
Singlet Exciton Lifetimes in Conjugated Polymer Films for Organic Solar Cells

 ,
,  , ,
, ,
Abstract
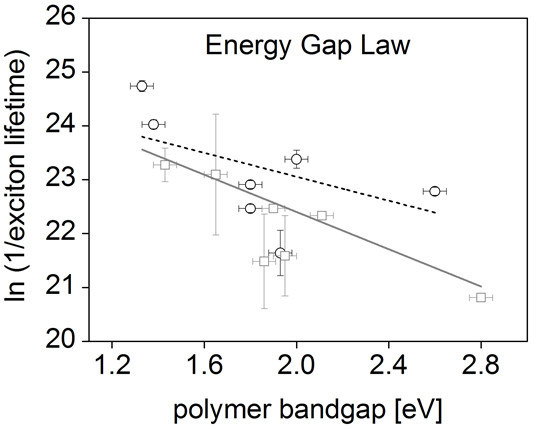
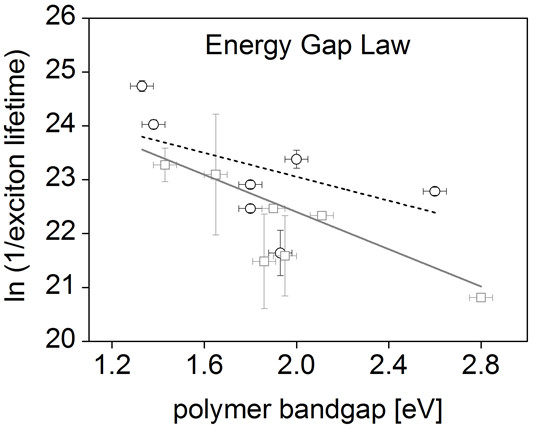
:
1. Introduction
1.1. Excited States in Conjugated Polymers

1.2. Exciton Diffusion in Polymer Films
1.3. Exciton Lifetime
2. Materials and Methods
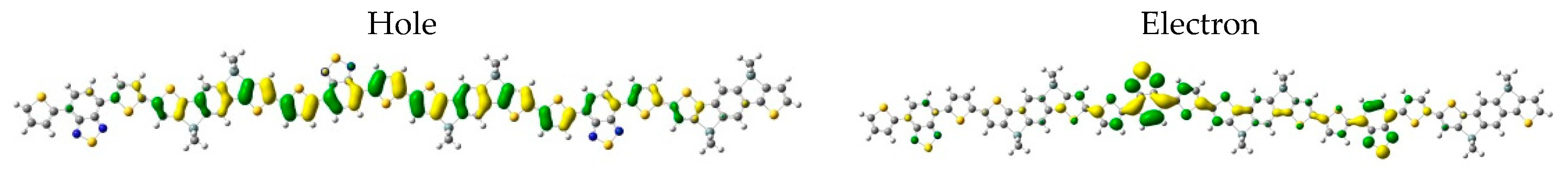
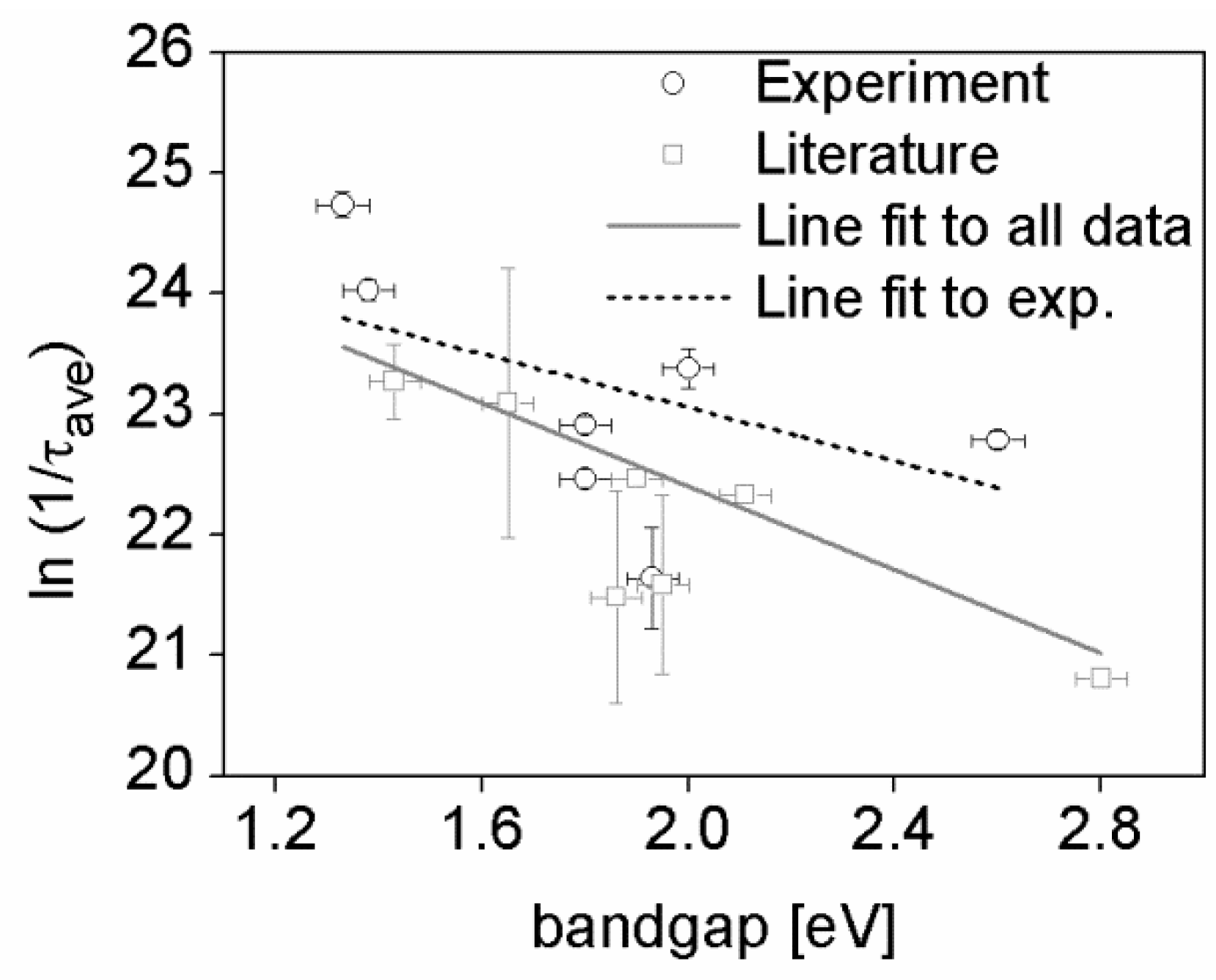
3. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Optical bandgap (eV) | Exciton lifetime (ps) |
|---|---|---|
| BTT-DPP | 1.33 | 18 ± 0.9 a |
| DPP-TT-T | 1.38 | 36.8 ± 1.5 a |
| SiIDT-BT | 1.80 | 112 ± 4 a |
| SiIDT-2FBT | 1.80 | 175 ± 7 a |
| APFO-3 | 1.93 | 400 ± 83 a |
| SiIDT-TPD | 2.00 | 70 ± 6 a |
| TTP | 2.60 | 127 ± 5 a |
| PCPDTBT | 1.43 | 78 b |
| PTB7 | 1.65 | 93 ± 48 b |
| PCDTBT | 1.86 | 463 ± 193 b |
| PBTTT | 1.90 | 175 b |
| P3HT | 1.95 | 422 ± 150 b |
| MEH-PPV | 2.11 | 210 ± 79 b |
| PFO | 2.80 | 430 b |

| Polymer name | Optical bandgap (eV) | Exciton lifetime (ps) |
|---|---|---|
| BTT-DPP 90 kg·mol−1 | 1.37 | 17.1 ± 1.5 |
| BTT-DPP 73 kg·mol−1 | 1.34 | 16.4 ± 1.0 |
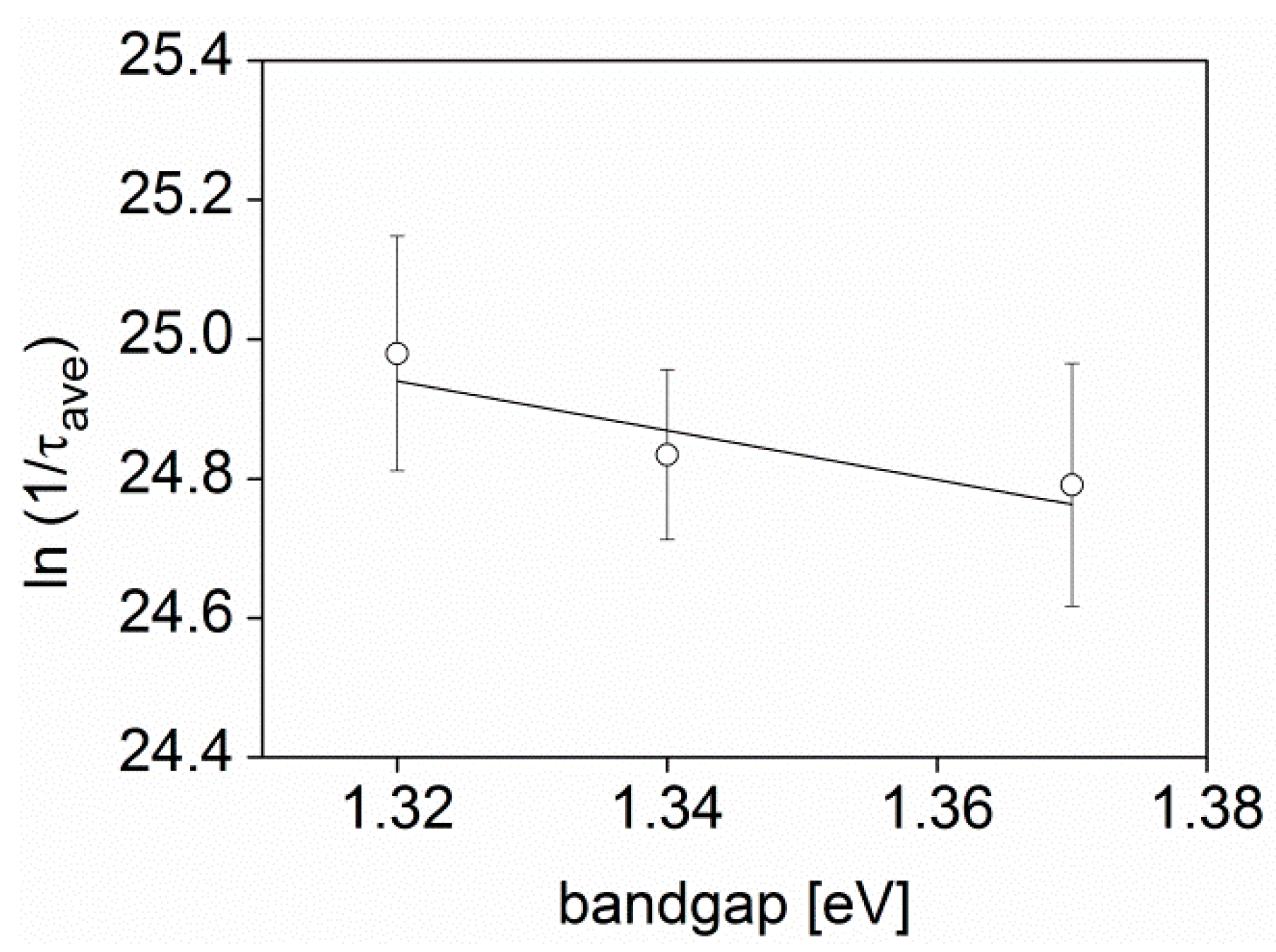
| BTT-DPP 22 kg·mol−1 | 1.32 | 14.2 ± 1.2 |

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tang, C.W. Two-layer organic photovoltaic cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, K.; Yang, G.; Liu, J.; Li, Z.; Lin, H.; Liu, Y.; Zhao, J.; Zhang, J.; Huang, F.; et al. Terthiophene-based D–A polymer with an asymmetric arrangement of alkyl chains that enables efficient polymer solar cells. J. Am. Chem. Soc. 2015, 137, 14149–14157. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells. Nat. Commun. 2014, 5, 5293. [Google Scholar] [CrossRef] [PubMed]
- Reid, O.G.; Pensack, R.D.; Song, Y.; Scholes, G.D.; Rumbles, G. Charge photogeneration in neat conjugated polymers. Chem. Mat. 2014, 26, 561–575. [Google Scholar] [CrossRef]
- Sajjad, M.T.; Ward, A.J.; Kästner, C.; Ruseckas, A.; Hoppe, H.; Samuel, I.D.W. Controlling exciton diffusion and fullerene distribution in photovoltaic blends by side chain modification. J. Phys. Chem. Lett. 2015, 6, 3054–3060. [Google Scholar] [CrossRef] [PubMed]
- Mikhnenko, O.V.; Blom, P.W.M.; Nguyen, T.-Q. Exciton diffusion in organic semiconductors. Energy Environ. Sci. 2015, 8, 1867–1888. [Google Scholar] [CrossRef]
