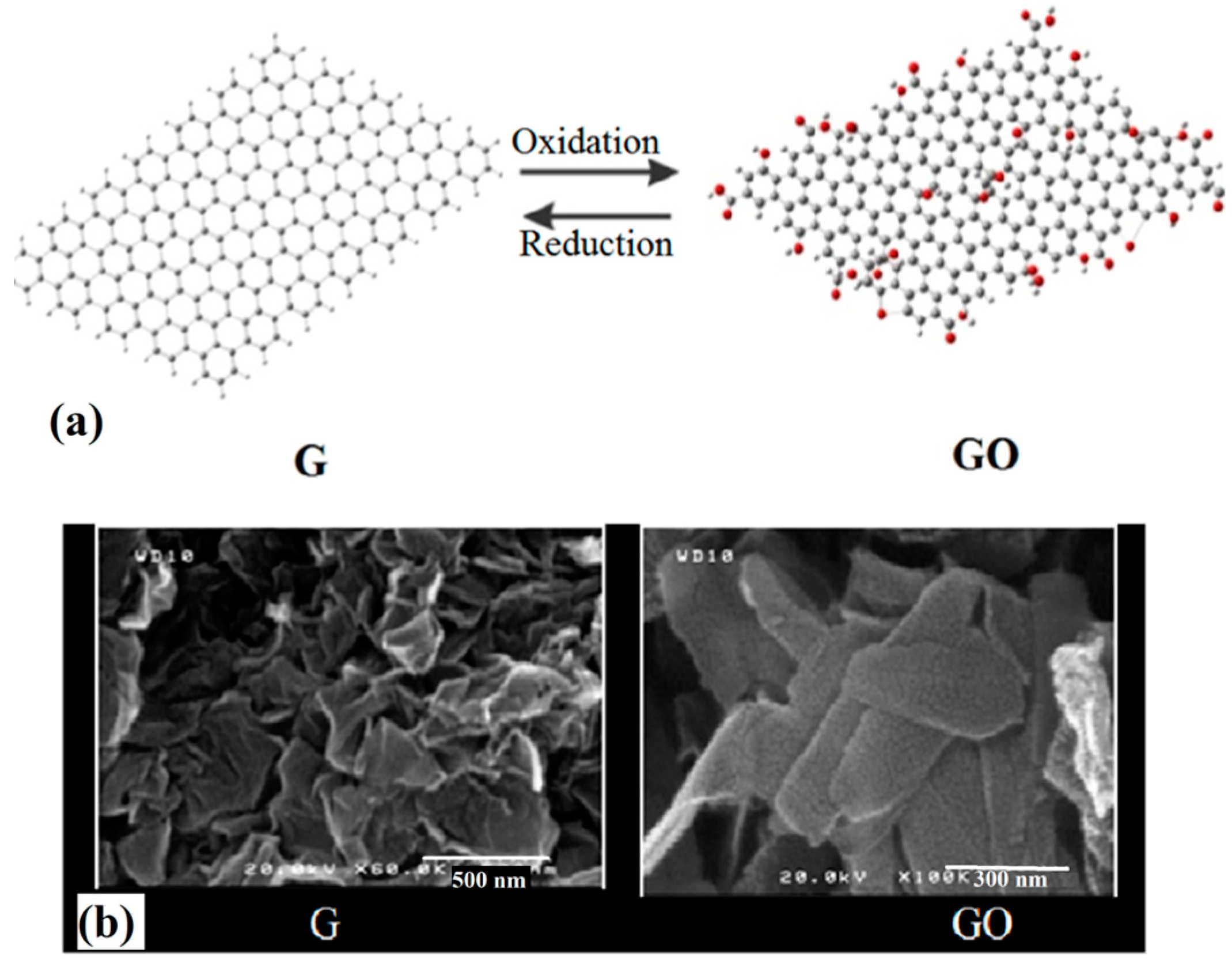
3.1. Morphology and Crystallographic Studies
Pristine and composite PVDF nanofibers were successfully synthesized with the morphologies that have been depicted in
Figure 3. The FESEM micrographs demonstrate that addition of G in the PVDF precursor solution significantly influence the nanofibers morphology and thicknesses. The diameters of the nanofibers were increased in the presence of G and the increasing of its concentration in the precursor solution, cause the diameter of the produced fibers to increase. From
Figure 3a, pristine PVDF nanofibrous film (produced by electrospinning of PVDF 10.0 wt% precursor solution), contains the fibers with diameters in the range of 100–250 nm. Fibers’ surfaces are very smooth, without any observable roughness. In the case of PVDF/G film (produced by electrospinning of PVDF/G, 10:1, precursor solution), as shown in
Figure 3b, the synthesized fibers’ diameters span the range of 350 nm–1 µm. In addition, the fibers’ surface roughening, due to G interposing, is depicted. The bead formation on the surface of the fibers is also observed, which is related to those G flakes that could not interpose inside the fibers during the electrospinning. Synthesized G flakes have with range of sizes (100–500 nm, from
Figure 1b), However, during the PVDF/G solution preparation, as well as electrospinning process, G flakes likely get ruptured, which results in their size reduction.
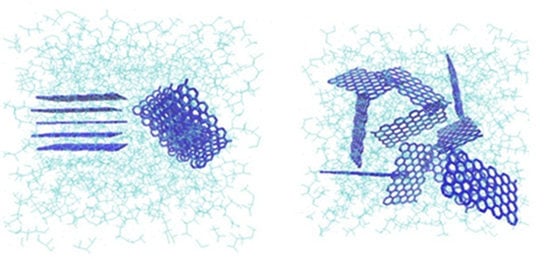
Proper arrangements of the G flakes inside the fibers plays an important role in the produced fibers final characteristics. In the presence of the strong electrical field that is applied for electrospinning, G flakes are likely to arrange parallel to the fiber axis (
Figure 4a) and they rarely interpose into the fiber’s cross section, which is not favored from a mechanical strength point of view.
Figure 3.
FESEM images of the prepared films: (a) M0; (b) M4; (c) M'0, (M0 and M4 represent the prepared electrospun membranes respectively with 0.0 and 1.0 wt% of grapheme; M'0 is for the pristine membrane prepared via phase separation method).
