer. Expert Rev. Anticancer Ther. 2007, 7, 415–422. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; Terret, C.; Howell, S.B.; Baud, C.M.; de Boer, R.F.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.; Droz, J.P. A phase pharmacological and I study of the platinum polymer AP5280 given as an intravenous infusion once every 3 weeks in patients with solid tumors. Clin. Cancer Res. 2004, 10, 3386–3395. [Google Scholar] [CrossRef]
- Campone, M.; Rademaker-Lakhai, J.M.; Bennouna, J.; Howell, S.B.; Nowotnik, D.P.; Beijnen, J.H.; Schellens, J.H. Phase I and pharmacokinetic trial of AP5346, a DACH-platinum-polymer conjugate, administered weekly for three out of every 4 weeks to advanced solid tumor patients. Cancer Chemother Pharmacol. 2007, 60, 523–533. [Google Scholar]
- Terwogt, J.M.M.; Bokkel Huinink, W.W.; Schellens, J.H.; Schot, M.; Mandjes, I.A.; Zurlo, M.G.; Rocchetti, M.; Rosing, H.; Koopman, F.J.; Beijnen, J.H. Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs 2001, 12, 315–323. [Google Scholar] [CrossRef]
- Schoemaker, N.E.; van Kesteren, C.; Rosing, H.; Jansen, S.; Swart, M.; Lieverst, J.; Fraier, D.; Breda, M.; Pellizzoni, C.; Spinelli, R.; et al. A phase I and pharmacokinetic study of MAG-CPT, a water-soluble polymer conjugate of camptothecin. Br. J. Cancer 2002, 87, 608–614. [Google Scholar] [CrossRef]
- Bissett, D.; Cassidy, J.; de Bono, J.S.; Muirhead, F.; Main, M.; Robson, L.; Fraier, D.; Magnè, M.L.; Pellizzoni, C.; Porro, M.G.; et al. Phase I and pharmacokinetic (PK) study of MAG-CPT (PNU 166148): A polymeric derivative of camptothecin (CPT). Br. J. Cancer 2004, 91, 50–55. [Google Scholar] [CrossRef]
- Wachters, F.M.; Groen, H.J.; Maring, J.G.; Gietema, J.A.; Porro, M.; Dumez, H.; de Vries, E.G.; van Oosterom, A.T. A phase I study with MAG-camptothecin intravenously administered weekly for 3 weeks in a 4-week cycle in adult patients with solid tumours. Br. J. Cancer 2004, 90, 2261–2267. [Google Scholar]
- Rowinsky, E.K.; Rizzo, J.; Ochoa, L.; Takimoto, C.H.; Forouzesh, B.; Schwartz, G.; Hammond, L.A.; Patnaik, A.; Kwiatek, J.; Goetz, A.; et al. A phase I and pharmacokinetic study of pegylatedcamptothecin as a 1-h infusion every 3 weeks in patients with advanced solid malignancies. J. Clin. Oncol. 2003, 21, 148–157. [Google Scholar] [CrossRef]
- Posey, J.A; Saif, M.W.; Carlisle, R.; Goetz, A.; Rizzo, J.; Stevenson, S.; Rudoltz, M.S.; Kwiatek, J.; Simmons, P.; Rowinsky, E.K.; et al. Phase 1 study of weekly polyethylene glycol-camptothecin in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2005, 11, 7866–7871. [Google Scholar] [CrossRef]
- Scott, L.C.; Yao, J.C.; Thomas, A.L.; Falk, S.; Mena, R.R.; Picus, J.; Wright, J.; Mulcahy, M.F.; Ajani, J.A.; Evans, T.R. A phase II study of pegylated-camptothecin (pegamotecan) in the treatment of locally advanced and metastatic gastric and gastro-oesophageal junction adenocarcinoma. Cancer Chemother. Pharmacol. 2009, 63, 363–370. [Google Scholar] [CrossRef]
- Beeram, M.; Rowinsky, E.K.; Hammond, L.A.; Patnaik, A.; Schwartz, G.H.; de Bono, J.S.; Forero, L.; Forouzesh, B.; Berg, K.E.; Rubin, E.H.; et al. A phase I pharmacokinetic (PK) study of PEG-paclitaxel in patients with advanced solid tumors. Proc. Am. Soc. Clin. Oncol. 2002, 21, 405. [Google Scholar]
