
Multi-Scale Simulation of Hyperbranched Polymers


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Model and Simulation Method
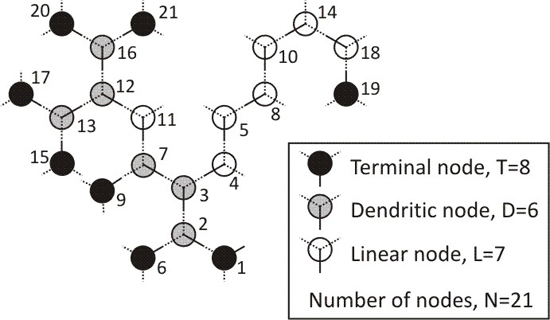
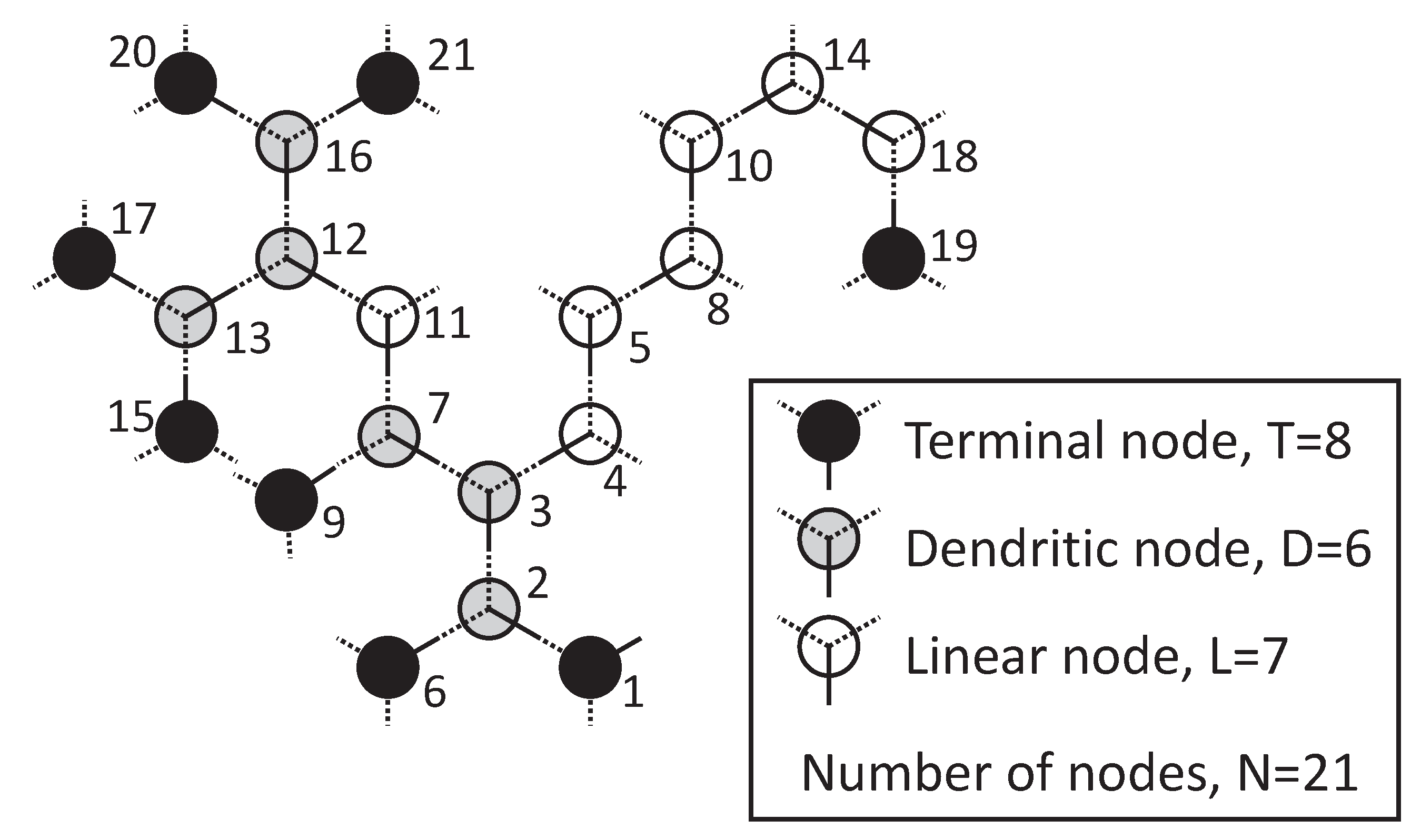
2.1. Topology
- (1)
- End or terminal nodes (T ) if they are connected to just 1 other node,
- (2)
- Linear nodes (L) if they are connected to 2 other nodes, and
- (3)
- Branching or dendritic nodes (D) if they are connected to f nodes, being f any number from 3 to F.