- Tamai, Y.; Ohkita, H.; Benten, H.; Ito, S. Exciton diffusion in conjugated polymers: From fundamental understanding to improvement in photovoltaic conversion efficiency. J. Phys. Chem. Lett. 2015, 6, 3417–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, R.A.J.; Nelson, J. Factors limiting device efficiency in organic photovoltaics. Adv. Mater. 2013, 25, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Englman, R.; Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar] [CrossRef]
- Siebrand, W. On the relation between radiative and nonradiative transitions in molecules. Chem. Phys. Lett. 1971, 9, 157–159. [Google Scholar] [CrossRef]
- Ohkita, H.; Cook, S.; Astuti, Y.; Duffy, W.; Tierney, S.; Zhang, W.; Heeney, M.; McCulloch, I.; Nelson, J.; Bradley, D.D.C.; Durrant, J.R. Charge carrier formation in polythiophene/fullerene blend films studied by transient absorption spectroscopy. J. Am. Chem. Soc. 2008, 130, 3030–3042. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Chow, P.C.Y.; Gelinas, S.; Schlenker, C.W.; Li, C.-Z.; Yip, H.-L.; Jen, A.K.Y.; Ginger, D.S.; Friend, R.H. The role of spin in the kinetic control of recombination in organic photovoltaics. Nature 2013, 500, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, S.D.; Wheeler, S.; Niedzialek, D.; Schroeder, B.C.; Utzat, H.; Frost, J.M.; Yao, J.; Gillett, A.; Tuladhar, P.S.; McCulloch, I.; et al. Polaron pair mediated triplet generation in polymer/fullerene blends. Nat. Commun. 2015, 6, 6501. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, D.W.; Howard, I.A.; Laquai, F. Charge carrier generation followed by triplet state formation, annihilation, and carrier recreation in PBDTTT-C/PC60BM photovoltaic blends. J. Phys. Chem. C 2015, 119, 13509–13515. [Google Scholar] [CrossRef]
- Kohler, A.; dos Santos, D.A.; Beljonne, D.; Shuai, Z.; Bredas, J.L.; Holmes, A.B.; Kraus, A.; Mullen, K.; Friend, R.H. Charge separation in localized and delocalized electronic states in polymeric semiconductors. Nature 1998, 392, 903–906. [Google Scholar]
- Rothberg, L.J.; Yan, M.; Papadimitrakopoulos, F.; Galvin, M.E.; Kwock, E.W.; Miller, T.M. Photophysics of phenylenevinylene polymers. Synth. Met. 1996, 80, 41–58. [Google Scholar] [CrossRef]
- Sariciftci, N.S. Front matter. In Primary Photoexcitations in Conjugated Polymers: Molecular Exciton Versus Semiconductor Band Model; World Scientific: London, UK, 2013. [Google Scholar]