- Patnaik, A.; Papadopoulos, K.P.; Tolcher, A.W.; Beeram, M.; Urien, S.; Schaaf, L.J.; Tahiri, S.; Bekaii-Saab, T.; Lokiec, F.M.; Rezaï, K.; et al. Phase I dose-escalation study of EZN-2208 (PEG-SN38), a novel conjugate of poly(ethylene) glycol and SN38, administered weekly in patients with advanced cancer. Cancer Chemother. Pharmacol. 2013, 71, 1499–1506. [Google Scholar]
- Garrett, C.R.; Bekaii-Saab, T.S.; Ryan, T.; Fisher, G.A.; Clive, S.; Kavan, P.; Shacham-Shmueli, E.; Buchbinder, A.; Goldberg, R.M. Randomized phase 2 study of pegylated SN-38 (EZN-2208) or irinotecan plus cetuximab in patients with advanced colorectal cancer. Cancer 2013, 119, 4223–4230. [Google Scholar]
- Von Hoff, D.D.; Jameson, G.S.; Borad, M.J.; Rosen, L.S.; Utz, J.; Basche, M.; Alemany, C.; Dhar, S.; Acosta, L.; Barker, T.; et al. First phase I trial of NKTR-102 (PEG-irinotecan) reveals early evidence of broad anti-tumor activity in three different schedules. Available online: https://www.nektar.com/pdf/pipeline/NKTR-102/NKTR-102_poster_595.pdf (accessed on 3 November 2013).
- Vergote, I.B.; Garcia, A.; Micha, J.; Pippitt, C.; Bendell, J.; Spitz, D.; Reed, N.; Dark, G.; Fracasso, P.M.; Ibrahim, E.N.; et al. Randomized multicenter phase II trial comparing two schedules of etirinotecan pegol (NKTR-102) in women with recurrent platinum-resistant/refractory epithelial ovarian cancer. J. Clin. Oncol. 2013, 31, 4060–4066. [Google Scholar] [CrossRef]
- Awada, A.; Garcia, A.A.; Chan, S.; Jerusalem, G.H.; Coleman, R.E.; Huizing, M.T.; Mehdi, A.; O’Reilly, S.M.; Hamm, J.T.; Barrett-Lee, P.J.; et al. Two schedules of etirinotecan pegol (NKTR-102) in patients with previously treated metastatic breast cancer: A randomised phase 2 study. Lancet Oncol. 2013, 14, 1216–1225. [Google Scholar] [CrossRef]
- Etirinotecan pegol (NKTR-102). Available online: http://www.nektar.com/product_pipeline/oncology_nktr-102.html (accessed on 15 May 2014).
- Calvo, E.; Hoch, U.; Maslyar, D.J.; Tolcher, A.W. Dose-escalation phase I study of NKTR-105, a novel pegylated form of docetaxel. J. Clin. Oncol. 2010, 28, 15. [Google Scholar] [CrossRef]
- Sausville, E.A.; Garbo, L.; Weiss, G.J.; Shkolny, D.; Yurkovetskiy, A.V.; Bethune, C.; Ramanathan, R.K.; Fram, R.J. Phase 1 study of XMT-1001, a novel water soluble camptothecin conjugate, given as an intravenous infusion once every three weeks to patients with advanced solid tumors. Mol. Cancer Ther. 2009, 8, 12. [Google Scholar]
- Weiss, G.J.; Chao, J.; Neidhart, J.D.; Ramanathan, R.K.; Bassett, D.; Neidhart, J.A.; Choi, C.H.; Chow, W.; Chung, V.; Forman, S.J.; et al. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymer-camptothecinnanopharmaceutical in patients with advanced solid tumor malignancies. Invest. New Drugs 2013, 31, 986–1000. [Google Scholar] [CrossRef]
- Bassi, P.F.; Volpe, A.; D’Agostino, D.; Palermo, G.; Renier, D.; Franchini, S.; Rosato, A.; Racioppi, M. Paclitaxel-hyaluronic acid for intravesical therapy of bacillus Calmette-Guérin refractory carcinoma in situ of the bladder: Results of a phase I study. J. Urol. 2011, 185, 445–449. [Google Scholar] [CrossRef]
- Danhauser-Riedl, S.; Hausmann, E.; Schick, H.D.; Bender, R.; Dietzfelbinger, H.; Rastetter, J.; Hanauske, A.R. Phase I clinical and pharmacokinetic trial of dextran-conjugated doxorubicin (AD-70, DOX-O.XD). Invest. New Drugs 1993, 11, 187–195. [Google Scholar] [CrossRef]
- Duncan, R. Development of HPMA copolymer-anticancer conjugates: Clinical experience and lessons learnt. Adv. Drug Deliv. Rev. 2009, 61, 1131–1148. [Google Scholar] [CrossRef]