2.1.1. Degree of Branching
2.1.2. Generation of Chain Topology
2.2. Coarse-Grained Model
2.3. Monte Carlo Simulation and Hydrodynamics
2.4. Atomic-Level Calculations
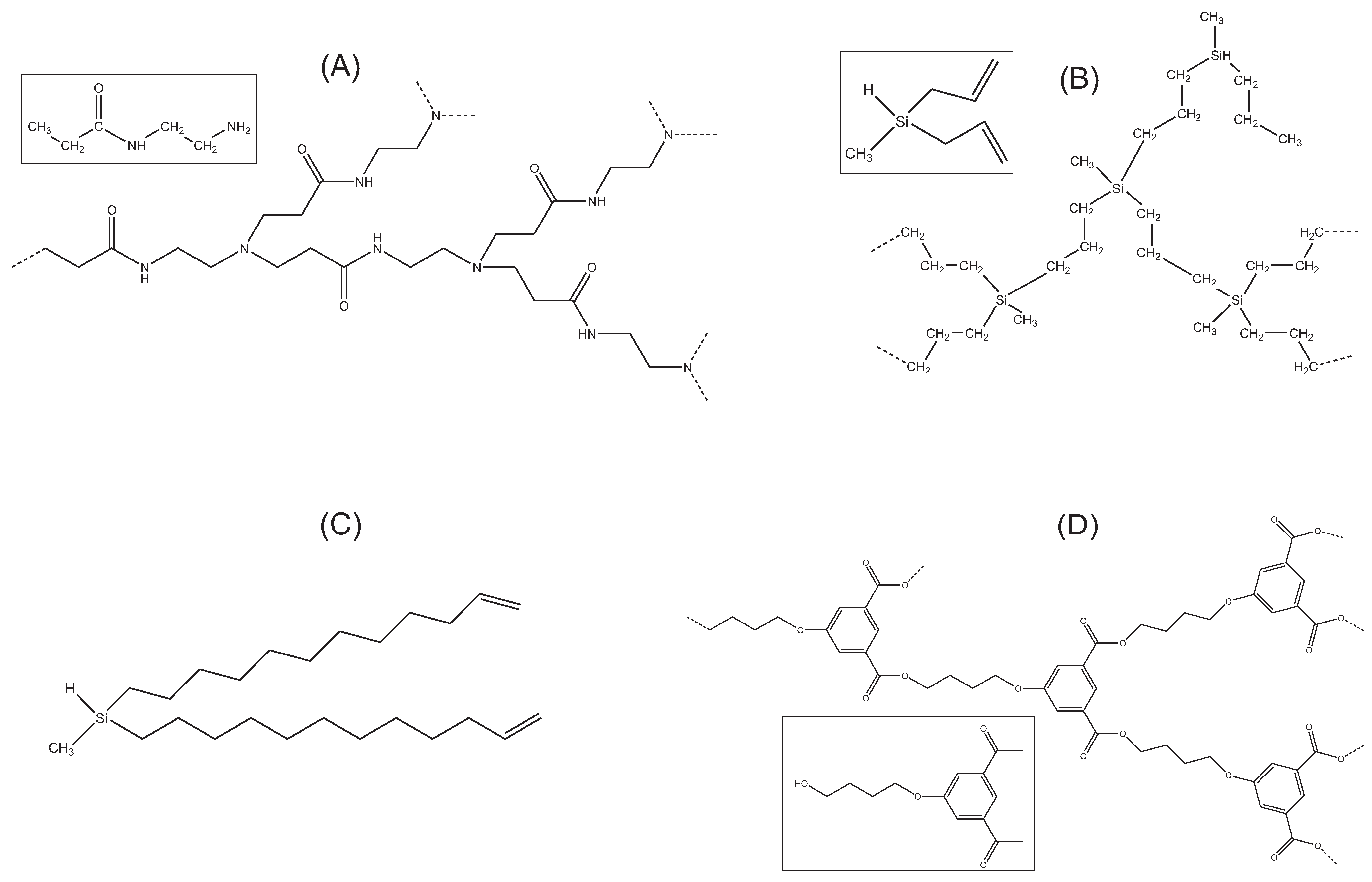
- Beads:The atomic-level simulation with HYPERCHEM allows to sweep the conformational space of the monomeric unit represented by a bead. From the atomic coordinates, the hydrodynamic Stokes radius (where ft is the friction coefficient of the particle and η0 the solvent viscosity), and the equivalent radius to radius of gyration , are computed for each conformation by using our public domain program HYDROPRO [22] (see http://leonardo.inf.um.es/macromol/programs/programs.htm). Regarding the possible influence of solvation on the effective hydrodynamic radius of the atoms, we follow previous experience [35] that, in the case of small molecular entities, such effective radius can be equated to the Van der Waals radius, of typically 1.8 Å [36]. The results are the averages over a sample of conformations. Because Rh turns out to be similar to RG, we take as the hydrodynamic radius of the bead representing the monomer unit their mean a = (RG + Rh)/2.In order to avoid free parameters in the model, the radius of the beads in the hard spheres EV potential was taken equal to a so that the contact distance becomes σHS = 2a. This choice seems convenient because it makes neighboring beads almost tangent and prevent “phantom” crosses of the connectors.
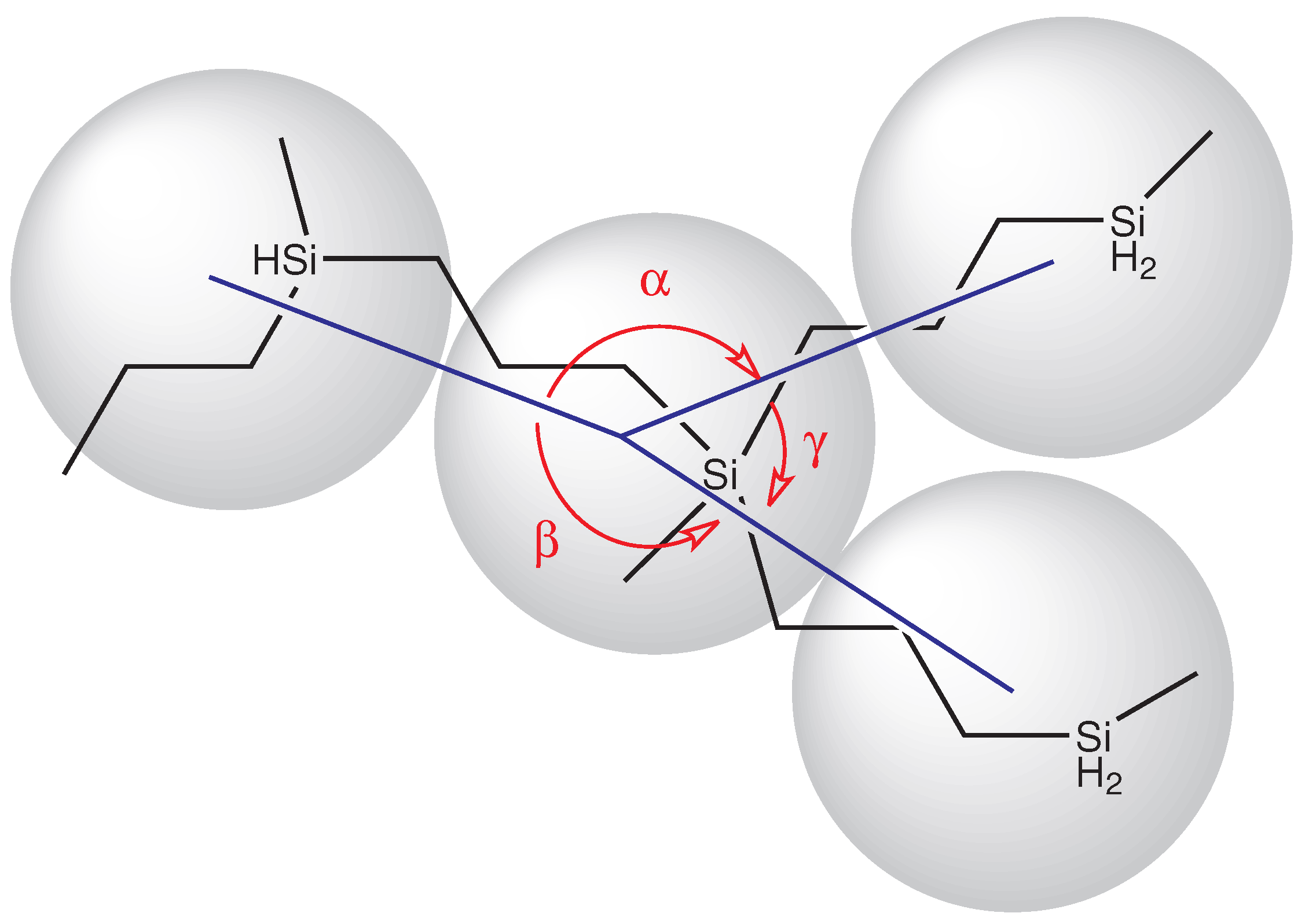
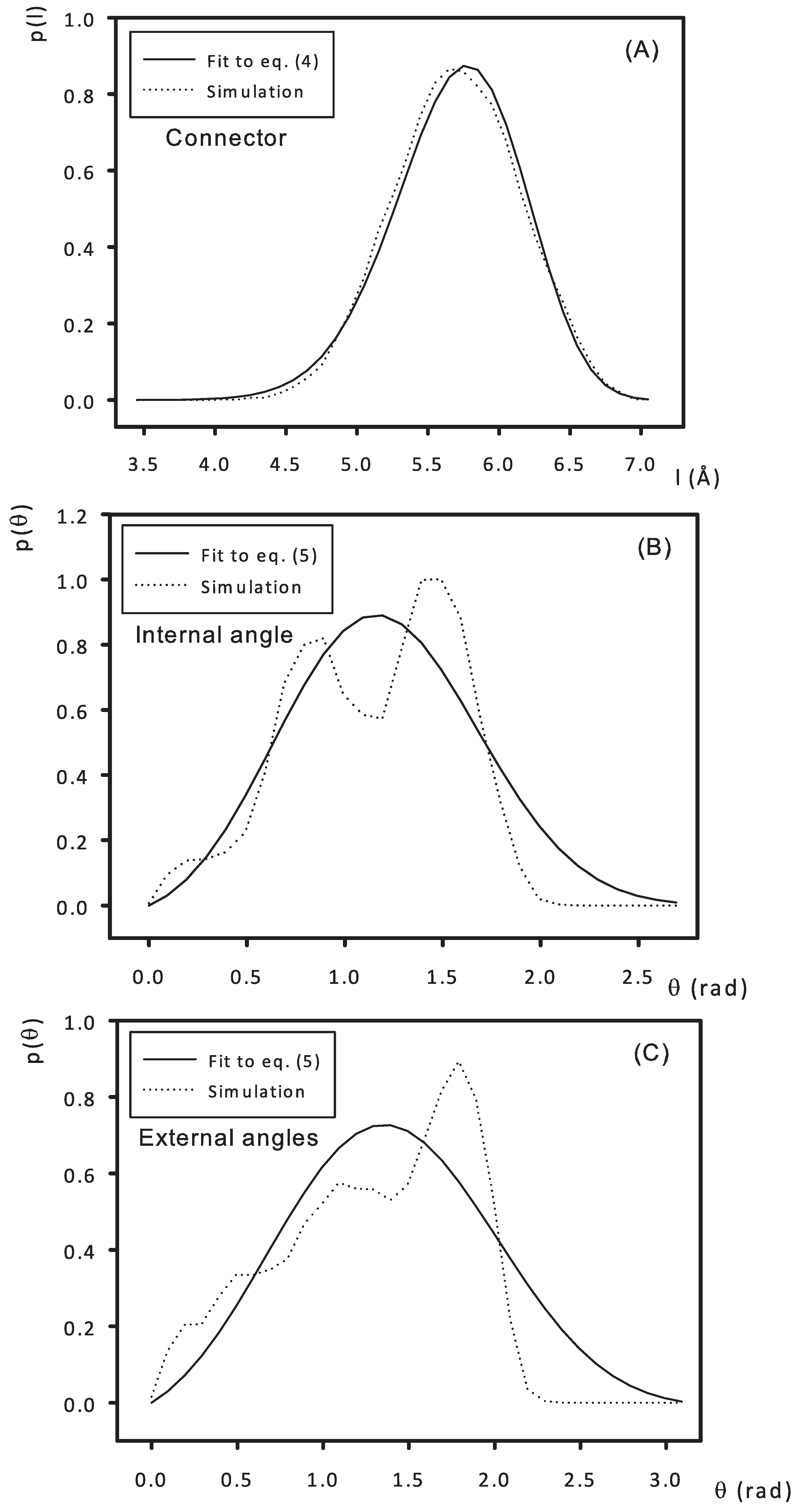