Figure 3.
FESEM images of the prepared films: (a) M0; (b) M4; (c) M'0, (M0 and M4 represent the prepared electrospun membranes respectively with 0.0 and 1.0 wt% of grapheme; M'0 is for the pristine membrane prepared via phase separation method).
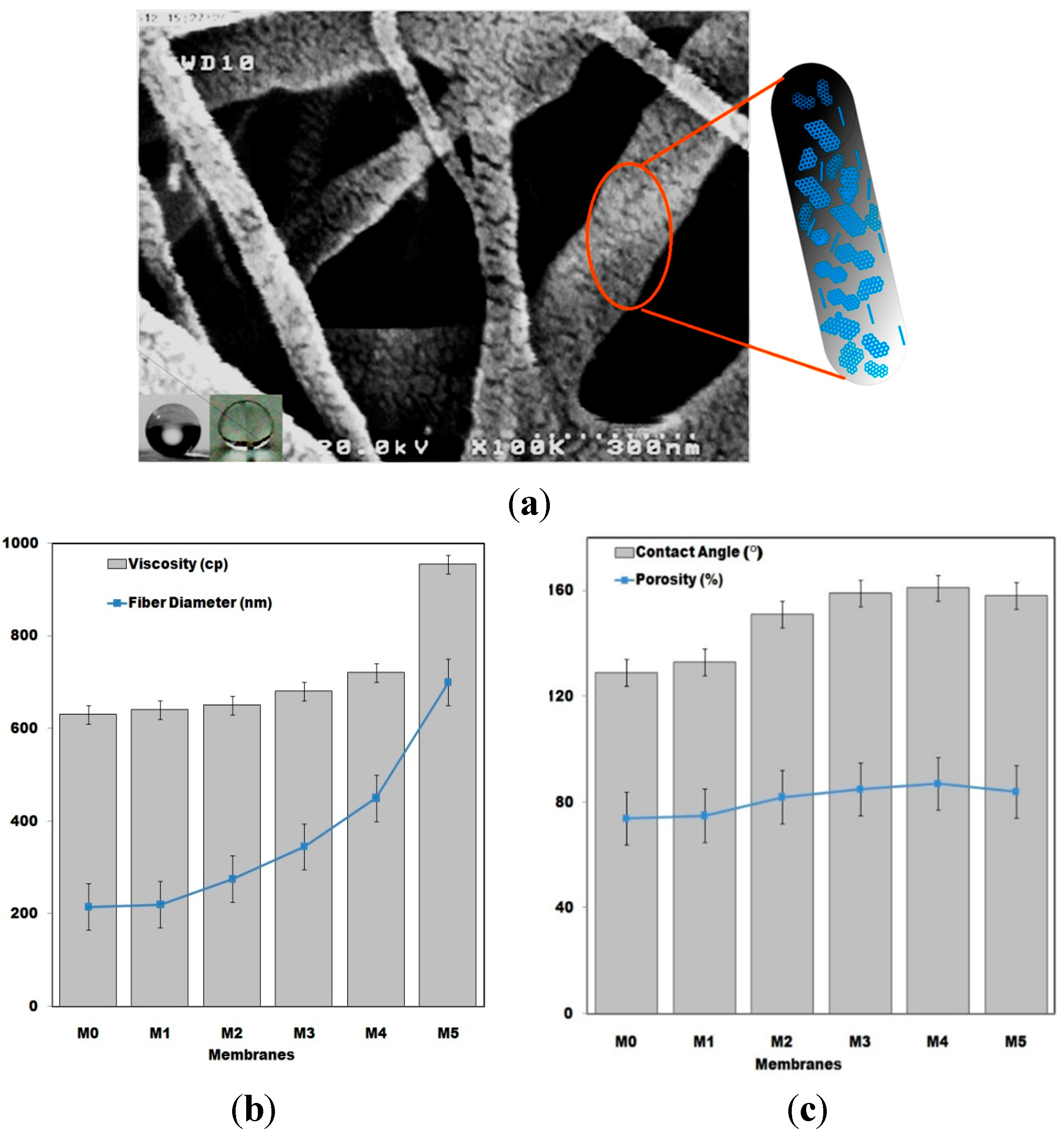
Fabricated pristine and composite PVDF fiber thickness variation, as a function of G amount in various films, as well as the viscosity change of the polymeric precursor solution are represented in
Figure 4b. It was observed that the viscosity of the precursor solution increases with G addition, which directly influences the synthesized fibers diameters. However, the average diameter for the produced PVDF nanofibers, M1, were 150 nm, whereas for the PVDF/G, M4, the average diameter of about 400 nm was obtained. It seems that viscosity increases in the PVDF/G solution could not be the sole reason of fiber diameters increasing. G flakes’ entrance into the PVDF fiber structure affects the fiber diameters by acting as reinforcement and nucleation agents. In addition, G addition affects the PVDF crystallization process that result in the changes of produced film hydrophobicity and porosity (
Figure 4c).
Figure 4.
(a) FE-SEM photograph of the PVDF/G nanofibrous membrane (M4). Water droplet image on the film surface and obtained wettability measurement image are also depicted. The schematic representation of G flakes interposed inside the PVDF fibers is shown at the right side of the image; (b) Precursor solution viscosity and fiber diameter changes of the prepared membranes; (c) Porosity and contact angle tests results for the prepared membranes. (M0, M1, M2, M3, M4 and M5 represent the membranes which were prepared via electrospinning with grapheme content of 0.0, 0.01, 0.03, 0.50, 1.0 and 1.5 wt % respectively).
Figure 4.
(a) FE-SEM photograph of the PVDF/G nanofibrous membrane (M4). Water droplet image on the film surface and obtained wettability measurement image are also depicted. The schematic representation of G flakes interposed inside the PVDF fibers is shown at the right side of the image; (b) Precursor solution viscosity and fiber diameter changes of the prepared membranes; (c) Porosity and contact angle tests results for the prepared membranes. (M0, M1, M2, M3, M4 and M5 represent the membranes which were prepared via electrospinning with grapheme content of 0.0, 0.01, 0.03, 0.50, 1.0 and 1.5 wt % respectively).