- Pasut, G.; Veronese, F.M. PEG conjugates in clinical development or use as anticancer agents: An overview. Adv. Drug Deliv. Rev. 2009, 61, 1177–1188. [Google Scholar] [CrossRef]
- Podgorski, I.; Sloane, B.F. Cathepsin B and its role(s) in cancer progression. Biochem. Soc. Symp. 2003, 70, 263–276. [Google Scholar]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar]
- Sloane, B.F.; Dunn, J.R.; Honn, K.V. Lysosomal cathepsin B: Correlation with metastatic potential. Science 1981, 212, 1151–1153. [Google Scholar]
- Turk, V.; Kos, J.; Turk, B. Cysteine cathepsins (proteases)—On the main stage of cancer? Cancer Cell 2004, 5, 409–410. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Duncan, R.; Kopecková-Rejmanová, P.; Strohalm, J.; Hume, I.; Cable, H.C.; Pohl, J.; Lloyd, J.B.; Kopecek, J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of daunomycin and puromycin conjugates in vitro. Br. J. Cancer 1987, 55, 165–174. [Google Scholar] [CrossRef]
- Putnam, D.; Shiah, J.G.; Kopecek, J. Intra cellularly biorecognizable derivatives of 5-fluorouracil: Implications of targetable delivery in the human condition. Biochem. Pharmacol. 1996, 52, 957–962. [Google Scholar] [CrossRef]
- Chen, Q.; Fei, J.; Wu, L.; Jiang, Z.; Wu, Y.; Zheng, Y.; Lu, G. Detection of cathepsin B, cathepsin L, cystatin C, urokinase plasminogen activator and urokinase plasminogen activator receptor in the sera of lung cancer patients. Oncol. Lett. 2011, 2, 693–699. [Google Scholar]
- Smith, V.; Wirth, G.J.; Fiebig, H.H.; Burger, A.M. Tissue microarrays of human tumorxenografts: Characterization of proteins involved in migration and angiogenesis for applications in the development of targeted anticancer agents. Cancer Genomics Proteomics 2008, 5, 263–273. [Google Scholar]
- Ledakis, P.; Tester, W.T.; Rosenberg, N.; Romero-Fischmann, D.; Daskal, I.; Lah, T.T. Cathepsins D, B, and L in malignant human lung tissue. Clin. Cancer Res. 1996, 2, 561–568. [Google Scholar]
- Kos, J.; Sekirnik, A.; Kopitar, G.; Cimerman, N.; Kayser, K.; Stremmer, A.; Fiehn, W.; Werle, B. Cathepsin S in tumours, regional lymph nodes and sera of patients with lung cancer: Relation to prognosis. Br. J. Cancer 2001, 85, 1193–1200. [Google Scholar] [CrossRef]
- Schweiger, A.; Staib, A.; Werle, B.; Krasovec, M.; Lah, T.T.; Ebert, W.; Turk, V.; Kos, J. Cysteine proteinase cathepsin H in tumours and sera of lung cancer patients: Relation to prognosis and cigarette smoking. Br. J. Cancer 2000, 82, 782–788. [Google Scholar] [CrossRef]
- Li, C.; Chen, L.; Wang, J.; Zhang, L.; Tang, P.; Zhai, S.; Guo, W.; Yu, N.; Zhao, L.; Liu, M.; et al. Expression and clinical significance of cathepsin B and stefin A in laryngeal cancer. Oncol. Rep. 2011, 26, 869–875. [Google Scholar]
- Saleh, Y.; Wnukiewicz, J.; Andrzejak, R.; Trziszka, T.; Siewinski, M.; Ziolkowski, P.; Kopec, W. Cathepsin B and cysteine protease inhibitors in human tongue cancer: Correlation with tumor staging and in vitro inhibition of cathepsin B by chicken cystatin. J. Cancer Mol. 2006, 2, 67–72. [Google Scholar]
- Shuja, S.; Cai, J.; Iacobuzio-Donahue, C.; Zacks, J.; Beazley, R.M.; Kasznica, J.M.; O’Hara, C.J.; Heimann, R.; Murnane, M.J. Cathepsin B activity and protein levels in thyroid carcinoma, Graves’ disease, and multinodular goiters. Thyroid 1999, 9, 569–577. [Google Scholar] [CrossRef]
- Demchik, L.L.; Sameni, M.; Nelson, K.; Mikkelsen, T.; Sloane, B.F. Cathepsin B and glioma invasion. Int. J. Dev. Neurosci. 1999, 17, 483–494. [Google Scholar] [CrossRef]