- Hestand, N.J.; Spano, F.C. The effect of chain bending on the photophysical properties of conjugated polymers. J. Phys. Chem. B 2014, 118, 8352–8363. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Huang, F.; Cao, Y. Recent development of push-pull conjugated polymers for bulk-heterojunction photovoltaics: Rational design and fine tailoring of molecular structures. J. Mater. Chem. 2012, 22, 10416–10434. [Google Scholar] [CrossRef]
- Kirkpatrick, J.; Nielsen, C.B.; Zhang, W.; Bronstein, H.; Ashraf, R.S.; Heeney, M.; McCulloch, I. A systematic approach to the design optimization of light-absorbing indenofluorene polymers for organic photovoltaics. Adv. Energy Mater. 2012, 2, 260–265. [Google Scholar] [CrossRef]
- Bronstein, H.; Chen, Z.Y.; Ashraf, R.S.; Zhang, W.M.; Du, J.P.; Durrant, J.R.; Tuladhar, P.S.; Song, K.; Watkins, S.E.; Geerts, Y.; et al. Thieno 3,2-b thiophene-diketopyrrolopyrrole-containing polymers for high-performance organic field-effect transistors and organic photovoltaic devices. J. Am. Chem. Soc. 2011, 133, 3272–3275. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yu, L. Understanding low bandgap polymer PTB7 and optimizing polymer solar cells based on it. Adv. Mater. 2014, 26, 4413–4430. [Google Scholar] [CrossRef] [PubMed]
- Kroon, R.; Lenes, M.; Hummelen, J.C.; Blom, P.W.M.; de Boer, B. Small bandgap polymers for organic solar cells (polymer material development in the last 5 years). Polym. Rev. 2008, 48, 531–582. [Google Scholar] [CrossRef]
- Li, W.; Roelofs, W.S.C.; Wienk, M.M.; Janssen, R.A.J. Enhancing the photocurrent in diketopyrrolopyrrole-based polymer solar cells via energy level control. J. Am. Chem. Soc. 2012, 134, 13787–13795. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.H.; Chen, H.Y.; Zhang, S.Q.; Chen, R.I.; Yang, Y.; Wu, Y.; Li, G. Synthesis of a low band gap polymer and its application in highly efficient polymer solar cells. J. Am. Chem. Soc. 2009, 131, 15586–15587. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C. The spectral signatures of frenkel polarons in H- and J-aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Davydov, A.S. Theory of Molecular Excitons; Springer: New York, NY, USA, 1971. [Google Scholar]
- Clark, J.; Chang, J.-F.; Spano, F.C.; Friend, R.H.; Silva, C. Determining exciton bandwidth and film microstructure in polythiophene films using linear absorption spectroscopy. Appl. Phys. Lett. 2009, 94, 163306. [Google Scholar] [CrossRef]