Another aspect is that G acting as the nucleation agent in polymer matrix improves the crystallization process in the formation of PVDF nanofibers. The surface roughness of PVDF/G nanofibers is higher than PVDF nanofibers, which directly affects the wettability properties of the nanofibers. XRD crystallographic analyses indicate that PVDF chains arrange in various crystallization forms in PVDF/G rather than pristine PVDF nanofibers. From
Figure 5, increases in the intensities of the achieved diffraction peaks at 2θ = 18° and 20° is due to the enhancement of the crystallization zones in PVDF/G. In a similar study about the morphology of pure PVDF and PVDF/G composite films, which were prepared via phase inversion method, the same observations have been reported [
8]. The diffraction peak near 2θ = 25° is characteristic of the PVDF α phase crystal structure. G addition affects polymorphism of the PVDF in favor of β phase. The disappearance of the diffraction peak at 2θ = 25° for PVDF/G, is due to formation of β phase in PVDF/G crystallization zones [
30,
31,
32].
Figure 5.
XRD patterns of (a) PVDF/G composite nanofibers (M4) and (b) pristine PVDF nanofibers (M0).
Figure 5.
XRD patterns of (a) PVDF/G composite nanofibers (M4) and (b) pristine PVDF nanofibers (M0).
Based on previous studies, the morphology of the PVDF film is also dependent on its formation mechanisms, where the existence of G in PVDF/G nanocomposite changes the morphology to assemble nanofibers with a diameter around 250 nm. Due to the good affinity of PVDF to carbon surfaces, like G sheets, the immobilization of PVDF chains on G sheets takes place, which act as the crystallization nuclei for the polymer chains. Previous studies also reported the similar phenomenon in the compositing of PVDF with carbon nanomaterials, such as carbon nanotubes [
32]. G improves not only the morphology of the synthesized film but also affects the porosity of the prepared films (
Figure 4c). The PVDF/G films likely have large pore-sizes and surface area in comparison with pure PVDF porous materials. As shown in
Figure 3a and
Figure 4a, in the cellular structure of the prepared mats different sizes of fibers span the range from 100 nm to a few micrometers, with observable surface roughness. This is because of the different sizes of G flakes that interposed inside the produced fibers’ structures and, hence, change their thicknesses in the wide range. Simultaneous presence of the nano and micro fibers is one of the novel properties of the synthesized PVDF/G films that promotes the film properties in some aspects of porosity, hydrophobicity,
etc. [
33,
34,
35,
36,
37].
SEM images from M4 and M1 films indicate that the net-like nanoporous structure is achievable for both; however, the nanofibers’ thicknesses in the case of M4 are greater than M1. Additionally, thicker PVDF/G nanofibers, in combination with better crystallization, cause changes to the effectiveness of their surface nanostructured morphology. This kind of structure could trap the air when it makes contact with water drops, enforcing the upward micro-pressures which strengthen the hydrophobicity.
3.2. Spectroscopic Analyses Results
For better understanding of the prepared film’s chemical structure, the FT-IR and micro-Raman studies have been performed.
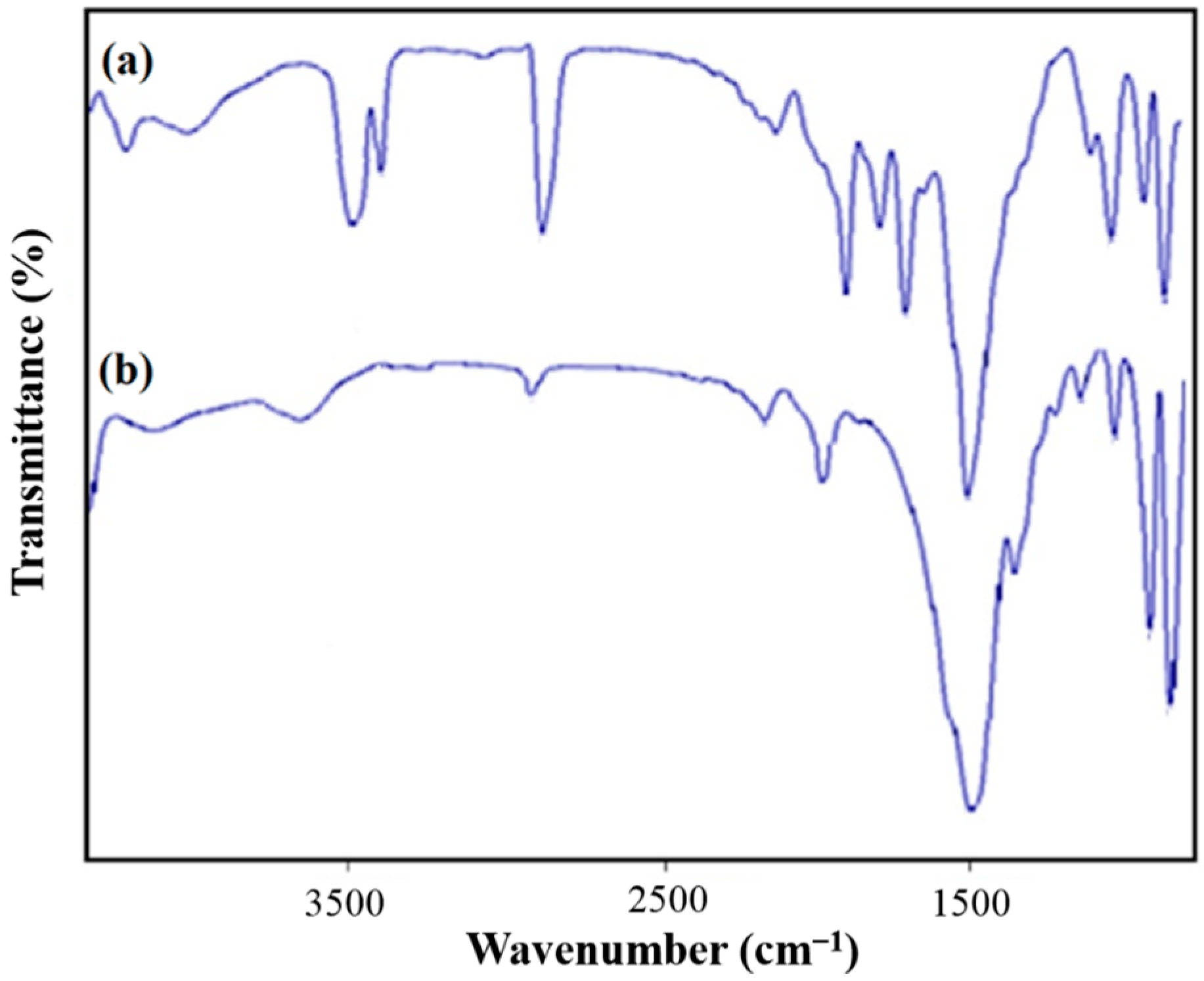
Figure 6 shows the obtained FT-IR spectra for the synthesized PVDF, PVDF/G electrospun films. FT-IR analysis was carried out for surface characterization of PVDF nanofibrous films in the range of 400–3800 cm
−1. In the case of PVDF film, three strong peaks at about 850, 1400 and 1200 cm
−1 were observed. The former two peaks were due to the C–F stretching vibration, and the latter one was due to the C–C bond of PVDF and the two weak bands appeared at 510 and 486 cm
−1 were assigned to the CF
2 bending vibration [
37,
38].