- Rempel, S.A.; Rosenblum, M.L.; Mikkelsen, T.; Yan, P.S.; Ellis, K.D.; Golembieski, W.A.; Sameni, M.; Rozhin, J.; Ziegler, G.; Sloane, B.F. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994, 54, 6027–6031. [Google Scholar]
- Gole, B.; Huszthy, P.C.; Popović, M.; Jeruc, J.; Ardebili, Y.S.; Bjerkvig, R.; Lah, T.T. The regulation of cysteine cathepsins and cystatins in human gliomas. Int. J. Cancer 2012, 131, 1779–1789. [Google Scholar]
- Fan, K.; Zhang, Y.; Song, D.; Zhang, Y.; Ma, J. Relation of cystatin C and cathepsin B expression to the pathological grade and invasion of human gliomas. Chin. J. Clin. Oncol. 2007, 4, 303–306. [Google Scholar] [CrossRef]
- Sivaparvathi, M.; Sawaya, R.; Chintala, S.K.; Go, Y.; Gokaslan, Z.L.; Rao, J.S. Expression of cathepsin D during the progression of human gliomas. Neurosci. Lett. 1996, 208, 171–174. [Google Scholar] [CrossRef]
- Sivaparvathi, M.; Sawaya, R.; Gokaslan, Z.L.; Chintala, S.K.; Rao, J.S. Expression and the role of cathepsin H in human glioma progression and invasion. Cancer Lett. 1996, 104, 121–126. [Google Scholar]
- Plebani, M.; Herszènyi, L.; Cardin, R.; Roveroni, G.; Carraro, P.; Paoli, M.D.; Rugge, M.; Grigioni, W.F.; Nitti, D.; Naccarato, R. Cysteine and serine proteases in gastric cancer. Cancer 1995, 76, 367–375. [Google Scholar]
- Herszényi, L.; István, G.; Cardin, R.; de Paoli, M.; Plebani, M.; Tulassay, Z.; Farinati, F. Serum cathepsin B and plasma urokinase-type plasminogen activator levels in gastrointestinal tract cancers. Eur. J. Cancer Prev. 2008, 17, 438–445. [Google Scholar] [CrossRef]
- Herszènyi, L.; Plebani, M.; Carraro, P.; de Paoli, M.; Roveroni, G.; Cardin, R.; Tulassay, Z.; Naccarato, R.; Farinati, F. The role of cysteine and serine proteases in colorectal carcinoma. Cancer 1999, 86, 1135–1142. [Google Scholar]
- Doxakis, A.; Maria, A.; Savvas, P.; Zafiroula, I.K. Assessment of the Roles of Cathepsins B, H and L in the progression of colorectal cancer. J. Cancer Ther. 2013, 4, 1–7. [Google Scholar]
- Hazen, L.G.; Bleeker, F.E.; Lauritzen, B.; Bahns, S.; Song, J.; Jonker, A.; van Driel, B.E.; Lyon, H.; Hansen, U.; Köhler, A.; et al. Comparitive localization of cathepsin B protein and activity in colorectal cancer. J. Histochem. Cytochem. 2000, 48, 1421–1430. [Google Scholar] [CrossRef]
- Vizin, T.; Christensen, I.J.; Nielsen, H.J.; Kos, J. Cathepsin X in serum from patients with colorectal cancer: Relation to prognosis. Radiol. Oncol. 2012, 46, 207–212. [Google Scholar]
- Bremer, C.; Tung, C.H.; Bogdanov, A.; Weissleder, R., Jr. Imaging of differential protease expression in breast cancers for detection of aggressive tumor phenotypes. Radiology 2002, 222, 814–818. [Google Scholar]
- Hulkower, K.I.; Butler, C.C.; Linebaugh, B.E.; Klaus, J.L.; Keppler, D.; Giranda, V.L.; Sloane, B.F. Fluorescent microplate assay for cancer cell-associated cathepsin B. Eur. J. Biochem. 2000, 267, 4165–4170. [Google Scholar] [CrossRef]
- Ruibal, A.; Herranz, M.; Arias, J.I. Clinical and biological significance of cathepsin D levels in breast cancer cytosol in women over 70 years. Biomark. Cancer 2012, 4, 1–6. [Google Scholar]
- Pečar Fonović, U.; Jevnikar, Z.; Rojnik, M.; Doljak, B.; Fonović, M.; Jamnik, P.; Kos, J. Profilin 1 as a target for cathepsin X activity in tumor cells. PLoS One 2013, 8, e53918. [Google Scholar]
