
Fabrication of Alkoxyamine-Functionalized Magnetic Core-Shell Microspheres via Reflux Precipitation Polymerization for Glycopeptide Enrichment

Abstract
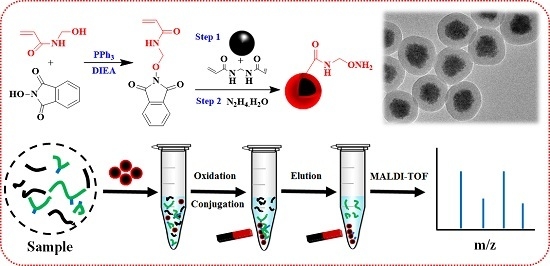
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrument and Analysis
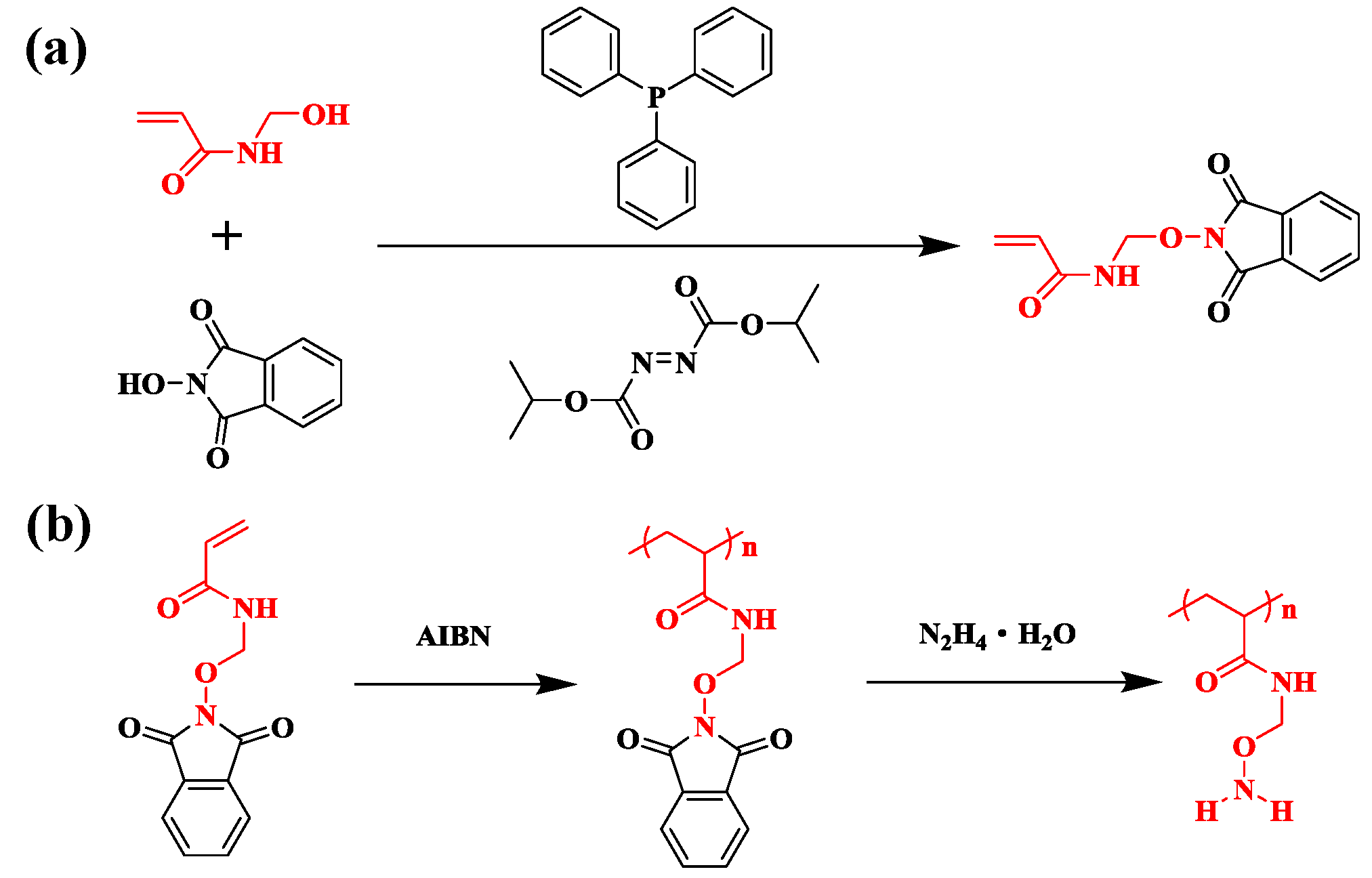
2.3. Synthesis of N-Aminooxy Methylacylimde-p (NAMAm-p)
2.4. Preparation of PNAMAm-p Polymer Chain and Deprotection for PNAMAm
2.5. Preparation of Monodisperse Crosslinked PNAMAm-p Microspheres and Deprotection Study
2.6. Preparation of MPS Modified MSPs (MSP-m)
2.7. Preparation of MSP@PNAMAm-p Magnetic Hybrid Microspheres and Deprotection for MSP@PNAMAm
2.8. Enrichment of Model N-glycoprotein and N-glycopeptides with MSP@PNAMAm Microspheres
2.9. Enrichment of N-glycopeptides from Human Serum with MSP@PNAMAm
2.10. Data Analysis
3. Results and Discussion
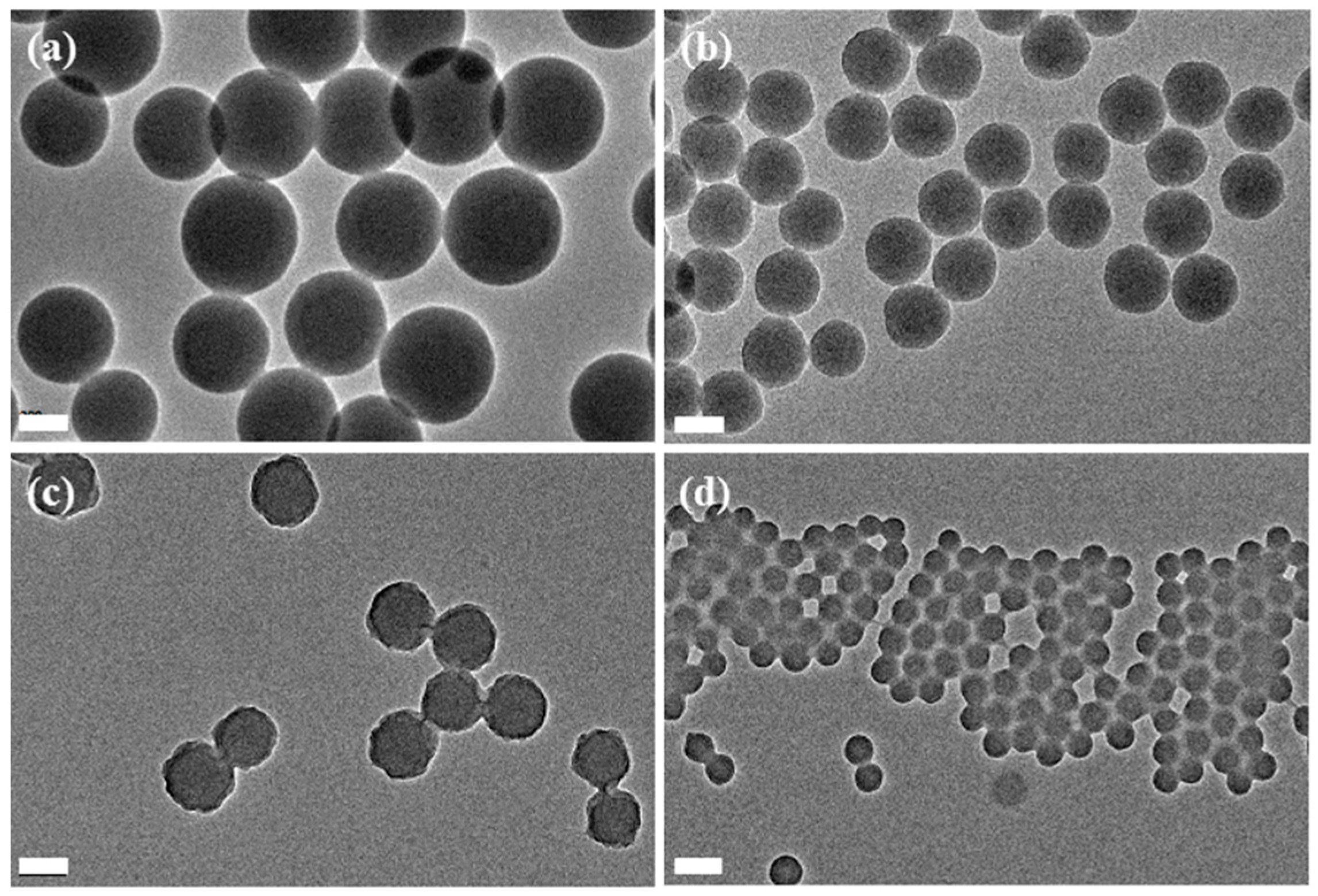
3.1. Polymerization of PNAMAm-p and the Deprotection Study
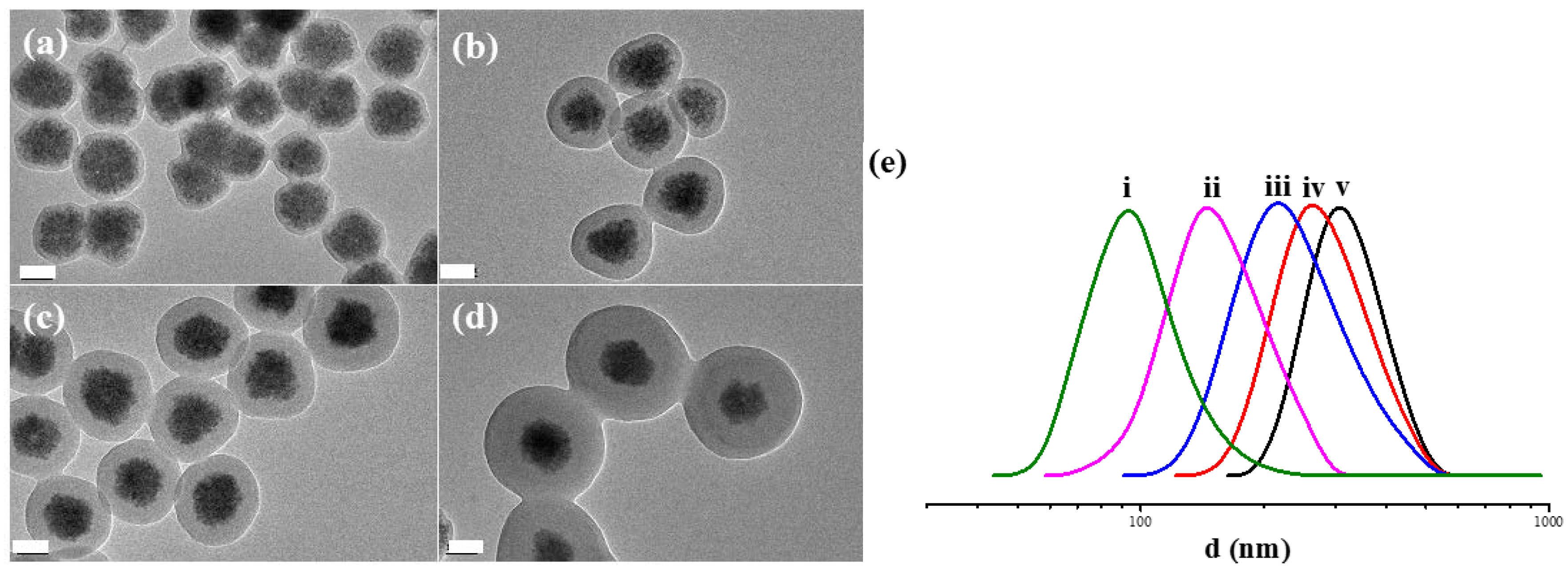
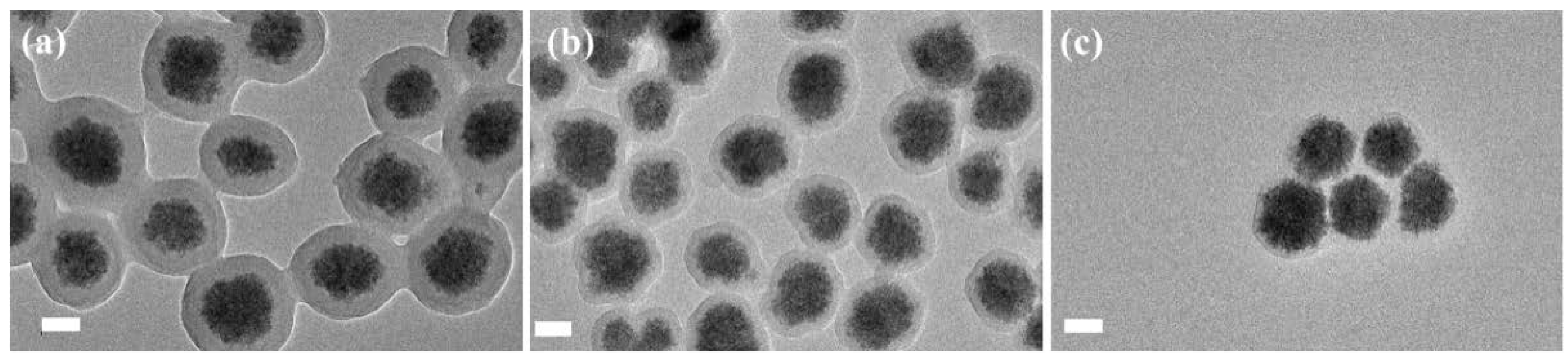
3.2. Preparation of PNAMAm Microspheres and MSP@PNAMAm Core-Shell Microspheres
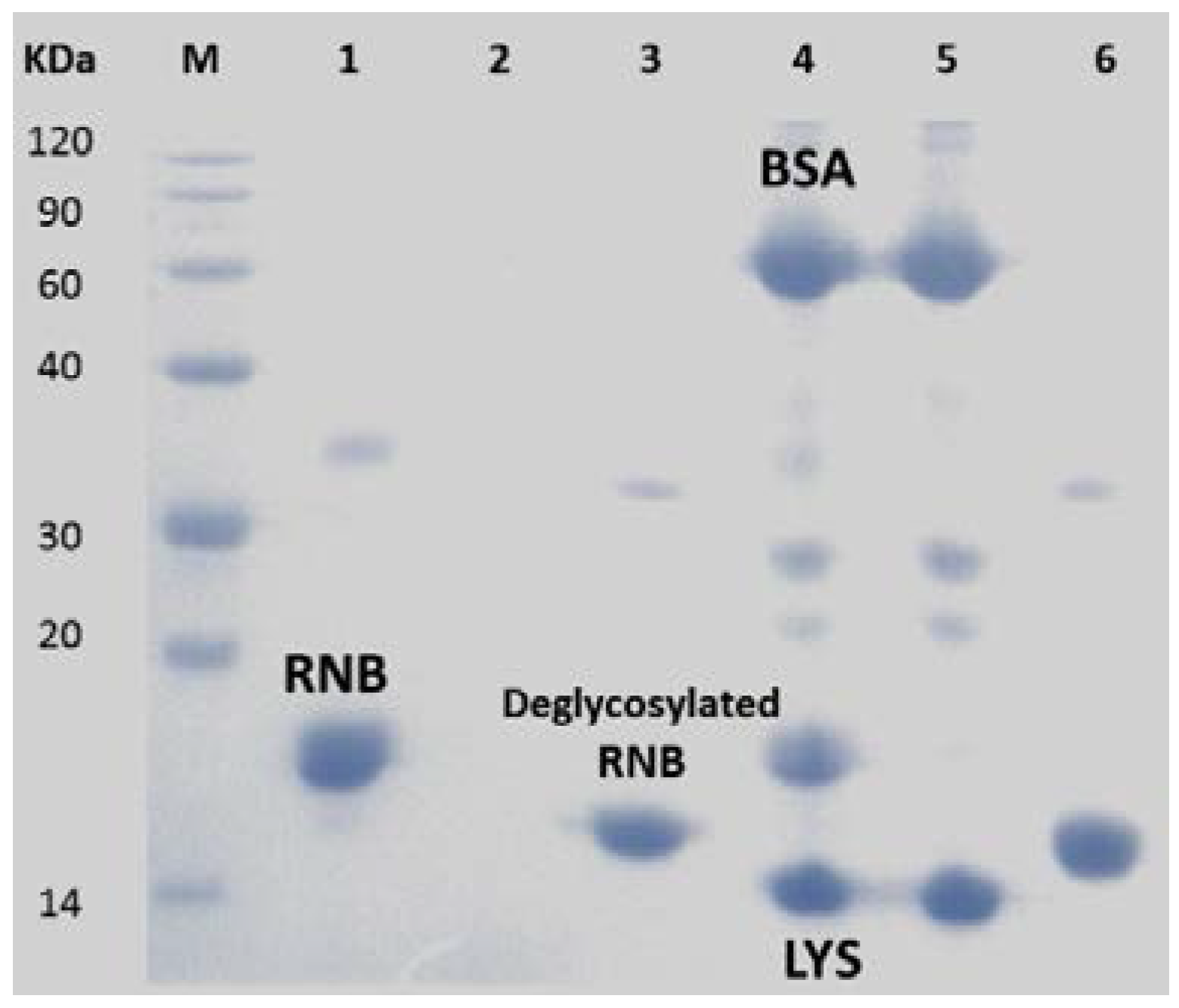
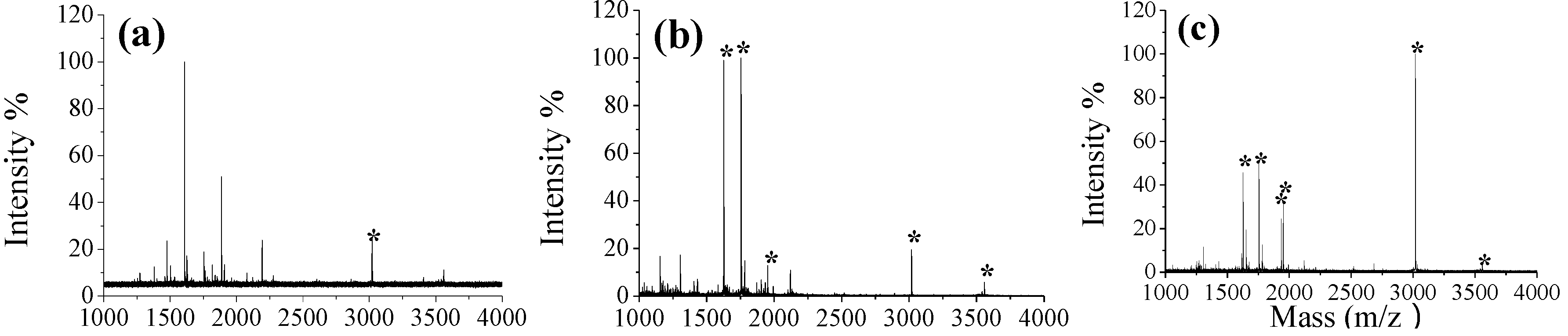
3.3. Selective Enrichment of Glycoproteins and Glycopeptides
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, G.L.; Möhwald, H.; Shchukin, D.G. Precipitation polymerization for fabrication of complex core–shell hybrid particles and hollow structures. Chem. Soc. Rev. 2013, 42, 3628–3646. [Google Scholar] [CrossRef] [PubMed]