Figure 6.
FT-IR spectra of (a) PVDF/G nanofibers (M4) and (b) pristine PVDF nanofibers (M0).
Figure 6.
FT-IR spectra of (a) PVDF/G nanofibers (M4) and (b) pristine PVDF nanofibers (M0).
For PVDF/G composite film, G affects the infrared absorption spectra with the appearance of absorption peaks at about 1700, 2000 and 3000 cm
−1. The absorption bands around 1700 cm
−1 show the existence of C–O bond, which is related to the bonds on the surface of G flakes. The strengthening of the broad peak around 3000 cm
−1 confirms the formation of hydrogen bonds between the hydroxyl groups of G flakes and the fluorine of PVDF. In fact, this confirms that there are yet some hydroxyl, epoxide and carboxylic groups on the edges of G planes, which could bond to PVDF chains. For PVDF/GO composites, this peak is broadening in the wider range than PVDF/G due to numerous functional groups on the basal planes and edges of GO. Interactions between G flakes and PVDF chains could be helpful to enhance dispersion of G in the polymer matrix during the electrospinning process, in which G flakes are likely to move from the polymer matrix to the surface of the formed nanofibers [
34,
35]. There is also an increase in the intensity of the peak at around 1000 cm
−1, with loading the G and GO into the PVDF, which corresponds to the increasing of the PVDF β phase [
27,
31].
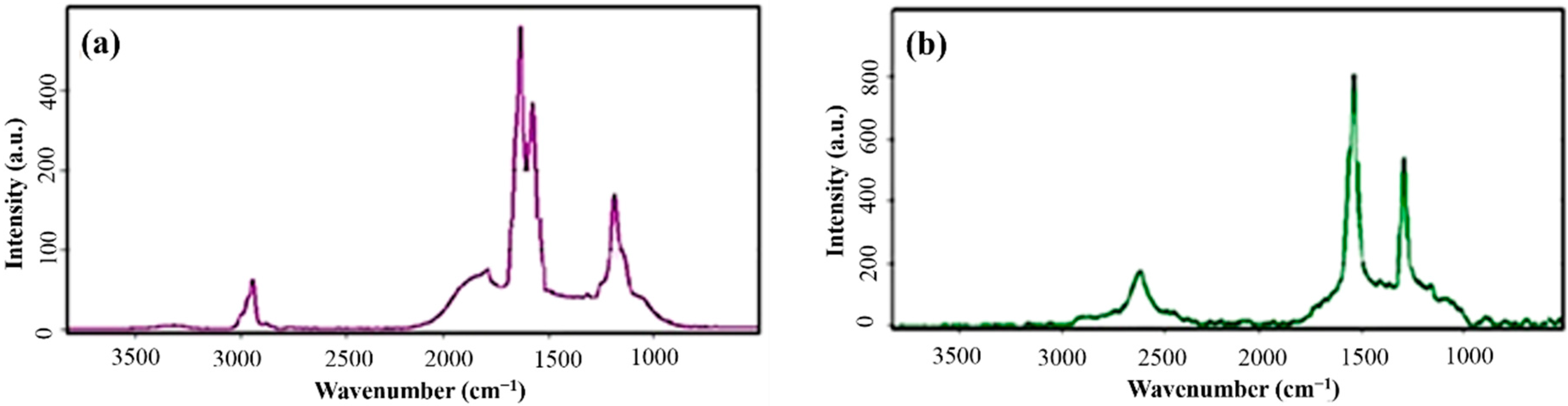
Raman spectroscopy was used for further study of the changes of G layers in combination with polymeric chains and the correspond influence on the PVDF structure. Micro-Raman spectra of the synthesized films for PVDF/G electrospun nanocomposite film and pure G powder are shown in
Figure 7. Introduction of G nano-sheets into the polymer matrix, as well as intercalating of the polymer chains among G layers confirmed by the changes, could be proven by both of the appearance of the peaks and shifts. The three peaks of PVDF/G at 1234, 1589 and 2915 cm
−1 correspond to D (defect) - band, G-band and 2D-band, respectively.
Figure 7.
Micro-Raman spectra of (a) PVDF/G composite membrane and (b) synthesized pure graphene.
Figure 7.
Micro-Raman spectra of (a) PVDF/G composite membrane and (b) synthesized pure graphene.