- Jenekhe, S.A.; Osaheni, J.A. Excimers and exciplexes of conjugated polymers. Science 1994, 265, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.B. Excimers and exciplexes. Nature 1967, 214, 1187–1190. [Google Scholar] [CrossRef]
- Sheng, C.X.; Tong, M.; Singh, S.; Vardeny, Z.V. Experimental determination of the charge/neutral branching ratio η in the photoexcitation of π-conjugated polymers by broadband ultrafast spectroscopy. Phys. Rev. B 2007, 75, 085206. [Google Scholar] [CrossRef]
- Jakubiak, R.; Collison, C.J.; Wan, W.C.; Rothberg, L.J.; Hsieh, B.R. Aggregation quenching of luminescence in electroluminescent conjugated polymers. J. Phys. Chem.A 1999, 103, 2394–2398. [Google Scholar] [CrossRef]
- Reid, O.G.; Malik, J.A.N.; Latini, G.; Dayal, S.; Kopidakis, N.; Silva, C.; Stingelin, N.; Rumbles, G. The influence of solid-state microstructure on the origin and yield of long-lived photogenerated charge in neat semiconducting polymers. J. Polym. Sci. Polym. Phys. 2012, 50, 27–37. [Google Scholar] [CrossRef]
- Huijser, A.; Savenije, T.J.; Meskers, S.C.J.; Vermeulen, M.J.W.; Siebbeles, L.D.A. The mechanism of long-range exciton diffusion in a nematically organized porphyrin layer. J. Am. Chem. Soc. 2008, 130, 12496–12500. [Google Scholar] [CrossRef] [PubMed]
- Masri, Z.; Ruseckas, A.; Emelianova, E.V.; Wang, L.; Bansal, A.K.; Matheson, A.; Lemke, H.T.; Nielsen, M.M.; Nguyen, H.; Coulembier, O.; et al. Molecular weight dependence of exciton diffusion in poly(3-hexylthiophene). Adv. Energy Mater. 2013, 3, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.D.A.; Mikhnenko, O.V.; Chen, J.; Masri, Z.; Ruseckas, A.; Mikhailovsky, A.; Raab, R.P.; Liu, J.; Blom, P.W.M.; Loi, M.A.; et al. Systematic study of exciton diffusion length in organic semiconductors by six experimental methods. Mater. Horiz. 2014, 1, 280–285. [Google Scholar] [CrossRef]
- Clarke, T.M.; Durrant, J.R. Charge photogeneration in organic solar cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, N.; Lou, S.J.; Smith, J.; Tice, D.B.; Hennek, J.W.; Ortiz, R.P.; Navarrete, J.T.L.; Li, S.; Strzalka, J.; et al. Polymer solar cells with enhanced fill factors. Nat. Photon. 2013, 7, 825–833. [Google Scholar] [CrossRef]