- Connector and angles:Parameters of the potentials associated to connectors and angles were estimated from atomic-level simulations of the minimal atomic structure that defines those connectors and angles, as illustrated in Figure 3 for a PCS3. From the atomic trajectories generated with program HYPERCHEM, the distribution functions for connector length (taken as the distance between the centers of mass of two connected monomers/beads) and the angle subtended by those connectors were obtained.We distinguish between two types of angles: “internal” angles subtended between the two B groups of a monomer and “external” angles subtended between the groups A and B of the monomer (see Figure 3). In terms of the formation process of the bead-and-connector model, the “internal” angle is that subtended between the two free connectors emerging from a newly incorporated node, and the “external” angle is that subtended between any one of those free connectors and the connector that links that new node to the chain structure. Then, the connector length distribution and the two angles distributions were fit, respectively, to the Boltzmann exponentials associated to the connector-spring potential and the angle-spring potentials (see Figure 4):p(l) = All2 exp[−V(l)/kBT]where Al and Aθ are normalization constants.p(θ) = Aθ sinθ exp[−V (θ)/kBT]


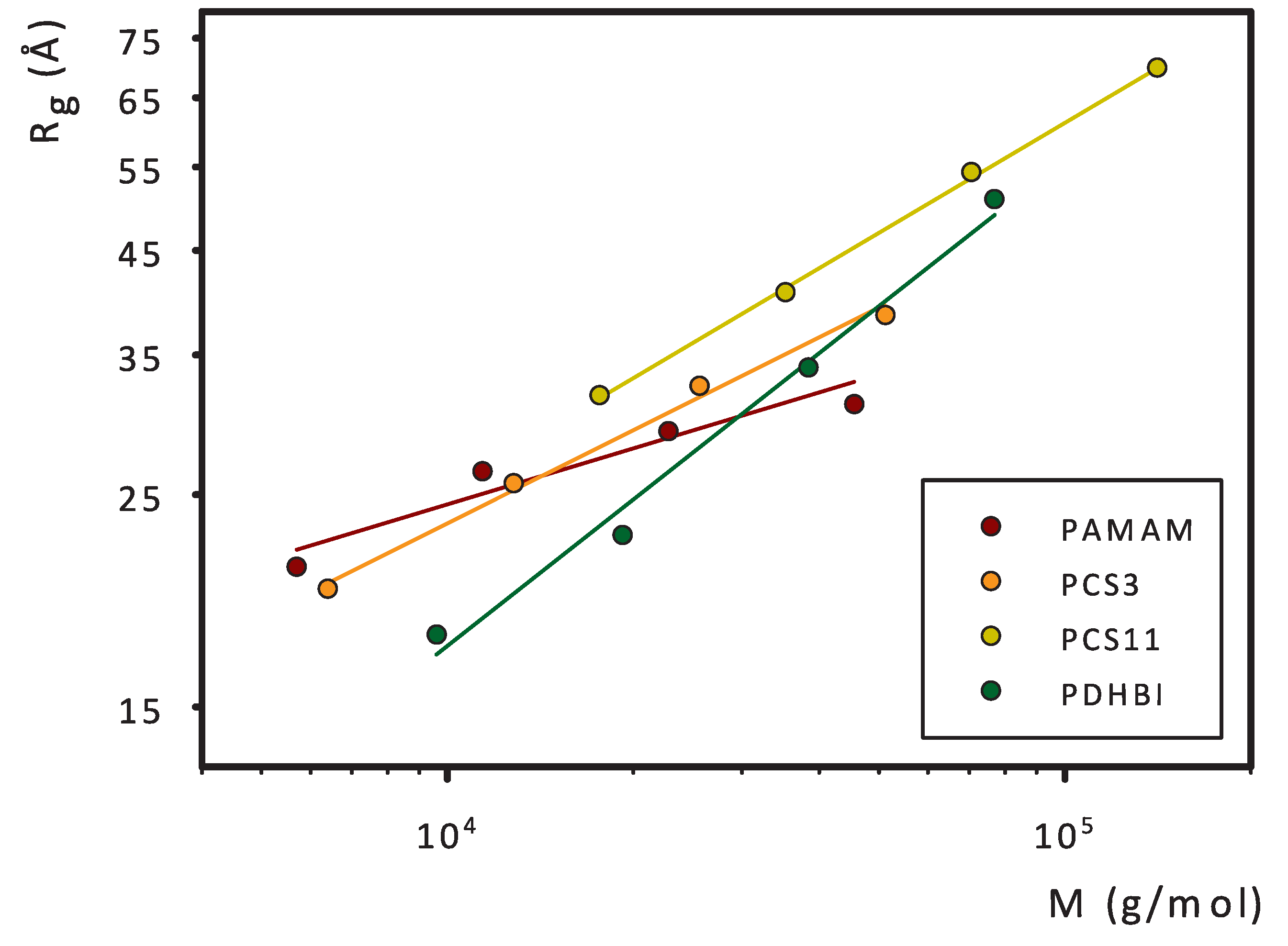
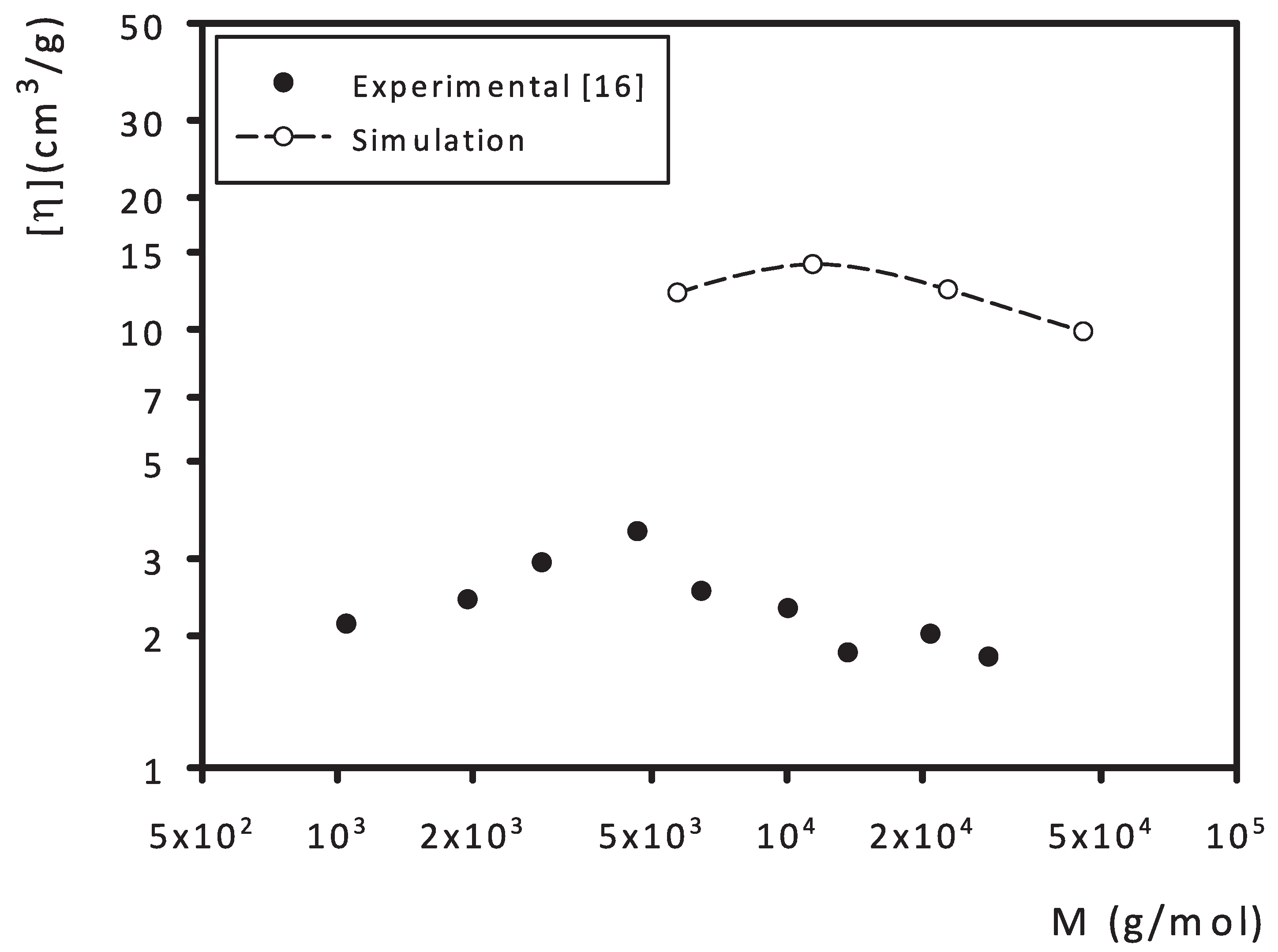
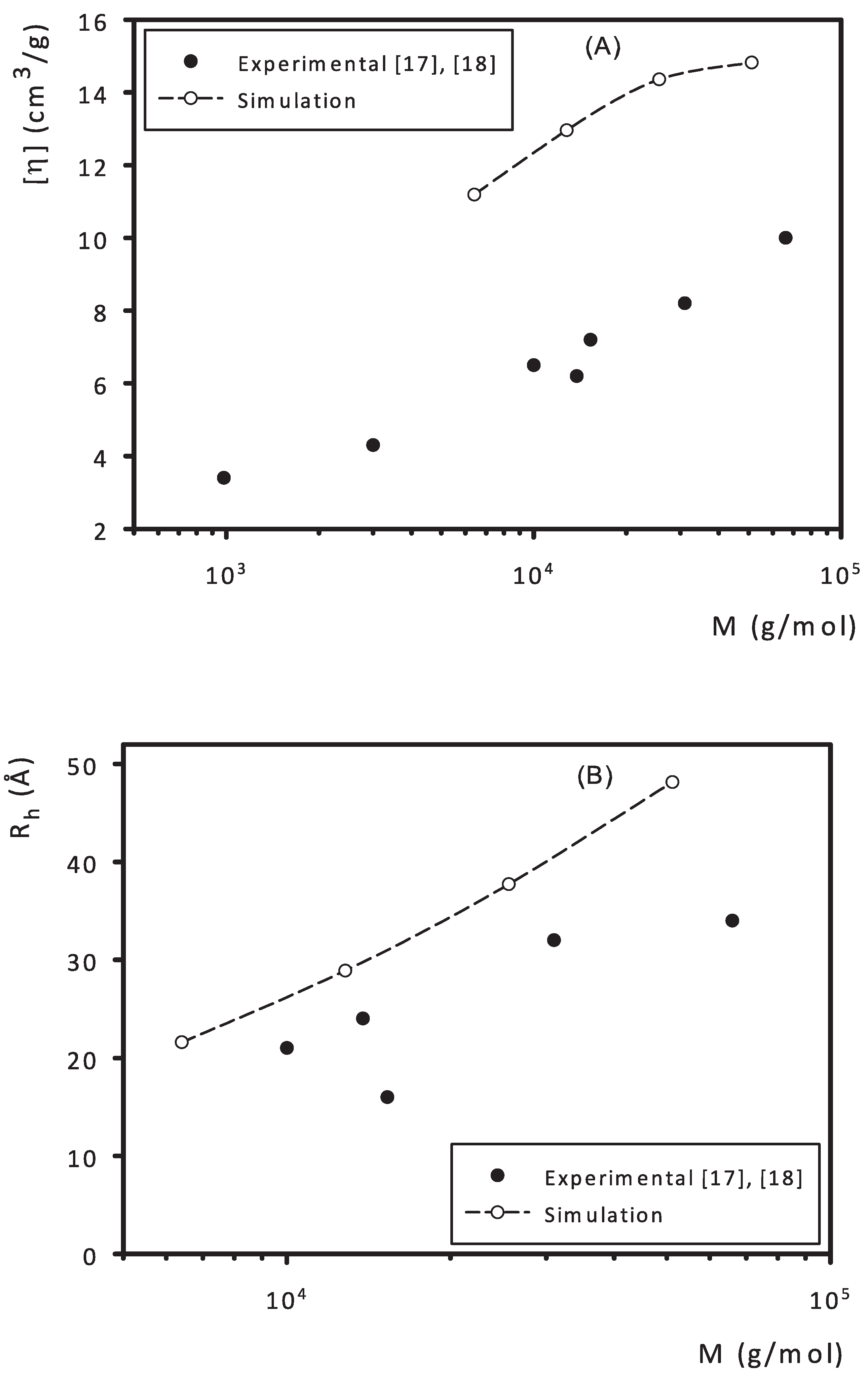
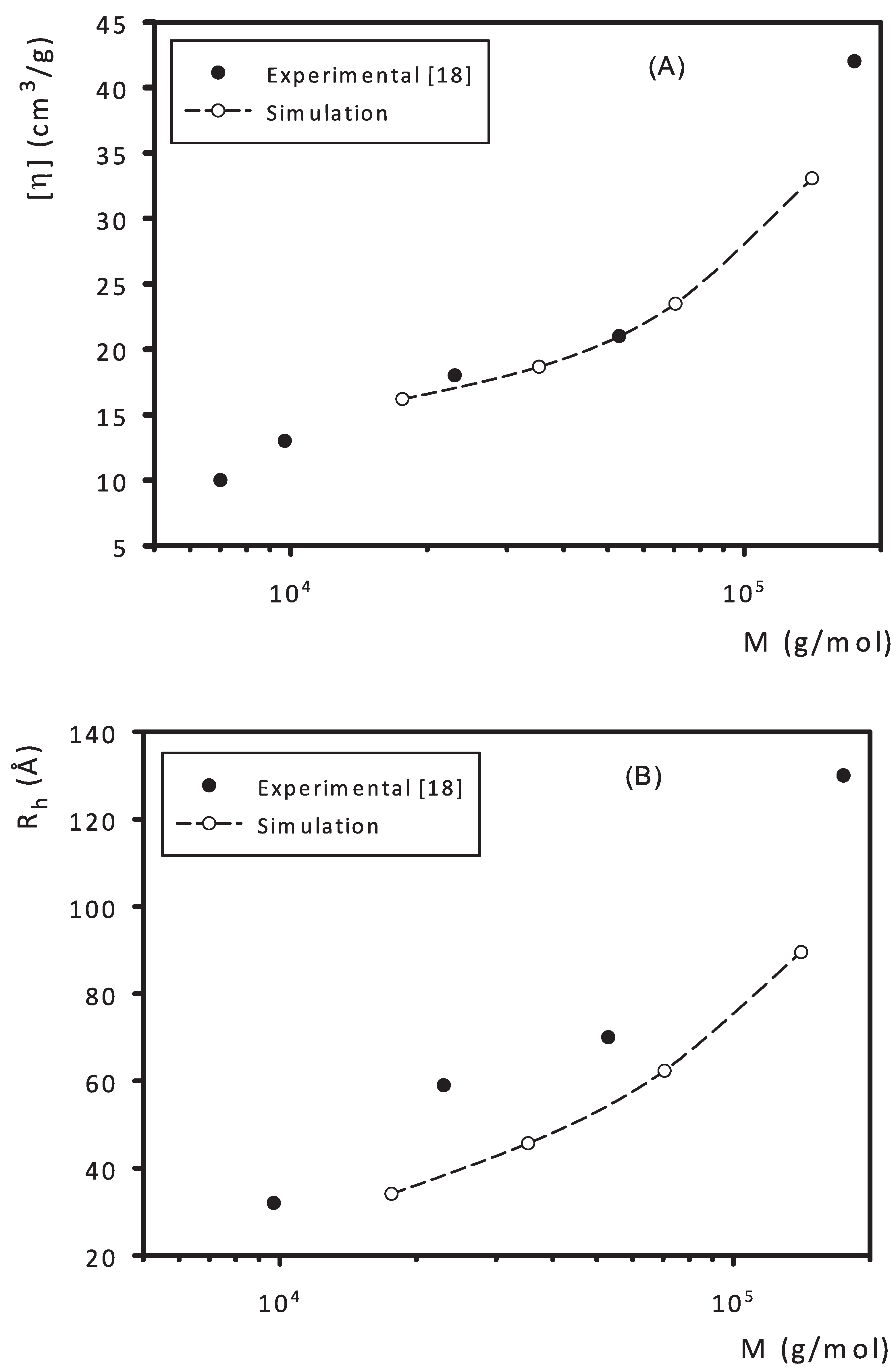
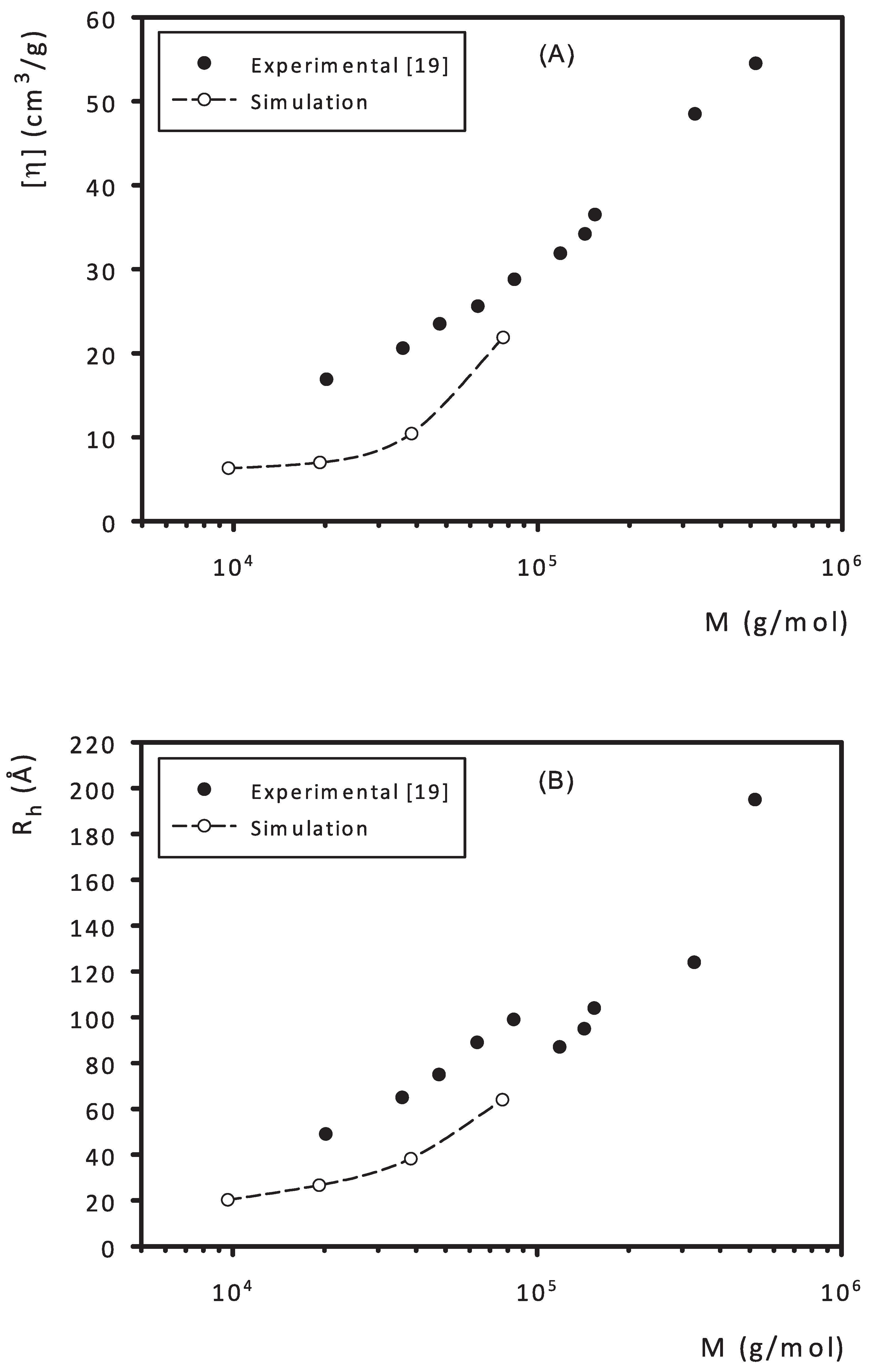
3. Results and Discussion





4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seiler, M. Hyperbranched polymers: Phase behavior and new applications in the field of chemical engineering. Fluid Phase Equilib. 2006, 241, 155–174. [Google Scholar] [CrossRef]