In comparison with the G band at 1564 cm
−1 of a few layers of pure graphene, this peak splits to two distinct peaks at 1531 and 1589 cm
−1 in PVDF/G nanocomposite electrospun film. The splitting in the G-bond occurs due to the compositing of G by fluorinated compounds, such as PVDF, and has been reported in some earlier studies, especially about the molecular-decorated G by F
2 [
33], G interaction with aromatic molecules [
34] or G doping by fluorinated hydrocarbons [
35] in a plasma environment. The observation of G-bond splitting in prepared PVDF/G electrospun nanocomposite film may refer to the interaction between PVDF polymer chains and G flake surfaces.
Raman shift increasing by blending of the G with PVDF, related to intercalating of polymer between G platelets and their segregation. The observed red-shift in D-bond from 2600 cm
−1 in pure G to 2915 cm
−1 in PVDF/G electrospun nanocomposite film and also decreasing of its intensity, both indicating that G flakes exfoliated in PVDF polymer matrix which provides their good integration with polymer chains in the prepared electrospun films [
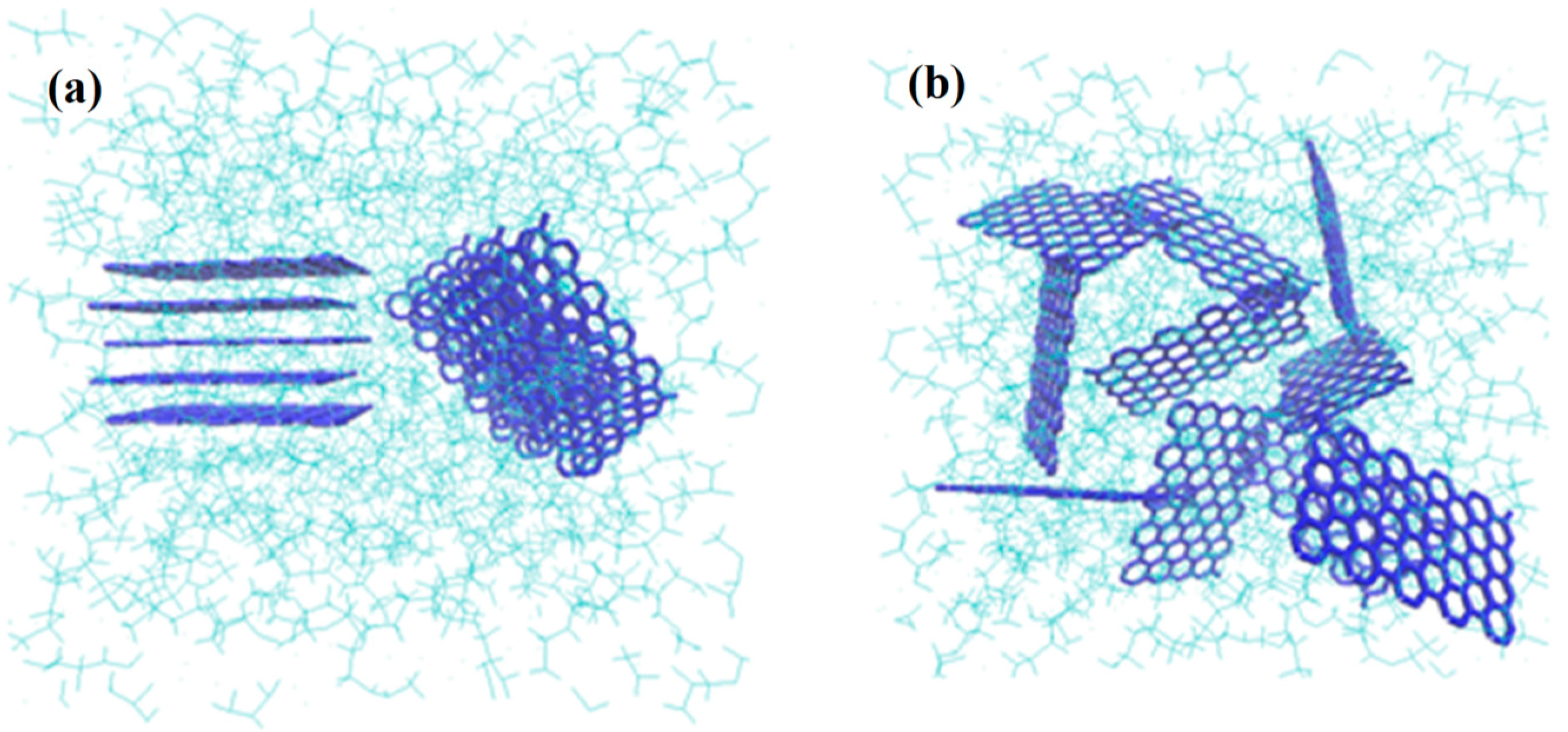
37]. The intercalation of G layers that is schematically shown in
Figure 8a improves its exfoliation and dispersion in the polymer solution and enriches as depicted in
Figure 8b. G flake exfoliation inside the PVDF matrix plays the important role in the observed morphological properties changes.
Figure 8.
PVDF chains interposing among the G flakes: (a) intercalation and (b) exfoliation of the G layers (Green colored structure represents the polymer matrix wherein the graphene platelets with blue color are dispersed).
Figure 8.
PVDF chains interposing among the G flakes: (a) intercalation and (b) exfoliation of the G layers (Green colored structure represents the polymer matrix wherein the graphene platelets with blue color are dispersed).