- Müller, C.; Ferenczi, T.A.M.; Campoy-Quiles, M.; Frost, J.M.; Bradley, D.D.C.; Smith, P.; Stingelin-Stutzmann, N.; Nelson, J. Binary organic photovoltaic blends: A simple rationale for optimum compositions. Adv. Mater. 2008, 20, 3510–3515. [Google Scholar] [CrossRef]
- Keivanidis, P.E.; Clarke, T.M.; Lilliu, S.; Agostinelli, T.; Macdonald, J.E.; Durrant, J.R.; Bradley, D.D.C.; Nelson, J. Dependence of charge separation efficiency on film microstructure in poly(3-hexylthiophene-2,5-diyl):[6,6]-phenyl-C61 butyric acid methyl ester blend films. J. Phys. Chem. Lett. 2010, 1, 734–738. [Google Scholar] [CrossRef]
- Turro, N.J. Modern Molecular Photochemistry; University Science Books: Mill Valley, CA, USA, 1991. [Google Scholar]
- Cook, S.; Furube, A.; Katoh, R. Analysis of the excited states of regioregular polythiophene P3HT. Energy Environ. Sci. 2008, 1, 294–299. [Google Scholar] [CrossRef]
- Greenham, N.C.; Samuel, I.D.W.; Hayes, G.R.; Phillips, R.T.; Kessener, Y.A.R.R.; Moratti, S.C.; Holmes, A.B.; Friend, R.H. Measurement of absolute photoluminescence quantum efficiencies in conjugated polymers. Chem. Phys. Lett. 1995, 241, 89–96. [Google Scholar] [CrossRef]
- Hedley, G.J.; Ward, A.J.; Alekseev, A.; Howells, C.T.; Martins, E.R.; Serrano, L.A.; Cooke, G.; Ruseckas, A.; Samuel, I.D.W. Determining the optimum morphology in high-performance polymer-fullerene organic photovoltaic cells. Nat. Commun. 2013, 4, 2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariu, M.; Lidzey, D.G.; Sims, M.; Cadby, A.J.; Lane, P.A.; Bradley, D.D.C. The effect of morphology on the temperature-dependent photoluminescence quantum efficiency of the conjugated polymer poly(9,9-dioctylfluorene). J. Phys. Condens. Matter 2002, 14, 9975. [Google Scholar] [CrossRef]
- Pope, M.; Swenberg, C.E. Electronic Processes in Organic Crystals and Polymers; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Bixon, M.; Jortner, J.; Cortes, J.; Heitele, H.; Michel-Beyerle, M.E. Energy gap law for nonradiative and radiative charge transfer in isolated and in solvated supermolecules. J. Phys. Chem. 1994, 98, 7289–7299. [Google Scholar] [CrossRef]
- Wilson, J.S.; Chawdhury, N.; Al-Mandhary, M.R.A.; Younus, M.; Khan, M.S.; Raithby, P.R.; Köhler, A.; Friend, R.H. The energy gap law for triplet states in Pt-containing conjugated polymers and monomers. J. Am. Chem. Soc. 2001, 123, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.D.; Eisenberg, R. Tuning the excited-state properties of platinum(ii) diimine dithiolate complexes. J. Am. Chem. Soc. 1996, 118, 1949–1960. [Google Scholar] [CrossRef]