- Hawker, C.J.; Fréchet, J.M.J. Control of surface functionality in the synthesis of dendritic macromolecules using the convergent-growth approach. Macromolecules 1990, 23, 4726–4729. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular size distribution in three dimensional polymers. VI. Branched polymers containing A–R–Bf−1 type units. J. Am. Chem. Soc. 1952, 74, 2718–2723. [Google Scholar] [CrossRef]
- Zhang, B.; Yu, H.; Schlüter, A.D.; Halperin, A.; Kröger, M. Synthetic regimes due to packing constraints in dendritic molecules confirmed by labelling experiments. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Kröger, M.; Schlüter, A.D.; Halperin, A. Branching defects in dendritic molecules: Coupling efficiency and congestion effects. Macromolecules 2013, 46, 7550–7564. [Google Scholar] [CrossRef]
- Aerts, J. Prediction of intrinsic viscosities of dendritic, hyperbranched and branched polymers. Comput. Theor. Polym. Sci. 1998, 8, 49–54. [Google Scholar] [CrossRef]
- Widmann, A.H.; Davies, G.R. Simulation of the intrinsic viscosity of hyperbranched polymers built by sequential addition. Comput. Theor. Polym. Sci. 1998, 8, 191–199. [Google Scholar] [CrossRef]
- Lyulin, A.V.; Adolf, D.B.; Davies, G.R. Computer simulations of hyperbranched polymers in shear flows. Macromolecules 2001, 31, 3783–3789. [Google Scholar] [CrossRef]
- Sheridan, P.F.; Adolf, D.B.; Lyulin, A.V.; Neelov, I.; Davies, G.R. Computer simulations of hyperbranched polymers: The influence of the Wiener index on the intrinsic viscosity and radius of gyration. J. Chem. Phys. 2002, 117, 7802–7812. [Google Scholar] [CrossRef]