3.3. Mechanical Strength Analyses Results
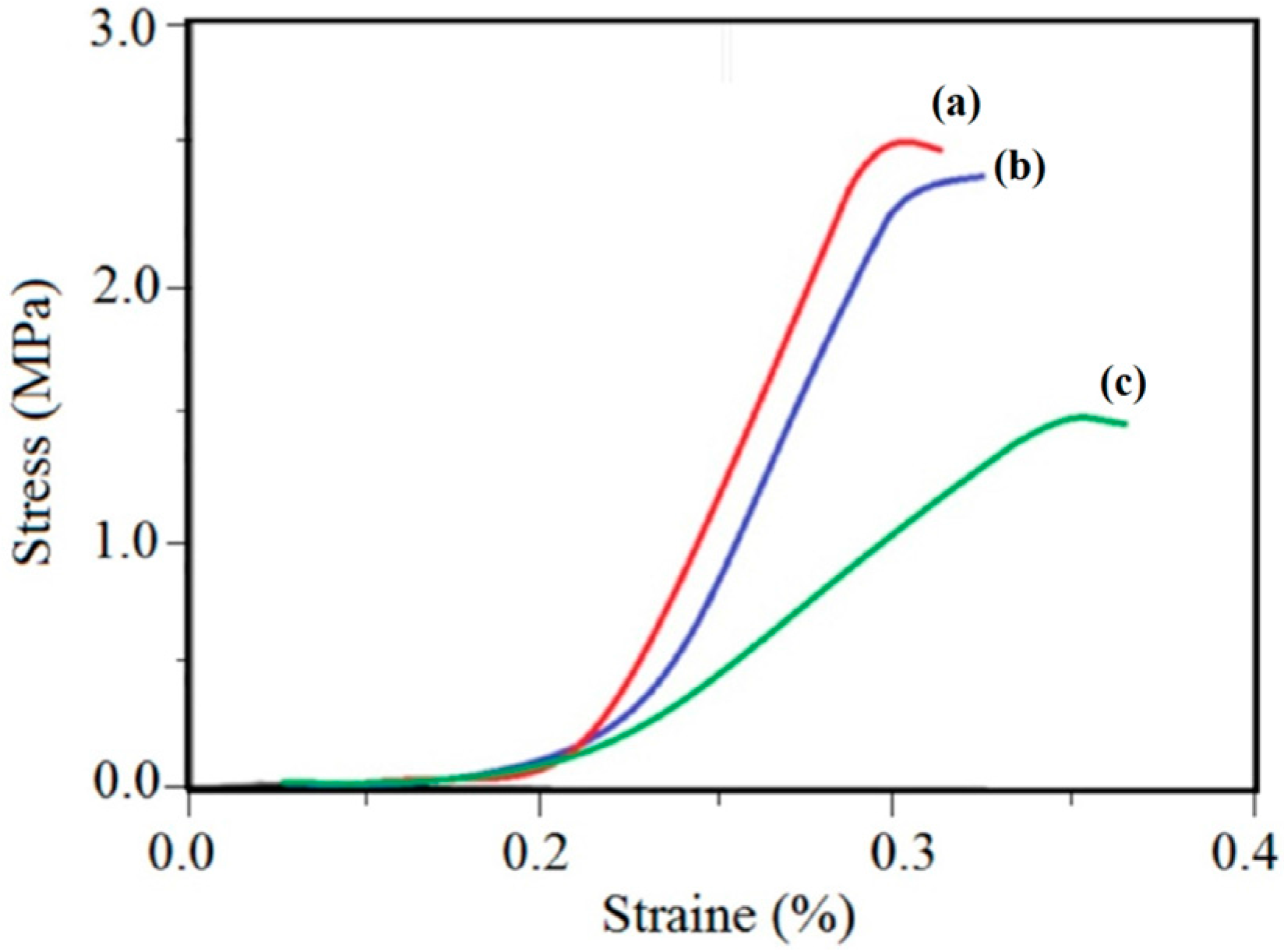
Figure 9 shows typical stress–strain curves of nanofibrous PVDF/G (M4, M5) and pristine PVDF (M0) films. It is noticeable that the tensile strength of nanofibrous PVDF film remarkably enhanced after G introducing. However, for the M5 membrane there is no sharp increasing in mechanical properties in compassion to M4.
The resulted stress-strain plot shows, that electrospun PVDF/G film exhibited higher tensile strength, tensile modulus and lower elongation and breaking point than PVDF films. The tensile strength of M4 nanofibrous film shows a peak value of 2.50 MPa, where for the electrospun M0 films a peak value is 0.70 MPa, therefore 3.5 times greater strength for produced electrospun films could be achieved by compositing of PVDF with G in the 100:1 weight ratio. In the case of M5 nanofibrous films, better mechanical properties are achieved in comparison with PVDF/G. However, considering the amount of the G introduced in M5 (1.5 wt%), the observed changes are not noticeable. The obtained mechanical strength analyses results of synthesized pristine and composite films have been represented in
Table 2.
Figure 9.
Stress-strain test results of the prepared membranes (a) M5, (b) M4 and (c) M0. (M0, M4 and M5 represent the prepared electrospun membranes respectively with 0.0, 1.0 and 1.5 wt% of geaphene).
Figure 9.
Stress-strain test results of the prepared membranes (a) M5, (b) M4 and (c) M0. (M0, M4 and M5 represent the prepared electrospun membranes respectively with 0.0, 1.0 and 1.5 wt% of geaphene).
Table 2.
Comparative analyses results for various prepared membranes.
Table 2.
Comparative analyses results for various prepared membranes.
| Film | LEP (kPa) | Contact angle (°) | Porosity (%) | Mean Pore Size (nm) | Young Modulus (MPa) |
|---|
| M0 | 66 ± 4 | 124 ± 2 | 73 ± 3 | 450 ± 15 | 410 ± 50 |
| M1 | 70 ± 5 | 135 ± 2 | 72 ± 3 | 445 ± 10 | 450 ± 80 |
| M2 | 84 ± 5 | 135 ± 3 | 74 ± 3 | 450 ± 10 | 480 ± 40 |
| M3 | 110 ± 4 | 155 ± 3 | 85 ± 3 | 500 ± 15 | 560 ± 50 |
| M4 | 122 ± 3 | 161 ± 3 | 84 ± 3 | 580 ± 20 | 720 ± 60 |
| M5 | 130 ± 5 | 160 ± 2 | 84 ± 3 | 590 ± 20 | 740 ± 80 |
| M'0 | 56 ± 2 | 95 ± 3 | 54 ± 3 | 200 ± 10 | 420 ± 50 |
| M'1 | 58 ± 2 | 100 ± 5 | 55 ± 3 | 190 ± 5 | 490 ± 50 |
Explored crystalline zones in the PVDF/G nanofibers structure and intercalating of polymer chains between the G layers are two main reasons for improving the mechanical properties of nanocomposite PVDF/G films [
8,
30,
33]. In addition, as mentioned earlier the simultaneous presence of nanofibers and microfibers, depicted in
Figure 3b, generates a novel structure for nanocomposite films that significantly improves the mechanical properties of the PVDF/G composite films.