- Asahi, T.; Ohkohchi, M.; Mataga, N. Energy gap dependences of charge recombination processes of ion pairs produced by excitation of charge-transfer complexes: Solvent polarity effects. J. Phys. Chem. 1993, 97, 13132–13137. [Google Scholar] [CrossRef]
- Siebrand, W. Radiationless transitions in polyatomic molecules. II. Triplet-ground-state transitions in aromatic hydrocarbons. J. Chem. Phys. 1967, 47, 2411–2422. [Google Scholar] [CrossRef]
- Dimitrov, S.D.; Nielsen, C.B.; Shoaee, S.; Tuladhar, P.S.; Du, J.; McCulloch, I.; Durrant, J.R. Efficient charge photogeneration by the dissociation of PC70BM excitons in polymer/fullerene solar cells. J. Phys. Chem. Lett. 2012, 3, 140–144. [Google Scholar] [CrossRef]
- Ashraf, R.S.; Chen, Z.; Leem, D.S.; Bronstein, H.; Zhang, W.; Schroeder, B.; Geerts, Y.; Smith, J.; Watkins, S.; Anthopoulos, T.D.; et al. Silaindacenodithiophene semiconducting polymers for efficient solar cells and high-mobility ambipolar transistors. Chem. Mat. 2011, 23, 768–770. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Huang, Z.; Ashraf, R.S.; Smith, J.; D’Angelo, P.; Watkins, S.E.; Anthopoulos, T.D.; Durrant, J.R.; McCulloch, I. Silaindacenodithiophene-based low band gap polymers—The effect of fluorine substitution on device performances and film morphologies. Adv. Funct. Mater. 2012, 22, 1663–1670. [Google Scholar] [CrossRef]
- Fei, Z.; Kim, Y.; Smith, J.; Domingo, E.B.; Stingelin, N.; McLachlan, M.A.; Song, K.; Anthopoulos, T.D.; Heeney, M. Comparative optoelectronic study between copolymers of peripherally alkylated dithienosilole and dithienogermole. Macromolecules 2012, 45, 735–742. [Google Scholar] [CrossRef]
- Andernach, R.; Utzat, H.; Dimitrov, S.D.; McCulloch, I.; Heeney, M.; Durrant, J.R.; Bronstein, H. Synthesis and exciton dynamics of triplet sensitized conjugated polymers. J. Am. Chem. Soc. 2015, 137, 10383–10390. [Google Scholar] [CrossRef] [PubMed]
- Gaab, K.M.; Bardeen, C.J. Anomalous exciton diffusion in the conjugated polymer MEH−PPV measured using a three-pulse pump−dump−probe anisotropy experiment. J. Phys. Chem. A 2004, 108, 10801–10806. [Google Scholar] [CrossRef]