- Zhang, W.; Hu, X.X.; Yang, X.Z.; Wang, Q.; Cheng, H.; Wang, S.M.; Hu, Y.L.; Yang, Z.J.; Li, L. Combined detection of serum matrix metalloproteinase 9, acetyl heparinase and cathepsin L in diagnosis of ovarian cancer. Chin. J. Cancer Res. 2012, 24, 67–71. [Google Scholar] [CrossRef]
- Gopinathan, A.; Denicola, G.M.; Frese, K.K.; Cook, N.; Karreth, F.A.; Mayerle, J.; Lerch, M.M.; Reinheckel, T.; Tuveson, D.A. Cathepsin B promotes the progression of pancreatic ductal adenocarcinoma in mice. Gut 2012, 61, 877–884. [Google Scholar]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar]
- Kartz, F.; Senter, P.; Steinhagam, H. Drug Delivery in Oncology; Wiley-VCH-Verlag & Co. KGaA: Weinheim, Germany, 2012; pp. 875–876. [Google Scholar]
- Maeda, H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. B Phys. Biol. Sci. 2012, 88, 53–71. [Google Scholar]
- Maki, S.; Konno, T.; Maeda, H. Image enhancement in computerized tomography for sensitive diagnosis of liver cancer and semiquantitation of tumor selective drug targeting with oily contrast medium. Cancer 1985, 56, 751–757. [Google Scholar]
- Nagamitsu, A.; Greish, K.; Maeda, H. Elevating blood pressure as a strategy to increase tumortargeted delivery of macromolecular drug SMANCS: Cases of advanced solid tumors. Jpn. J. Clin. Oncol. 2009, 39, 756–766. [Google Scholar] [CrossRef]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylatedliposomes. Clin. Cancer Res. 2001, 7, 243–254. [Google Scholar]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Desai, N.; Trieu, V.; Yao, Z.; Louie, L.; Ci, S.; Yang, A.; Tao, C.; de, T.; Beals, B.; Dykes, D.; et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 2006, 12, 1317–1324. [Google Scholar] [CrossRef]
- Pimm, M.V.; Perkins, A.C.; Strohalm, J.; Ulbrich, K.; Duncan, R. Gamma scintigraphy of the biodistribution of 123I-labelled N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in mice with transplanted melanoma and mammary carcinoma. J. Drug Target 1996, 3, 375–383. [Google Scholar] [CrossRef]
- Sapra, P.; Zhao, H.; Mehlig, M.; Malaby, J.; Kraft, P.; Longley, C.; Greenberger, L.M.; Horak, I.D. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin. Cancer Res. 2008, 14, 1888–1986. [Google Scholar]
- Li, C.J.; Miyamoto, Y.; Kojima, Y.; Maeda, H. Augmentation of tumour delivery of macromolecular drugs with reduced bone marrow delivery by elevating blood pressure. Br. J. Cancer 1993, 67, 975–980. [Google Scholar] [CrossRef]
- Kommareddy, S.; Amiji, M. Preparation and evaluation of thiol-modified gelatin nanoparticles for intracellular DNA delivery in response to glutathione. Bioconjug. Chem. 2005, 16, 1423–1432. [Google Scholar] [CrossRef]
- Cabrala, H.; Murakamia, M.; Hojob, H.; Teradac, Y.; Kanod, M.R.; Chunga, U.; Nishiyamae, N.; Kataokaa, K. Targeted therapy of spontaneous murine pancreatic tumors by polymeric micelles prolongs survival and prevents peritoneal metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 11397–11402. [Google Scholar]
- Koukourakis, M.I.; Koukouraki, S.; Giatromanolaki, A.; Archimandritis, S.C.; Skarlatos, J.; Beroukas, K.; Bizakis, J.G.; Retalis, G.; Karkavitsas, N.; Helidonis, E.S. Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer. J. Clin. Oncol. 1999, 17, 3512–3521. [Google Scholar]
- Gabizon, A.; Price, D.C.; Huberty, J.; Bresalier, R.S.; Papahadjopoulos, D. Effect of liposome composition and other factors on the targeting of liposomes to experimental tumors: Biodistribution and imaging studies. Cancer Res. 1990, 50, 6371–6378. [Google Scholar]