The observed increase in the fiber diameters due to graphene interposing at the same electrospinning conditions is one of the reasons for mechanical strength enhancement. For the fibers whose their matrixes include graphene, the fiber diameters increase up to micrometer sizes, while the fiber diameters in which the graphene could not interpose, or interpose in low amounts, do not increase and span in the nanometer range. Therefore, we encounter with the fiber-network structure the simultaneous presence of nanometer and micrometer fibers, which improve the tensile strength. Another reason that we used XRD experiments is the graphene influence on the PVDF crystallinity. The improvement in the crystalline zones due to graphene existence in the polymer matrix somehow affects the fibers’ mechanical strength. From
Figure 9, an increase is observed in the Young modulus of the composite membrane, however the decline in the value of elongation at the break point is shown, which is the direct result of the enhancement in the crystalline zones in the PVDF/G film. Therefore, the fibers’ diameters and diameter distribution are increased by graphene addition that results in the mechanical strength improving. Since we applied the standard tensile test in all strain-stress experiments the sizes, thicknesses and weights of the examined specimens were equal, and the mass of the electrospun mat did not affect the results [
38,
39,
40,
41].
3.4. Hydrophobicity Test Results
In general, the hydrophobicity is evaluated by water contact angle, and higher hydrophobic films exhibits larger contact angle. The contact angle images of the prepared nanofibrous films are shown in
Figure 10.
Figure 10.
(a) Schematic representation of the surface structure effect (black colored) on the contact angle of the water droplet (blue colored) (b) Resulted images (black colored circles) and angles ( green colored lines) of water contact angle measurements (c) SEM images of related films. Respectively from right to left: M'0, M'1, M0 and M4. (M0, M4 represent the prepared electrospun membranes respectively with 0.0 and 1.0 wt% of geaphene. M'0, M'1 are for the membranes prepared via phase inversion method with graphene content of 0.0 and 1.0 wt%.)
Figure 10.
(a) Schematic representation of the surface structure effect (black colored) on the contact angle of the water droplet (blue colored) (b) Resulted images (black colored circles) and angles ( green colored lines) of water contact angle measurements (c) SEM images of related films. Respectively from right to left: M'0, M'1, M0 and M4. (M0, M4 represent the prepared electrospun membranes respectively with 0.0 and 1.0 wt% of geaphene. M'0, M'1 are for the membranes prepared via phase inversion method with graphene content of 0.0 and 1.0 wt%.)
As introduced, fluoropolymers usually exhibit low surface energy, which may be attributed to intensive electronegative characteristics of the element fluorine. Thus, its attractive force to other substances is weak [
3,
36]. Due to its fluorinated composition, the PVDF films naturally exhibit high water contact angle values of around 85° to 130°, which confirm its inherent hydrophobicity. M'0 membrane which was prepared via phase inversion method the contact angle measurements results obtain the mean value of about 95° (
Figure 10b). The water contact angle of about 128° was also reported, regarding to the preparation conditions [
8,
30]. However, the achieved hydrophobicity mainly depends to the film preparation method and the achieved morphology. As FESEM micrographs illustrate (
Figure 3c), the prepared non-fibrous PVDF films possess the skin layer with the smoother and less porous surface.
Hydrophobicity is also improved by introducing G into the polymer matrix whereby the contact angles of the nanocomposite PVDF films were increased in comparison with the pristine PVDF films. From
Figure 10b, the contact angle measurement for the non-fibrous PVDF/G film (M'1), results in the value of about 120°. Using electrospinning to prepare the fibrous films has the noticeable influence on the film wettability property, as the water contact angle of about 130° achieved for M0 (
Figure 10b). In the case of electrospun PVDF/G film (M4), the water contact angle measured to be near 160°. The observed promotion in hydrophobicity suggests that the incorporation of G significantly affects the morphology and surface properties of the prepared nanofibers.
The SEM images of each tested film are represented under the related contact angle image in
Figure 10c. The correspondent schematic representations in
Figure 10a show how various surface structures influence the hydrophobicity of the engineered film. As shown, PVDF/G nanofibrous structures with the large surface roughness and area have larger water contact angles in comparison with virgin PVDF film and PVDF/G nanocomposite flat film. Additionally, adding a small amount of G to the PVDF precursor electrospun blend enhances the water contact angle significantly, where the 1.00 wt% of PVDF/G electrospun film we could achieve the superhydrophobicity with the water contact angle of 160°. Obtained hierarchical structures for PVDF/G is the result of three different structural stairs in the composition of prepared nanofibrous films. First hierarchy stair is formed by PP nonwoven sheets that we used as the membrane support, the second hierarchy is formed on it in the form of the fiber network, and the last one is the roughness of the fibers surfaces due to G presence.
To confirm and better comprehend the static contact angle results, dynamic contact angle measurements were performed.