- Barahona, F.; Turiel, E.; Cormack, P.A.G.; Martín-Esteban, A. Chromatographic performance of molecularly imprinted polymers: Core–shell microspheres by precipitation polymerization and grafted mip films via iniferter-modified silica beads. J. Polym. Sci. Polym. Chem. 2010, 48, 1058–1066. [Google Scholar] [CrossRef]
- Qin, W.W.; Silvestre, M.E.; Kirschhöfer, F.; Brenner-Weiss, G.; Franzreb, M. Insights into chromatographic separation using core–shell metal–organic frameworks: Size exclusion and polarity effects. J. Chromatogr. A 2015, 1411, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Mhlanga, N.; Sinha Ray, S.; Lemmer, Y.; Wesley-Smith, J. Polylactide-based magnetic spheres as efficient carriers for anticancer drug delivery. ACS Appl. Mater. Interfaces 2015, 7, 22692–22701. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Guo, W.M.; Ho, V.H.B.; Saha, A.; Chong, H.C.; Tan, N.S.; Widjaja, E.; Tan, E.Y.; Loo, S.C.J. Inhibition of 3-D tumor spheroids by timed-released hydrophilic and hydrophobic drugs from multilayered polymeric microparticles. Small 2014, 10, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Sun, Y.F.; Zheng, J.; Yang, W.L. Boronic acid-functionalized core–shell–shell magnetic composite microspheres for the selective enrichment of glycoprotein. ACS Appl. Mater. Interfaces 2013, 5, 8351–8358. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.P.; Hu, Y.X.; Zhang, T.R.; Yin, Y.D. Superparamagnetic composite colloids with anisotropic structures. J. Am. Chem. Soc. 2007, 129, 8974–8975. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qin, H.; Hu, Z.; Zhang, Y.; Wu, R.A.; Zou, H. A poly(ethylene glycol)-brush decorated magnetic polymer for highly specific enrichment of phosphopeptides. Chem. Sci. 2012, 3, 2828–2838. [Google Scholar] [CrossRef]