- Kim, S.C.; Kim, D.W.; Shim, Y.H.; Bang, J.S.; Oh, H.S.; Wan Kim, S.; Seo, M.H. In vivo evaluation of polymeric micellar paclitaxel formulation: Toxicity and efficacy. J. Control Release 2001, 191–202. [Google Scholar]
- Veronese, F.M.; Schiavon, O.; Pasut, G.; Mendichi, R.; Andersson, L.; Tsirk, A.; Ford, J.; Wu, G.; Kneller, S.; Davies, J.; et al. PEG-doxorubicin conjugates: Influence of polymer structure on drug release, in vitro cytotoxicity, biodistribution, and antitumor activity. Bioconjug. Chem. 2005, 16, 775–784. [Google Scholar] [CrossRef]
- Sano, K.; Nakajima, T.; Choyke, P.L.; Kobayashi, H. Markedly enhanced permeability and retention effects induced by photo-immunotherapy of tumors. ACS Nano 2013, 7, 717–724. [Google Scholar]
- Gianasi, E.; Wasil, M.; Evagorou, E.G.; Keddle, A.; Wilson, G.; Duncan, R. HPMA copolymer platinates as novel antitumour agents: In vitro properties, pharmacokinetics and antitumour activity in vivo. Eur. J. Cancer 1999, 35, 994–1002. [Google Scholar] [CrossRef]
- Rice, J.R.; Gerberas, J.L.; Nowotnik, D.P.; Howell, S.B. Preclinical efficacy and pharmacokinetics of AP5346, a novel diaminocyclohexane-platinum tumor-targeting drug delivery system. Clin. Cancer Res. 2006, 12, 2248–2254. [Google Scholar] [CrossRef]
- Li, C.; Ke, S.; Wu, Q.P.; Tansey, W.; Hunter, N.; Buchmiller, L.M.; Milas, L.; Charnsangavej, C.; Wallace, S. Tumor irradiation enhances the tumor-specific distribution of poly(l-glutamic acid)-conjugated paclitaxel and its antitumor efficacy. Clin. Cancer Res. 2000, 6, 2829–2834. [Google Scholar]
- Li, C.; Newman, R.A.; Wu, Q.P.; Ke, S.; Chen, W.; Hutto, T.; Kan, Z.; Brannan, M.D.; Charnsangavej, C.; Wallace, S. Biodistribution of paclitaxel and poly(l-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother. Pharmacol. 2000, 46, 416–422. [Google Scholar] [CrossRef]
- Sadekar, S.; Ray, A.; Janàt-Amsbury, M.; Peterson, C.M.; Ghandehari, H. Comparative biodistribution of PAMAM dendrimers and HPMA copolymers in ovarian-tumor-bearing mice. Biomacromolecules 2011, 12, 88–96. [Google Scholar]
- Shiah, J.G.; Dvorák, M.; Kopecková, P.; Sun, Y.; Peterson, C.M.; Kopecek, J. Biodistribution and antitumour efficacy of long-circulating N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in nude mice. Eur. J. Cancer 2001, 37, 131–139. [Google Scholar]
- Eliasof, S.; Lazarus, D.; Peters, C.G.; Case, R.I.; Cole, R.O.; Hwang, J.; Schluep, T.; Chao, J.; Lin, J.; Yen, Y.; et al. Correlating preclinical animal studies and human clinical trials of a multifunctional, polymeric nanoparticle. Proc. Natl. Acad. Sci. USA 2013, 110, 15127–15132. [Google Scholar] [CrossRef]
- Kano, M.R.; Bae, Y.; Iwata, C.; Morishita, Y.; Yashiro, M.; Oka, M.; Fujii, T.; Komuro, A.; Kiyono, K.; Kaminishi, M.; et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 3460–3465. [Google Scholar] [CrossRef]
- Sarapa, N.; Britto, M.R.; Speed, W.; Jannuzzo, M.; Breda, M.; James, C.A.; Porro, M.; Rocchetti, M.; Wanders, A.; Mahteme, H.; et al. Assessment of normal and tumor tissue uptake of MAG-CPT, a polymer-bound prodrug of camptothecin, in patients undergoing elective surgery for colorectal carcinoma. Cancer Chemother. Pharmacol. 2003, 52, 424–430. [Google Scholar] [CrossRef]