- Lewis, A.J.; Ruseckas, A.; Gaudin, O.P.M.; Webster, G.R.; Burn, P.L.; Samuel, I.D.W. Singlet exciton diffusion in MEH-PPV films studied by exciton–exciton annihilation. Org. Electron. 2006, 7, 452–456. [Google Scholar] [CrossRef]
- Hayes, G.R.; Samuel, I.D.W.; Phillips, R.T. Exciton dynamics in electroluminescent polymers studied by femtosecond time-resolved photoluminescence spectroscopy. Phys. Rev. B 1995, 52, R11569–R11572. [Google Scholar] [CrossRef]
- Piris, J.; Dykstra, T.E.; Bakulin, A.A.; Loosdrecht, P.H.M.V.; Knulst, W.; Trinh, M.T.; Schins, J.M.; Siebbeles, L.D.A. Photogeneration and ultrafast dynamics of excitons and charges in P3HT/PCBM blends. J. Phys. Chem. C 2009, 113, 14500–14506. [Google Scholar] [CrossRef]
- Guo, J.; Ohkita, H.; Benten, H.; Ito, S. Near-IR femtosecond transient absorption spectroscopy of ultrafast polaron and triplet exciton formation in polythiophene films with different regioregularities. J. Am. Chem. Soc. 2009, 131, 16869–16880. [Google Scholar] [CrossRef] [PubMed]
- Scarongella, M.; de Jonghe-Risse, J.; Buchaca-Domingo, E.; Causa’, M.; Fei, Z.; Heeney, M.; Moser, J.-E.; Stingelin, N.; Banerji, N. A close look at charge generation in polymer: Fullerene blends with microstructure control. J. Am. Chem. Soc. 2015, 137, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Etzold, F.; Howard, I.A.; Forler, N.; Cho, D.M.; Meister, M.; Mangold, H.; Shu, J.; Hansen, M.R.; Muellen, K.; Laquai, F. The effect of solvent additives on morphology and excited-state dynamics in PCPDTBT:PCBM photovoltaic blends. J. Am. Chem. Soc. 2012, 134, 10569–10583. [Google Scholar] [CrossRef] [PubMed]
- Gieseking, B.; Jäck, B.; Preis, E.; Jung, S.; Forster, M.; Scherf, U.; Deibel, C.; Dyakonov, V. Excitation dynamics in low band gap donor–acceptor copolymers and blends. Adv. Energy Mater. 2012, 2, 1477–1482. [Google Scholar] [CrossRef]
- Etzold, F.; Howard, I.A.; Mauer, R.; Meister, M.; Kim, T.D.; Lee, K.S.; Baek, N.S.; Laquai, F. Ultrafast exciton dissociation followed by nongeminate charge recombination in PCDTBT:PCBM photovoltaic blends. J. Am. Chem. Soc. 2011, 133, 9469–9479. [Google Scholar] [CrossRef] [PubMed]
- Banerji, N.; Cowan, S.; Leclerc, M.; Vauthey, E.; Heeger, A.J. Exciton formation, relaxation, and decay in PCDTBT. J. Am. Chem. Soc. 2010, 132, 17459–17470. [Google Scholar] [CrossRef] [PubMed]