- Qin, W.; Song, Z.; Fan, C.; Zhang, W.; Cai, Y.; Zhang, Y.; Qian, X. Trypsin immobilization on hairy polymer chains hybrid magnetic nanoparticles for ultra fast, highly efficient proteome digestion, facile 18O labeling and absolute protein quantification. Anal. Chem. 2012, 84, 3138–3144. [Google Scholar] [CrossRef] [PubMed]
- Grignard, B.; Jérôme, C.; Calberg, C.; Jérôme, R.; Wang, W.; Howdle, S.M.; Detrembleur, C. Copper bromide complexed by fluorinated macroligands: Towards microspheres by ATRP of vinyl monomers in sc CO2. Chem. Commun. 2008, 44, 314–316. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, D.; Zhao, T.; Wei, X.; Mcmahon, S.; Ahern, J.O.; Wang, W.; Greiser, U.; Rodriguez, B.J.; Wang, W. Intramolecular cyclization dominating homopolymerization of multivinyl monomers toward single-chain cyclized/knotted polymeric nanoparticles. Macromolecules 2015, 48, 6882–6889. [Google Scholar] [CrossRef]
- Gonzato, C.; Courty, M.; Pasetto, P.; Haupt, K. Magnetic molecularly imprinted polymer nanocomposites via surface-initiated RAFT polymerization. Adv. Funct. Mater. 2011, 21, 3947–3953. [Google Scholar] [CrossRef]
- Li, X.; Bao, M.M.; Weng, Y.Y.; Yang, K.; Zhang, W.D.; Chen, G.J. Glycopolymer-coated iron oxide nanoparticles: Shape-controlled synthesis and cellular uptake. J. Mater. Chem. B 2014, 2, 5569–5575. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Ma, W.F.; Li, D.; Yu, M.; Guo, J.; Wang, C.C. Benzoboroxole-functionalized magnetic core/shell microspheres for highly specifi c enrichment of glycoproteins under physiological conditions. Small 2014, 10, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ma, C.J.; Sun, Y.F.; Pan, M.R.; Li, L.; Hu, X.J.; Yang, W.L. Maltodextrin-modified magnetic microspheres for selective enrichment of maltose binding proteins. ACS Appl. Mater. Interfaces 2014, 6, 3568–3574. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Li, Y.P.; Sun, Y.F.; Yang, Y.K.; Ding, Y.; Lin, Y.; Yang, W.L. A generic magnetic microsphere platform with “clickable” ligands for purification and immobilization of targeted proteins. ACS Appl. Mater. Interfaces 2015, 7, 7241–7250. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Shu, Q.; Wang, L.Y.; Wu, H.; Wang, A.Y.; Mao, H. Layer-by-layer assembled milk protein coated magnetic nanoparticle enabled oral drug delivery with high stability in stomach and enzyme-responsive release in small intestine. Biomaterials 2015, 39, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, J.N.; Cao, Q.Y.; Deng, H.D.; Shao, G.; Deng, D.Y.B.; Zhou, W.Y. One-step synthesis of magnetic hollow mesoporous silica (MHMS) nanospheres for drug delivery nanosystems via electrostatic self-assembly templated approach. Chem. Commun. 2015, 51, 2357–2360. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liang, H.; Racha, C.A.G.; Hamilton, L.; Fraylich, M.; Shakesheff, K.M.; Saunders, B.; Alexander, C. Biodegradable thermoresponsive microparticle dispersions for injectable cell delivery prepared using a single–step process. Adv. Mater. 2009, 21, 1809–1813. [Google Scholar] [CrossRef]