- Chow, T.H.; Lin, Y.Y.; Hwang, J.J.; Wang, H.E.; Tseng, Y.L.; Wang, S.J.; Liu, R.S.; Lin, W.J.; Yang, C.S.; Ting, G. Improvement of biodistribution and therapeutic index via increase of polyethylene glycol on drug-carrying liposomes in an HT-29/lucxenografted mouse model. Anticancer Res. 2009, 29, 2111–2120. [Google Scholar]
- Maeda, N.; Takeuchi, Y.; Takada, M.; Sadzuka, Y.; Namba, Y.; Oku, N. Anti-neovascular therapy by use of tumorneovasculature-targeted long-circulating liposome. J. Control. Release 2004, 100, 41–52. [Google Scholar] [CrossRef]
- Kwon, G.; Suwa, S.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Enhanced tumour accumulation and prolonged circulation times of micelle-forming poly(ethylene oxide-aspartate) block copolymer-adriamycin conjugates. J. Control. Release 1994, 29, 17–23. [Google Scholar]
- Schluep, T.; Cheng, J.; Khin, K.T.; Davis, M.E. Pharmacokinetics and biodistribution of the camptothecin-polymer conjugate IT-101 in rats and tumor-bearing mice. Cancer Chemother. Pharmacol. 2006, 57, 654–662. [Google Scholar] [CrossRef]
- Conover, C.D.; Greenwald, R.B.; Pendri, A.; Gilbert, C.W.; Shum, K.L. Camptothecin delivery systems: Enhanced efficacy and tumour accumulation of camptothecin following its conjugation to polyethylene glycol via a glycine linker. Cancer Chemother. Pharmacol. 1998, 42, 407–414. [Google Scholar] [CrossRef]
- Deshan, Y.; Peng, P.; Dharap, S.S.; Wang, Y.; Mehlig, M.; Chandna, P.; Zhao, H.; Filpula, D.; Yang, K.; Borowski, V.; et al. Antitumor activity of poly(ethylene glycol)-camptothecin conjugate: The inhibition of tumor growth in vivo. J. Control. Release 2005, 110, 90–102. [Google Scholar] [CrossRef]
- Caiolfa, V.R.; Zamai, M.; Fiorino, A.; Frigerio, E.; Pellizzoni, C.; Argy, R.; Ghiglieri, A.; Castelli, M.G.; Farao, M.; Pesenti, E.; et al. Polymer-bound camptothecin: Initial biodistribution and antitumour activity studies. J. Control. Release 2000, 65, 105–119. [Google Scholar] [CrossRef]
- Hollis, C.P.; Weiss, H.L.; Leggas, M.; Evers, B.M.; Gemeinhart, R.A.; Li, T. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: Lessons learned of the EPR effect and image-guided drug delivery. J. Control. Release 2013, 172, 12–21. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Koukouraki, S.; Fezoulidis, I.; Kelekis, N.; Kyrias, G.; Archimandritis, S.; Karkavitsas, N. High intratumoural accumulation of stealth® liposomal doxorubicin (Caelyx®) in glioblastomas and in metastatic brain tumours. Br. J. Cancer 2000, 83, 1281–1286. [Google Scholar] [CrossRef]
- Stapleton, S.; Allen, C.; Pintilie, M.; Jaffray, D.A. Tumour perfusion imaging predicts the intra-tumoral accumulation of liposomes. J. Control. Release 2013, 172, 351–357. [Google Scholar] [CrossRef]
- Greish, K.; Nagamitsu, A.; Fang, J.; Maeda, H. Copoly(styrene-maleic acid)-pirarubicin micelles: High tumor-targeting efficiency with little toxicity. Bioconjugate Chem. 2005, 16, 230–236. [Google Scholar] [CrossRef]
- Greish, K.; Sawa, T.; Fang, J.; Akaike, T.; Maeda, H. SMA-doxorubicin, a new polymeric micellar drug for effective targeting to solid tumours. J. Control. Release 2004, 97, 219–230. [Google Scholar] [CrossRef]
- Noguchi, Y.; Wu, J.; Duncan, R.; Strohalm, J.; Ulbrich, K.; Akaike, T.; Maeda, H. Early phase tumor accumulation of macromolecules: A great difference in clearance rate between tumor and normal tissues. Jpn. J. Cancer Res. 1998, 89, 307–314. [Google Scholar] [CrossRef]
- Kumazawa, E.; Ochi, Y. DE-310, a novel macromolecular carrier system for the camptothecin analog DX-8951f: Potent antitumor activities in various murine tumor models. Cancer Sci. 2004, 95, 168–175. [Google Scholar]