- Mulder, T.; Lyulin, A.V.; van der Schoot, P.; Michels, M.A.J. Architecture and conformation of uncharged and charged hyperbranched polymers: Computer simulation and mean-field theory. Macromolecules 2005, 38, 996–1006. [Google Scholar] [CrossRef]
- Wiener, H. Structural determination of paraffin boiling points. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Hölter, D.; Burgath, A.; Frey, H. Degree of branching in hyperbranched polymers. Acta Polym. 1997, 48, 30–35. [Google Scholar] [CrossRef]
- Del Río Echenique, G.; Hernández Cifre, J.G.; Rodríguez, E.; Rubio, A.; Freire, J.J.; García de la Torre, J. Multi-scale simulation of the conformation and dynamics of dendrimeric macromolecules. Chem. Macromol. Symp. 2007, 245, 386–389. [Google Scholar]
- Freire, J.J. Realistic numeric simulations of dendrimer molecules. Soft Matter 2008, 4, 2139–2143. [Google Scholar] [CrossRef]
- Del Río Echenique, G.; Rodríguez Schmidt, R.; Freire, J.J.; Hernández Cifre, J.G.; García de la Torre, J. A multiscale scheme for the simulation of conformational and solution properties of different dendrimer molecules. J. Am. Chem. Soc. 2009, 131, 8548–8556. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Schmidt, R.; Pamies, R.; Kjøniksen, A.L.; Zhu, K.; Hernández Cifre, J.G.; Nyström, B.; García de la Torre, J. Single-molecule behavior of asymmetric thermoresponsive amphiphilic copolymers in dilute solution. J. Phys. Chem. B 2010, 114, 8887–8893. [Google Scholar] [CrossRef] [PubMed]
- Maleki, A.; Zhu, K.; Pamies, R.; Rodríguez Schmidt, R.; Kjøniksen, A.L.; Karlsson, G.; Hernández Cifre, J.G.; García de la Torre, J.; Nyström, B. Effect of polyethylene Glycol (PEG) length on the association properties of temperature-sensitive amphiphilic triblock copolymers (PNIPAAMm-b-PEGn-b-PNIPAAMm) in aqueous solution. Soft Matter 2011, 7, 8111–8119. [Google Scholar] [CrossRef]