- Huang, L.; Ao, L.J.; Wang, W.; Hu, D.H.; Sheng, Z.H.; Su, W. Multifunctional magnetic silica nanotubes for mr imaging and targeted drug delivery. Chem. Commun. 2015, 51, 3923–3926. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sugihara, F.; Matsushita, H.; Yoshioka, Y.; Mizukami, S.; Kikuchi, K. Mesoporous silica nanoparticles for 19F magnetic resonance imaging, fluorescence imaging, and drug delivery. Chem. Sci. 2015, 6, 1986–1990. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.T.; Li, R.M.; Guo, J.; Wang, C.C.; Tang, C.B. Selective capture and quick detection of targeting cells with sers-coding microsphere suspension chip. Small 2015, 11, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Kim, M.; Kang, S.M.; Lim, K.T.; Kim, T.S.; Kang, J.Y. Magnetic bead droplet immunoassay of oligomer amyloid beta for the diagnosis of alzheimer’s disease using micro-pillars to enhance the stability of the oil-water interface. Biosens. Bioelectron. 2015, 67, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, Y.; Yang, P.; Jin, S.; Yu, M.; Guo, J.; Wang, C. Fabrication of polymeric microgels using reflux-precipitation polymerization and its application for phosphoprotein enrichment. J. Mater. Chem. B 2014, 2, 2575–2582. [Google Scholar] [CrossRef]
- Chen, Y.; Xiong, Z.; Zhang, L.; Zhao, J.; Zhang, Q.; Peng, L.; Zhang, W.; Ye, M.; Zou, H. Facile synthesis of zwitterionic polymer-coated core–shell magnetic nanoparticles for highly specific capture of N-linked glycopeptides. Nanoscale 2015, 7, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Deng, C.; Yan, L.; Ying, D.; Yang, P.; Zhang, X. A facile synthesis approach to c 8 -functionalized magnetic carbonaceous polysaccharide microspheres for the highly efficient and rapid enrichment of peptides and direct maldi-tof-ms analysis. Adv. Mater. 