- Seymour, L.W.; Miyamoto, Y.; Maeda, H.; Brereton, M.; Strohalm, J.; Ulbrich, K.; Duncan, R. Influence of molecular weight on passive tumour accumulation of a soluble macromolecular drug carrier. Eur. J. Cancer 1995, 31, 766–770. [Google Scholar]
- Liu, T.-J.; Liu, S.; Hu, X.-L.; Sheng, S.-H.; Huang, Y.-B.; Jing, X.-B. EPR effect of amphiphilic copolymer micelles observed by fluorescent imaging. Chem. Res. Chin. Univ. 2011, 27, 628–634. [Google Scholar]
- Iwai, K.; Maeda, H.; Konno, T. Use of oily contrast medium for selective drug targeting to tumor: Enhanced therapeutic effect and X-ray image. Cancer Res. 1984, 44, 2115–2121. [Google Scholar]
- Iwai, K.; Maeda, H.; Konno, T.; Matsumura, Y.; Yamashita, R.; Yamasaki, K.; Hirayama, S.; Miyauchi, Y. Tumor targeting by arterial administration of lipids: Rabbit model with VX2 carcinoma in the liver. Anticancer Res. 1987, 7, 321–327. [Google Scholar]
- Etrych, T.; Kovář, L.; Strohalm, J.; Chytil, P.; Ríhová, B.; Ulbrich, K. Biodegradable star HPMA polymer-drug conjugates: Biodegradability, distribution and anti-tumor efficacy. J. Control Release 2011, 154, 241–248. [Google Scholar] [CrossRef]
- Heneweer, C.; Holland, J.P.; Divilov, V.; Carlin, S.; Lewi, J.S. Magnitude of enhanced permeability and retention effect in tumors with different phenotypes: 89Zr-albumin as a model system. J. Nucl. Med. 2011, 52, 625–633. [Google Scholar] [CrossRef]
- Borgman, M.P.; Aras, O.; Geyser-Stoops, S.; Sausville, E.A.; Ghandehari, H. Biodistribution of HPMA copolymer-aminohexylgeldanamycin-RGDfK conjugates for prostate cancer drug delivery. Mol. Pharm. 2009, 6, 1836–1847. [Google Scholar] [CrossRef]
- Lammers, T.; Kühnlein, R.; Kissel, M.; Subr, V.; Etrych, T.; Pola, R.; Pechar, M.; Ulbrich, K.; Storm, G.; Huber, P.; et al. Effect of physicochemical modification on the biodistribution and tumor accumulation of HPMA copolymers. J. Control. Release 2005, 110, 103–118. [Google Scholar] [CrossRef]
- Lammers, T.; Subr, V.; Peschke, P.; Kühnlein, R.; Hennink, W.E.; Ulbrich, K.; Kiessling, F.; Heimann, M.; Debus, J.; Huber, P.E.; et al. Image-guided and passively tumour-targeted polymeric nanomedicines for radiochemotherapy. Br. J. Cancer 2008, 99, 900–910. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rajora, A.K.; Ravishankar, D.; Osborn, H.M.I.; Greco, F. Impact of the Enhanced Permeability and Retention (EPR) Effect and Cathepsins Levels on the Activity of Polymer-Drug Conjugates. Polymers 2014, 6, 2186-2220. https://doi.org/10.3390/polym6082186
Rajora AK, Ravishankar D, Osborn HMI, Greco F. Impact of the Enhanced Permeability and Retention (EPR) Effect and Cathepsins Levels on the Activity of Polymer-Drug Conjugates. Polymers. 2014; 6(8):2186-2220. https://doi.org/10.3390/polym6082186
Chicago/Turabian StyleRajora, Amit K., Divyashree Ravishankar, Helen M. I. Osborn, and Francesca Greco. 2014. "Impact of the Enhanced Permeability and Retention (EPR) Effect and Cathepsins Levels on the Activity of Polymer-Drug Conjugates" Polymers 6, no. 8: 2186-2220. https://doi.org/10.3390/polym6082186
APA StyleRajora, A. K., Ravishankar, D., Osborn, H. M. I., & Greco, F. (2014). Impact of the Enhanced Permeability and Retention (EPR) Effect and Cathepsins Levels on the Activity of Polymer-Drug Conjugates. Polymers, 6(8), 2186-2220. https://doi.org/10.3390/polym6082186