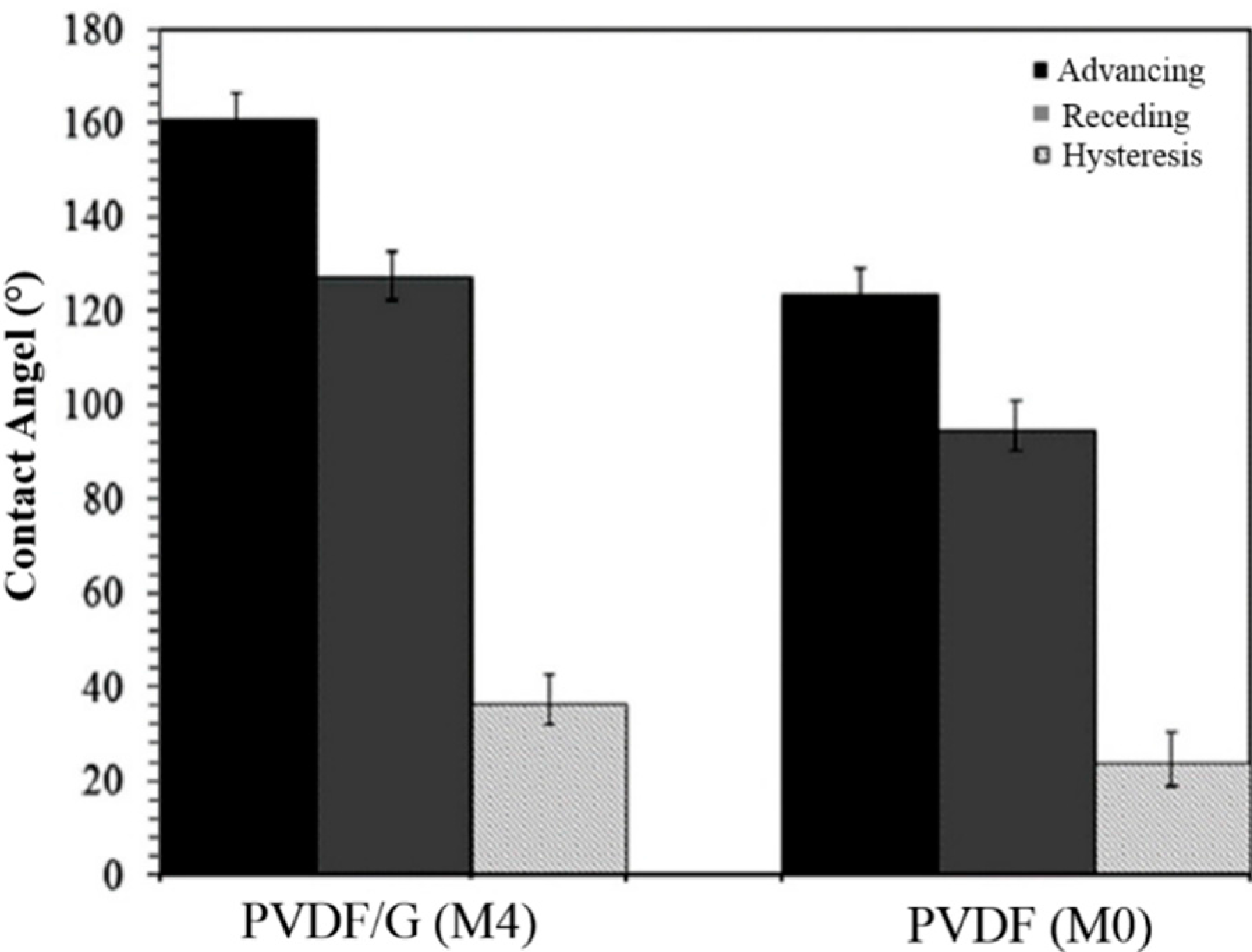
Figure 11 shows the obtained results for dynamic tests of the wetting phenomena. Herein, we employed droplet volume changing technique to measure the advancing and receding contact angles, so-called low rate dynamic contact angles. The graph of
Figure 11a indicates the contact radiuses of the drop on the surface of PVDF/G film that were determined as a function of injection time. The graph of
Figure 11b displays the dynamic contact angle as a function of the contact radius of the growing drop in triangle markers and the decreasing drop in squares. As can be seen, there is a linear relation between the droplet contact radius and time, which indicate a regime of constant speed of volume and contact line change. The average dynamic contact angles of PVDF/G nanofibrous film (M4) are obtained at the mentioned regime and the obtained values are represented in
Figure 12 in comparison with the results of pristine PVDF nanofibrous film (M0). PVDF/G presents an average advancing contact angle of 161.2° ± 3.4° and the receding one is 127.5° ± 2.6° with a mean hysteresis of 34.3° ± 6°. In contrast, PVDF exhibits a mean advancing contact angle of 124.6° ± 2.5° and a receding one of 95.5° ± 2.3° with a mean hysteresis of 24.0° ± 5°. The highly hydrophobic characteristics of PVDF/G are again confirmed with the higher values of advancing and receding contact angles compared to those of PVDF. In addition the more hysteretic behavior of PVDF/G electrospun film probably due to a higher surface roughness.
Figure 11.
The low-rate dynamic contact angles of the water on (a,b) composite M4 and (c,d) pristine M0 electrospun films. Triangle markers (∆) correspond to the advancing front of the sessile drop and squared markers (□) stand for the receding one; (a,c) Droplet contact radius as a function of injection and extraction time; and (b,d) Contact angle values variations with contact radius of dynamic drop are represented.
Figure 11.
The low-rate dynamic contact angles of the water on (a,b) composite M4 and (c,d) pristine M0 electrospun films. Triangle markers (∆) correspond to the advancing front of the sessile drop and squared markers (□) stand for the receding one; (a,c) Droplet contact radius as a function of injection and extraction time; and (b,d) Contact angle values variations with contact radius of dynamic drop are represented.
Figure 12.
Experimental dynamic contact angles of sessile water drops in the advancing, receding states and the related hysteresis for PVDF/G (M4) and PVDF (M0) films surfaces.
Figure 12.
Experimental dynamic contact angles of sessile water drops in the advancing, receding states and the related hysteresis for PVDF/G (M4) and PVDF (M0) films surfaces.
The other reason for increasing of hydrophobicity by G addition into the PVDF fibrous mats is the increase in the prepared film surface porosity. Due to the elimination of the skin layer of the membranes by using electrospinning, in spite of phase inversion method, the obtained porosities for M0–M5 membranes were higher than M'0 and M'1 membranes (
Table 2). In addition, as explained earlier, the membrane’s porosity increases with the amount of G nano-sheets added in the precursor PVDF solution. Higher contact angle values directly results in higher LEP values, which is an important factor for using the membranes in many separation processes, such as membrane distillation processes. Obtained results for LEP measurements are also represented in
Table 2.
In the presented approach, the amount of used G is very little and it seems that the developed method is economic for achieving superhydrophobicity with wide range of applications. We will aim to employ the prepared PVDF/G films as the highly hydrophobic membranes in the membrane distillation process for separation purposes.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