- Jarzab, D.; Cordella, F.; Gao, J.; Scharber, M.; Egelhaaf, H.-J.; Loi, M.A. Low-temperature behaviour of charge transfer excitons in narrow-bandgap polymer-based bulk heterojunctions. Adv. Energy Mater. 2011, 1, 604–609. [Google Scholar] [CrossRef]
- Kouhei, Y.; Hayato, K.; Takeshi, Y.; Liyuan, H.; Yutaka, M. Exciton-to-carrier conversion processes in a low-band-gap organic photovoltaic. Jpn. J. Appl. Phys. 2013, 52, 062405. [Google Scholar]
- Kouhei, Y.; Hayato, K.; Takeshi, Y.; Liyuan, H.; Yutaka, M. Fast carrier formation from acceptor exciton in low-gap organic photovotalic. Appl. Phys. Express 2012, 5, 042302. [Google Scholar]
- Dimitrov, S.D.; Huang, Z.; Deledalle, F.; Nielsen, C.B.; Schroeder, B.C.; Ashraf, R.S.; Shoaee, S.; McCulloch, I.; Durrant, J.R. Towards optimisation of photocurrent from fullerene excitons in organic solar cells. Energy Environ. Sci. 2014, 7, 1037–1043. [Google Scholar] [CrossRef]
- Sweetnam, S.; Graham, K.R.; Ngongang Ndjawa, G.O.; Heumüller, T.; Bartelt, J.A.; Burke, T.M.; Li, W.; You, W.; Amassian, A.; McGehee, M.D. Characterization of the polymer energy landscape in polymer:fullerene bulk heterojunctions with pure and mixed phases. J. Am. Chem. Soc. 2014, 136, 14078–14088. [Google Scholar] [CrossRef] [PubMed]
- Armin, A.; Kassal, I.; Shaw, P.E.; Hambsch, M.; Stolterfoht, M.; Lyons, D.M.; Li, J.; Shi, Z.; Burn, P.L.; Meredith, P. Spectral dependence of the internal quantum efficiency of organic solar cells: Effect of charge generation pathways. J. Am. Chem. Soc. 2014, 136, 11465–11472. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitrov, S.D.; Schroeder, B.C.; Nielsen, C.B.; Bronstein, H.; Fei, Z.; McCulloch, I.; Heeney, M.; Durrant, J.R. Singlet Exciton Lifetimes in Conjugated Polymer Films for Organic Solar Cells. Polymers 2016, 8, 14. https://doi.org/10.3390/polym8010014
Dimitrov SD, Schroeder BC, Nielsen CB, Bronstein H, Fei Z, McCulloch I, Heeney M, Durrant JR. Singlet Exciton Lifetimes in Conjugated Polymer Films for Organic Solar Cells. Polymers. 2016; 8(1):14. https://doi.org/10.3390/polym8010014
Chicago/Turabian StyleDimitrov, Stoichko D., Bob C. Schroeder, Christian B. Nielsen, Hugo Bronstein, Zhuping Fei, Iain McCulloch, Martin Heeney, and James R. Durrant. 2016. "Singlet Exciton Lifetimes in Conjugated Polymer Films for Organic Solar Cells" Polymers 8, no. 1: 14. https://doi.org/10.3390/polym8010014