- Hobson, L.J.; Feast, W.J. Poly(amidoamine) hyperbranched systems: Synthesis, structure and characterization. Polymer 1999, 40, 1279–1297. [Google Scholar] [CrossRef]
- Tarabukina, E.B.; Shpyrkov, A.A.; Potapova, D.V.; Filippov, A.P.; Shumilkina, N.A.; Muzafarov, A.M. Hydrodynamic and conformational properties of a hyperbranched polymethylallylcarbosilane in dilute solutions. Polym. Sci. Ser. A 2006, 48, 974–980. [Google Scholar] [CrossRef]
- Tarabukina, E.B.; Shpyrkov, A.A.; Tarasova, E.V.; Amirova, A.I.; Filippov, A.P.; Sheremet’eva, N.A.; Muzafarov, A.M. Effect of the length of branches on hydrodynamic and conformational properties of hyperbranched polycarbosilanes. Polym. Sci. Ser. A 2009, 51, 150–160. [Google Scholar] [CrossRef]
- De Luca, E.; Richards, R.W. Molecular characterization of a hyperbranched polyester. I. Dilute solution properties. J. Polym. Sci. B 2003, 41, 1339–1351. [Google Scholar] [CrossRef]
- García de la Torre, J.; Huertas, M.L.; Carrasco, B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structures. Biophys. J. 2000, 78, 719–730. [Google Scholar] [CrossRef] [PubMed]
- García de la Torre, J.; Pérez Sánchez, H.E.; Ortega, A.; Hernández Cifre, J.G.; Fernandes, M.X.; Díaz Baños, F.G.; López Martínez, M.C. Calculation of the solution properties of flexible macromolecules: Methods and applications. Eur. Biophys. J. 2003, 32, 477–486. [Google Scholar] [CrossRef] [PubMed]