2009, 21, 2200–2205. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Yang, Y.K.; Ma, W.F.; Guo, J.; Lin, Y.; Wang, C.C. Uniform magnetic core/shell microspheres functionalized with Ni2+-iminodiacetic acid for one step purification and immobilization of his-tagged enzymes. ACS Appl. Mater. Interfaces 2013, 5, 2626–2633. [Google Scholar] [CrossRef] [PubMed]
- Layer, R.W. The chemistry of imines. Chem. Rev. 1962, 63, 489–510. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, M.; Zhang, C.; Ma, W.F.; Zhang, Y.T.; Wang, C.C.; Lu, H.J. Highly selective and ultra fast solid-phase extraction of N-glycoproteome by oxime click chemistry using aminooxy-functionalized magnetic nanoparticles. Anal. Chem. 2014, 86, 7920–7924. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tang, J.; Wei, C.; Guo, J.; Wang, S.L.; Chaudhary, D.; Wang, C.C. Doxorubicin-conjugated mesoporous magnetic colloidal nanocrystal clusters stabilized by polysaccharide as a smart anticancer drug vehicle. Small 2012, 8, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Hili, R.; Liu, D.R. Enzyme-free translation of DNA into sequence-defined synthetic polymers structurally unrelated to nucleic acids. Nat. Chem. 2013, 5, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.N.; Lam, J.; Nguyen, T.H.; Segura, T.; Maynard, H.D. Biocompatible hydrogels by oxime click chemistry. Biomacromolecules 2012, 13, 3013–3017. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Francis, M.B. Recyclable thermoresponsive polymer–cellulase bioconjugates for biomass depolymerization. J. Am. Chem. Soc. 2013, 135, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Wendeler, M.; Grinberg, L.; Wang, X.Y.; Dawson, P.E.; Baca, M. Enhanced catalysis of oxime-based bioconjugations by substituted anilines. Bioconjugate Chem. 2014, 25, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, M.B.; Munch, H.; Sauer, J.; Clo, E.; Jorgensen, M.R.; Hindsgaul, O.; Jensen, K.J. Nucleophilic catalysis of carbohydrate oxime formation by anilines. J. Org. Chem. 2010, 75, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Loskot, S.A.; Zhang, J.J.; Langenhan, J.M. Nucleophilic catalysis of meon-neoglycoside formation by aniline derivatives. J. Org. Chem. 2013, 78, 12189–12193. [Google Scholar] [CrossRef] [PubMed]
- Dirksen, A.; Hackeng, T.M.; Dawson, P.E. Nucleophilic catalysis of oxime ligation. Angew. Chem. 2006, 45, 7581–7584. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, M.B.; Sorensen, K.K.; Clo, E.; Jensen, K.J. Direct chemoselective synthesis of glyconanoparticles from unprotected reducing glycans and glycopeptide aldehydes. Chem. Commun. 2009, 6367–6369. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, H.; Kuramoto, H.; Furukawa, J.-I.; Miura, Y.; Kurogochi, M.; Kita, Y.; Hinou, H.; Shinohara, Y.; Nishimura, S.-I. One-pot solid-phase glycoblotting and probing by transoximization for high-throughput glycomics and glycoproteomics. Chem. A Eur. J. 2007, 13, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.R.; Mukherjee, S.; Costanzo, P.J.; Sumerlin, B.S. Modular oxime functionalization of well-defined alkoxyamine-containing polymers. Polym. Chem. 2012, 3, 1758–1762. [Google Scholar] [CrossRef]
- Kumara Swamy, K.C.; Bhuvan Kumar, N.N.; Balaraman, E.; Pavan Kumar, K.V.P. Mitsunobu and related reactions advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, M.; Zhang, Y.; Wang, C.; Lu, H. Hydrazide functionalized core–shell magnetic nanocomposites for highly specific enrichment of N-glycopeptides. ACS Appl. Mater. Interfaces 2014, 6, 7823–7832. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | m(MSP) mg | m(NAMAm-p) mg | m(MBA) mg | W1 | W2 | D mmol/g |
|---|---|---|---|---|---|---|
| MSP@PNAMAm-1 | 25 | 80 | 20 | 78.30% | 65.40% | 1.49 |
| MSP@PNAMAm-2 | 25 | 75 | 30 | 72.40% | 63.10% | 1.12 |
| MSP@PNAMAm-3 | 25 | 60 | 40 | 76.50% | 67.96% | 0.95 |
| MSP@PNAMAm-4 | 25 | 50 | 50 | 73.80% | 67.00% | 0.77 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, M.; Di, Y.; Zhang, Y.; Zhang, Y.; Guo, J.; Lu, H.; Wang, C. Fabrication of Alkoxyamine-Functionalized Magnetic Core-Shell Microspheres via Reflux Precipitation Polymerization for Glycopeptide Enrichment. Polymers 2016, 8, 74. https://doi.org/10.3390/polym8030074
Yu M, Di Y, Zhang Y, Zhang Y, Guo J, Lu H, Wang C. Fabrication of Alkoxyamine-Functionalized Magnetic Core-Shell Microspheres via Reflux Precipitation Polymerization for Glycopeptide Enrichment. Polymers. 2016; 8(3):74. https://doi.org/10.3390/polym8030074
Chicago/Turabian StyleYu, Meng, Yi Di, Ying Zhang, Yuting Zhang, Jia Guo, Haojie Lu, and Changchun Wang. 2016. "Fabrication of Alkoxyamine-Functionalized Magnetic Core-Shell Microspheres via Reflux Precipitation Polymerization for Glycopeptide Enrichment" Polymers 8, no. 3: 74. https://doi.org/10.3390/polym8030074