- García de la Torre, J.; Ortega, A.; Pérez Sánchez, H.E.; Hernández Cifre, J.G. MULTIHYDRO and MONTEHYDRO: Conformational search and Monte Carlo calculation of solution properties of rigid and flexible macromolecular models. Biophys. Chem. 2005, 116, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; An, L.; Wang, Z.-G. Intrinsic viscosity of polymers: General theory based on partially permeable sphere model. Macromolecules 2013, 46, 5731–5740. [Google Scholar] [CrossRef]
- Bird, R.B.; Curtiss, C.F.; Armstrong, R.C.; Hassager, O. Dynamics of Polymeric Liquids, Kinetic Theory, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1987; Volume 2. [Google Scholar]
- Zimm, B.H. Chain molecule hydrodynamics by the Monte-Carlo method and the validity of the Kirkwood–Riseman approximation. Macromolecules 1980, 13, 592–602. [Google Scholar] [CrossRef]
- García de la Torre, J.; Jiménez, A.; Freire, J.J. Monte Carlo calculation of hydrodynamic properties of freely jointed, freely rotating and real polymethylene chains. Macromolecules 1982, 15, 148–154. [Google Scholar] [CrossRef]
- García de la Torre, J.; Freire, J.J. Intrinsic viscosities and translational diffusion coefficients of n-alkanes in solution. Macromolecules 1982, 15, 155–159. [Google Scholar] [CrossRef]
- Kirkwood, J.G.; Riseman, J. The intrinsic viscosities and diffusion constants of flexible macromolecules in solution. J. Chem. Phys. 1948, 16, 565–573. [Google Scholar] [CrossRef]
- Kirkwood, J.G. The general theory of irreversible processes in solutions of macromolecules. J. Polym. Sci. 1954, 12, 1–14. [Google Scholar] [CrossRef]
- Carrasco, B.; Garcia de la Torre, J. Intrinsic viscosity and rotational diffusion of bead models for rigid macromolecules and bioparticles. Eur. Biophys. J. 1998, 27, 549–557. [Google Scholar] [CrossRef]
- Rodríguez, E.; Freire, J.J.; Del Río Echenique, G.; Hernández Cifre, J.G.; García de la Torre, J. Improved simulation method for the calculation of the intrinsic viscosity of some dendrimer molecules. Polymer 2007, 48, 1155–1163. [Google Scholar] [CrossRef]
- García de la Torre, J.; del Río Echenique, G.; Ortega, A. Improved calculation of rotational diffusion and intrinsic viscosity of bead models for macromolecules and nanoparticles. J. Phys. Chem. B 2007, 111, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, P.; García de la Torre, J. Theoretical prediction of translational diffusion coefficients of small rigid molecules. J. Phys. Chem. 1987, 91, 3612–3616. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Wang, L.; He, X. Investigation of ABn (n = 2, 4) type hyperbranched polymerization with cyclization and steric factors: Influences of monomer concentration, reactivity, and substitution effect. J. Polym. Sci. Pol. Chem. 2009, 47, 523–533. [Google Scholar] [CrossRef]
- Karatasos, K.; Adolf, D.B.; Davies, G.R. Statics and dynamics of model dendrimers as studied by molecular dynamics simulations. J. Chem. Phys. 2001, 115, 5310–5318. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, R.R.; Hernández Cifre, J.G.; De la Torre, J.G. Multi-Scale Simulation of Hyperbranched Polymers. Polymers 2015, 7, 610-628. https://doi.org/10.3390/polym7040610
Schmidt RR, Hernández Cifre JG, De la Torre JG. Multi-Scale Simulation of Hyperbranched Polymers. Polymers. 2015; 7(4):610-628. https://doi.org/10.3390/polym7040610
Chicago/Turabian StyleSchmidt, Ricardo Rodríguez, José Ginés Hernández Cifre, and José García De la Torre. 2015. "Multi-Scale Simulation of Hyperbranched Polymers" Polymers 7, no. 4: 610-628. https://doi.org/10.3390/polym7040610